

Curcumin-Dichloroacetate Hybrid Molecule as an Antitumor Oral Drug against Multidrug-Resistant Advanced Bladder Cancers

 ,
,  ,
, 
Abstract
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Reagents and Chemicals
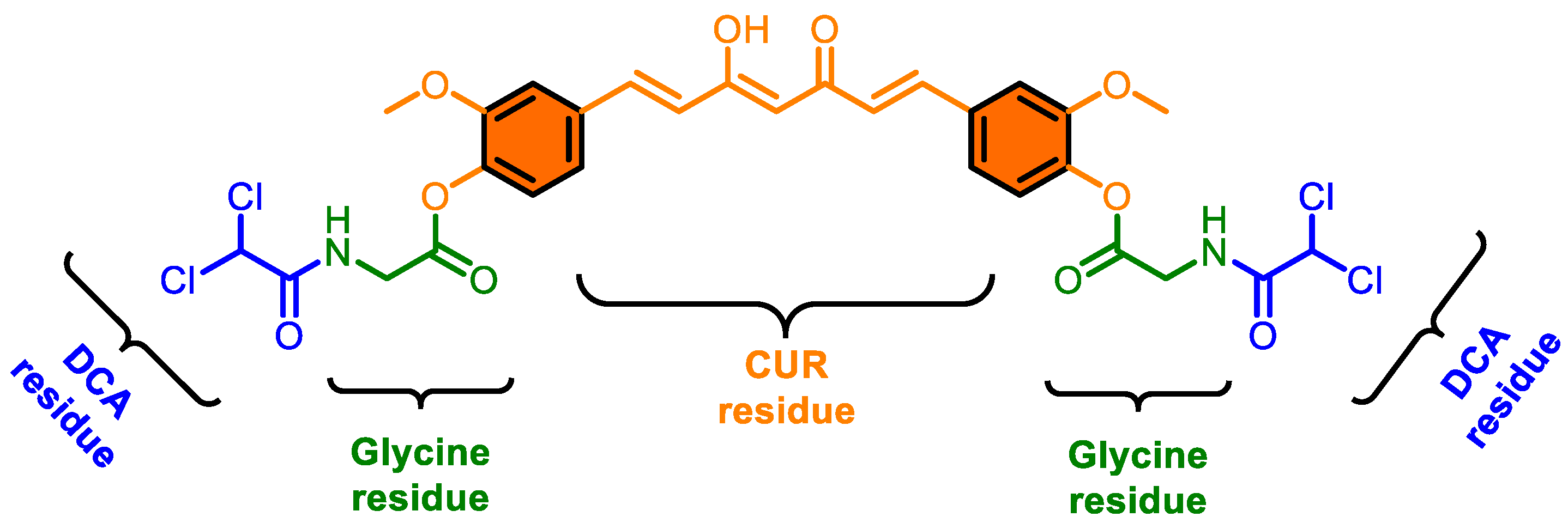
2.2. Synthesis of CMC-2
2.3. Procedures Related to Cell Culture, Maintenance, and Cell Line Authentication
2.4. Establishing Stable Multidrug-Resistant BCa Cell Lines
2.5. Cell Viability Assessment
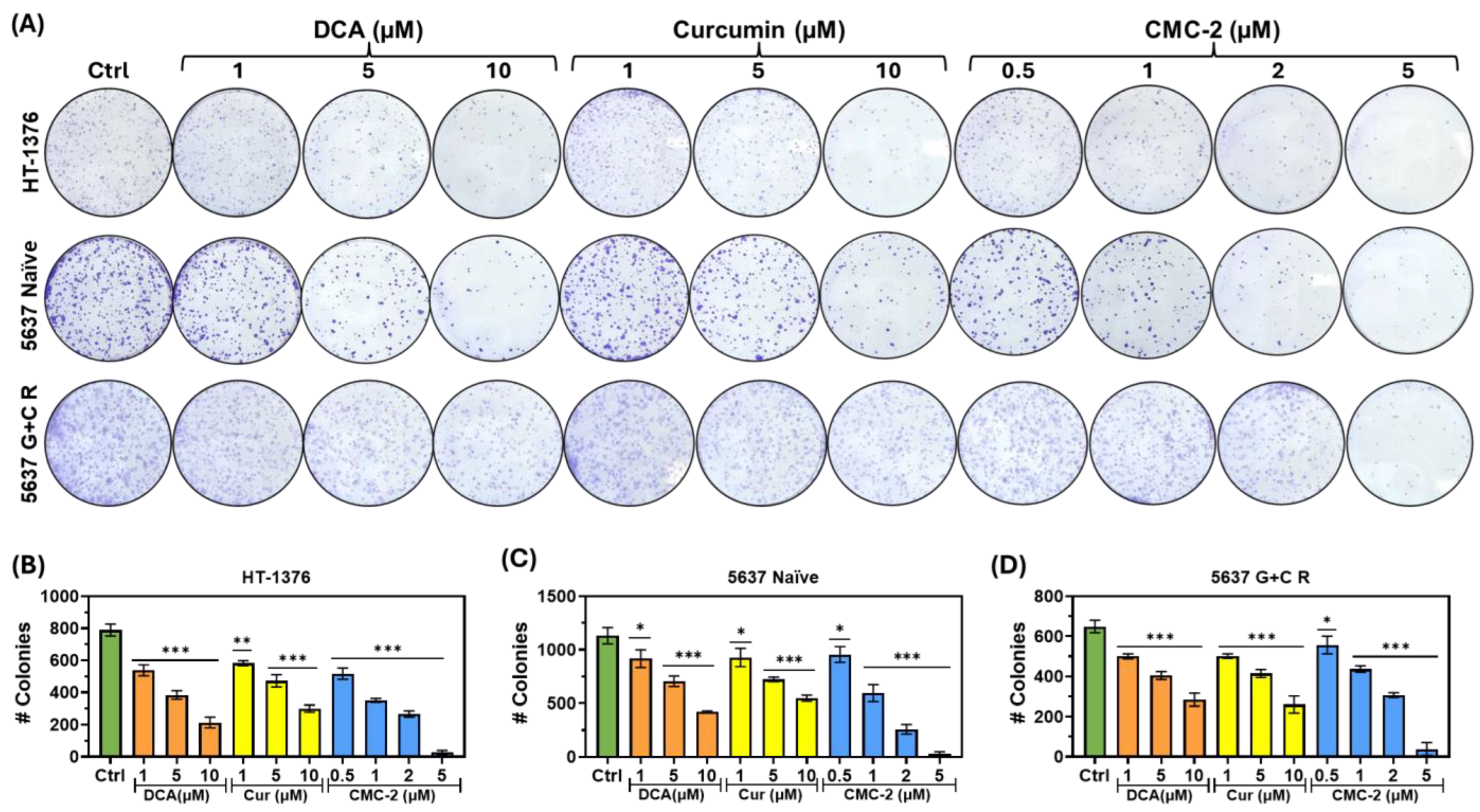
2.6. Colony Forming Assay
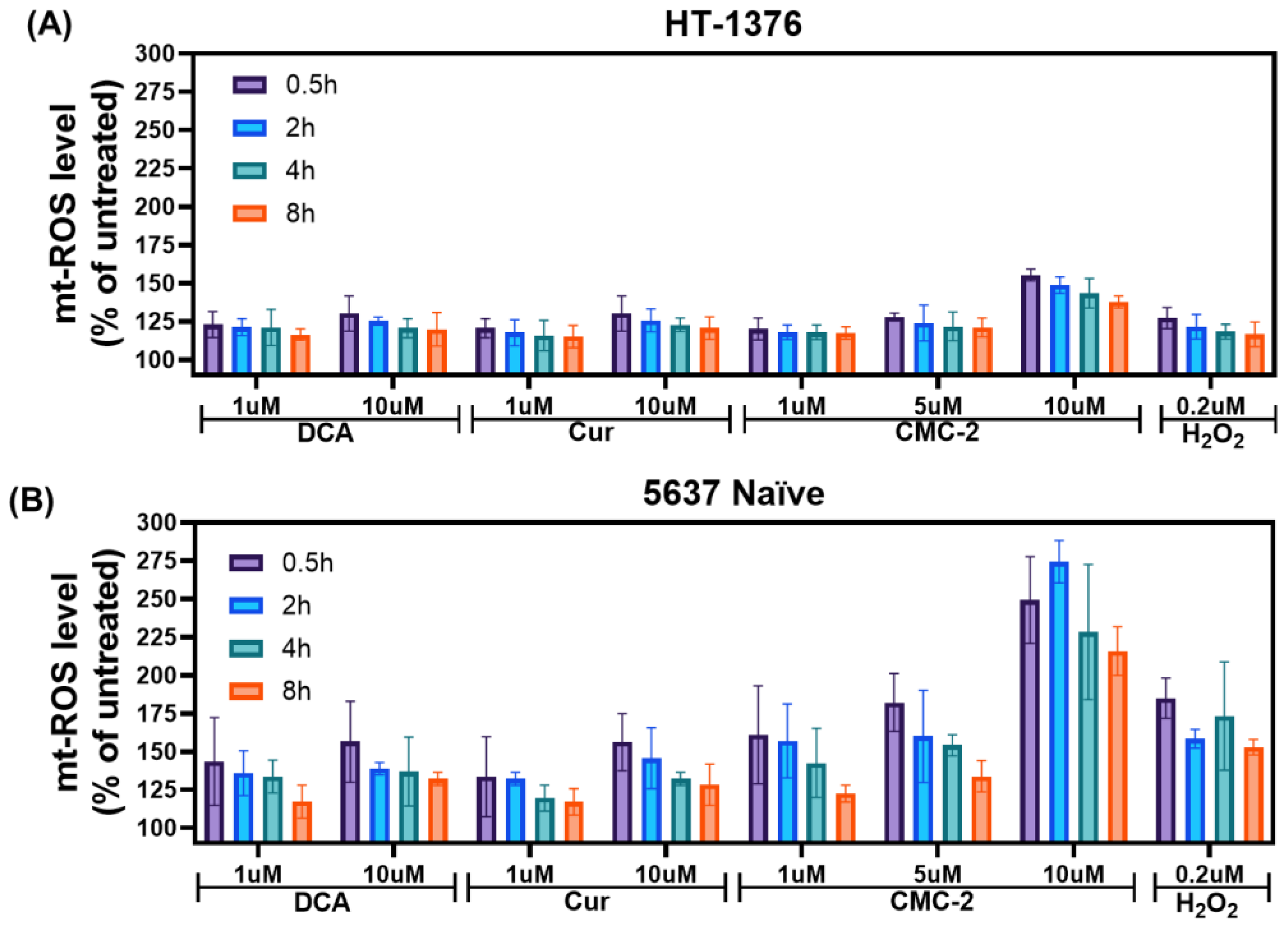
2.7. Estimating Reactive Oxygen Species (ROS)
2.8. Estimation of Mitochondrial Trans-Membrane Potential (∆ψM)
2.9. Measuring Changes in Cell Cycle Phase Fractions
2.10. Detection of Apoptosis
2.11. Protein Immunoblotting (Western Blotting)
2.12. In Vivo Studies to Test Antitumor Activities of DCA, Cur, and CMC-2
2.13. Statistical Analysis
3. Results
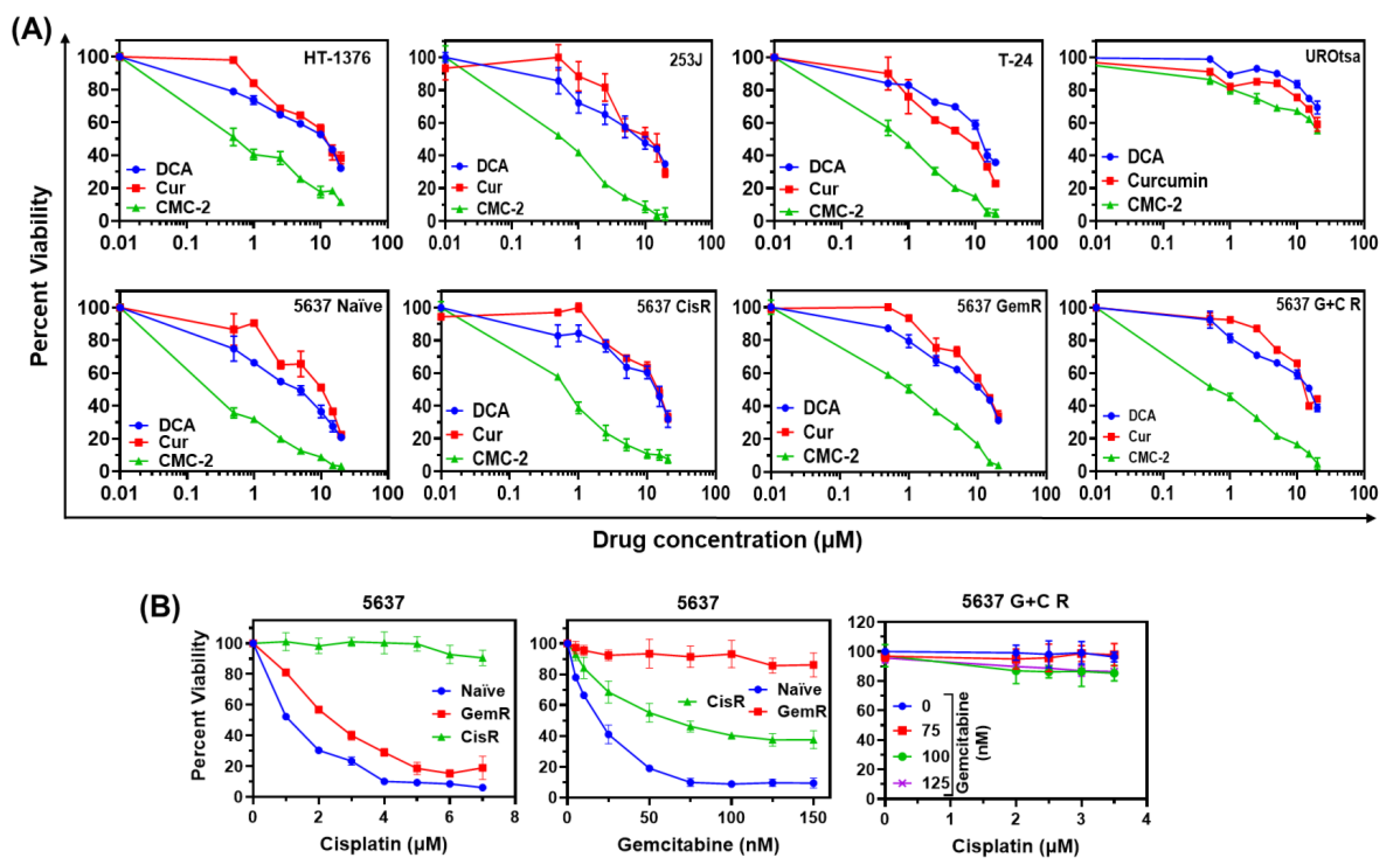
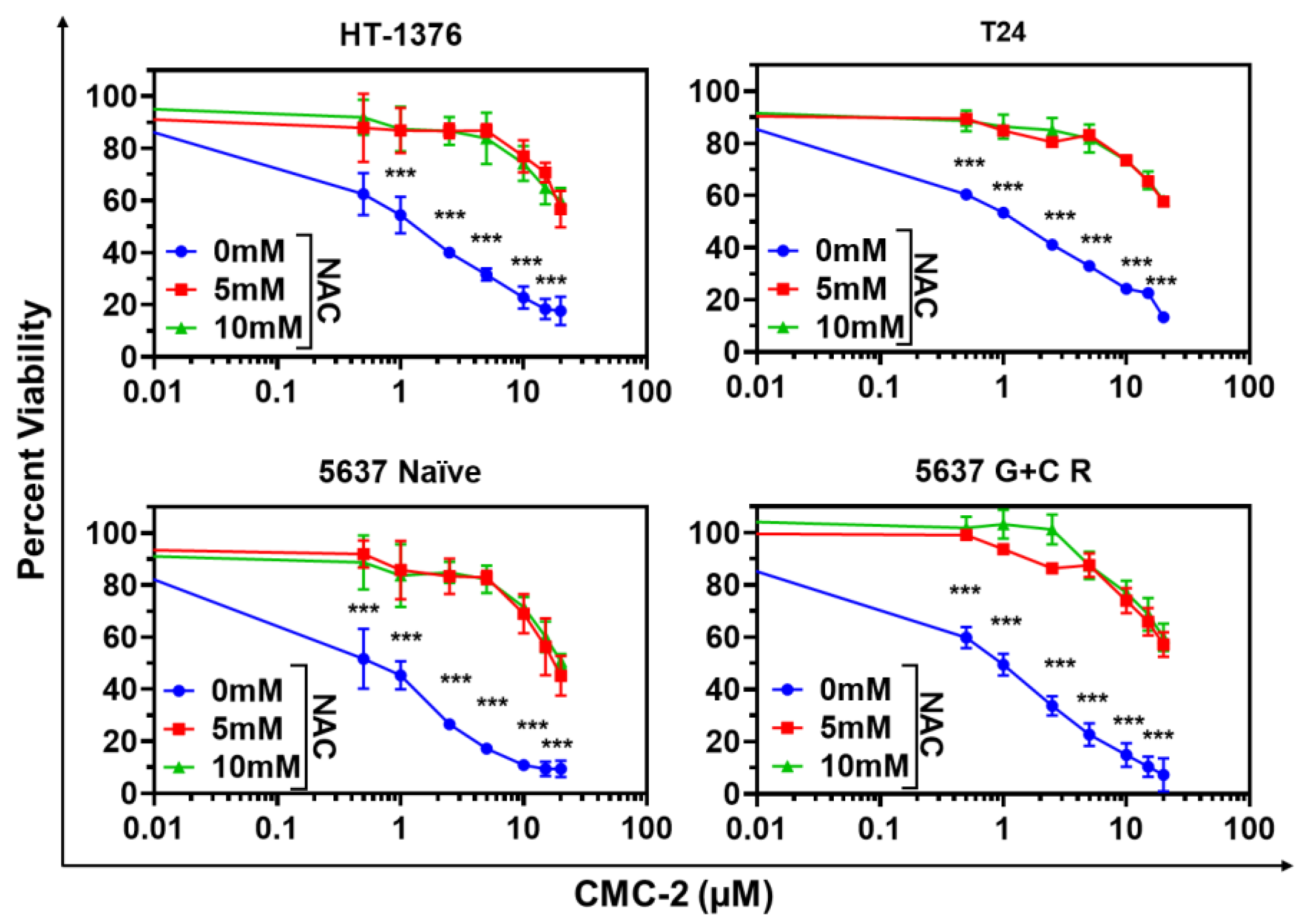
3.1. Cytotoxicity of DCA, Cur, and CMC-2 on BCa Cells
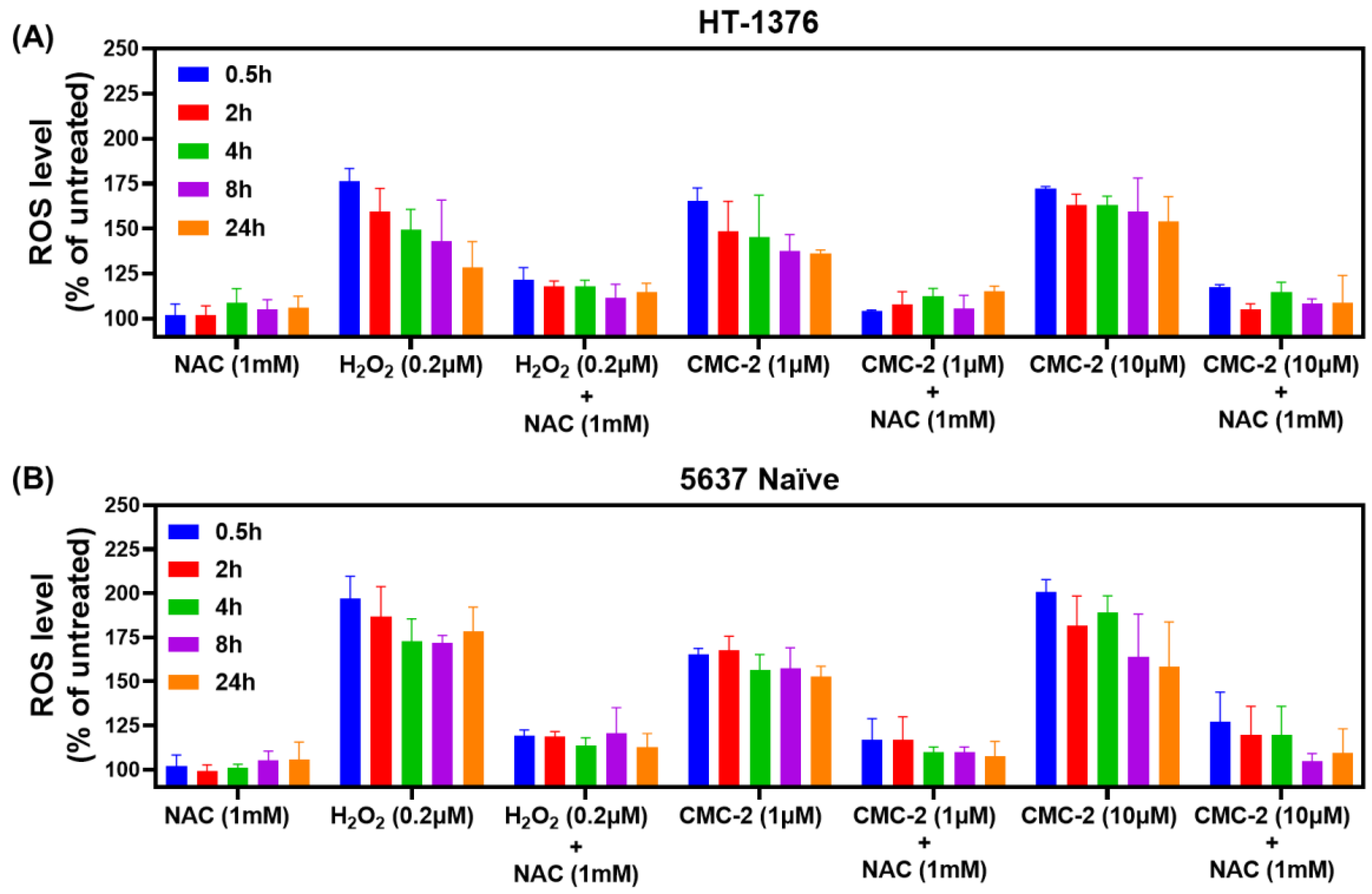
3.2. Mechanism of DCA, Cur, and CMC-2 Cytotoxicity
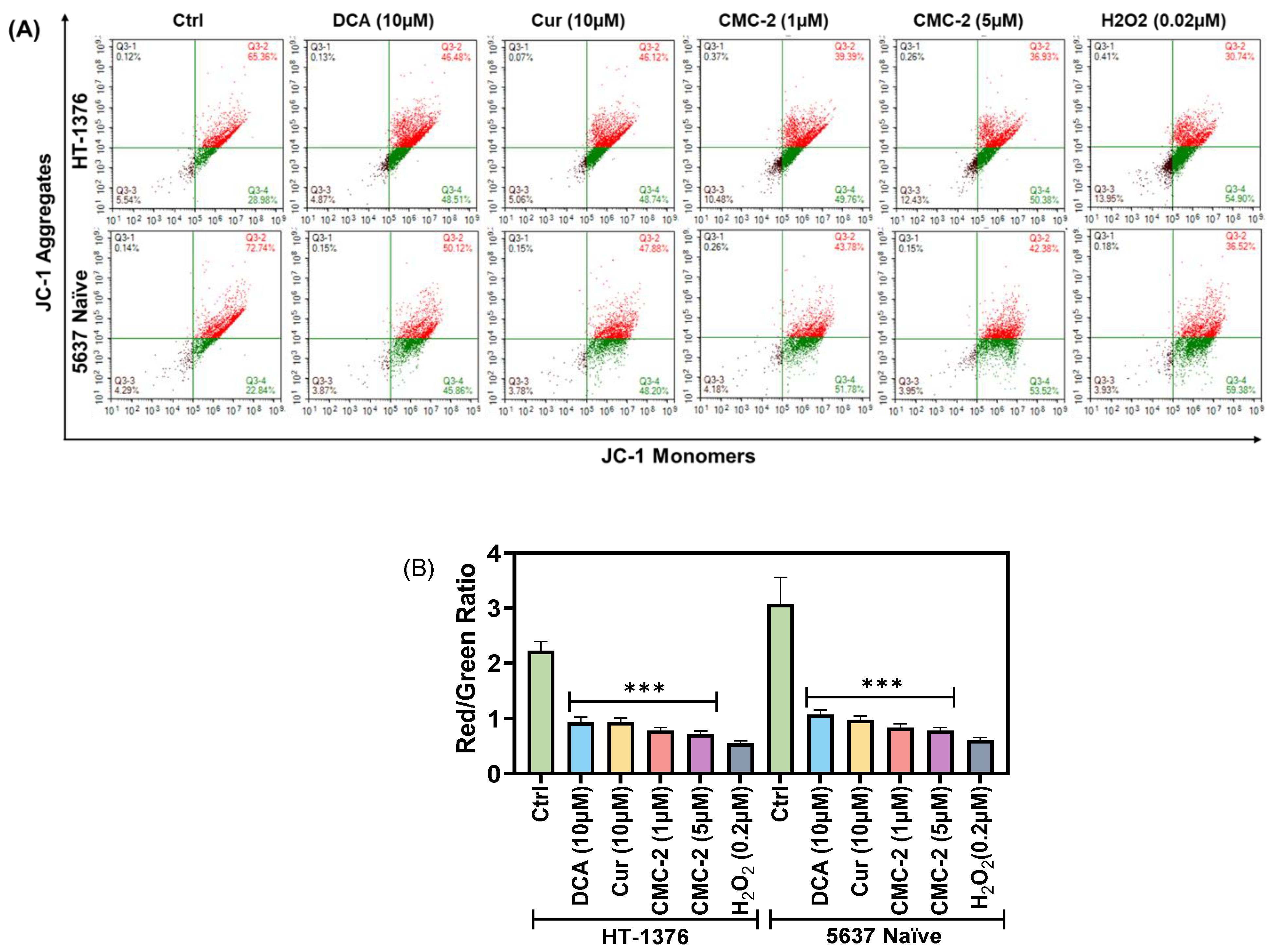
3.3. Mitochondrial Transmembrane Potential (MTP; ψM) Is Significantly Decreased in CMC-2-Treated BC Cells
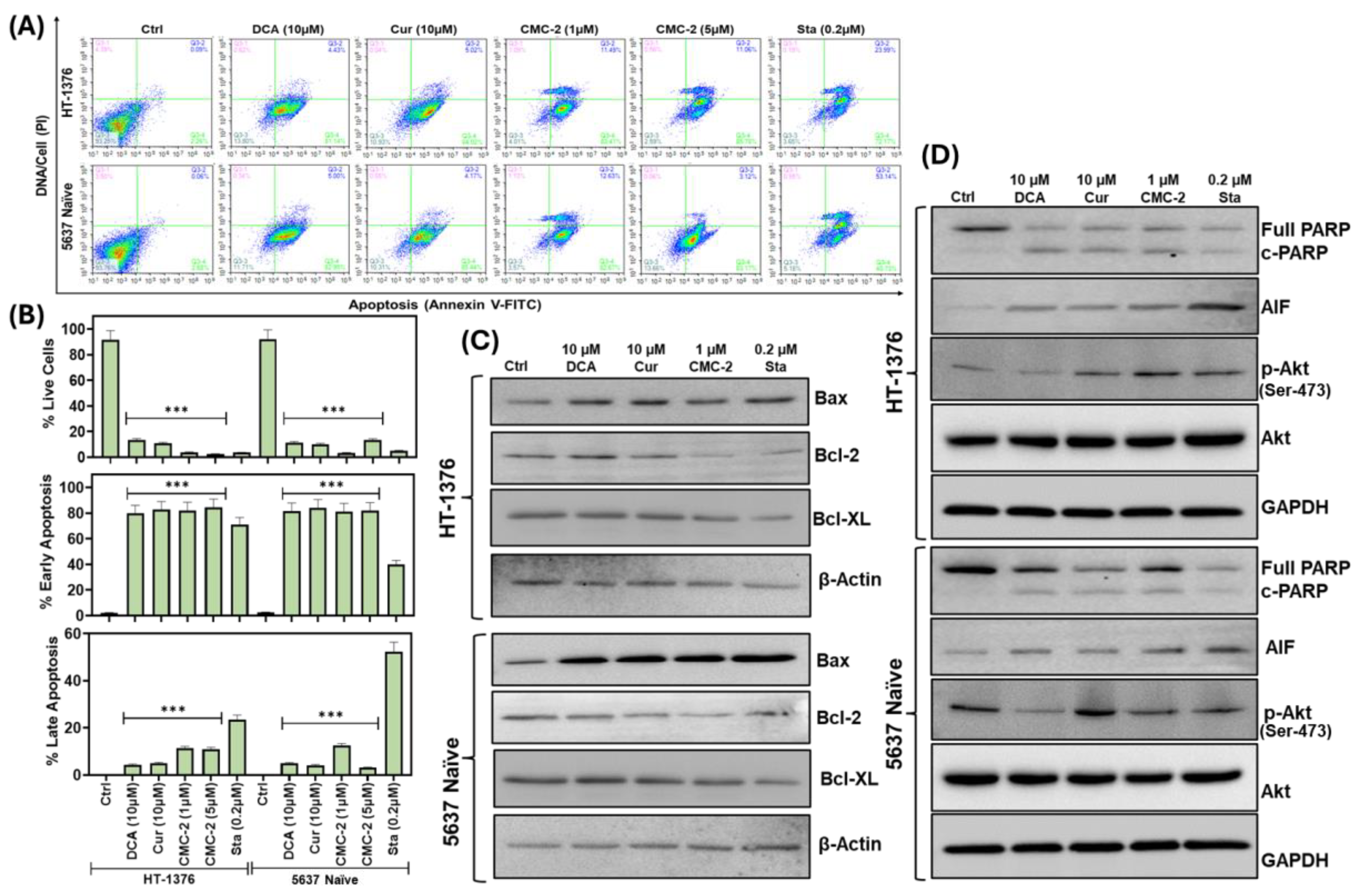
3.4. CMC-2 Treatment Induces Apoptotic Cell Death in the BCa Cells
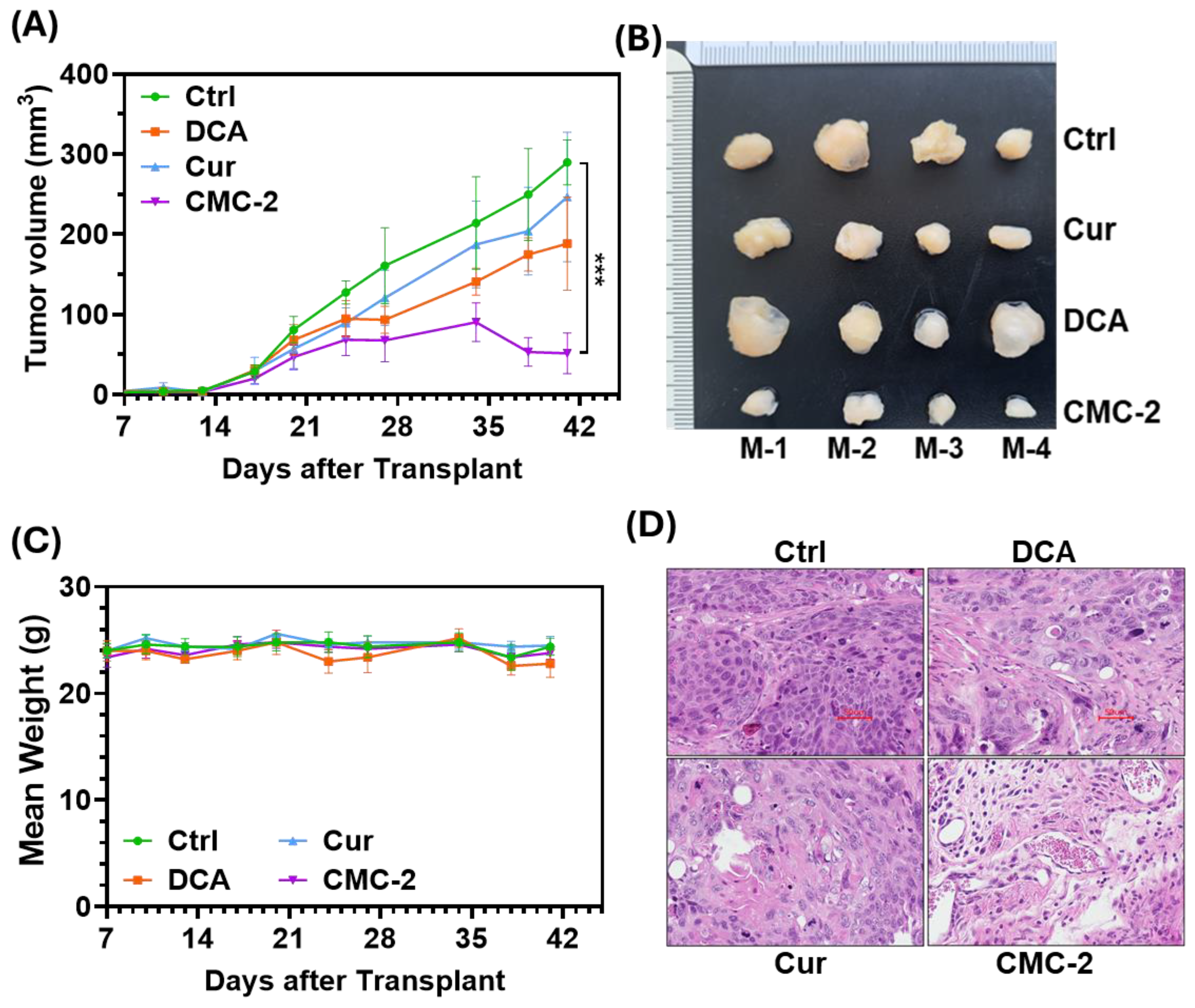
3.5. CMC-2 Significantly Inhibits the Growth of Tumors In Vivo
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Priyadarsini, K.I. The chemistry of curcumin: From extraction to therapeutic agent. Molecules 2014, 19, 20091–20112. [Google Scholar] [CrossRef]
- Authority, E.F.S. Refined exposure assessment for curcumin (E 100). EFSA J. 2014, 12, 3876. [Google Scholar] [CrossRef]
- Kamat, A.M.; Sethi, G.; Aggarwal, B.B. Curcumin potentiates the apoptotic effects of chemotherapeutic agents and cytokines through down-regulation of nuclear factor-kappaB and nuclear factor-kappaB-regulated gene products in IFN-alpha-sensitive and IFN-alpha-resistant human bladder cancer cells. Mol. Cancer Ther. 2007, 6, 1022–1030. [Google Scholar] [CrossRef] [PubMed]
- Kunnumakkara, A.B.; Guha, S.; Krishnan, S.; Diagaradjane, P.; Gelovani, J.; Aggarwal, B.B. Curcumin potentiates antitumor activity of gemcitabine in an orthotopic model of pancreatic cancer through suppression of proliferation, angiogenesis, and inhibition of nuclear factor-kappaB-regulated gene products. Cancer Res. 2007, 67, 3853–3861. [Google Scholar] [CrossRef]
- Banik, U.; Parasuraman, S.; Adhikary, A.K.; Othman, N.H. Curcumin: The spicy modulator of breast carcinogenesis. J. Exp. Clin. Cancer Res. 2017, 36, 98. [Google Scholar] [CrossRef] [PubMed]
- Dei Cas, M.; Ghidoni, R. Dietary Curcumin: Correlation between Bioavailability and Health Potential. Nutrients 2019, 11, 2147. [Google Scholar] [CrossRef]
- Qin, H.; Zheng, G.; Li, Q.; Shen, L. Metabolic reprogramming induced by DCA enhances cisplatin sensitivity through increasing mitochondrial oxidative stress in cholangiocarcinoma. Front. Pharmacol. 2023, 14, 1128312. [Google Scholar] [CrossRef]
- Bonnet, S.; Archer, S.L.; Allalunis-Turner, J.; Haromy, A.; Beaulieu, C.; Thompson, R.; Lee, C.T.; Lopaschuk, G.D.; Puttagunta, L.; Bonnet, S.; et al. A mitochondria-K+ channel axis is suppressed in cancer and its normalization promotes apoptosis and inhibits cancer growth. Cancer Cell 2007, 11, 37–51. [Google Scholar] [CrossRef]
- Madhok, B.M.; Yeluri, S.; Perry, S.L.; Hughes, T.A.; Jayne, D.G. Dichloroacetate induces apoptosis and cell-cycle arrest in colorectal cancer cells. Br. J. Cancer 2010, 102, 1746–1752. [Google Scholar] [CrossRef]
- Parczyk, J.; Ruhnau, J.; Pelz, C.; Schilling, M.; Wu, H.; Piaskowski, N.N.; Eickholt, B.; Kühn, H.; Danker, K.; Klein, A. Dichloroacetate and PX-478 exhibit strong synergistic effects in a various number of cancer cell lines. BMC Cancer 2021, 21, 481. [Google Scholar] [CrossRef]
- Florio, R.; De Lellis, L.; Veschi, S.; Verginelli, F.; di Giacomo, V.; Gallorini, M.; Perconti, S.; Sanna, M.; Mariani-Costantini, R.; Natale, A.; et al. Effects of dichloroacetate as single agent or in combination with GW6471 and metformin in paraganglioma cells. Sci. Rep. 2018, 8, 13610. [Google Scholar] [CrossRef] [PubMed]
- Tomeh, M.A.; Hadianamrei, R.; Zhao, X. A Review of Curcumin and Its Derivatives as Anticancer Agents. Int. J. Mol. Sci. 2019, 20, 1033. [Google Scholar] [CrossRef] [PubMed]
- Agrawal, D.K.; Mishra, P.K. Curcumin and its analogues: Potential anticancer agents. Med. Res. Rev. 2010, 30, 818–860. [Google Scholar] [CrossRef]
- Panda, S.S.; Tran, Q.L.; Rajpurohit, P.; Pillai, G.G.; Thomas, S.J.; Bridges, A.E.; Capito, J.E.; Thangaraju, M.; Lokeshwar, B.L. Design, Synthesis, and Molecular Docking Studies of Curcumin Hybrid Conjugates as Potential Therapeutics for Breast Cancer. Pharmaceuticals 2022, 15, 451. [Google Scholar] [CrossRef] [PubMed]
- Rossi, M.R.; Masters, J.R.; Park, S.; Todd, J.H.; Garrett, S.H.; Sens, M.A.; Somji, S.; Nath, J.; Sens, D.A. The immortalized UROtsa cell line as a potential cell culture model of human urothelium. Environ. Health Perspect. 2001, 109, 801–808. [Google Scholar] [CrossRef]
- Kallifatidis, G.; Smith, D.K.; Morera, D.S.; Gao, J.; Hennig, M.J.; Hoy, J.J.; Pearce, R.F.; Dabke, I.R.; Li, J.; Merseburger, A.S.; et al. β-Arrestins Regulate Stem Cell-Like Phenotype and Response to Chemotherapy in Bladder Cancer. Mol. Cancer Ther. 2019, 18, 801–811. [Google Scholar] [CrossRef]
- Seremak, J.R.; Gupta, K.B.; Bonigala, S.; Liu, E.; Marshall, B.; Zhi, W.; Bokhtia, R.M.; Panda, S.S.; Lokeshwar, V.B.; Lokeshwar, B.L. Targeting Chemoresistance in Advanced Bladder Cancers with a Novel Adjuvant Strategy. Mol. Cancer Ther. 2024, Of1–Of15. [Google Scholar] [CrossRef]
- Shamaladevi, N.; Lyn, D.A.; Escudero, D.O.; Lokeshwar, B.L. CXC receptor-1 silencing inhibits androgen-independent prostate cancer. Cancer Res. 2009, 69, 8265–8274. [Google Scholar] [CrossRef]
- Gupta, K.B.; Mantha, A.K.; Dhiman, M. Mitigation of Gliadin-Induced Inflammation and Cellular Damage by Curcumin in Human Intestinal Cell Lines. Inflammation 2021, 44, 873–889. [Google Scholar] [CrossRef]
- Ng, N.S.; Ooi, L. A Simple Microplate Assay for Reactive Oxygen Species Generation and Rapid Cellular Protein Normalization. Bio-Protocol 2021, 11, e3877. [Google Scholar] [CrossRef]
- Lokeshwar, B.L.; Selzer, M.G.; Zhu, B.Q.; Block, N.L.; Golub, L.M. Inhibition of cell proliferation, invasion, tumor growth and metastasis by an oral non-antimicrobial tetracycline analog (COL-3) in a metastatic prostate cancer model. Int. J. Cancer 2002, 98, 297–309. [Google Scholar] [CrossRef] [PubMed]
- Logue, S.E.; Elgendy, M.; Martin, S.J. Expression, purification and use of recombinant annexin V for the detection of apoptotic cells. Nat. Protoc. 2009, 4, 1383–1395. [Google Scholar] [CrossRef]
- Woo, J.H.; Kim, Y.H.; Choi, Y.J.; Kim, D.G.; Lee, K.S.; Bae, J.H.; Min, D.S.; Chang, J.S.; Jeong, Y.J.; Lee, Y.H.; et al. Molecular mechanisms of curcumin-induced cytotoxicity: Induction of apoptosis through generation of reactive oxygen species, down-regulation of Bcl-XL and IAP, the release of cytochrome c and inhibition of Akt. Carcinogenesis 2003, 24, 1199–1208. [Google Scholar] [CrossRef] [PubMed]
- Kerksick, C.; Willoughby, D. The antioxidant role of glutathione and N-acetyl-cysteine supplements and exercise-induced oxidative stress. J. Int. Soc. Sports Nutr. 2005, 2, 38–44. [Google Scholar] [CrossRef] [PubMed]
- Fagundes, R.; Teixeira, L.K. Cyclin E/CDK2: DNA Replication, Replication Stress and Genomic Instability. Front. Cell Dev. Biol. 2021, 9, 774845. [Google Scholar] [CrossRef]
- Hu, Y.; Cheng, L.; Du, S.; Wang, K.; Liu, S. Antioxidant curcumin induces oxidative stress to kill tumor cells (Review). Oncol. Lett. 2024, 27, 67. [Google Scholar] [CrossRef]
- Liu, X.; Li, J.; Huang, Q.; Jin, M.; Huang, G. Ginsenoside Rh2 shifts tumor metabolism from aerobic glycolysis to oxidative phosphorylation through regulating the HIF1-α/PDK4 axis in non-small cell lung cancer. Mol. Med. 2024, 30, 56. [Google Scholar] [CrossRef]
- Tataranni, T.; Piccoli, C. Dichloroacetate (DCA) and Cancer: An Overview towards Clinical Applications. Oxidative Med. Cell. Longev. 2019, 2019, 8201079. [Google Scholar] [CrossRef]
- Stacpoole, P.W.; Martyniuk, C.J.; James, M.O.; Calcutt, N.A. Dichloroacetate-induced peripheral neuropathy. Int. Rev. Neurobiol. 2019, 145, 211–238. [Google Scholar]
- Siegel, R.L.; Giaquinto, A.N.; Jemal, A. Cancer statistics, 2024. CA Cancer J. Clin. 2024, 74, 12–49. [Google Scholar] [CrossRef]
- Teoh, J.Y.; Huang, J.; Ko, W.Y.; Lok, V.; Choi, P.; Ng, C.F.; Sengupta, S.; Mostafid, H.; Kamat, A.M.; Black, P.C.; et al. Global Trends of Bladder Cancer Incidence and Mortality, and Their Associations with Tobacco Use and Gross Domestic Product Per Capita. Eur. Urol. 2020, 78, 893–906. [Google Scholar] [CrossRef]
- Amin, M.B.; McKenney, J.K.; Paner, G.P.; Hansel, D.E.; Grignon, D.J.; Montironi, R.; Lin, O.; Jorda, M.; Jenkins, L.C.; Soloway, M.; et al. ICUD-EAU International Consultation on Bladder Cancer 2012: Pathology. Eur. Urol. 2013, 63, 16–35. [Google Scholar] [CrossRef] [PubMed]
- Lokeshwar, S.D.; Lopez, M.; Sarcan, S.; Aguilar, K.; Morera, D.S.; Shaheen, D.M.; Lokeshwar, B.L.; Lokeshwar, V.B. Molecular Oncology of Bladder Cancer from Inception to Modern Perspective. Cancers 2022, 14, 2578. [Google Scholar] [CrossRef] [PubMed]
- Lenis, A.T.; Lec, P.M.; Chamie, K.; Mshs, M.D. Bladder Cancer: A Review. JAMA 2020, 324, 1980–1991. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Yang, W.; Xue, X.; Li, Y.; Jin, Z.; Ji, Z. Neoadjuvant immunotherapy and chemoimmunotherapy for stage II-III muscle invasive bladder cancer. Front. Immunol. 2022, 13, 986359. [Google Scholar] [CrossRef] [PubMed]
- Tripathi, A.; MacDougall, K.; Sonpavde, G.P. Therapeutic Landscape Beyond Immunotherapy in Advanced Urothelial Carcinoma: Moving Past the Checkpoint. Drugs 2022, 82, 1649–1662. [Google Scholar] [CrossRef]
- Abdollah, F.; Gandaglia, G.; Thuret, R.; Schmitges, J.; Tian, Z.; Jeldres, C.; Passoni, N.M.; Briganti, A.; Shariat, S.F.; Perrotte, P.; et al. Incidence, survival and mortality rates of stage-specific bladder cancer in United States: A trend analysis. Cancer Epidemiol. 2013, 37, 219–225. [Google Scholar] [CrossRef]
- BCAN. Statement on American Cancer Society’s Estimated Bladder Cancer Cases in 2023. 2023. Available online: https://bcan.org/bladder-cancer-advocacy-network-statement-on-american-cancer-societys-estimated-bladder-cancer-cases-in-2023 (accessed on 3 August 2024).
- Pilala, K.M.; Papadimitriou, M.A.; Panoutsopoulou, K.; Barbarigos, P.; Levis, P.; Kotronopoulos, G.; Stravodimos, K.; Scorilas, A.; Avgeris, M. Epigenetic regulation of MIR145 core promoter controls miR-143/145 cluster in bladder cancer progression and treatment outcome. Mol. Ther. Nucleic Acids. 2022, 30, 311–322. [Google Scholar] [CrossRef]
- Lee, D.Y.; Hou, Y.C.; Yang, J.S.; Lin, H.Y.; Chang, T.Y.; Lee, K.H.; Kuo, S.C.; Hsieh, M.T. Synthesis, Anticancer Activity, and Preliminary Pharmacokinetic Evaluation of 4,4-Disubstituted Curcuminoid 2,2-bis(Hydroxymethyl)Propionate Derivatives. Molecules 2020, 25, 479. [Google Scholar] [CrossRef]
- Hackler, L., Jr.; Ózsvári, B.; Gyuris, M.; Sipos, P.; Fábián, G.; Molnár, E.; Marton, A.; Faragó, N.; Mihály, J.; Nagy, L.I.; et al. The Curcumin Analog C-150, Influencing NF-κB, UPR and Akt/Notch Pathways Has Potent Anticancer Activity In Vitro and In Vivo. PLoS ONE 2016, 11, e0149832. [Google Scholar] [CrossRef]
- Almalki, S.G. The pathophysiology of the cell cycle in cancer and treatment strategies using various cell cycle checkpoint inhibitors. Pathol. Res. Pract. 2023, 251, 154854. [Google Scholar] [CrossRef] [PubMed]
- Luo, D.; Yu, C.; Yu, J.; Su, C.; Li, S.; Liang, P. p53-mediated G1 arrest requires the induction of both p21 and Killin in human colon cancer cells. Cell Cycle 2022, 21, 140–151. [Google Scholar] [CrossRef]
- McIlwain, D.R.; Berger, T.; Mak, T.W. Caspase functions in cell death and disease. Cold Spring Harb. Perspect. Biol. 2013, 5, a008656. [Google Scholar] [CrossRef] [PubMed]
- Chen, Q.; Ma, K.; Liu, X.; Chen, S.H.; Li, P.; Yu, Y.; Leung, A.K.L.; Yu, X. Truncated PARP1 mediates ADP-ribosylation of RNA polymerase III for apoptosis. Cell Discov. 2022, 8, 3. [Google Scholar] [CrossRef]
- Lopez, J.; Tait, S.W. Mitochondrial apoptosis: Killing cancer using the enemy within. Br. J. Cancer 2015, 112, 957–962. [Google Scholar] [CrossRef] [PubMed]
- Susin, S.A.; Lorenzo, H.K.; Zamzami, N.; Marzo, I.; Snow, B.E.; Brothers, G.M.; Mangion, J.; Jacotot, E.; Costantini, P.; Loeffler, M.; et al. Molecular characterization of mitochondrial apoptosis-inducing factor. Nature 1999, 397, 441–446. [Google Scholar] [CrossRef]
- Singh, R.; Letai, A.; Sarosiek, K. Regulation of apoptosis in health and disease: The balancing act of BCL-2 family proteins. Nat. Rev. Mol. Cell Biol. 2019, 20, 175–193. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Name of Chemicals and Kits | Manufacturers with Catalog No. |
|---|---|
| 2′,7′-Dichlorodihydrofluorescein diacetate (DCFDA) | Sigma Aldrich (St. Louis, MO, USA) # D6883 |
| Apoptosis Detection Kit | eBioscience™ Annexin V Apoptosis Detection Kits; Invitrogen (Pittsburgh, PA, USA) # 88-8005-72 |
| JC-1 dye | G-Biosciences (St. Louis, MO, USA) #786-1322 |
| MitoSOX™ | Thermo Fisher Scientific (Eugene, OR, USA); #M36008 |
| MTT | Alfa Aesar (Ward Hill, MA, USA) #L11939 |
| Propidium Iodide | Sigma Aldrich (St. Louis, MO, USA) #P4170 |
| Name of Antibody | Manufacturers with Catalog No. | RRID | Dilution |
|---|---|---|---|
| AKT | Cell Signaling Technology #9272 | AB_329827 | 1:1000 |
| p-AKT (Ser-473) | Cell Signaling Technology #4060 | AB_2315049 | 1:1500 |
| Cyclin-E1 | Epitomics #3327-1 | AB_10640935 | 1:1000 |
| PARP | Cell Signaling Technology #9542 | AB_2160739 | 1:1000 |
| AIF | Cell Signaling Technology #4642 | AB_2224542 | 1:1000 |
| BAX | Cell Signaling Technology # 2772 | AB_10695870 | 1:1000 |
| Bcl-2 | Cell Signaling Technology #3498 | AB_1903907 | 1:1000 |
| Bcl-XL | Cell Signaling Technology #2762 | AB_10694844 | 1:1000 |
| GAPDH | Cell Signaling Technology #2118 | AB_561053 | 1:2000 |
| β-Actin | Proteintech #HRP-60008 | AB_2819183 | 1:5000 |
| Anti-rabbit Secondary | Cell Signaling Technology #7074 | AB_2099233 | 1:5000 |
| Cell Lines | DCA | Cur | CMC-2 |
|---|---|---|---|
| HT-1376 | 8.25 ± 0.02 | 10.59 ± 0.11 | 0.80 ± 0.12 |
| T24 | 10.22 ± 1.24 | 10.47 ± 0.18 | 0.89 ± 0.08 |
| 253J | 7.17 ± 1.36 | 9.37 ± 0.39 | 0.62 ± 0.01 |
| 5637 naïve | 6.96 ± 0.26 | 7.91 ± 0.10 | 0.48 ± 0.31 |
| 5637 GemR | 7.99 ± 1.33 | 9.88 ± 2.67 | 0.93 ± 0.07 |
| 5637 CisR | 9.46 ± 2.6 | 10.57 ± 2.96 | 0.82 ± 0.25 |
| 5637 G+C R | 12.65 ± 1.15 | 12.12 ± 3.05 | 0.59 ± 0.13 |
| UROtsa | 62.81 ± 17.59 | 66.31 ± 25.42 | 44.65 ± 6.86 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gupta, K.B.; Taylor, T.L.; Panda, S.S.; Thangaraju, M.; Lokeshwar, B.L. Curcumin-Dichloroacetate Hybrid Molecule as an Antitumor Oral Drug against Multidrug-Resistant Advanced Bladder Cancers. Cancers 2024, 16, 3108. https://doi.org/10.3390/cancers16173108
Gupta KB, Taylor TL, Panda SS, Thangaraju M, Lokeshwar BL. Curcumin-Dichloroacetate Hybrid Molecule as an Antitumor Oral Drug against Multidrug-Resistant Advanced Bladder Cancers. Cancers. 2024; 16(17):3108. https://doi.org/10.3390/cancers16173108
Chicago/Turabian StyleGupta, Kunj Bihari, Truett L. Taylor, Siva S. Panda, Muthusamy Thangaraju, and Bal. L. Lokeshwar. 2024. "Curcumin-Dichloroacetate Hybrid Molecule as an Antitumor Oral Drug against Multidrug-Resistant Advanced Bladder Cancers" Cancers 16, no. 17: 3108. https://doi.org/10.3390/cancers16173108
APA StyleGupta, K. B., Taylor, T. L., Panda, S. S., Thangaraju, M., & Lokeshwar, B. L. (2024). Curcumin-Dichloroacetate Hybrid Molecule as an Antitumor Oral Drug against Multidrug-Resistant Advanced Bladder Cancers. Cancers, 16(17), 3108. https://doi.org/10.3390/cancers16173108