
cfDNA Chimerism and Somatic Mutation Testing in Early Prediction of Relapse After Allogeneic Stem Cell Transplantation for Myeloid Malignancies

 , , , , ,
, , , , ,
Simple Summary
Abstract
1. Introduction
2. Methods
2.1. Bone Marrow and Peripheral Blood Analysis
2.2. Post-Transplant Consolidation Therapy
2.3. Diagnosis of Relapse
2.4. DNA and RNA Extraction and Sequencing
2.5. Statistical Analysis
3. Results
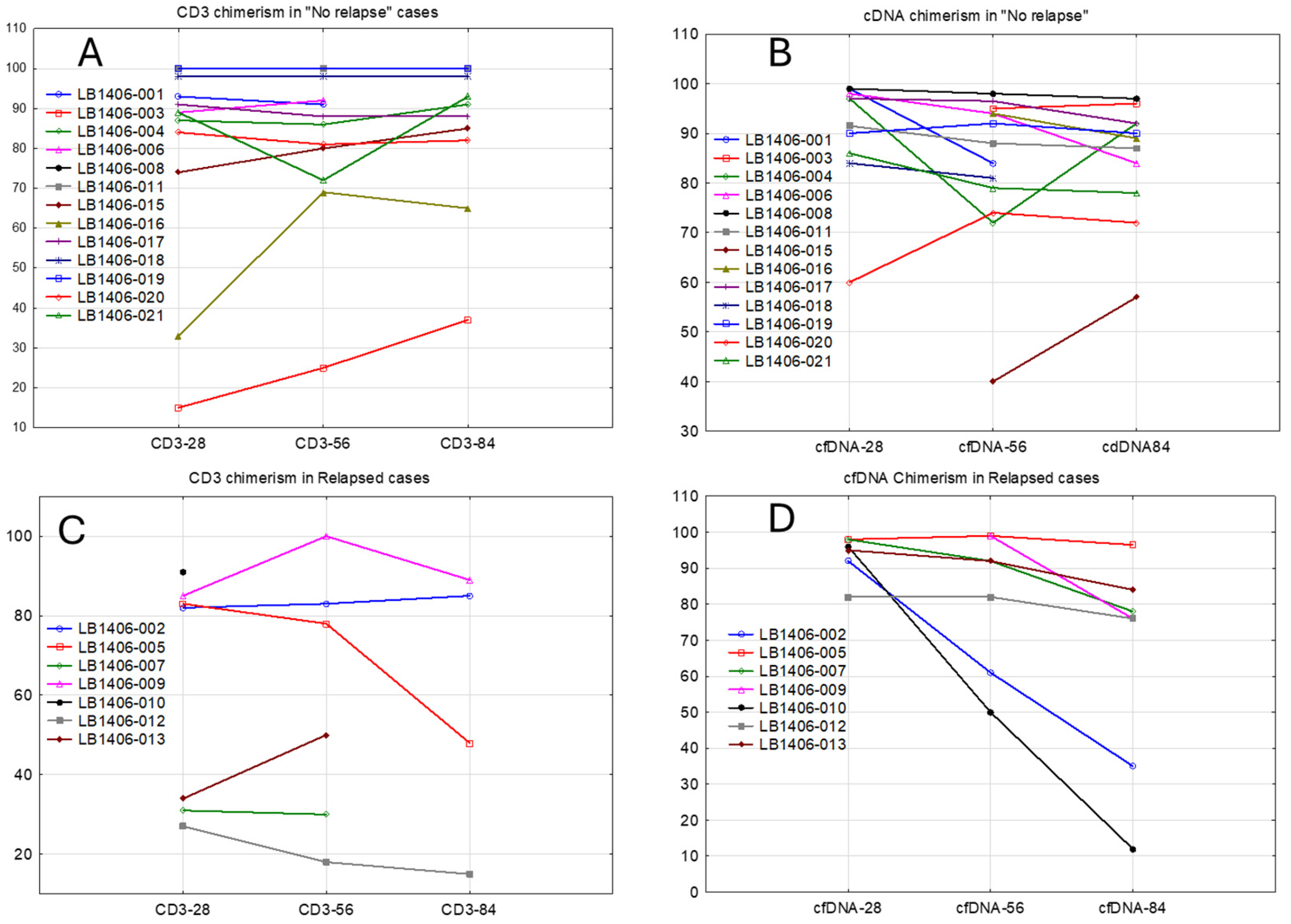
3.1. Concordance of cfDNA and Bone Marrow Samples
3.2. Correlation of cfDNA Detection with Relapse
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Dillon, L.W.; Gui, G.; Page, K.M.; Ravindra, N.; Wong, Z.C.; Andrew, G.; Mukherjee, D.; Zeger, S.L.; El Chaer, F.; Spellman, S.; et al. DNA Sequencing to detect residual disease in adults with acute myeloid leukemia prior to hematopoietic cell transplant. JAMA 2023, 329, 745–755. [Google Scholar] [CrossRef]
- Buckley, S.A.; Wood, B.L.; Othus, M.; Hourigan, C.S.; Ustun, C.; Linden, M.A.; DeFor, T.E.; Malagola, M.; Anthias, C.; Valkova, V.; et al. Minimal residual disease prior to allogeneic hematopoietic cell transplantation in acute myeloid leukemia: A meta-analysis. Haematologica 2017, 102, 865–873. [Google Scholar] [CrossRef]
- Moukalled, N.; Labopin, M.; Versluis, J.; Socié, G.; Blaise, D.; Salmenniemi, U.; Rambaldi, A.; Gedde-Dahl, T.; Tholouli, E.; Kröger, N.; et al. Complex karyotype but not other cytogenetic abnormalities is associated with worse posttransplant survival of patients with nucleophosmin 1-mutated acute myeloid leukemia: A study from the European Society for Blood and Marrow Transplantation Acute Leukemia Working Party. Am. J. Hematol. 2024, 99, 360–369. [Google Scholar]
- Dillon, L.W.; Gui, G.; Ravindra, N.; Andrew, G.; Mukherjee, D.; Wong, Z.C.; Huang, Y.; Gerhold, J.; Holman, M.; D’Angelo, J.; et al. Measurable residual flt3 internal tandem duplication before allogeneic transplant for acute myeloid leukemia. JAMA Oncol. 2024, 10, 1104–1110. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Tsirigotis, P.; Byrne, M.; Schmid, C.; Baron, F.; Ciceri, F.; Esteve, J.; Gorin, N.C.; Giebel, S.; Mohty, M.; Savani, B.N.; et al. Relapse of AML after hematopoietic stem cell transplantation: Methods of monitoring and preventive strategies. A review from the ALWP of the EBMT. Bone Marrow Transplant. 2016, 51, 1431–1438. [Google Scholar] [CrossRef]
- DeFilipp, Z.; Chen, Y.B. How I treat with maintenance therapy after allogeneic HCT. Blood 2023, 141, 39–48. [Google Scholar] [CrossRef]
- Biederstädt, A.; Rezvani, K. How I treat high-risk acute myeloid leukemia using preemptive adoptive cellular immunotherapy. Blood 2023, 141, 22–38. [Google Scholar] [CrossRef]
- Chen, K.; Zhao, H.; Shi, Y.; Yang, F.; Wang, L.T.; Kang, G.; Nie, Y.; Wang, J. Perioperative Dynamic Changes in Circulating Tumor DNA in Patients with Lung Cancer (DYNAMIC). Clin. Cancer Res. 2019, 25, 7058–7067. [Google Scholar] [CrossRef]
- Cescon, D.W.; Bratman, S.V.; Chan, S.M.; Siu, L.L. Circulating tumor DNA and liquid biopsy in oncology. Nat. Cancer 2020, 1, 276–290. [Google Scholar] [CrossRef]
- Crowley, E.; Di Nicolantonio, F.; Loupakis, F.; Bardelli, A. Liquid biopsy: Monitoring cancer-genetics in the blood. Nat. Rev. Clin. Oncol. 2013, 10, 472–484. [Google Scholar] [CrossRef]
- Waldeck, S.; Mitschke, J.; Wiesemann, S.; Rassner, M.; Andrieux, G.; Deuter, M.; Mutter, J.; Lüchtenborg, A.M.; Kottmann, D.; Titze, L.; et al. Early assessment of circulating tumor DNA after curative-intent resection predicts tumor recurrence in early-stage and locally advanced non-small-cell lung cancer. Mol. Oncol. 2022, 16, 527–537. [Google Scholar] [CrossRef] [PubMed]
- Pellini, B.; Chaudhuri, A.A. Circulating tumor DNA minimal residual disease detection of non-small-cell lung cancer treated with curative intent. J. Clin. Oncol. 2022, 40, 567–575. [Google Scholar] [CrossRef] [PubMed]
- Picard, C.; Frassati, C.; Cherouat, N.; Maioli, S.; Moskovtchenko, P.; Cherel, M.; Chiaroni, J.; Pedini, P. New methods for the quantification of mixed chimerism in transplantation. Front. Immunol. 2023, 14, 1023116. [Google Scholar] [CrossRef] [PubMed]
- Sargas, C.; Ayala, R.; Larráyoz, M.J.; Chillón, M.C.; Carrillo-Cruz, E.; Bilbao-Sieyro, C.; Prados de la Torre, E.; Martínez-Cuadrón, D.; Rodríguez-Veiga, R.; Boluda, B.; et al. Molecular Landscape and Validation of New Genomic Classification in 2668 Adult AML Patients: Real Life Data from the PETHEMA Registry. Cancers 2023, 15, 438. [Google Scholar] [CrossRef]
- Döhner, H.; Wei, A.H.; Appelbaum, F.R.; Craddock, C.; DiNardo, C.D.; Dombret, H.; Bert, B.L.; Fenaux, P.; Godley, L.A.; Hasserjian, R.P.; et al. Diagnosis and management of AML in adults: 2022 recommendations from an international expert panel on behalf of the ELN. Blood 2022, 140, 1345–1377. [Google Scholar] [CrossRef] [PubMed]
- Tazi, Y.; Arango-Ossa, J.E.; Zhou, Y.; Bernard, E.; Thomas, I.; Gilkes, A.; Freeman, S.; Pradat, Y.; Johnson, S.J.; Hills, R.; et al. Unified classification and risk-stratification in Acute Myeloid Leukemia. Nat. Commun. 2022, 13, 4622. [Google Scholar] [CrossRef]
- Tobiasson, M.; Pandzic, T.; Illman, J.; Nilsson, L.; Weström, S.; Ejerblad, E.; Olesen, G.; Björklund, A.; Olsnes Kittang, A.; Werlenius, O.; et al. Patient-specific measurable residual disease markers predict outcome in patients with myelodysplastic syndrome and related diseases after hematopoietic stem-cell transplantation. J. Clin. Oncol. 2024, 42, 1378–1390. [Google Scholar] [CrossRef] [PubMed]
- Pasca, S.; Guo, M.Z.; Wang, S.; Stokvis, K.; Shedeck, A.; Pallavajjala, A.; Shams, C.; Pallavajjala, R.; DeZern, A.E.; Varadhan, R.; et al. Cell-free DNA measurable residual disease as a predictor of postallogeneic hematopoietic cell transplant outcomes. Blood Adv. 2023, 7, 4660–4670. [Google Scholar] [CrossRef]
- Murdock, H.M.; Kim, H.T.; Denlinger, N.; Vachhani, P.; Hambley, B.; Manning, B.S.; Gier, S.; Cho, C.; Tsai, H.K.; McCurdy, S.; et al. Impact of diagnostic genetics on remission MRD and transplantation outcomes in older patients with AML. Blood 2022, 139, 3546–3557. [Google Scholar] [CrossRef]
- Bernard, E.; Tuechler, H.; Greenberg, P.L.; Hasserjian, R.P.; Arango Ossa, J.E.; Nannya, Y.; Devlin, S.M.; Creignou, M.; Pinel, P.; Monnier, L.; et al. Molecular International Prognostic Scoring System for myelodysplastic syndromes. NEJM Evid. 2022, 1, EVIDoa2200008. [Google Scholar] [CrossRef] [PubMed]
- Austin, R.J.; Straube, J.; Halder, R.; Janardhanan, Y.; Bruedigam, C.; Witkowski, M.; Cooper, L.; Porter, A.; Braun, M.; Souza-Fonseca-Guimaraes, F.; et al. Oncogenic drivers dictate immune control of acute myeloid leukemia. Nat. Commun. 2023, 14, 2155. [Google Scholar] [CrossRef] [PubMed]
- Gibson, C.J.; Kim, H.T.; Zhao, L.; Murdock, H.M.; Hambley, B.; Ogata, A.; Madero-Marroquin, R.; Wang, S.; Green, L.; Fleharty, M.; et al. Donor clonal hematopoiesis and recipient outcomes after transplantation. J. Clin. Oncol. 2022, 40, 189–201. [Google Scholar] [CrossRef] [PubMed]
- Vago, L.; Gojo, I. Immune escape and immunotherapy of acute myeloid leukemia. J. Clin. Investig. 2020, 130, 1552–1564. [Google Scholar] [CrossRef] [PubMed]
- Tettamanti, S.; Pievani, A.; Biondi, A.; Dotti, G.; Serafini, M. Catch me if you can: How AML and its niche escape immunotherapy. Leukemia 2022, 36, 13–22. [Google Scholar] [CrossRef]
- Limongello, R.; Marra, A.; Mancusi, A.; Bonato, S.; Hoxha, E.; Ruggeri, L.; Hui, S.; Velardi, A.; Pierini, A. Novel immune cell-based therapies to eradicate high-risk acute myeloid leukemia. Front. Immunol. 2021, 12, 695051. [Google Scholar] [CrossRef] [PubMed]
- Frick, M.; Chan, W.; Arends, C.M.; Hablesreiter, R.; Halik, A.; Heuser, M.; Michonneau, D.; Blau, O.; Hoyer, K.; Christen, F.; et al. Role of donor clonal hematopoiesis in allogeneic hematopoietic stem-cell transplantation. J. Clin. Oncol. 2019, 37, 375–385. [Google Scholar] [CrossRef]
- Newell, L.F.; Williams, T.; Liu, J.; Yu, Y.; Chen, Y.; Booth, G.C.; Knight, R.J.; Goslee, K.R.; Cook, R.J.; Leonard, J.; et al. Engrafted donor-derived clonal hematopoiesis after allogenic hematopoietic cell transplantation is associated with chronic graft-versus-host disease requiring immunosuppressive therapy, but no adverse impact on overall survival or relapse. Transplant. Cell Ther. 2021, 27, 662.e1–662.e9. [Google Scholar] [CrossRef]
- Boettcher, S.; Wilk, C.M.; Singer, J.; Beier, F.; Burcklen, E.; Beisel, C.; Ventura Ferreira, M.S.; Gourri, E.; Gassner, C.; Frey, B.M.; et al. Clonal hematopoiesis in donors and long-term survivors of related allogeneic hematopoietic stem cell transplantation. Blood 2020, 135, 1548–1559. [Google Scholar] [CrossRef]
- Jansko-Gadermeir, B.; Leisch, M.; Gassner, F.J.; Zaborsky, N.; Dillinger, T.; Hutter, S.; Risch, A.; Melchardt, T.; Egle, A.; Drost, M.; et al. Myeloid NGS analyses of paired samples from bone marrow and peripheral blood yield concordant results: A prospective cohort analysis of the AGMT Study Group. Cancers 2023, 15, 2305. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Nahas, G.; Gandhi, A.P.; Donato, M.L.; Nyirenda, T.; Vesole, D.H.; Pecora, A.L.; Goldberg, S.; Goy, A.; Feldman, T.; Mato, A.R.; et al. Donor CD3 chimerism after allogeneic transplantation predicts probability of relapse for patients treated for acute myelogenous leukemia (AML), acute lymphoblastic leukemia (ALL), and non-Hodgkin’s lymphoma (NHL). Blood 2013, 122, 4545. [Google Scholar]
- Huisman, C.; de Weger, R.A.; de Vries, L.; Tilanus, M.G.; Verdonck, L.F. Chimerism analysis within 6 months of allogeneic stem cell transplantation predicts relapse in acute myeloid leukemia. Bone Marrow Transplant. 2007, 39, 285–291. [Google Scholar] [CrossRef] [PubMed]
- Rowley, S.D.; Gunning, T.S.; Pelliccia, M.; Della Pia, A.; Lee, A.; Behrmann, J.; Bangolo, A.; Jandir, P.; Zhang, H.; Kaur, S.; et al. Using targeted transcriptome and machine learning of pre- and post-transplant bone marrow samples to predict acute graft-versus-host disease and overall survival after allogeneic stem cell transplantation. Cancers 2024, 16, 1357. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Bacigalupo, A.; Ballen, K.; Rizzo, D.; Giralt, S.; Lazarus, H.; Ho, V.; Apperley, J.; Slavin, S.; Pasquini, M.; Sandmaier, B.M.; et al. Defining the intensity of conditioning regimens: Working definitions. Biol. Blood Marrow Transplant. 2009, 15, 1628–1633. [Google Scholar] [CrossRef] [PubMed]
- Khoury, J.D.; Solary, E.; Abla, O.; Akkari, Y.; Alaggio, R.; Apperley, J.F.; Bejar, R.; Berti, E.; Busque, L.; Chan, J.K.; et al. The 5th edition of the World Health Organization Classification of Haematolymphoid Tumours: Myeloid and Histiocytic/Dendritic Neoplasms. Leukemia 2022, 36, 1703–1719. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
| Subject Age, Median (Range) | 59 (27–77) | Donor Age, Median (Range) | 25 (16–56) |
| Subject Sex (N) | Donor Sex (N) | ||
| Male | 13 | Male | 11 |
| Female | 7 | Female | 9 |
| Transplant Diagnosis (N) | Donor (N) | ||
| Primary AML | 6 | HLA-Matched Sibling | 2 |
| Secondary AML | 6 | Haploidentical | 3 |
| MDS | 7 | URD Matched | 12 |
| CML, Blast Crisis | 1 | URD Mismatched | 3 |
| ABO (N) | CMV (Recipient or Donor, N) | ||
| Match | 11 | +/+ | 3 |
| Minor Mismatch | 3 | +/− | 4 |
| Major or Bidirectional Mismatch | 6 | −/+ | 4 |
| −/− | 9 | ||
| HSC Source (N) | Conditioning Regimen (N) | ||
| PBSC | 20 | MA | 6 |
| BM | 0 | RIC | 10 |
| Non-MA | 4 | ||
| GvHD Regimen (N) | |||
| Tac + MTX | 3 | Post-Transplant Consolidation (N) | |
| Tac, MTX + abatacept | 11 | Yes | 9 |
| PTCy | 2 | No | 11 |
| PTCy or abatacept | 4 |
| Sub No. | DX | Donor/HLA Match | CondReg | GvHD Reg | Post-Transplant Consolidation | Relapse | Current Status | Number of Mutations/Geometric Mean of VAF | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Regimen | Day Started | Y/N | Day | Pre-Transplant BM | Pre cfDNA | Day 28 | Day 56 | Day 84 | Post-Transplant BM | ||||||
| 001 | AML | URD 10/10 | MA + ATG | MTX | NA | N | Expired, RRT, day 59 | 3/4.58 | 2/20.92 | 5/0.1 | 3/0.51 | ND | ND | ||
| 002 | t-MDS | URD 10/10 | MA + ATG | MTX + Abat | NA | Y | 170 | Alive, >day 365 | 2/34.42 | 2/42.72 | 3/2.4 | 3/4.02 | 3/2.77 | 2/1.13 | |
| 003 | MDS | URD 8/10 | NMA | PTCy + Abat | NA | N | Alive, >day 365 | 2/11.04 | 0/NE | 0/NE | 1/2.31 | 0/NE | 0/NE | ||
| 004 | AML | URD 10/10 | MA + ATG | MTX + Abat | Sorafenib +HMA | 51 | N | Expired, infection, day 357 | 1/44.48 | 1/44.87 | 1/0.5 | 1/0.26 | 0/NE | 1/0.28 | |
| 005 | t-MDS | URD 10/10 | RIC + ATG | MTX + Abat | HMA +DLI | 121 | N | Alive, >day 365 | 5/3.54 | 16/0.97 | 2/0.54 | 2/0.30 | 2/0.34 | 5/0.37 | |
| 006 | 2nd-AML (CMML) | URD 10/10 | RIC + ATG | MTX + Abat | NA | N | Alive, >day 365 | 4/37.62 | 4/35.91 | 0/NE | 0/NE | 2/0.11 | 1/2.55 | ||
| 007 | 2nd-AML (MDS) | URD 10/10 | NMA | PTCy + Abat | NA | Y | 62 | Expired, relapse, day 250 | 8/22.97 | 9/18.72 | 1/0.66 | 6/1.07 | 7/1.52 | 10/4.29 | |
| 008 | AML | Haplo | RIC | PTCy | Crenolinib +sorafenib | 91 | N | Alive, >day 365 | 0/NE | 0/NE | 0/NE | 0/NE | 0/NE | 0/NE | |
| 009 | AML | RD 10/10 | RIC + ATG | MTX | HMA +MT401 DLI Group 2 | 76 | Y | 168 | Expired, relapse, day 285 | 2/0.66 | 6/0.84 | 0/NE | 0/NE | 7/0.9 | 4/0.24 |
| 010 | 2nd-AML | Haplo | NMA | PTCy + Abat | NA | Y | 52 | Expired, relapse, day 125 | 3/8.14 | 3/23.98 | 3/0.64 | 2/19.28 | 2/33.48 | 4/5.92 | |
| 011 | 2nd-AML (MDS) | URD 9/10 | RIC | PTCy + Abat | HMA | 88 | N | Alive, >day 365 | 3/19.83 | 3/9.66 | 1/1.59 | 1/3.40 | 1/6.32 | 0/NE | |
| 012 | MDS | URD 10/10 | MA + ATG | MTX + Abat | NA | Y | 167 | Alive, relapse, day 347 | 4/7.67 | 11/5.68 | 5/0.62 | 3/0.04 | 7/0.36 | 4/0.46 | |
| 013 | MDS | URD 10/10 | RIC + ATG | MTX + Abat | NA | Y | 139 | Alive, >day 365 | 1/48.78 | 5/2.50 | 1/0.26 | 2/1.63 | 2/1.60 | 2/4.23 | |
| 015 | AML | RD 10/10 | MA | MTX | Midostaurin + gilteritinib | 35 | N | Alive, >day 365 | 0/NE | 0/NE | 1/36.07 | 1/30.3 | 1/31.2 | 2/0.94 | |
| 016 | MDS | URD 10/10 | RIC + ATG | MTX + Abat | HMA | 89 | N | Expired, sepsis, day 268 | 1/33.18 | 5/1.46 | 1/0.98 | 1/0.03 | 1/0.06 | 0/NE | |
| 017 | CML | URD 10/10 | RIC | MTX + Abat | Imatinib | 52 | N | Alive, >day 365 | 1/6.66 | 3/3.78 | 0/NE | 1/0.21 | 0/NE | 0/NE | |
| 018 | 2nd-AML (BrCa) | URD 10/10 | RIC | MTX + Abat | NA | N | Alive, >day 365 | 6/1.42 | 8/0.63 | 2/0.57 | 0/NE | 0/NE | ND | ||
| 019 | 2nd AML (MF) | Haplo | NMA | PTCy | NA | N | Alive, >day 365 | 9/5.79 | 6/9.43 | 3/0.18 | 5/0.54 | 2/0.23 | 1/0.06 | ||
| 020 | AML | URD 10/10 | MA + ATG | MTX + Abat | NA | N | Alive, >day 365 | 5/4.11 | 7/1.71 | 1/0.97 | 1/0.37 | 0/NE | 1/0.37 | ||
| 021 | MDS | URD 10/10 | RIC + ATG | MTX + Abat | Sorafenib | 77 | N | Alive, >day 365 | 2/0.69 | 4/1.20 | 0/NE | 1/0.11 | 1/0.11 | 0/NE | |
| Sub No | Relapse (Y/N) | Pre-Transplant BM | VAF | Pre-Transplant cfDNA | VAF | Day 84 BM | VAF | Day 84 cfDNA | VAF |
|---|---|---|---|---|---|---|---|---|---|
| 001 | N | DNMT3a SRSF2 IDH1 | 15.85 4.66 1.3 | DNMT3A SRSF2 | 28.57 15.38 | ND | ND | ||
| 002 | Y | TP53 HNF1A | 48.34 24.54 | TP53 HNF1A | 63.57 28.71 | TP53 HNF1A | 2.5 0.51 | TP53 HNF1A | 3.19 0.84 |
| 003 | N | BCOR KMT2D | 13.85 8.8 | Neg | Neg | Neg | |||
| 004 | N | DNMT3A | 44.48 | DNMT3A | 44.87 | DNMT3A | 0.28 | Neg | |
| 005 | N | TP53 PPM1D CHEK2 NOTCH3 PPM1D | 5.2 2.18 7.53 3.95 1.66 | TP53 PPM1D CHEK2 NOTCH3 PPM1D BRAF PPM1D CARD11 KMT2A PMS1 CDK12 PBRM1 TP53 KEAP1 CARD11 KMT2C | 5.53 5.13 4.76 2.79 0.67 5.97 3.23 0.76 0.7 0.58 0.56 0.52 0.35 0.21 0.18 0.11 | KMT2A GATA3 MAP3Ki4 TET2 KRCC2 | 0.19 0.95 0.47 0.37 0.21 | TP53 PPM1D | 0.37 0.32 |
| 006 | N | TET2 EZH2 ASXL1 TET2 | 52.71 47.1 33.71 23.92 | TET2 EZH2 ASXL1 TET2 | 49.13 36.36 34 27.4 | ASXL1 | 2.55 | TET2 TET2 | 0.25 0.05 |
| 007 | Y | SRSF2 ASXL1 NRAS MTOR MTOR MTOR KDM6A ARAF | 55.44 50.62 41.9 31.37 30.0 27.27 6.15 4.17 | SRSF2 ASXL1 NRAS MTOR MTOR MTOR FH GNAQ IRF4 | 45.55 48.27 48.59 40.26 38.3 36.09 2.71 4.17 4.11 | SRSF2 ASXL1 NRAS MTOR MTOR MTOR GNAS GNAS GNAS BCL6 | 9.4 10.24 7.63 0/73 0.48 0.8 10.41 8.46 8.67 13.43 | SRSF2 ASXL1 NRAS MTOR MTOR FH DNMT3A | 5.62 8.29 4.46 0.55 0.31 1.85 0.27 |
| 008 | N | Neg | Neg | Neg | Neg | ||||
| 009 | Y | TET2 NRAS | 0.28 1.54 | TET2 TET2 WT1 NFKBIA NRAS FLT3-ITD | 5.78 1.38 0.83 0.6 0.39 0.23 | TET2 WT1 NFKBIA FLT3-ITD | 0.15 0.69 0.25 0.14 | TET2 WT1 NFKBIA NRAS FLT3-ITD WT1 WT1 | 2.73 3.02 3.77 0.13 2.4 0.12 0.41 |
| 010 | Y | SRSF2 MPL IDH2 | 18.93 4.31 6.61 | SRSF2 MPL IDH2 | 37.89 23.93 15.2 | SRSF2 MPL IDH2 KMT2C | 36.08 0.54 16.24 3.87 | SRSF2 IDH2 | 31.04 36.11 |
| 011 | N | TP53 TET2 PDGFRB | 31.54 16.72 14.79 | TP53 TET2 PDGFRB | 5.26 8.62 19.86 | Neg | PDGFRB | 6.32 | |
| 012 | Y | AXIN1 SF3B1 ASXL1 ASXL1 AMER1 | 16.83 17.7 6.4 4 8.05 | AXIN1 SF3B1 ASXL1 ASXL1 AMER1 H3F3A EGFR RUNX1 KMT2C ASXL1 | 34.8 33.02 3.03 3.01 2.14 17.7 3.46 2.61 1.79 1.4 | AXIN1 SF3B1 RUNX1 KMT2B | 0.25 0.49 0.28 1.68 | AXIN1 SF3B1 AMER1 EGFR RUNX1 KMT2C KMT2C | 0.5 0.35 0.05 0.36 0.28 0.48 1.86 |
| 013 | Y | Neg | TP53 TET2 NOTCH1 TET2 | 43.38 0.78 0.47 0.19 | NF2 | 0.37 | TP53 | 0/37 | |
| 015 | N | Neg | Neg | KMT2B DNMT3A | 1.25 0.7 | Neg | |||
| 016 | N | SF3B1 | 33.18 | SF3B1 TNFRSF14 KMT2D DNMT3A MAP3K1 | 43.33 1.48 0.83 0.33 0.26 | Neg | SF3B1 | 0.06 | |
| 017 | N | ASXL1 | 6.66 | ASXL1 CEBPA DNMT3A | 12.81 5.2 0.81 | Neg | Neg | ||
| 018 | N | ALK SRSF2 TET2 SRSF2 DDX41 | 0.97 0.8 0.51 0.29 1.82 | ALK SRSF2 TET2 SF3B1 SF3B1 ALK FGFR4 | 1.46 0.17 0.2 0.21 0.34 0.44 0.35 | ND | Neg | ||
| 019 | N | CALR U2AF1 ASXL1 GNAS KRAS RUNX1 GALNT12 TP31 GRIN2A | 39.23 38.39 34.88 20.25 20.11 2.04 0.87 0.6 0.32 | CALR U2AF1 ASXL1 GNAS KRAS RUNX1 | 17.76 15.04 19.57 12.98 12.92 0.8 | ASXL1 | 0.06 | CALR U2AF1 | 0.29 0.18 |
| 020 | N | DMNT3A. NF1 ASXL1 TET2 SMC3 | 32.74 4.4 2.52 3.72 0.87 | DMNT3A. NF1 ASXL1 TET2 SMC3 PDGFRB KMT2C | 33.87 4.27 3.64 2.2 0.4 0.4 0.23 | DNMT3A | 0.37 | DNMT3A | 0.36 |
| 021 | N | DNMT3A DNMT3A | 1.02 0.47 | DNMT3A DNMT3A CBL NFI I | 1.38 1.01 1.03 1.46 | Neg | CBL | 0.11 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rowley, S.D.; Albitar, M.; Baker, M.F.; Ali, A.; Kaur, S.; Suh, H.C.; Goy, A.; Donato, M.L. cfDNA Chimerism and Somatic Mutation Testing in Early Prediction of Relapse After Allogeneic Stem Cell Transplantation for Myeloid Malignancies. Cancers 2025, 17, 625. https://doi.org/10.3390/cancers17040625
Rowley SD, Albitar M, Baker MF, Ali A, Kaur S, Suh HC, Goy A, Donato ML. cfDNA Chimerism and Somatic Mutation Testing in Early Prediction of Relapse After Allogeneic Stem Cell Transplantation for Myeloid Malignancies. Cancers. 2025; 17(4):625. https://doi.org/10.3390/cancers17040625
Chicago/Turabian StyleRowley, Scott D., Maher Albitar, Melissa F. Baker, Alaa Ali, Sukhdeep Kaur, Hyung C. Suh, Andre Goy, and Michele L. Donato. 2025. "cfDNA Chimerism and Somatic Mutation Testing in Early Prediction of Relapse After Allogeneic Stem Cell Transplantation for Myeloid Malignancies" Cancers 17, no. 4: 625. https://doi.org/10.3390/cancers17040625
APA StyleRowley, S. D., Albitar, M., Baker, M. F., Ali, A., Kaur, S., Suh, H. C., Goy, A., & Donato, M. L. (2025). cfDNA Chimerism and Somatic Mutation Testing in Early Prediction of Relapse After Allogeneic Stem Cell Transplantation for Myeloid Malignancies. Cancers, 17(4), 625. https://doi.org/10.3390/cancers17040625