

Emerging Targeted Therapies for HER2-Positive Breast Cancer


Abstract
Simple Summary
Abstract
1. Introduction
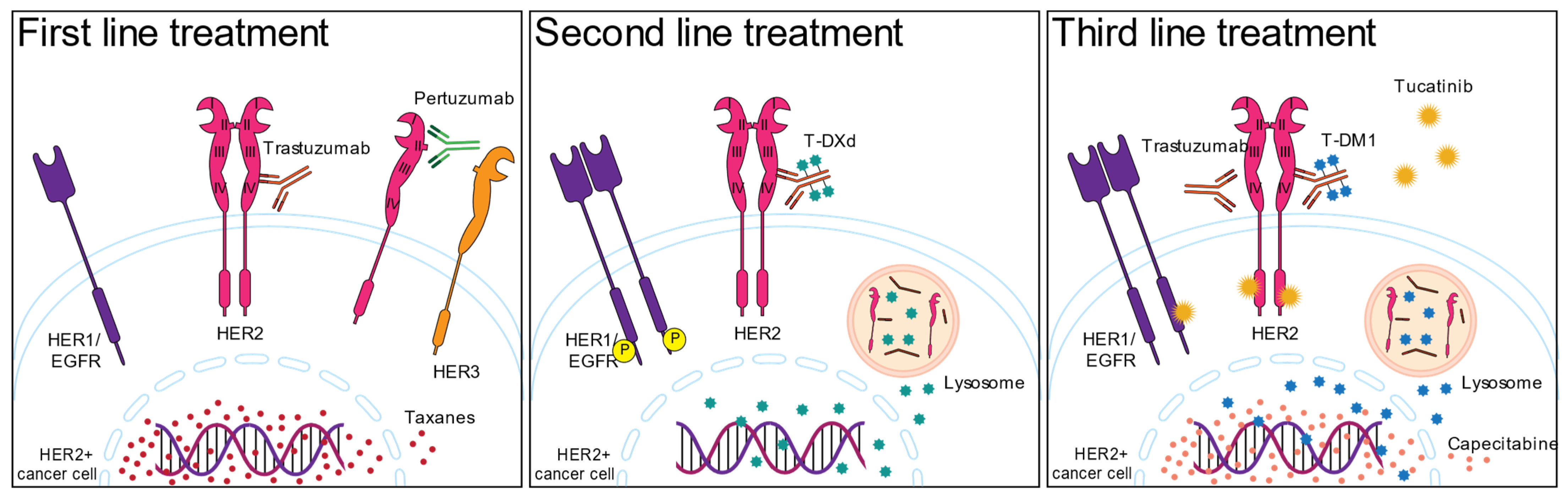
2. HER2-Targeted Therapies
2.1. Antibodies
2.1.1. Monoclonal Antibodies
2.1.2. Bispecific Antibodies
HER2/HER2 BsAb
HER2/HER3 BsAb

{kind=link}
| Drug | Clinical Trial Identifier | In Combination with | Population | Reference |
|---|---|---|---|---|
| Trastuzumab/Pertuzumab | ||||
| MBS301 | NCT03842085 | Malignant HER2-expressing solid tumors | [44] | |
| Zanidatamab (ZW25) | NCT02892123 | Chemotherapy | HER2-expressing solid tumors | [49,50] |
| NCT05035836 | Early HER2+ breast cancer | |||
| NCT04224272 | Palbociclib and fulvestrant | Advanced HER2+ breast cancer | ||
| NCT05027139 | Anti-CD47 | Solid HER2+ tumors including the HER2-low breast cancer | ||
| KN026 | NCT04881929 | Chemotherapy | HER2+ breast cancer | |
| NCT04521179 NCT04040699 | KN046 (bispecific antibody against PD-1 and CTLA-4) | Locally advanced HER2+ solid tumors and HER2+ solid tumor | [54] | |
| NCT04778982 | Palbociclib and fulvestrant | Advanced breast cancer | ||
| HER2/HER3 | ||||
| Zenocutuzumab (MCLA-128) | NCT03321981 | Trastuzumab and chemotherapy or trastuzumab and vinorelbine | HER2-low breast cancer and metastatic HER2+ breast cancer that progressed to T-DM1 treatment | [61] |
| MM-111 | NCT01097460 | Trastuzumab | Advanced HER2 amplified and heregulin-positive breast cancer | |
| NCT00911898 | Advanced, refractory HER2 A\amplified and heregulin-positive cancers | |||
HER2 and CD3 BsAb
HER2 and CD16 BsAb
2.1.3. Antibody-Drug Conjugates (ADCs)
| Drug | Payload | Drug-to-Antibody Ratio | Clinical Trial Identifyer | In Combination with | Population | Reference |
|---|---|---|---|---|---|---|
| T-DXd | Deruxtecan (topoisomerase I inhibitor) | ~8 | NCT04784715 | Pertuzumab | HER2+ metastatic breast cancer | |
| NCT04538742 | Durvalumab (anti-PD-L1,) | HER2+ metastatic breast cancer | ||||
| NCT04538742, NCT04539938 | Tucatinib | HER2+ breast cancer or HER2+ metastatic breast cancer | ||||
| NCT04556773 | Durvalumab, paclitaxel, capivasertib, anastrozole, fulvestrant, or capecitabine | HER2-low advanced or metastatic breast cancer | [104] | |||
| Trastuzumab-duocarmycine (SYD985) | Duocarmycine (DNA alkylating agent) | 2.8 | NCT03262935 | HER2+ locally advanced or metastatic breast cancer | [109] | |
| NCT01042379 (I-SPY) | Chemotherapy | Breast cancer | ||||
| NCT04602117 (ISPY-P1.01) | Paclitaxel | Metastatic cancer | ||||
| NCT04235101 | Niraparib (PARP inhibitor) | Solid tumors | ||||
| ARX788 | Amberstatin 269 (microtubule inhibitor) | 2 | NCT01042379 | HER2+ breast cancer | ||
| NCT04829604, NCT02512237 | HER2+ metastatic breast cancer | |||||
| NCT05041972 | HER2-mutated or HER2-amplified tumors | |||||
| NCT05018676 | HER2-low breast cancer | |||||
| NCT05018702 | Breast cancer patients with brain metastasis | |||||
| NCT03255070 | HER2+ solid tumors | |||||
| Disitamab vedotin (RC48) | Monomethyl auristatin E (microtubule inhibitor) | 4 | NCT02881190 | Advanced or metastatic HER2+ tumors | [121] | |
| NCT05134519 | HER2+ breast cancer | |||||
| NCT04400695 | Locally advanced or metastatic HER2-low breast cancer | |||||
| NCT05331326 | HER2-expression metastatic breast cancer with abnormal activation of PAM pathway | |||||
| NCT03052634 | Advanced breast cancer | |||||
| NCT05726175 | Penpulimab (AK105) | HER2-low breast cancer | ||||
| NCT03500380 | HER2+ metastatic breast cancer with or without liver metastases | |||||
| A166 | Duo-5 (microtubule inhibitor) | 2.8 | NCT03602079 | Relapsed/refractory cancers wxpressing HER2 antigen or amplified HER2 gene | [123,124,125] | |
| MRG002 | Monomethyl auristatin E (microtubule inhibitor) | ~3.8 | NCT05263869 | HER2+ advanced breast cancer | ||
| NCT04924699 | HER2+ metastatic tumors | |||||
| NCT04742153 | HER2-low locally advanced metastatic breast cancer | |||||
| Zanidatamab zovodotin (ZW49) | Auristatin based (microtubule inhibitor) | 2 | NCT03821233 | Metastatic HER2+ tumors | ||
| BDC-1001 | TLR7/8 agonist | Not reported | NCT04278144 | Nivolumab | Advanced HER2-expressing solid tumors | [128] |
| ALT-P7 | Monomethyl auristatin E (microtubule inhibitor) | 2 | NCT03281824 | HER2+ breast cancer | [130] | |
| XMT-1522 | Auristatin derivative (AF-HPA) | 12 | NCT02952729 | Advanced HER2+ breast cancer patients | [133] | |
| PF-06804103 | Derivative of auristatin | 4 | NCT03284723 | HER2+ breast cancer | [136] | |
| Targeted thorium-227 conjugates (TTCs)/BAY2701439 | Thorium-227 (cytotoxic alpha radiation) | Not reported | NCT04147819 | Advanced HER2-expressing cancer |
2.2. TKIs
| Drug | Description | In Combination with | Clinical Trial Identifyer | Population | Reference |
|---|---|---|---|---|---|
| Tucatinib | Selective and reversible HER2 inhibitor with minimal inhibition of EGFR/HER1 | T-DM1 | NCT04457596, NCT03975647, NCT01983501, NCT05323955 | HER2+ breast cancer | |
| T-DXd | NCT04539938, NCT04538742 | HER2+ breast cancer | |||
| Pyrotinib | Irreversible pan-HER inhibitor | NCT01937689 | HER2+ metastatic breast cancer | [178] | |
| Capecitabine | NCT02361112 | HER2+ metastatic breast cancer | [180] | ||
| Poziotinib | Irreversible pan-HER inhibitor | T-DM1 | NCT03429101 | HER2+ breast cancer | |
| Epertinib (S-222611) | Reversible pan-HER inhibitor | 2013-003894-87 | HER2+ tumors | [192,193,194] | |
| DZD1516 | Selective HER2 inhibitor | Trastuzumab and capecitabine or T-DM1 | NCT04509596 | Metastatic HER2+ breast cancer | [196] |
3. HER2-Targeted Therapies in the Era of Immunotherapy
3.1. PD-1/PD-L1 Antibodies
3.2. Immunotherapy-Enhancing ADCC and ADCP
3.3. Immunotherapy Enhancing Adaptive Immune Response
4. Cell Therapies
4.1. CAR-T Cells
4.2. CAR-NK
4.3. CAR-M
5. Anti-Cancer Vaccines
6. Exosomes
7. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef]
- Slamon, D.J.; Clark, G.M.; Wong, S.G.; Levin, W.J.; Ullrich, A.; McGuire, W.L. Human Breast Cancer: Correlation of Relapse and Survival with Amplification of the HER-2/neu Oncogene. Science 1987, 235, 177–182. [Google Scholar] [CrossRef]
- Chua, T.C.; Merrett, N.D. Clinicopathologic Factors Associated with HER2-Positive Gastric Cancer and Its Impact on Survival Outcomes-A Systematic Review. Int. J. Cancer 2012, 130, 2845–2856. [Google Scholar] [CrossRef]
- Slamon, D.J.; Godolphin, W.; Ullrich, A.; Michael, F. Press Studies of the HER-2/neu Proto-Oncogene in Human Breast Cancer. Cancer Genet. Cytogenet. 1989, 41, 219. [Google Scholar] [CrossRef]
- Howlader, N.; Cronin, K.A.; Kurian, A.W.; Andridge, R. Differences in Breast Cancer Survival by Molecular Subtypes in the United States. CancerEpidemiol. Biomark. Prev. 2018, 27, 619–626. [Google Scholar] [CrossRef]
- Arteaga, C.L.; Engelman, J.A. ERBB Receptors: From Oncogene Discovery to Basic Science to Mechanism-Based Cancer Therapeutics. Cancer Cell 2014, 25, 282–303. [Google Scholar] [CrossRef]
- Yarden, Y.; Sliwkowski, M.X. Untangling the ErbB Signalling Network. Nat. Rev. Mol. Cell Biol. 2001, 2, 127–137. [Google Scholar] [CrossRef]
- Yarden, Y. Biology of HER2 and Its Importance in Breast Cancer. Oncology 2001, 61, 1–13. [Google Scholar] [CrossRef]
- Cogswell, P.C.; Guttridge, D.C.; Funkhouser, W.K.; Baldwin, A.S., Jr. Selective Activation of NF-Kappa B Subunits in Human Breast Cancer: Potential Roles for NF-Kappa B2/p52 and for Bcl-3. Oncogene 2000, 19, 1123–1131. [Google Scholar] [CrossRef]
- Lemmon, M.A.; Schlessinger, J. Cell Signaling by Receptor Tyrosine Kinases. Cell 2010, 141, 1117–1134. [Google Scholar] [CrossRef]
- Hynes, N.E.; Lane, H.A. ERBB Receptors and Cancer: The Complexity of Targeted Inhibitors. Nat. Rev. Cancer 2005, 5, 341–354. [Google Scholar] [CrossRef]
- Slamon, D.J.; Leyland-Jones, B.; Shak, S.; Fuchs, H.; Paton, V.; Bajamonde, A.; Fleming, T.; Eiermann, W.; Wolter, J.; Pegram, M.; et al. Use of Chemotherapy plus a Monoclonal Antibody against HER2 for Metastatic Breast Cancer That Overexpresses HER2. N. Engl. J. Med. 2001, 344, 783–792. [Google Scholar] [CrossRef]
- Montagna, E.; Colleoni, M. Hormonal Treatment Combined with Targeted Therapies in Endocrine-Responsive and HER2-Positive Metastatic Breast Cancer. Ther. Adv. Med. Oncol. 2019, 11, 1758835919894105. [Google Scholar] [CrossRef]
- Hua, X.; Bi, X.-W.; Zhao, J.-L.; Shi, Y.-X.; Lin, Y.; Wu, Z.-Y.; Zhang, Y.-Q.; Zhang, L.-H.; Zhang, A.-Q.; Huang, H.; et al. Trastuzumab Plus Endocrine Therapy or Chemotherapy as First-Line Treatment for Patients with Hormone Receptor-Positive and HER2-Positive Metastatic Breast Cancer (SYSUCC-002). Clin. Cancer Res. 2022, 28, 637–645. [Google Scholar] [CrossRef]
- Perez, E.A.; Romond, E.H.; Suman, V.J.; Jeong, J.-H.; Sledge, G.; Geyer, C.E., Jr.; Martino, S.; Rastogi, P.; Gralow, J.; Swain, S.M.; et al. Trastuzumab plus Adjuvant Chemotherapy for Human Epidermal Growth Factor Receptor 2-Positive Breast Cancer: Planned Joint Analysis of Overall Survival from NSABP B-31 and NCCTG N9831. J. Clin. Oncol. 2014, 32, 3744–3752. [Google Scholar] [CrossRef]
- Gianni, L.; Eiermann, W.; Semiglazov, V.; Lluch, A.; Tjulandin, S.; Zambetti, M.; Moliterni, A.; Vazquez, F.; Byakhov, M.J.; Lichinitser, M.; et al. Neoadjuvant and Adjuvant Trastuzumab in Patients with HER2-Positive Locally Advanced Breast Cancer (NOAH): Follow-up of a Randomised Controlled Superiority Trial with a Parallel HER2-Negative Cohort. Lancet Oncol. 2014, 15, 640–647. [Google Scholar] [CrossRef]
- Junttila, T.T.; Akita, R.W.; Parsons, K.; Fields, C.; Lewis Phillips, G.D.; Friedman, L.S.; Sampath, D.; Sliwkowski, M.X. Ligand-Independent HER2/HER3/PI3K Complex Is Disrupted by Trastuzumab and Is Effectively Inhibited by the PI3K Inhibitor GDC-0941. Cancer Cell 2009, 15, 429–440. [Google Scholar] [CrossRef]
- Park, S.; Jiang, Z.; Mortenson, E.D.; Deng, L.; Radkevich-Brown, O.; Yang, X.; Sattar, H.; Wang, Y.; Brown, N.K.; Greene, M.; et al. The Therapeutic Effect of Anti-HER2/neu Antibody Depends on Both Innate and Adaptive Immunity. Cancer Cell 2010, 18, 160–170. [Google Scholar] [CrossRef]
- Shi, Y.; Fan, X.; Deng, H.; Brezski, R.J.; Rycyzyn, M.; Jordan, R.E.; Strohl, W.R.; Zou, Q.; Zhang, N.; An, Z. Trastuzumab Triggers Phagocytic Killing of High HER2 Cancer Cells In Vitro and In Vivo by Interaction with Fcγ Receptors on Macrophages. J. Immunol. 2015, 194, 4379–4386. [Google Scholar] [CrossRef]
- Hurvitz, S.A.; Martin, M.; Symmans, W.F.; Jung, K.H.; Huang, C.-S.; Thompson, A.M.; Harbeck, N.; Valero, V.; Stroyakovskiy, D.; Wildiers, H.; et al. Neoadjuvant Trastuzumab, Pertuzumab, and Chemotherapy versus Trastuzumab Emtansine plus Pertuzumab in Patients with HER2-Positive Breast Cancer (KRISTINE): A Randomised, Open-Label, Multicentre, Phase 3 Trial. Lancet Oncol. 2018, 19, 115–126. [Google Scholar] [CrossRef]
- Scheuer, W.; Friess, T.; Burtscher, H.; Bossenmaier, B.; Endl, J.; Hasmann, M. Strongly Enhanced Antitumor Activity of Trastuzumab and Pertuzumab Combination Treatment on HER2-Positive Human Xenograft Tumor Models. Cancer Res. 2009, 69, 9330–9336. [Google Scholar] [CrossRef]
- Perez, E.A.; López-Vega, J.M.; Petit, T.; Zamagni, C.; Easton, V.; Kamber, J.; Restuccia, E.; Andersson, M. Safety and Efficacy of Vinorelbine in Combination with Pertuzumab and Trastuzumab for First-Line Treatment of Patients with HER2-Positive Locally Advanced or Metastatic Breast Cancer: VELVET Cohort 1 Final Results. Breast Cancer Res. 2016, 18, 126. [Google Scholar] [CrossRef]
- Miles, D.; Ciruelos, E.; Schneeweiss, A.; Puglisi, F.; Peretz-Yablonski, T.; Campone, M.; Bondarenko, I.; Nowecki, Z.; Errihani, H.; Paluch-Shimon, S.; et al. Final Results from the PERUSE Study of First-Line Pertuzumab plus Trastuzumab plus a Taxane for HER2-Positive Locally Recurrent or Metastatic Breast Cancer, with a Multivariable Approach to Guide Prognostication. Ann. Oncol. 2021, 32, 1245–1255. [Google Scholar] [CrossRef]
- Wang, R.; Smyth, L.M.; Iyengar, N.M.; Modi, S.; Chandarlapaty, S.; Patil, S.; Norton, L.; Baselga, J.; Hudis, C.A.; Dang, C.T. Longer Follow-up on Clinical Outcomes of Weekly Paclitaxel with Trastuzumab and Pertuzumab in Patients with HER2 Overexpressing Metastatic Breast Cancer. J. Clin. Oncol. 2018, 36, e13005. [Google Scholar] [CrossRef]
- Woodward, N.; De Boer, R.H.; Redfern, A.; White, M.; Young, J.; Truman, M.; Beith, J. Results from the First Multicenter, Open-Label, Phase IIIb Study Investigating the Combination of Pertuzumab with Subcutaneous Trastuzumab and a Taxane in Patients With HER2-Positive Metastatic Breast Cancer (SAPPHIRE). Clin. Breast Cancer 2019, 19, 216–224. [Google Scholar] [CrossRef]
- Lemery, S.J.; Ricci, M.S.; Keegan, P.; McKee, A.E.; Pazdur, R. FDA’s Approach to Regulating Biosimilars. Clin. Cancer Res. 2017, 23, 1882–1885. [Google Scholar] [CrossRef]
- O’Callaghan, J.; Barry, S.P.; Bermingham, M.; Morris, J.M.; Griffin, B.T. Regulation of Biosimilar Medicines and Current Perspectives on Interchangeability and Policy. Eur. J. Clin. Pharmacol. 2019, 75, 1–11. [Google Scholar] [CrossRef]
- Triantafyllidi, E.; Triantafillidis, J.K. Systematic Review on the Use of Biosimilars of Trastuzumab in HER2+ Breast Cancer. Biomedicines 2022, 10, 2045. [Google Scholar] [CrossRef]
- Royce, M.; Osgood, C.L.; Amatya, A.K.; Fiero, M.H.; George Chang, C.J.; Ricks, T.K.; Shetty, K.A.; Kraft, J.; Qiu, J.; Song, P.; et al. FDA Approval Summary: Margetuximab plus Chemotherapy for Advanced or Metastatic HER2-Positive Breast Cancer. Clin. Cancer Res. 2022, 28, 1487–1492. [Google Scholar] [CrossRef]
- Nordstrom, J.L.; Gorlatov, S.; Zhang, W.; Yang, Y.; Huang, L.; Burke, S.; Li, H.; Ciccarone, V.; Zhang, T.; Stavenhagen, J.; et al. Anti-Tumor Activity and Toxicokinetics Analysis of MGAH22, an Anti-HER2 Monoclonal Antibody with Enhanced Fcγ Receptor Binding Properties. Breast Cancer Res. 2011, 13, R123. [Google Scholar] [CrossRef]
- Rugo, H.S.; Im, S.-A.; Cardoso, F.; Cortés, J.; Curigliano, G.; Musolino, A.; Pegram, M.D.; Wright, G.S.; Saura, C.; Escrivá-de-Romaní, S.; et al. Efficacy of Margetuximab vs Trastuzumab in Patients With Pretreated ERBB2-Positive Advanced Breast Cancer: A Phase 3 Randomized Clinical Trial. JAMA Oncol. 2021, 7, 573–584. [Google Scholar] [CrossRef]
- Rugo, H.S.; Im, S.-A.; Cardoso, F.; Cortes, J.; Curigliano, G.; Musolino, A.; Pegram, M.D.; Bachelot, T.; Wright, G.S.; Saura, C.; et al. Margetuximab Versus Trastuzumab in Patients With Previously Treated HER2-Positive Advanced Breast Cancer (SOPHIA): Final Overall Survival Results From a Randomized Phase 3 Trial. J. Clin. Oncol. 2023, 41, 198–205. [Google Scholar] [CrossRef]
- Mandó, P.; Rivero, S.G.; Rizzo, M.M.; Pinkasz, M.; Levy, E.M. Targeting ADCC: A Different Approach to HER2 Breast Cancer in the Immunotherapy Era. Breast 2021, 60, 15–25. [Google Scholar] [CrossRef]
- Ko, B.-K.; Lee, S.-Y.; Lee, Y.-H.; Hwang, I.-S.; Persson, H.; Rockberg, J.; Borrebaeck, C.; Park, D.; Kim, K.-T.; Uhlen, M.; et al. Combination of Novel HER2-Targeting Antibody 1E11 with Trastuzumab Shows Synergistic Antitumor Activity in HER2-Positive Gastric Cancer. Mol. Oncol. 2015, 9, 398–408. [Google Scholar] [CrossRef]
- Espelin, C.W.; Leonard, S.C.; Geretti, E.; Wickham, T.J.; Hendriks, B.S. Dual HER2 Targeting with Trastuzumab and Liposomal-Encapsulated Doxorubicin (MM-302) Demonstrates Synergistic Antitumor Activity in Breast and Gastric Cancer. Cancer Res. 2016, 76, 1517–1527. [Google Scholar] [CrossRef]
- Vivekanandhan, S.; Knutson, K.L. Resistance to Trastuzumab. Cancers 2022, 14, 5115. [Google Scholar] [CrossRef]
- Derakhshani, A.; Rezaei, Z.; Safarpour, H.; Sabri, M.; Mir, A.; Sanati, M.A.; Vahidian, F.; Gholamiyan Moghadam, A.; Aghadoukht, A.; Hajiasgharzadeh, K.; et al. Overcoming Trastuzumab Resistance in HER2-Positive Breast Cancer Using Combination Therapy. J. Cell. Physiol. 2020, 235, 3142–3156. [Google Scholar] [CrossRef]
- Nagy, P.; Friedländer, E.; Tanner, M.; Kapanen, A.I.; Carraway, K.L.; Isola, J.; Jovin, T.M. Decreased Accessibility and Lack of Activation of ErbB2 in JIMT-1, a Herceptin-Resistant, MUC4-Expressing Breast Cancer Cell Line. Cancer Res. 2005, 65, 473–482. [Google Scholar] [CrossRef]
- Mercogliano, M.F.; De Martino, M.; Venturutti, L.; Rivas, M.A.; Proietti, C.J.; Inurrigarro, G.; Frahm, I.; Allemand, D.H.; Deza, E.G.; Ares, S.; et al. TNFα-Induced Mucin 4 Expression Elicits Trastuzumab Resistance in HER2-Positive Breast Cancer. Clin. Cancer Res. 2017, 23, 636–648. [Google Scholar] [CrossRef]
- Mercogliano, M.F.; De Martino, M.; Bruni, S.; Venturutti, L.; Rivas, M.; Amasino, M.; Proietti, C.J.; Elizalde, P.V.; Schillaci, R. Abstract 1195: TNFα Induces Multiresistance to HER2-Targeted TNFα Induces Multiresistance to HER2-Targeted Therapies in HER2-Positive Breast Cancer. Cancer Res. 2017, 77, 1195. [Google Scholar] [CrossRef]
- Steed, P.M.; Tansey, M.G.; Zalevsky, J.; Zhukovsky, E.A.; Desjarlais, J.R.; Szymkowski, D.E.; Abbott, C.; Carmichael, D.; Chan, C.; Cherry, L.; et al. Inactivation of TNF Signaling by Rationally Designed Dominant-Negative TNF Variants. Science 2003, 301, 1895–1898. [Google Scholar] [CrossRef] [PubMed]
- Bruni, S.; Mauro, F.L.; Proietti, C.J.; Cordo-Russo, R.I.; Rivas, M.A.; Inurrigarro, G.; Dupont, A.; Rocha, D.; Fernández, E.A.; Deza, E.G.; et al. Blocking soluble TNFα sensitizes HER2-positive breast cancer to trastuzumab through MUC4 downregulation and subverts immunosuppression. J. Immunother. Cancer 2023, 11, e005325. [Google Scholar] [CrossRef] [PubMed]
- Liguori, L.; Polcaro, G.; Nigro, A.; Conti, V.; Sellitto, C.; Perri, F.; Ottaiano, A.; Cascella, M.; Zeppa, P.; Caputo, A.; et al. Bispecific Antibodies: A Novel Approach for the Treatment of Solid Tumors. Pharmaceutics 2022, 14, 2442. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.; Li, F.; Liu, H.; Ye, P.; Fan, X.; Yuan, X.; Wu, Z.; Chen, J.; Jin, C.; Shen, B.; et al. Structural and Functional Characterization of MBS301, an Afucosylated Bispecific Anti-HER2 Antibody. MAbs 2018, 10, 864–875. [Google Scholar] [CrossRef]
- Weisser, N.; Wickman, G.; Davies, R.; Rowse, G. Abstract 31: Preclinical Development of a Novel Biparatopic HER2 Antibody with Activity in Low to High HER2 Expressing Cancers. Cancer Res. 2017, 77, 31. [Google Scholar] [CrossRef]
- Meric-Bernstam, F.; Hamilton, E.P.; Beeram, M.; Hanna, D.L.; El-Khoueiry, A.B.; Kang, Y.-K.; Lee, K.W.; Lee, J.; Rha, S.Y.; Chaves, J.M.; et al. Zanidatamab (ZW25) in HER2-Expressing Gastroesophageal Adenocarcinoma (GEA): Results from a Phase I Study. J. Clin. Oncol. 2021, 39, 164. [Google Scholar] [CrossRef]
- Weisser, N.E.; Wickman, G.; Abraham, L.; O’Toole, J.; Harbourne, B.; Guedia, J.; Cheng, C.W.; Chan, P.; Browman, D.; Gold, M.R.; et al. Abstract 1005: The Bispecific Antibody Zanidatamab’s (ZW25′s) Unique Mechanisms of Action and Durable Anti-Tumor Activity in HER2-Expressing Cancers. Cancer Res. 2021, 81, 1005. [Google Scholar] [CrossRef]
- Meric-Bernstam, F.; Beeram, M.; Hamilton, E.; Oh, D.-Y.; Hanna, D.L.; Kang, Y.-K.; Elimova, E.; Chaves, J.; Goodwin, R.; Lee, J.; et al. Zanidatamab, a Novel Bispecific Antibody, for the Treatment of Locally Advanced or Metastatic HER2-Expressing or HER2-Amplified Cancers: A Phase 1, Dose-Escalation and Expansion Study. Lancet Oncol. 2022, 23, 1558–1570. [Google Scholar] [CrossRef]
- Meric-Bernstam, F.; Hanna, D.; Beeram, M.; Lee, K.-W.; Kang, Y.-K.; Chaves, J.; Lee, J.; Goodwin, R.; Vaklavas, C.; Oh, D.-Y.; et al. Safety, Anti-Tumour Activity, and Biomarker Results of the HER2-Targeted Bispecific Antibody ZW25 in HER2-Expressing Solid Tumours. Ann. Oncol. 2019, 30, v167–v168. [Google Scholar] [CrossRef]
- Bedard, P.L.; Im, S.-A.; Elimova, E.; Rha, S.Y.; Goodwin, R.; Ferrario, C.; Lee, K.-W.; Hanna, D.; Meric-Bernstam, F.; Mayordomo, J.; et al. Abstract P2-13-07: Zanidatamab (ZW25), a HER2-Targeted Bispecific Antibody, in Combination with Chemotherapy (chemo) for HER2-Positive Breast Cancer (BC): Results from a Phase 1 Study. Cancer Res. 2022, 82, P2–P13. [Google Scholar] [CrossRef]
- Proctor, J.R.; Gartner, E.M.; Gray, T.E.; Davies, R.H. Population Pharmacokinetics of Zanidatamab, an Anti-HER2 Biparatopic Antibody, in Patients with Advanced or Metastatic Cancer. Cancer Chemother. Pharmacol. 2022, 90, 399–408. [Google Scholar] [CrossRef]
- Wei, H.; Cai, H.; Jin, Y.; Wang, P.; Zhang, Q.; Lin, Y.; Wang, W.; Cheng, J.; Zeng, N.; Xu, T.; et al. Structural Basis of a Novel Heterodimeric Fc for Bispecific Antibody Production. Oncotarget 2017, 8, 51037–51049. [Google Scholar] [CrossRef]
- Zhang, J.; Ji, D.; Cai, L.; Yao, H.; Yan, M.; Wang, X.; Shen, W.; Du, Y.; Pang, H.; Lai, X.; et al. First-in-Human HER2-Targeted Bispecific Antibody KN026 for the Treatment of Patients with HER2-Positive Metastatic Breast Cancer: Results from a Phase I Study. Clin. Cancer Res. 2022, 28, 618–628. [Google Scholar] [CrossRef]
- Gong, J.; Dong, Z.; Liu, D.; Xu, J.; Yang, J.; Yang, Y.; Qi, Y.; Men, J.; Kong, P.; Xu, T.; et al. 339 Preliminary Safety, Tolerability and Efficacy Results of KN026 (a HER2-Targeted Bispecific Antibody) in Combination with KN046 (an Anti-PD-L1/CTLA-4 Bispecific Antibody) in Patients (pts) with HER2 Aberrated Solid Tumors. Regul. Young Investig. Award. Abstr. 2020, 8. [Google Scholar] [CrossRef]
- Maussang-Detaille, D.; de Nardis, C.; Hendriks, L.; Bartelink-Clements, C.; Rovers, E.; Gallenne, T.; Doornbos, R.; Bakker, L.; de Kruif, J.; Logtenberg, T.; et al. Abstract 33: The Binding Mode of the Bispecific Anti-HER2xHER3 Antibody MCLA-128 Is Responsible for Its Potent Inhibition of HRG-Driven Tumorigenesis. Cancer Res. 2017, 77, 33. [Google Scholar] [CrossRef]
- Geuijen, C.A.W.; De Nardis, C.; Maussang, D.; Rovers, E.; Gallenne, T.; Hendriks, L.J.A.; Visser, T.; Nijhuis, R.; Logtenberg, T.; de Kruif, J.; et al. Unbiased Combinatorial Screening Identifies a Bispecific IgG1 That Potently Inhibits HER3 Signaling via HER2-Guided Ligand Blockade. Cancer Cell 2021, 39, 1163–1164. [Google Scholar] [CrossRef]
- Schram, A.M.; Odintsov, I.; Espinosa-Cotton, M.; Khodos, I.; Sisso, W.J.; Mattar, M.S.; Lui, A.J.W.; Vojnic, M.; Shameem, S.H.; Chauhan, T.; et al. Zenocutuzumab, a HER2xHER3 Bispecific Antibody, Is Effective Therapy for Tumors Driven by NRG1 Gene Rearrangements. Cancer Discov. 2022, 12, 1233–1247. [Google Scholar] [CrossRef]
- Calvo, E.; Alsina, M.; Schellens, J.H.M.; Huitema, A.D.R.; Tabernero, J.; de Vries-Schultink, A.; Boni, V.; Doger, B.; Geuijen, C.; Doornbos, R.; et al. Abstract CT050: A Phase I/II Study of MCLA-128, a Full Length IgG1 Bispecific Antibody Targeting HER2 and HER3, in Patients with Solid Tumors. Cancer Res. 2016, 76, CT050. [Google Scholar] [CrossRef]
- Geuijen, C.; Rovers, E.; Nijhuis, R.; den Blanken-Smit, R.; Visser, T.; Bartelink, W.; Kramer, A.; der Zande, V.Z.; Clements, C.; Kaldenberg, L.; et al. Preclinical Activity of MCLA-128, an ADCC Enhanced Bispecific IgG1 Antibody Targeting the HER2:HER3 Heterodimer. J. Clin. Oncol. 2014, 32, 560. [Google Scholar] [CrossRef]
- Alsina, M.; Boni, V.; Schellens, J.H.M.; Moreno, V.; Bol, K.; Westendorp, M.; Andres Sirulnik, L.; Tabernero, J.; Calvo, E. First-in-Human Phase 1/2 Study of MCLA-128, a Full Length IgG1 Bispecific Antibody Targeting HER2 and HER3: Final Phase 1 Data and Preliminary Activity in HER2 Metastatic Breast Cancer (MBC). J. Clin. Oncol. 2017, 35, 2522. [Google Scholar] [CrossRef]
- Hamilton, E.P.; Petit, T.; Pistilli, B.; Goncalves, A.; Ferreira, A.A.; Dalenc, F.; Cardoso, F.; Mita, M.M.; Dezentjé, V.O.; Manso, L.; et al. Clinical Activity of MCLA-128 (zenocutuzumab), Trastuzumab, and Vinorelbine in HER2 Amplified Metastatic Breast Cancer (MBC) Patients (pts) Who Had Progressed on Anti-HER2 ADCs. J. Clin. Oncol. 2020, 38, 3093. [Google Scholar] [CrossRef]
- McDonagh, C.F.; Huhalov, A.; Harms, B.D.; Adams, S.; Paragas, V.; Oyama, S.; Zhang, B.; Luus, L.; Overland, R.; Nguyen, S.; et al. Antitumor Activity of a Novel Bispecific Antibody That Targets the ErbB2/ErbB3 Oncogenic Unit and Inhibits Heregulin-Induced Activation of ErbB3. Mol. Cancer Ther. 2012, 11, 582–593. [Google Scholar] [CrossRef]
- Richards, D.A.; Braiteh, F.S.; Garcia, A.A.; Denlinger, C.S.; Conkling, P.R.; Edenfield, W.J.; Anthony, S.P.; Hellerstedt, B.A.; Raju, R.N.; Becerra, C.; et al. A Phase 1 Study of MM-111, a Bispecific HER2/HER3 Antibody Fusion Protein, Combined with Multiple Treatment Regimens in Patients with Advanced HER2-Positive Solid Tumors. J. Clin. Oncol. 2014, 32, 651. [Google Scholar] [CrossRef]
- Kiewe, P.; Hasmüller, S.; Kahlert, S.; Heinrigs, M.; Rack, B.; Marmé, A.; Korfel, A.; Jäger, M.; Lindhofer, H.; Sommer, H.; et al. Phase I Trial of the Trifunctional Anti-HER2 X Anti-CD3 Antibody Ertumaxomab in Metastatic Breast Cancer. Clin. Cancer Res. 2006, 12, 3085–3091. [Google Scholar] [CrossRef]
- Jäger, M.; Schoberth, A.; Ruf, P.; Hess, J.; Lindhofer, H. The Trifunctional Antibody Ertumaxomab Destroys Tumor Cells That Express Low Levels of Human Epidermal Growth Factor Receptor 2. Cancer Res. 2009, 69, 4270–4276. [Google Scholar] [CrossRef]
- Haense, N.; Atmaca, A.; Pauligk, C.; Steinmetz, K.; Marmé, F.; Haag, G.M.; Rieger, M.; Ottmann, O.G.; Ruf, P.; Lindhofer, H.; et al. A Phase I Trial of the Trifunctional Anti Her2 × Anti CD3 Antibody Ertumaxomab in Patients with Advanced Solid Tumors. BMC Cancer 2016, 16, 420. [Google Scholar] [CrossRef]
- Rius Ruiz, I.; Vicario, R.; Morancho, B.; Morales, C.B.; Arenas, E.J.; Herter, S.; Freimoser-Grundschober, A.; Somandin, J.; Sam, J.; Ast, O.; et al. p95HER2-T Cell Bispecific Antibody for Breast Cancer Treatment. Sci. Transl. Med. 2018, 10, eaat1445. [Google Scholar] [CrossRef]
- Wermke, M.; Alt, J.; Kauh, J.S.; Back, J.; Salhi, Y.; Reddy, V.; Bayever, E.; Ochsenreither, S. Preliminary Biomarker and Pharmacodynamic Data from a Phase I Study of Single-Agent Bispecific Antibody T-Cell Engager GBR 1302 in Subjects with HER2-Positive Cancers. J. Clin. Oncol. 2018, 36, 69. [Google Scholar] [CrossRef]
- Deng, W.; Liu, J.; Pan, H.; Li, L.; Zhou, C.; Wang, X.; Shu, R.; Dong, B.; Cao, D.; Li, Q.; et al. A Bispecific Antibody Based on Pertuzumab Fab Has Potent Antitumor Activity. J. Immunother. 2018, 41, 1–8. [Google Scholar] [CrossRef]
- Turini, M.; Chames, P.; Bruhns, P.; Baty, D.; Kerfelec, B. A FcγRIII-Engaging Bispecific Antibody Expands the Range of HER2-Expressing Breast Tumors Eligible to Antibody Therapy. Oncotarget 2014, 5, 5304–5319. [Google Scholar] [CrossRef]
- Li, A.; Xing, J.; Li, L.; Zhou, C.; Dong, B.; He, P.; Li, Q.; Wang, Z. A Single-Domain Antibody-Linked Fab Bispecific Antibody Her2-S-Fab Has Potent Cytotoxicity against Her2-Expressing Tumor Cells. AMB Express 2016, 6, 32. [Google Scholar] [CrossRef] [PubMed]
- Oberg, H.H.; Kellner, C.; Gonnermann, D.; Sebens, S.; Bauerschlag, D.; Gramatzki, M.; Kabelitz, D.; Peipp, M.; Wesch, D. Tribody [(HER2)2xCD16] Is More Effective Than Trastuzumab in Enhancing γδ T Cell and Natural Killer Cell Cytotoxicity Against HER2-Expressing Cancer Cells. Front. Immunol. 2018, 9, 814. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Lam, L.; Nagai, Y.; Zhu, Z.; Chen, X.; Ji, M.Q.; Greene, M.I. A Targeted Immunotherapy Approach for HER2/neu Transformed Tumors by Coupling an Engineered Effector Domain with Interferon-γ. Oncoimmunology 2018, 7, e1300739. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Yi, M.; Zhu, S.; Wang, H.; Wu, K. Recent Advances and Challenges of Bispecific Antibodies in Solid Tumors. Exp. Hematol. Oncol. 2021, 10, 56. [Google Scholar] [CrossRef] [PubMed]
- Teachey, D.T.; Rheingold, S.R.; Maude, S.L.; Zugmaier, G.; Barrett, D.M.; Seif, A.E.; Nichols, K.E.; Suppa, E.K.; Kalos, M.; Berg, R.A.; et al. Cytokine Release Syndrome after Blinatumomab Treatment Related to Abnormal Macrophage Activation and Ameliorated with Cytokine-Directed Therapy. Blood 2013, 121, 5154–5157, Erratum in Blood 2016, 128, 1441. [Google Scholar] [CrossRef]
- Thomas, A.; Teicher, B.A.; Hassan, R. Antibody–drug Conjugates for Cancer Therapy. Lancet Oncol. 2016, 17, e254–e262. [Google Scholar] [CrossRef]
- Fu, Z.; Li, S.; Han, S.; Shi, C.; Zhang, Y. Antibody Drug Conjugate: The “biological Missile” for Targeted Cancer Therapy. Signal Transduct. Target. Ther. 2022, 7, 93. [Google Scholar] [CrossRef]
- Amiri-Kordestani, L.; Blumenthal, G.M.; Xu, Q.C.; Zhang, L.; Tang, S.W.; Ha, L.; Weinberg, W.C.; Chi, B.; Candau-Chacon, R.; Hughes, P.; et al. FDA Approval: Ado-Trastuzumab Emtansine for the Treatment of Patients with HER2-Positive Metastatic Breast Cancer. Clin. Cancer Res. 2014, 20, 4436–4441. [Google Scholar] [CrossRef]
- Wang, Y.; Xu, H.; Han, Y.; Wu, Y.; Wang, J. Comparative Efficacy of Tyrosine Kinase Inhibitors and Antibody-Drug Conjugates in HER2-Positive Metastatic Breast Cancer Patients with Brain Metastases: A Systematic Review and Network Meta-Analysis. Cancers 2022, 14, 3372. [Google Scholar] [CrossRef]
- Li, L.; Zhang, D.; Liu, B.; Lv, D.; Zhai, J.; Guan, X.; Yi, Z.; Ma, F. Antibody-Drug Conjugates in HER2-Positive Breast Cancer. Chin. Med. J. 2021, 135, 261–267. [Google Scholar] [CrossRef]
- Verma, S.; Miles, D.; Gianni, L.; Krop, I.E.; Welslau, M.; Baselga, J.; Pegram, M.; Oh, D.-Y.; Diéras, V.; Guardino, E.; et al. Trastuzumab Emtansine for HER2-Positive Advanced Breast Cancer. N. Engl. J. Med. 2012, 367, 1783–1791. [Google Scholar] [CrossRef] [PubMed]
- Modi, S.; Saura, C.; Yamashita, T.; Park, Y.H.; Kim, S.-B.; Tamura, K.; Andre, F.; Iwata, H.; Ito, Y.; Tsurutani, J.; et al. Trastuzumab Deruxtecan in Previously Treated HER2-Positive Breast Cancer. N. Engl. J. Med. 2020, 382, 610–621. [Google Scholar] [CrossRef]
- van der Lee, M.M.C.; Groothuis, P.G.; Ubink, R.; van der Vleuten, M.A.J.; van Achterberg, T.A.; Loosveld, E.M.; Damming, D.; Jacobs, D.C.H.; Rouwette, M.; Egging, D.F.; et al. The Preclinical Profile of the Duocarmycin-Based HER2-Targeting ADC SYD985 Predicts for Clinical Benefit in Low HER2-Expressing Breast Cancers. Mol. Cancer Ther. 2015, 14, 692–703. [Google Scholar] [CrossRef] [PubMed]
- Banerji, U.; van Herpen, C.M.L.; Saura, C.; Thistlethwaite, F.; Lord, S.; Moreno, V.; Macpherson, I.R.; Boni, V.; Rolfo, C.; de Vries, E.G.E.; et al. Trastuzumab Duocarmazine in Locally Advanced and Metastatic Solid Tumours and HER2-Expressing Breast Cancer: A Phase 1 Dose-Escalation and Dose-Expansion Study. Lancet Oncol. 2019, 20, 1124–1135. [Google Scholar] [CrossRef]
- Saura, C.; Thistlethwaite, F.; Banerji, U.; Lord, S.; Moreno, V.; MacPherson, I.; Boni, V.; Rolfo, C.D.; de Vries, E.G.E.; Van Herpen, C.M.L.; et al. A Phase I Expansion Cohorts Study of SYD985 in Heavily Pretreated Patients with HER2-Positive or HER2-Low Metastatic Breast Cancer. J. Clin. Oncol. 2018, 36, 1014. [Google Scholar] [CrossRef]
- von Minckwitz, G.; Huang, C.-S.; Mano, M.S.; Loibl, S.; Mamounas, E.P.; Untch, M.; Wolmark, N.; Rastogi, P.; Schneeweiss, A.; Redondo, A.; et al. Trastuzumab Emtansine for Residual Invasive HER2-Positive Breast Cancer. N. Engl. J. Med. 2019, 380, 617–628. [Google Scholar] [CrossRef]
- Mamounas, E.P.; Untch, M.; Mano, M.S.; Huang, C.-S.; Geyer, C.E., Jr.; von Minckwitz, G.; Wolmark, N.; Pivot, X.; Kuemmel, S.; DiGiovanna, M.P.; et al. Adjuvant T-DM1 versus Trastuzumab in Patients with Residual Invasive Disease after Neoadjuvant Therapy for HER2-Positive Breast Cancer: Subgroup Analyses from KATHERINE. Ann. Oncol. 2021, 32, 1005–1014. [Google Scholar] [CrossRef]
- Isakoff, S.J.; Baselga, J. Trastuzumab-DM1: Building a Chemotherapy-Free Road in the Treatment of Human Epidermal Growth Factor Receptor 2-Positive Breast Cancer. J. Clin. Oncol. 2011, 29, 351–354. [Google Scholar] [CrossRef]
- Peddi, P.F.; Hurvitz, S.A. Ado-Trastuzumab Emtansine (T-DM1) in Human Epidermal Growth Factor Receptor 2 (HER2)-Positive Metastatic Breast Cancer: Latest Evidence and Clinical Potential. Ther. Adv. Med. Oncol. 2014, 6, 202–209. [Google Scholar] [CrossRef]
- Perez, E.A.; Barrios, C.; Eiermann, W.; Toi, M.; Im, Y.-H.; Conte, P.; Martin, M.; Pienkowski, T.; Pivot, X.; Burris, H.A.; et al. Trastuzumab Emtansine With or Without Pertuzumab Versus Trastuzumab Plus Taxane for Human Epidermal Growth Factor Receptor 2–Positive, Advanced Breast Cancer: Primary Results From the Phase III MARIANNE Study. J. Clin. Oncol. 2017, 35, 141–148. [Google Scholar] [CrossRef]
- Krop, I.E.; Lin, N.U.; Blackwell, K.; Guardino, E.; Huober, J.; Lu, M.; Miles, D.; Samant, M.; Welslau, M.; Diéras, V. Trastuzumab Emtansine (T-DM1) versus Lapatinib plus Capecitabine in Patients with HER2-Positive Metastatic Breast Cancer and Central Nervous System Metastases: A Retrospective, Exploratory Analysis in EMILIA. Ann. Oncol. 2015, 26, 113–119. [Google Scholar] [CrossRef] [PubMed]
- Montemurro, F.; Ellis, P.; Delaloge, S.; Wuerstlein, R.; Anton, A.; Button, P.; Lindegger, N.; Barrios, C. Abstract P1-12-10: Safety and Efficacy of Trastuzumab Emtansine (T-DM1) in 399 Patients with Central Nervous System Metastases: Exploratory Subgroup Analysis from the KAMILLA Study. Cancer Res. 2017, 77, P1–P12. [Google Scholar] [CrossRef]
- Krop, I.E.; Kim, S.-B.; Martin, A.G.; LoRusso, P.M.; Ferrero, J.-M.; Badovinac-Crnjevic, T.; Hoersch, S.; Smitt, M.; Wildiers, H. Trastuzumab Emtansine versus Treatment of Physician’s Choice in Patients with Previously Treated HER2-Positive Metastatic Breast Cancer (TH3RESA): Final Overall Survival Results from a Randomised Open-Label Phase 3 Trial. Lancet Oncol. 2017, 18, 743–754. [Google Scholar] [CrossRef]
- Hunter, F.W.; Barker, H.R.; Lipert, B.; Rothé, F.; Gebhart, G.; Piccart-Gebhart, M.J.; Sotiriou, C.; Jamieson, S.M.F. Mechanisms of Resistance to Trastuzumab Emtansine (T-DM1) in HER2-Positive Breast Cancer. Br. J. Cancer 2020, 122, 603–612. [Google Scholar] [CrossRef] [PubMed]
- Krop, I.E.; LoRusso, P.; Miller, K.D.; Modi, S.; Yardley, D.; Rodriguez, G.; Guardino, E.; Lu, M.; Zheng, M.; Girish, S.; et al. A Phase II Study of Trastuzumab Emtansine in Patients with Human Epidermal Growth Factor Receptor 2-Positive Metastatic Breast Cancer Who Were Previously Treated with Trastuzumab, Lapatinib, an Anthracycline, a Taxane, and Capecitabine. J. Clin. Oncol. 2012, 30, 3234–3241. [Google Scholar] [CrossRef] [PubMed]
- Ogitani, Y.; Aida, T.; Hagihara, K.; Yamaguchi, J.; Ishii, C.; Harada, N.; Soma, M.; Okamoto, H.; Oitate, M.; Arakawa, S.; et al. DS-8201a, A Novel HER2-Targeting ADC with a Novel DNA Topoisomerase I Inhibitor, Demonstrates a Promising Antitumor Efficacy with Differentiation from T-DM1. Clin. Cancer Res. 2016, 22, 5097–5108. [Google Scholar] [CrossRef]
- Ogitani, Y.; Hagihara, K.; Oitate, M.; Naito, H.; Agatsuma, T. Bystander Killing Effect of DS-8201a, a Novel Anti-Human Epidermal Growth Factor Receptor 2 Antibody-Drug Conjugate, in Tumors with Human Epidermal Growth Factor Receptor 2 Heterogeneity. Cancer Sci. 2016, 107, 1039–1046. [Google Scholar] [CrossRef]
- Doi, T.; Shitara, K.; Naito, Y.; Shimomura, A.; Fujiwara, Y.; Yonemori, K.; Shimizu, C.; Shimoi, T.; Kuboki, Y.; Matsubara, N.; et al. Safety, Pharmacokinetics, and Antitumour Activity of Trastuzumab Deruxtecan (DS-8201), a HER2-Targeting Antibody-Drug Conjugate, in Patients with Advanced Breast and Gastric or Gastro-Oesophageal Tumours: A Phase 1 Dose-Escalation Study. Lancet Oncol. 2017, 18, 1512–1522. [Google Scholar] [CrossRef]
- Iwata, H.; Tamura, K.; Doi, T.; Tsurutani, J.; Modi, S.; Park, H.; Krop, I.E.; Sagara, Y.; Redfern, C.H.; Murthy, R.K.; et al. Trastuzumab Deruxtecan (DS-8201a) in Subjects with HER2-Expressing Solid Tumors: Long-Term Results of a Large Phase 1 Study with Multiple Expansion Cohorts. J. Clin. Oncol. 2018, 36, 2501. [Google Scholar] [CrossRef]
- Tamura, K.; Tsurutani, J.; Takahashi, S.; Iwata, H.; Krop, I.E.; Redfern, C.; Sagara, Y.; Doi, T.; Park, H.; Murthy, R.K.; et al. Trastuzumab Deruxtecan (DS-8201a) in Patients with Advanced HER2-Positive Breast Cancer Previously Treated with Trastuzumab Emtansine: A Dose-Expansion, Phase 1 Study. Lancet Oncol. 2019, 20, 816–826. [Google Scholar] [CrossRef]
- Modi, S.; Saura, C.; Yamashita, T.; Park, Y.H.; Kim, S.-B.; Tamura, K.; Andre, F.; Iwata, H.; Ito, Y.; Tsurutani, J.; et al. Abstract PD3-06: Updated Results from DESTINY-breast01, a Phase 2 Trial of Trastuzumab Deruxtecan (T-DXd) in HER2 Positive Metastatic Breast Cancer. Cancer Res. 2021, 81, PD3–PD06. [Google Scholar] [CrossRef]
- Cortés, J.; Kim, S.-B.; Chung, W.-P.; Im, S.-A.; Park, Y.H.; Hegg, R.; Kim, M.H.; Tseng, L.-M.; Petry, V.; Chung, C.-F.; et al. Trastuzumab Deruxtecan versus Trastuzumab Emtansine for Breast Cancer. N. Engl. J. Med. 2022, 386, 1143–1154. [Google Scholar] [CrossRef] [PubMed]
- Hurvitz, S.A.; Hegg, R.; Chung, W.-P.; Im, S.-A.; Jacot, W.; Ganju, V.; Chiu, J.W.Y.; Xu, B.; Hamilton, E.; Madhusudan, S.; et al. Trastuzumab Deruxtecan versus Trastuzumab Emtansine in Patients with HER2-Positive Metastatic Breast Cancer: Updated Results from DESTINY-Breast03, a Randomised, Open-Label, Phase 3 Trial. Lancet 2023, 401, 105–117. [Google Scholar] [CrossRef]
- Modi, S.; Tsurutani, J.; Tamura, K.; Park, H.; Sagara, Y.; Murthy, R.; Iwata, H.; Krop, I.E.; Doi, T.; Redfern, C.; et al. Abstract P6-17-02: Trastuzumab Deruxtecan (DS-8201a) in Subjects with HER2-Low Expressing Breast Cancer: Updated Results of a Large Phase 1 Study. Cancer Res. 2019, 79, P6–P17. [Google Scholar] [CrossRef]
- Modi, S.; Park, H.; Murthy, R.K.; Iwata, H.; Tamura, K.; Tsurutani, J.; Moreno-Aspitia, A.; Doi, T.; Sagara, Y.; Redfern, C.; et al. Antitumor Activity and Safety of Trastuzumab Deruxtecan in Patients with HER2-Low–Expressing Advanced Breast Cancer: Results from a Phase Ib Study. J. Clin. Orthod. 2020, 38, 1887–1896. [Google Scholar] [CrossRef]
- Modi, S.; Jacot, W.; Yamashita, T.; Sohn, J.; Vidal, M.; Tokunaga, E.; Tsurutani, J.; Ueno, N.T.; Prat, A.; Chae, Y.S.; et al. Trastuzumab Deruxtecan in Previously Treated HER2-Low Advanced Breast Cancer. N. Engl. J. Med. 2022, 387, 9–20. [Google Scholar] [CrossRef] [PubMed]
- Dokter, W.; Ubink, R.; van der Lee, M.; van der Vleuten, M.; van Achterberg, T.; Jacobs, D.; Loosveld, E.; van den Dobbelsteen, D.; Egging, D.; Mattaar, E.; et al. Preclinical Profile of the HER2-Targeting ADC SYD983/SYD985: Introduction of a New Duocarmycin-Based Linker-Drug Platform. Mol. Cancer Ther. 2014, 13, 2618–2629. [Google Scholar] [CrossRef] [PubMed]
- Van Herpen, C.M.; Banerji, U.; Mommers, E.C.; Koper, N.P.; Goedings, P.; Lopez, J.; Awada, A.; Fiebrich, H.B.; Aftimos, P.G. 333 Phase I Dose-Escalation Trial with the DNA-Alkylating Anti-HER2 Antibody-Drug Conjugate SYD985. Eur. J. Cancer 2015, 51, S65. [Google Scholar] [CrossRef]
- Manich, C.S.; Saura Manich, C.; O’Shaughnessy, J.; Aftimos, P.G.; van den Tweel, E.; Oesterholt, M.; Escrivá-de-Romaní, S.I.; Quenel Tueux, N.; Tan, T.J.; Lim, J.S.; et al. LBA15 Primary Outcome of the Phase III SYD985.002/TULIP Trial Comparing [vic-]trastuzumab Duocarmazine to Physician’s Choice Treatment in Patients with Pre-Treated HER2-Positive Locally Advanced or Metastatic Breast Cancer. Ann. Oncol. 2021, 32, S1288. [Google Scholar] [CrossRef]
- Skidmore, L.; Sakamuri, S.; Knudsen, N.A.; Hewet, A.G.; Milutinovic, S.; Barkho, W.; Biroc, S.L.; Kirtley, J.; Marsden, R.; Storey, K.; et al. ARX788, a Site-Specific Anti-HER2 Antibody-Drug Conjugate, Demonstrates Potent and Selective Activity in HER2-Low and T-DM1-Resistant Breast and Gastric Cancers. Mol. Cancer Ther. 2020, 19, 1833–1843. [Google Scholar] [CrossRef]
- Abdollahpour-Alitappeh, M.; Lotfinia, M.; Gharibi, T.; Mardaneh, J.; Farhadihosseinabadi, B.; Larki, P.; Faghfourian, B.; Sepehr, K.S.; Abbaszadeh-Goudarzi, K.; Abbaszadeh-Goudarzi, G.; et al. Antibody-Drug Conjugates (ADCs) for Cancer Therapy: Strategies, Challenges, and Successes. J. Cell. Physiol. 2019, 234, 5628–5642. [Google Scholar] [CrossRef]
- Barok, M.; Le Joncour, V.; Martins, A.; Isola, J.; Salmikangas, M.; Laakkonen, P.; Joensuu, H. ARX788, a Novel Anti-HER2 Antibody-Drug Conjugate, Shows Anti-Tumor Effects in Preclinical Models of Trastuzumab Emtansine-Resistant HER2-Positive Breast Cancer and Gastric Cancer. Cancer Lett. 2020, 473, 156–163. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Ji, D.; Shen, W.; Xiao, Q.; Gu, Y.; O’Shaughnessy, J.; Hu, X. Phase I Trial of a Novel Anti-HER2 Antibody–Drug Conjugate, ARX788, for the Treatment of HER2-Positive Metastatic Breast Cancer. Clin. Cancer Res. 2022, 28, 4212–4221. [Google Scholar] [CrossRef] [PubMed]
- Ansell, S.M. Brentuximab Vedotin. Blood 2014, 124, 3197–3200. [Google Scholar] [CrossRef] [PubMed]
- Chari, R.V.J.; Miller, M.L.; Widdison, W.C. Antibody-Drug Conjugates: An Emerging Concept in Cancer Therapy. Angew. Chem. Int. Ed 2014, 53, 3796–3827. [Google Scholar] [CrossRef] [PubMed]
- Shi, F.; Liu, Y.; Zhou, X.; Shen, P.; Xue, R.; Zhang, M. Disitamab Vedotin: A Novel Antibody-Drug Conjugates for Cancer Therapy. Drug Deliv. 2022, 29, 1335–1344. [Google Scholar] [CrossRef] [PubMed]
- Deeks, E.D. Disitamab Vedotin: First Approval. Drugs 2021, 81, 1929–1935. [Google Scholar] [CrossRef]
- Yao, X.; Jiang, J.; Wang, X.; Huang, C.; Li, D.; Xie, K.; Xu, Q.; Li, H.; Li, Z.; Lou, L.; et al. A Novel Humanized Anti-HER2 Antibody Conjugated with MMAE Exerts Potent Anti-Tumor Activity. Breast Cancer Res. Treat. 2015, 153, 123–133. [Google Scholar] [CrossRef]
- Xu, Y.; Wang, Y.; Gong, J.; Zhang, X.; Peng, Z.; Sheng, X.; Mao, C.; Fan, Q.; Bai, Y.; Ba, Y.; et al. Phase I Study of the Recombinant Humanized Anti-HER2 Monoclonal antibody–MMAE Conjugate RC48-ADC in Patients with HER2-Positive Advanced Solid Tumors. Gastric Cancer 2021, 24, 913–925. [Google Scholar] [CrossRef]
- Wang, J.; Liu, Y.; Zhang, Q.; Feng, J.; Fang, J.; Chen, X.; Han, Y.; Li, Q.; Zhang, P.; Yuan, P.; et al. RC48-ADC, a HER2-Targeting Antibody-Drug Conjugate, in Patients with HER2-Positive and HER2-Low Expressing Advanced or Metastatic Breast Cancer: A Pooled Analysis of Two Studies. J. Clin. Oncol. 2021, 39, 1022. [Google Scholar] [CrossRef]
- Xu, B.; Wang, J.; Fang, J.; Chen, X.; Han, Y.; Li, Q.; Zhang, P.; Yuan, P.; Ma, F.; Luo, Y.; et al. Abstract PD4-06: Early Clinical Development of RC48-ADC in Patients with HER2 Positive Metastatic Breast Cancer. Cancer Res. 2020, 80, PD4–PD06. [Google Scholar] [CrossRef]
- Peng, Z.; Liu, T.; Wei, J.; Wang, A.; He, Y.; Yang, L.; Zhang, X.; Fan, N.; Luo, S.; Li, Z.; et al. Efficacy and Safety of a Novel anti-HER2 Therapeutic Antibody RC48 in Patients with HER2-overexpressing, Locally Advanced or Metastatic Gastric or Gastroesophageal Junction Cancer: A Single-arm Phase II Study. Cancer Commun. 2021, 41, 1173–1182. [Google Scholar] [CrossRef] [PubMed]
- Hu, X.; Zhang, J.; Liu, R.; Gao, S.; Qing, Y.; Yi, S.; Yuan, J.; Chen, H.; Fan, B.; Zheng, H.; et al. Phase I Study of A166 in Patients with HER2-Expressing Locally Advanced or Metastatic Solid Tumors. J. Clin. Oncol. 2021, 39, 1024. [Google Scholar] [CrossRef]
- Hu, X.; Zhang, J.; Liu, R.; Gao, S.; Wu, J.; Wang, Y.; Hao, Y.; Ge, J.; Qing, Y.; Yi, S.; et al. Updated Results and Biomarker Analyses from the Phase I Trial of A166 in Patients with HER2-Expressing Locally Advanced or Metastatic Solid Tumors. J. Clin. Oncol. 2022, 40, 1037. [Google Scholar] [CrossRef]
- Liu, Y.; Lian, W.; Zhao, X.; Qi, W.; Xu, J.; Xiao, L.; Qing, Y.; Xue, T.; Wang, J. A First in-Human Study of A166 in Patients with Locally Advanced/metastatic Solid Tumors Which Are HER2-Positive or HER2-Amplified Who Did Not Respond or Stopped Responding to Approved Therapies. J. Clin. Oncol. 2020, 38, 1049. [Google Scholar] [CrossRef]
- Li, H.; Zhang, X.; Xu, Z.; Li, L.; Liu, W.; Dai, Z.; Zhao, Z.; Xiao, L.; Li, H.; Hu, C. Preclinical Evaluation of MRG002, a Novel HER2-Targeting Antibody-Drug Conjugate with Potent Antitumor Activity against HER2-Positive Solid Tumors. Antib. Ther. 2021, 4, 175–184. [Google Scholar] [CrossRef]
- Hamblett, K.J.; Barnscher, S.D.; Davies, R.H.; Hammond, P.W.; Hernandez, A.; Wickman, G.R.; Fung, V.K.; Ding, T.; Garnett, G.; Galey, A.S.; et al. Abstract P6-17-13: ZW49, a HER2 Targeted Biparatopic Antibody Drug Conjugate for the Treatment of HER2 Expressing Cancers. Cancer Res. 2019, 79, P6–P17. [Google Scholar] [CrossRef]
- Dumbrava, E.I.; Sharma, M.R.; Carvajal, R.D.; Catenacci, D.; Emens, L.A.; Gadgeel, S.M.; Hanna, G.J.; Juric, D.; Kang, Y.-K.; Lee, J.; et al. Abstract OT-03-02: Phase 1/2 Study of a Novel HER2 Targeting TLR7/8 Immune-Stimulating Antibody Conjugate (ISAC), BDC-1001, as a Single Agent and in Combination with an Immune Checkpoint Inhibitor in Patients with Advanced HER2-Expressing Solid Tumors. Cancer Res. 2021, 81, OT-03. [Google Scholar] [CrossRef]
- Rinnerthaler, G.; Gampenrieder, S.P.; Greil, R. HER2 Directed Antibody-Drug-Conjugates beyond T-DM1 in Breast Cancer. Int. J. Mol. Sci. 2019, 20, 1115. [Google Scholar] [CrossRef]
- Park, Y.H.; Ahn, H.K.; Kim, J.-Y.; Ahn, J.S.; Im, Y.-H.; Kim, S.-H.; Lee, S.; Chung, H.-S.; Park, S.J. First-in-Human Phase I Study of ALT-P7, a HER2-Targeting Antibody-Drug Conjugate in Patients with HER2-Positive Advanced Breast Cancer. J. Clin. Oncol. 2020, 38, 3551. [Google Scholar] [CrossRef]
- Yurkovetskiy, A.; Gumerov, D.; Ter-Ovanesyan, E.; Conlon, P.; Devit, M.; Bu, C.; Bodyak, N.; Lowinger, T.; Bergstrom, D. Abstract 48: Non-Clinical Pharmacokinetics of XMT-1522, a HER2 Targeting Auristatin-Based Antibody Drug Conjugate. Cancer Res. 2017, 77, 48. [Google Scholar] [CrossRef]
- Le Joncour, V.; Martins, A.; Puhka, M.; Isola, J.; Salmikangas, M.; Laakkonen, P.; Joensuu, H.; Barok, M. A Novel Anti-HER2 Antibody-Drug Conjugate XMT-1522 for HER2-Positive Breast and Gastric Cancers Resistant to Trastuzumab Emtansine. Mol. Cancer Ther. 2019, 18, 1721–1730. [Google Scholar] [CrossRef] [PubMed]
- Hamilton, E.P.; Barve, M.A.; Bardia, A.; Beeram, M.; Bendell, J.C.; Mosher, R.; Hailman, E.; Bergstrom, D.A.; Burris, H.A.; Soliman, H.H. Phase 1 Dose Escalation of XMT-1522, a Novel HER2-Targeting Antibody-Drug Conjugate (ADC), in Patients (pts) with HER2-Expressing Breast, Lung and Gastric Tumors. J. Clin. Oncol. 2018, 36, 2546. [Google Scholar] [CrossRef]
- Sung, M.S.; Hopf, C.; Upeslacis, E.; Golas, J.; Kaplan, M.; Khandke, K.; Charati, M.; Kotch, F.; Loganzo, F.; Geles, K.; et al. Abstract 818: NG-HER2 ADC (PF-06804103) Is Superior to Trastuzumab Emtansine in a Mouse “Avatar” Head-to-Head Clinical Trial. Cancer Res. 2018, 78, 818. [Google Scholar] [CrossRef]
- Graziani, E.I.; Sung, M.; Ma, D.; Narayanan, B.; Marquette, K.; Puthenveetil, S.; Tumey, L.N.; Bikker, J.; Casavant, J.; Bennett, E.M.; et al. PF-06804103, A Site-Specific Anti-HER2 Antibody-Drug Conjugate for the Treatment of HER2-Expressing Breast, Gastric, and Lung Cancers. Mol. Cancer Ther. 2020, 19, 2068–2078. [Google Scholar] [CrossRef]
- Meric-Bernstam, F.; Calvo, E.; Moreno, V.; Chung, H.C.; Park, Y.H.; Bang, Y.-J.; Rosen, L.S.; Mita, M.M.; Garrido-Laguna, I.; Leung, A.C.F.; et al. A Phase I Dose Escalation Study Evaluating the Safety and Tolerability of a Novel Anti-HER2 Antibody-Drug Conjugate (PF-06804103) in Patients with HER2-Positive Solid Tumors. J. Clin. Oncol. 2020, 38, 1039. [Google Scholar] [CrossRef]
- Kang, J.C.; Sun, W.; Khare, P.; Karimi, M.; Wang, X.; Shen, Y.; Ober, R.J.; Ward, E.S. Engineering a HER2-Specific Antibody-Drug Conjugate to Increase Lysosomal Delivery and Therapeutic Efficacy. Nat. Biotechnol. 2019, 37, 523–526. [Google Scholar] [CrossRef]
- Hagemann, U.B.; Ellingsen, C.; Schuhmacher, J.; Kristian, A.; Mobergslien, A.; Cruciani, V.; Wickstroem, K.; Schatz, C.A.; Kneip, C.; Golfier, S.; et al. Mesothelin-Targeted Thorium-227 Conjugate (MSLN-TTC): Preclinical Evaluation of a New Targeted Alpha Therapy for Mesothelin-Positive Cancers. Clin. Cancer Res. 2019, 25, 4723–4734. [Google Scholar] [CrossRef]
- Hagemann, U.B.; Wickstroem, K.; Hammer, S.; Bjerke, R.M.; Zitzmann-Kolbe, S.; Ryan, O.B.; Karlsson, J.; Scholz, A.; Hennekes, H.; Mumberg, D.; et al. Advances in Precision Oncology: Targeted Thorium-227 Conjugates As a New Modality in Targeted Alpha Therapy. Cancer Biother. Radiopharm. 2020, 35, 497–510. [Google Scholar] [CrossRef]
- Schroeder, R.L.; Stevens, C.L.; Sridhar, J. Small Molecule Tyrosine Kinase Inhibitors of ErbB2/HER2/Neu in the Treatment of Aggressive Breast Cancer. Molecules 2014, 19, 15196–15212. [Google Scholar] [CrossRef]
- Ríos-Luci, C.; Díaz-Rodríguez, E.; Gandullo-Sánchez, L.; Díaz-Gil, L.; Ocaña, A.; Pandiella, A. Adaptive Resistance to Trastuzumab Impairs Response to Neratinib and Lapatinib through Deregulation of Cell Death Mechanisms. Cancer Lett. 2020, 470, 161–169. [Google Scholar] [CrossRef]
- Scaltriti, M.; Verma, C.; Guzman, M.; Jimenez, J.; Parra, J.L.; Pedersen, K.; Smith, D.J.; Landolfi, S.; Ramon y Cajal, S.; Arribas, J.; et al. Lapatinib, a HER2 Tyrosine Kinase Inhibitor, Induces Stabilization and Accumulation of HER2 and Potentiates Trastuzumab-Dependent Cell Cytotoxicity. Oncogene 2009, 28, 803–814. [Google Scholar] [CrossRef] [PubMed]
- Xia, W.; Mullin, R.J.; Keith, B.R.; Liu, L.-H.; Ma, H.; Rusnak, D.W.; Owens, G.; Alligood, K.J.; Spector, N.L. Anti-Tumor Activity of GW572016: A Dual Tyrosine Kinase Inhibitor Blocks EGF Activation of EGFR/erbB2 and Downstream Erk1/2 and AKT Pathways. Oncogene 2002, 21, 6255–6263. [Google Scholar] [CrossRef] [PubMed]
- Baselga, J.; Bradbury, I.; Eidtmann, H.; Di Cosimo, S.; de Azambuja, E.; Aura, C.; Gómez, H.; Dinh, P.; Fauria, K.; Van Dooren, V.; et al. Lapatinib with Trastuzumab for HER2-Positive Early Breast Cancer (NeoALTTO): A Randomised, Open-Label, Multicentre, Phase 3 Trial. Lancet 2012, 379, 633–640. [Google Scholar] [CrossRef] [PubMed]
- de Azambuja, E.; Holmes, A.P.; Piccart-Gebhart, M.; Holmes, E.; Di Cosimo, S.; Swaby, R.F.; Untch, M.; Jackisch, C.; Lang, I.; Smith, I.; et al. Lapatinib with Trastuzumab for HER2-Positive Early Breast Cancer (NeoALTTO): Survival Outcomes of a Randomised, Open-Label, Multicentre, Phase 3 Trial and Their Association with Pathological Complete Response. Lancet Oncol. 2014, 15, 1137–1146. [Google Scholar] [CrossRef]
- Goss, P.E.; Smith, I.E.; O’Shaughnessy, J.; Ejlertsen, B.; Kaufmann, M.; Boyle, F.; Buzdar, A.U.; Fumoleau, P.; Gradishar, W.; Martin, M.; et al. Adjuvant Lapatinib for Women with Early-Stage HER2-Positive Breast Cancer: A Randomised, Controlled, Phase 3 Trial. Lancet Oncol. 2013, 14, 88–96. [Google Scholar] [CrossRef]
- Piccart-Gebhart, M.; Holmes, E.; Baselga, J.; de Azambuja, E.; Dueck, A.C.; Viale, G.; Zujewski, J.A.; Goldhirsch, A.; Armour, A.; Pritchard, K.I.; et al. Adjuvant Lapatinib and Trastuzumab for Early Human Epidermal Growth Factor Receptor 2–Positive Breast Cancer: Results From the Randomized Phase III Adjuvant Lapatinib And/or Trastuzumab Treatment Optimization Trial. J. Clin. Oncol. 2016, 34, 1034–1042. [Google Scholar] [CrossRef]
- Chen, Z.-L.; Shen, Y.; Li, S.; Lv, M.; Yang, J.; Zhang, L.-X.; Li, C.-L.; Lin, Y.-Y.; Yang, J.; Wang, X. The Efficiency and Safety of Trastuzumab and Lapatinib Added to Neoadjuvant Chemotherapy in Her2-Positive Breast Cancer Patients: A Randomized Meta-Analysis. OncoTargets Ther. 2016, 9, 3233–3247. [Google Scholar] [CrossRef]
- Lin, N.U.; Diéras, V.; Paul, D.; Lossignol, D.; Christodoulou, C.; Stemmler, H.-J.; Roché, H.; Liu, M.C.; Greil, R.; Ciruelos, E.; et al. Multicenter Phase II Study of Lapatinib in Patients with Brain Metastases from HER2-Positive Breast Cancer. Clin. Cancer Res. 2009, 15, 1452–1459. [Google Scholar] [CrossRef]
- Bachelot, T.; Romieu, G.; Campone, M.; Diéras, V.; Cropet, C.; Dalenc, F.; Jimenez, M.; Le Rhun, E.; Pierga, J.-Y.; Gonçalves, A.; et al. Lapatinib plus Capecitabine in Patients with Previously Untreated Brain Metastases from HER2-Positive Metastatic Breast Cancer (LANDSCAPE): A Single-Group Phase 2 Study. Lancet Oncol. 2013, 14, 64–71. [Google Scholar] [CrossRef]
- Pivot, X.; Manikhas, A.; Żurawski, B.; Chmielowska, E.; Karaszewska, B.; Allerton, R.; Chan, S.; Fabi, A.; Bidoli, P.; Gori, S.; et al. CEREBEL (EGF111438): A Phase III, Randomized, Open-Label Study of Lapatinib Plus Capecitabine Versus Trastuzumab Plus Capecitabine in Patients With Human Epidermal Growth Factor Receptor 2-Positive Metastatic Breast Cancer. J. Clin. Oncol. 2015, 33, 1564–1573. [Google Scholar] [CrossRef] [PubMed]
- Khan, M.; Zhao, Z.; Arooj, S.; Zheng, T.; Liao, G. Lapatinib Plus Local Radiation Therapy for Brain Metastases From HER-2 Positive Breast Cancer Patients and Role of Trastuzumab: A Systematic Review and Meta-Analysis. Front. Oncol. 2020, 10, 576926. [Google Scholar] [CrossRef] [PubMed]
- Blackwell, K.L.; Burstein, H.J.; Storniolo, A.M.; Rugo, H.; Sledge, G.; Koehler, M.; Ellis, C.; Casey, M.; Vukelja, S.; Bischoff, J.; et al. Randomized Study of Lapatinib Alone or in Combination With Trastuzumab in Women With ErbB2-Positive, Trastuzumab-Refractory Metastatic Breast Cancer. J. Clin. Oncol. 2010, 28, 1124–1130. [Google Scholar] [CrossRef]
- Collins, D.M.; Conlon, N.T.; Kannan, S.; Verma, C.S.; Eli, L.D.; Lalani, A.S.; Crown, J. Preclinical Characteristics of the Irreversible Pan-HER Kinase Inhibitor Neratinib Compared with Lapatinib: Implications for the Treatment of HER2-Positive and HER2-Mutated Breast Cancer. Cancers 2019, 11, 737. [Google Scholar] [CrossRef] [PubMed]
- Collins, D.M.; Gately, K.; Hughes, C.; Edwards, C.; Davies, A.; Madden, S.F.; O’Byrne, K.J.; O’Donovan, N.; Crown, J. Tyrosine Kinase Inhibitors as Modulators of Trastuzumab-Mediated Antibody-Dependent Cell-Mediated Cytotoxicity in Breast Cancer Cell Lines. Cell. Immunol. 2017, 319, 35–42. [Google Scholar] [CrossRef]
- Dai, M.S.; Feng, Y.H.; Chen, S.W.; Masuda, N.; Yau, T.; Chen, S.T.; Lu, Y.S.; Yap, Y.S.; Ang, P.C.S.; Chu, S.C.; et al. Analysis of the Pan-Asian Subgroup of Patients in the NALA Trial: A Randomized Phase III NALA Trial Comparing Neratinib+capecitabine (N+C) vs Lapatinib+capecitabine (L+C) in Patients with HER2+metastatic Breast Cancer (mBC) Previously Treated with Two or More HER2-Directed Regimens. Breast Cancer Res. Treat. 2021, 189, 665–676. [Google Scholar] [CrossRef]
- Segovia-Mendoza, M.; González-González, M.E.; Barrera, D.; Díaz, L.; García-Becerra, R. Efficacy and Mechanism of Action of the Tyrosine Kinase Inhibitors Gefitinib, Lapatinib and Neratinib in the Treatment of HER2-Positive Breast Cancer: Preclinical and Clinical Evidence. Am. J. Cancer Res. 2015, 5, 2531–2561. [Google Scholar] [PubMed]
- Rabindran, S.K.; Discafani, C.M.; Rosfjord, E.C.; Baxter, M.; Brawner Floyd, M.; Golas, J.; Hallett, W.A.; Johnson, B.D.; Nilakantan, R.; Overbeek, E.; et al. Antitumor Activity of HKI-272, an Orally Active, Irreversible Inhibitor of the HER-2 Tyrosine Kinase. Cancer Res. 2004, 64, 3958–3965. [Google Scholar] [CrossRef]
- Chow, L.W.-C.; Xu, B.; Gupta, S.; Freyman, A.; Zhao, Y.; Abbas, R.; Vo Van, M.-L.; Bondarenko, I. Combination Neratinib (HKI-272) and Paclitaxel Therapy in Patients with HER2-Positive Metastatic Breast Cancer. Br. J. Cancer 2013, 108, 1985–1993. [Google Scholar] [CrossRef]
- Chan, A.; Delaloge, S.; Holmes, F.A.; Moy, B.; Iwata, H.; Harvey, V.J.; Robert, N.J.; Silovski, T.; Gokmen, E.; von Minckwitz, G.; et al. Neratinib after Trastuzumab-Based Adjuvant Therapy in Patients with HER2-Positive Breast Cancer (ExteNET): A Multicentre, Randomised, Double-Blind, Placebo-Controlled, Phase 3 Trial. Lancet Oncol. 2016, 17, 367–377. [Google Scholar] [CrossRef]
- Chan, A.; Moy, B.; Mansi, J.; Ejlertsen, B.; Holmes, F.A.; Chia, S.; Iwata, H.; Gnant, M.; Loibl, S.; Barrios, C.H.; et al. Final Efficacy Results of Neratinib in HER2-Positive Hormone Receptor-Positive Early-Stage Breast Cancer From the Phase III ExteNET Trial. Clin. Breast Cancer 2021, 21, 80–91.e7. [Google Scholar] [CrossRef]
- Barcenas, C.H.; Hurvitz, S.A.; Di Palma, J.A.; Bose, R.; Chien, A.J.; Iannotti, N.; Marx, G.; Brufsky, A.; Litvak, A.; Ibrahim, E.; et al. Improved Tolerability of Neratinib in Patients with HER2-Positive Early-Stage Breast Cancer: The CONTROL Trial. Ann. Oncol. 2020, 31, 1223–1230. [Google Scholar] [CrossRef]
- Martin, M.; Holmes, F.A.; Ejlertsen, B.; Delaloge, S.; Moy, B.; Iwata, H.; von Minckwitz, G.; Chia, S.K.L.; Mansi, J.; Barrios, C.H.; et al. Neratinib after Trastuzumab-Based Adjuvant Therapy in HER2-Positive Breast Cancer (ExteNET): 5-Year Analysis of a Randomised, Double-Blind, Placebo-Controlled, Phase 3 Trial. Lancet Oncol. 2017, 18, 1688–1700. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Wang, L.; Yu, Q.; Liu, Z.; Li, C.; Wang, F.; Yu, Z. The Effectiveness of Lapatinib in HER2-Positive Metastatic Breast Cancer Patients Pretreated With Multiline Anti-HER2 Treatment: A Retrospective Study in China. Technol. Cancer Res. Treat. 2021, 20, 15330338211037812. [Google Scholar] [CrossRef] [PubMed]
- Abraham, J.; Puhalla, S.; Sikov, W.M.; Montero, A.J.; Salkeni, M.A.; Razaq, W.; Beumer, J.H.; Kiesel, B.; Buyse, M.E.; Adamson, L.M.; et al. NSABP FB-10: Phase Ib Dose-Escalation Trial Evaluating Trastuzumab Emtansine (T-DM1) with Neratinib (N) in Women with Metastatic HER2 Breast Cancer (MBC). J. Clin. Oncol. 2018, 36, 1027. [Google Scholar] [CrossRef]
- Jhaveri, K.L.; Goldman, J.W.; Hurvitz, S.A.; Guerrero-Zotano, A.; Unni, N.; Brufsky, A.; Park, H.; Waisman, J.R.; Yang, E.S.-H.; Spanggaard, I.; et al. Neratinib plus Fulvestrant plus Trastzuzumab (N F T) for Hormone Receptor-Positive (HR), HER2-Negative, HER2-Mutant Metastatic Breast Cancer (MBC): Outcomes and Biomarker Analysis from the SUMMIT Trial. J. Clin. Oncol. 2022, 40, 1028. [Google Scholar] [CrossRef]
- Smyth, L.M.; Saura, C.; Piha-Paul, S.A.; Lu, J.; Mayer, I.A.; Brufksy, A.M.; Spanggaard, I.; Arnedos, M.; Cutler, R.E.; Hyman, D.M. Update on the Phase II SUMMIT Trial: Neratinib Fulvestrant for HER2-Mutant, HR-Positive, Metastatic Breast Cancer. Ann. Oncol. 2019, 30, iii10–iii11. [Google Scholar] [CrossRef]
- Awada, A.; Colomer, R.; Inoue, K.; Bondarenko, I.; Badwe, R.A.; Demetriou, G.; Lee, S.-C.; Mehta, A.O.; Kim, S.-B.; Bachelot, T.; et al. Neratinib Plus Paclitaxel vs Trastuzumab Plus Paclitaxel in Previously Untreated Metastatic ERBB2-Positive Breast Cancer: The NEfERT-T Randomized Clinical Trial. JAMA Oncol. 2016, 2, 1557–1564. [Google Scholar] [CrossRef]
- Saura, C.; Oliveira, M.; Feng, Y.-H.; Dai, M.-S.; Chen, S.-W.; Hurvitz, S.A.; Kim, S.-B.; Moy, B.; Delaloge, S.; Gradishar, W.; et al. Neratinib Plus Capecitabine Versus Lapatinib Plus Capecitabine in HER2-Positive Metastatic Breast Cancer Previously Treated With ≥ 2 HER2-Directed Regimens: Phase III NALA Trial. J. Clin. Oncol. 2020, 38, 3138–3149. [Google Scholar] [CrossRef] [PubMed]
- Freedman, R.A.; Gelman, R.S.; Anders, C.K.; Melisko, M.E.; Parsons, H.A.; Cropp, A.M.; Silvestri, K.; Cotter, C.M.; Componeschi, K.P.; Marte, J.M.; et al. TBCRC 022: A Phase II Trial of Neratinib and Capecitabine for Patients With Human Epidermal Growth Factor Receptor 2-Positive Breast Cancer and Brain Metastases. J. Clin. Oncol. 2019, 37, 1081–1089. [Google Scholar] [CrossRef]
- Saura, C.; Garcia-Saenz, J.A.; Xu, B.; Harb, W.; Moroose, R.; Pluard, T.; Cortés, J.; Kiger, C.; Germa, C.; Wang, K.; et al. Safety and Efficacy of Neratinib in Combination With Capecitabine in Patients With Metastatic Human Epidermal Growth Factor Receptor 2–Positive Breast Cancer. J. Clin. Oncol. 2014, 32, 3626–3633. [Google Scholar] [CrossRef] [PubMed]
- Kulukian, A.; Lee, P.; Taylor, J.; Rosler, R.; de Vries, P.; Watson, D.; Forero-Torres, A.; Peterson, S. Preclinical Activity of HER2-Selective Tyrosine Kinase Inhibitor Tucatinib as a Single Agent or in Combination with Trastuzumab or Docetaxel in Solid Tumor Models. Mol. Cancer Ther. 2020, 19, 976–987. [Google Scholar] [CrossRef]
- O’Brien, N.A.; Huang, H.K.T.; McDermott, M.S.J.; Madrid, A.M.; Luo, T.; Ayala, R.; Issakhanian, S.; Gong, K.W.; Lu, M.; Zhang, J.; et al. Tucatinib Has Selective Activity in HER2-Positive Cancers and Significant Combined Activity with Approved and Novel Breast Cancer–Targeted Therapies. Mol. Cancer Ther. 2022, 21, 751–761. [Google Scholar] [CrossRef]
- Murthy, R.; Borges, V.F.; Conlin, A.; Chaves, J.; Chamberlain, M.; Gray, T.; Vo, A.; Hamilton, E. Tucatinib with Capecitabine and Trastuzumab in Advanced HER2-Positive Metastatic Breast Cancer with and without Brain Metastases: A Non-Randomised, Open-Label, Phase 1b Study. Lancet Oncol. 2018, 19, 880–888. [Google Scholar] [CrossRef]
- Curigliano, G.; Mueller, V.; Borges, V.; Hamilton, E.; Hurvitz, S.; Loi, S.; Murthy, R.; Okines, A.; Paplomata, E.; Cameron, D.; et al. Tucatinib versus Placebo Added to Trastuzumab and Capecitabine for Patients with Pretreated HER2+ Metastatic Breast Cancer with and without Brain Metastases (HER2CLIMB): Final Overall Survival Analysis. Ann. Oncol. 2022, 33, 321–329. [Google Scholar] [CrossRef]
- Murthy, R.K.; Loi, S.; Okines, A.; Paplomata, E.; Hamilton, E.; Hurvitz, S.A.; Lin, N.U.; Borges, V.; Abramson, V.; Anders, C.; et al. Tucatinib, Trastuzumab, and Capecitabine for HER2-Positive Metastatic Breast Cancer. N. Engl. J. Med. 2020, 382, 597–609. [Google Scholar] [CrossRef] [PubMed]
- Lin, N.U.; Borges, V.; Anders, C.; Murthy, R.K.; Paplomata, E.; Hamilton, E.; Hurvitz, S.; Loi, S.; Okines, A.; Abramson, V.; et al. Intracranial Efficacy and Survival With Tucatinib Plus Trastuzumab and Capecitabine for Previously Treated HER2-Positive Breast Cancer With Brain Metastases in the HER2CLIMB Trial. J. Clin. Oncol. 2020, 38, 2610–2619. [Google Scholar] [CrossRef] [PubMed]
- Ma, F.; Li, Q.; Chen, S.; Zhu, W.; Fan, Y.; Wang, J.; Luo, Y.; Xing, P.; Lan, B.; Li, M.; et al. Phase I Study and Biomarker Analysis of Pyrotinib, a Novel Irreversible Pan-ErbB Receptor Tyrosine Kinase Inhibitor, in Patients With Human Epidermal Growth Factor Receptor 2-Positive Metastatic Breast Cancer. J. Clin. Oncol. 2017, 35, 3105–3112. [Google Scholar] [CrossRef]
- Li, X.; Yang, C.; Wan, H.; Zhang, G.; Feng, J.; Zhang, L.; Chen, X.; Zhong, D.; Lou, L.; Tao, W.; et al. Discovery and Development of Pyrotinib: A Novel Irreversible EGFR/HER2 Dual Tyrosine Kinase Inhibitor with Favorable Safety Profiles for the Treatment of Breast Cancer. Eur. J. Pharm. Sci. 2017, 110, 51–61. [Google Scholar] [CrossRef]
- Ma, F.; Li, Q.; Guan, X.; Chen, S.; Yi, Z.; Lan, B.; Xing, P.; Fan, Y.; Wang, J.; Luo, Y.; et al. Safety, Efficacy, and Biomarker Analysis of Pyrotinib in Combination with Capecitabine in HER2-Positive Metastatic Breast Cancer Patients: A Phase I Clinical Trial. J. Clin. Oncol. 2019, 37, 1035. [Google Scholar] [CrossRef]
- Ma, F.; Ouyang, Q.; Li, W.; Jiang, Z.; Tong, Z.; Liu, Y.; Li, H.; Yu, S.; Feng, J.; Wang, S.; et al. Pyrotinib or Lapatinib Combined With Capecitabine in HER2–Positive Metastatic Breast Cancer With Prior Taxanes, Anthracyclines, And/or Trastuzumab: A Randomized, Phase II Study. J. Clin. Oncol. 2019, 37, 2610–2619. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Z.; Yan, M.; Hu, X.; Zhang, Q.; Ouyang, Q.; Feng, J.; Yin, Y.; Sun, T.; Tong, Z.; Wang, X.; et al. Pyrotinib Combined with Capecitabine in Women with HER2 Metastatic Breast Cancer Previously Treated with Trastuzumab and Taxanes: A Randomized Phase III Study. J. Clin. Oncol. 2019, 37, 1001. [Google Scholar] [CrossRef]
- Wang, C.; Lin, Y.; Zhou, Y.; Mao, F.; Zhu, H.; Guan, J.; Zhang, X.; Shen, S.; Huang, X.; Chen, C.; et al. Pyrotinib with Trastuzumab and Aromatase Inhibitors as First-Line Treatment for HER2 Positive and Hormone Receptor Positive Metastatic or Locally Advanced Breast Cancer: Study Protocol of a Randomized Controlled Trial. BMC Cancer 2020, 20, 653. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.J.; Kim, H.-P.; Yoon, Y.-K.; Kim, M.-S.; Lee, G.-S.; Han, S.-W.; Im, S.-A.; Kim, T.-Y.; Oh, D.-Y.; Bang, Y.-J. Antitumor Activity of HM781-36B, a Pan-HER Tyrosine Kinase Inhibitor, in HER2-Amplified Breast Cancer Cells. Anticancer Drugs 2012, 23, 288–297. [Google Scholar] [CrossRef] [PubMed]
- Kim, T.M.; Lee, K.-W.; Oh, D.-Y.; Lee, J.-S.; Im, S.-A.; Kim, D.-W.; Han, S.-W.; Kim, Y.J.; Kim, T.-Y.; Kim, J.H.; et al. Phase 1 Studies of Poziotinib, an Irreversible Pan-HER Tyrosine Kinase Inhibitor in Patients with Advanced Solid Tumors. Cancer Res. Treat. 2018, 50, 835–842. [Google Scholar] [CrossRef]
- Robichaux, J.P.; Elamin, Y.Y.; Vijayan, R.S.K.; Nilsson, M.B.; Hu, L.; He, J.; Zhang, F.; Pisegna, M.; Poteete, A.; Sun, H.; et al. Pan-Cancer Landscape and Analysis of ERBB2 Mutations Identifies Poziotinib as a Clinically Active Inhibitor and Enhancer of T-DM1 Activity. Cancer Cell 2020, 37, 420. [Google Scholar] [CrossRef]
- Park, Y.H.; Lee, K.-H.; Sohn, J.H.; Lee, K.S.; Jung, K.H.; Kim, J.-H.; Lee, K.H.; Ahn, J.S.; Kim, T.-Y.; Kim, G.M.; et al. A Phase II Trial of the Pan-HER Inhibitor Poziotinib, in Patients with HER2-Positive Metastatic Breast Cancer Who Had Received at Least Two Prior HER2-Directed Regimens: Results of the NOV120101-203 Trial. Int. J. Cancer 2018, 143, 3240–3247. [Google Scholar] [CrossRef]
- Kim, J.-Y.; Lee, E.; Park, K.; Jung, H.H.; Park, W.-Y.; Lee, K.-H.; Sohn, J.; Lee, K.S.; Jung, K.H.; Kim, J.H.; et al. Molecular Alterations and Poziotinib Efficacy, a Pan-HER Inhibitor, in Human Epidermal Growth Factor Receptor 2 (HER2)-Positive Breast Cancers: Combined Exploratory Biomarker Analysis from a Phase II Clinical Trial of Poziotinib for Refractory HER2-Positive Breast Cancer Patients. Int. J. Cancer 2019, 145, 1669–1678. [Google Scholar] [CrossRef]
- Simmons, C.; Rayson, D.; Joy, A.A.; Henning, J.-W.; Lemieux, J.; McArthur, H.; Card, P.B.; Dent, R.; Brezden-Masley, C. Current and Future Landscape of Targeted Therapy in HER2-Positive Advanced Breast Cancer: Redrawing the Lines. Ther. Adv. Med. Oncol. 2022, 14, 17588359211066677. [Google Scholar] [CrossRef]
- Tanaka, H.; Hirata, M.; Shinonome, S.; Wada, T.; Iguchi, M.; Dohi, K.; Inoue, M.; Ishioka, Y.; Hojo, K.; Yamada, T.; et al. Preclinical Antitumor Activity of S-222611, an Oral Reversible Tyrosine Kinase Inhibitor of Epidermal Growth Factor Receptor and Human Epidermal Growth Factor Receptor 2. Cancer Sci. 2014, 105, 1040–1048. [Google Scholar] [CrossRef]
- Tanaka, Y.; Hirata, M.; Shinonome, S.; Torii, M.; Nezasa, K.-I.; Tanaka, H. Distribution Analysis of Epertinib in Brain Metastasis of HER2-Positive Breast Cancer by Imaging Mass Spectrometry and Prospect for Antitumor Activity. Sci. Rep. 2018, 8, 343. [Google Scholar] [CrossRef] [PubMed]
- Spicer, J.; Baird, R.; Suder, A.; Cresti, N.; Garcia Corbacho, J.; Hogarth, L.; Frenkel, E.; Matsumoto, S.; Kawabata, I.; Donaldson, K.; et al. Phase 1 Dose-Escalation Study of S-222611, an Oral Reversible Dual Tyrosine Kinase Inhibitor of EGFR and HER2, in Patients with Solid Tumours. Eur. J. Cancer 2015, 51, 137–145. [Google Scholar] [CrossRef] [PubMed]
- Arkenau, H.-T.; Italiano, A.; Mak, G.; Toulmonde, M.; Baird, R.D.; Garcia-Corbacho, J.; Plummer, R.; Flynn, M.; Forster, M.; Wilson, R.H.; et al. An Extended Phase Ib Study of Epertinib, an Orally Active Reversible Dual EGFR/HER2 Tyrosine Kinase Inhibitor, in Patients with Solid Tumours. Eur. J. Cancer 2018, 103, 17–23. [Google Scholar] [CrossRef] [PubMed]
- Macpherson, I.R.; Spiliopoulou, P.; Rafii, S.; Saggese, M.; Baird, R.D.; Garcia-Corbacho, J.; Italiano, A.; Bonneterre, J.; Campone, M.; Cresti, N.; et al. A Phase I/II Study of Epertinib plus Trastuzumab with or without Chemotherapy in Patients with HER2-Positive Metastatic Breast Cancer. Breast Cancer Res. 2020, 22, 1. [Google Scholar] [CrossRef]
- Zhang, J.; McAndrew, N.; Yu, W.; Pan, X.; Wang, M.; Hu, X. Abstract P2-13-43: Preclinical and Early Clinical Safety and Pharmacokinetics Data of DZD1516, an BBB-Penetrant Selective HER2 Inhibitor for the Treatment of HER2 Positive Metastatic Breast Cancer. Cancer Res. 2022, 82, P2–P13. [Google Scholar] [CrossRef]
- Hu, X.; Zhang, J.; McAndrew, N.P. Early Clinical Safety and Pharmacokinetics Data of DZD1516, an BBB-Penetrant Selective HER2 Inhibitor for the Treatment of HER2-Positive Metastatic Breast Cancer. J. Clin. Oncol. 2022, 40, 1038. [Google Scholar] [CrossRef]
- Cameron, D.; Piccart-Gebhart, M.J.; Gelber, R.D.; Procter, M.; Goldhirsch, A.; de Azambuja, E.; Castro, G.; Untch, M.; Smith, I.; Gianni, L.; et al. 11 Years’ Follow-up of Trastuzumab after Adjuvant Chemotherapy in HER2-Positive Early Breast Cancer: Final Analysis of the HERceptin Adjuvant (HERA) Trial. Lancet 2017, 389, 1195–1205. [Google Scholar] [CrossRef]
- Bianchini, G.; Gianni, L. The Immune System and Response to HER2-Targeted Treatment in Breast Cancer. Lancet Oncol. 2014, 15, e58–e68. [Google Scholar] [CrossRef]
- Petricevic, B.; Laengle, J.; Singer, J.; Sachet, M.; Fazekas, J.; Steger, G.; Bartsch, R.; Jensen-Jarolim, E.; Bergmann, M. Trastuzumab Mediates Antibody-Dependent Cell-Mediated Cytotoxicity and Phagocytosis to the Same Extent in Both Adjuvant and Metastatic HER2/neu Breast Cancer Patients. J. Transl. Med. 2013, 11, 307. [Google Scholar] [CrossRef]
- Clynes, R.A.; Towers, T.L.; Presta, L.G.; Ravetch, J.V. Inhibitory Fc Receptors Modulate in Vivo Cytotoxicity against Tumor Targets. Nat. Med. 2000, 6, 443–446. [Google Scholar] [CrossRef]
- Arnould, L.; Gelly, M.; Penault-Llorca, F.; Benoit, L.; Bonnetain, F.; Migeon, C.; Cabaret, V.; Fermeaux, V.; Bertheau, P.; Garnier, J.; et al. Trastuzumab-Based Treatment of HER2-Positive Breast Cancer: An Antibody-Dependent Cellular Cytotoxicity Mechanism? Br. J. Cancer 2006, 94, 259–267. [Google Scholar] [CrossRef] [PubMed]
- Stagg, J.; Loi, S.; Divisekera, U.; Ngiow, S.F.; Duret, H.; Yagita, H.; Teng, M.W.; Smyth, M.J. Anti-ErbB-2 mAb Therapy Requires Type I and II Interferons and Synergizes with Anti-PD-1 or Anti-CD137 mAb Therapy. Proc. Natl. Acad. Sci. USA 2011, 108, 7142–7147. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Lee, C.K.; Lin, C.-H.; Gassen, R.B.; Xu, X.; Huang, Z.; Xiao, C.; Bonorino, C.; Lu, L.-F.; Bui, J.D.; et al. PD-L1:CD80 Cis-Heterodimer Triggers the Co-Stimulatory Receptor CD28 While Repressing the Inhibitory PD-1 and CTLA-4 Pathways. Immunity 2019, 51, 1059–1073.e9. [Google Scholar] [CrossRef] [PubMed]
- Latif, F.; Bint Abdul Jabbar, H.; Malik, H.; Sadaf, H.; Sarfraz, A.; Sarfraz, Z.; Cherrez-Ojeda, I. Atezolizumab and Pembrolizumab in Triple-Negative Breast Cancer: A Meta-Analysis. Expert Rev. Anticancer Ther. 2022, 22, 229–235. [Google Scholar] [CrossRef] [PubMed]
- Loi, S.; Giobbie-Hurder, A.; Gombos, A.; Bachelot, T.; Hui, R.; Curigliano, G.; Campone, M.; Biganzoli, L.; Bonnefoi, H.; Jerusalem, G.; et al. Pembrolizumab plus Trastuzumab in Trastuzumab-Resistant, Advanced, HER2-Positive Breast Cancer (PANACEA): A Single-Arm, Multicentre, Phase 1b–2 Trial. Lancet Oncol. 2019, 20, 371–382. [Google Scholar] [CrossRef]
- Waks, A.G.; Keenan, T.E.; Li, T.; Tayob, N.; Wulf, G.M.; Richardson, E.T., 3rd; Attaya, V.; Anderson, L.; Mittendorf, E.A.; Overmoyer, B.; et al. Phase Ib Study of Pembrolizumab in Combination with Trastuzumab Emtansine for Metastatic HER2-Positive Breast Cancer. J. Immunother. Cancer 2022, 10, e005119. [Google Scholar] [CrossRef]
- Emens, L.A.; Esteva, F.J.; Beresford, M.; Saura, C.; De Laurentiis, M.; Kim, S.-B.; Im, S.-A.; Wang, Y.; Salgado, R.; Mani, A.; et al. Trastuzumab Emtansine plus Atezolizumab versus Trastuzumab Emtansine plus Placebo in Previously Treated, HER2-Positive Advanced Breast Cancer (KATE2): A Phase 2, Multicentre, Randomised, Double-Blind Trial. Lancet Oncol. 2020, 21, 1283–1295. [Google Scholar] [CrossRef]
- Huober, J.; Barrios, C.H.; Niikura, N.; Jarząb, M.; Chang, Y.-C.; Huggins-Puhalla, S.L.; Pedrini, J.; Zhukova, L.; Graupner, V.; Eiger, D.; et al. Atezolizumab With Neoadjuvant Anti-Human Epidermal Growth Factor Receptor 2 Therapy and Chemotherapy in Human Epidermal Growth Factor Receptor 2-Positive Early Breast Cancer: Primary Results of the Randomized Phase III IMpassion050 Trial. J. Clin. Oncol. 2022, 40, 2946–2956. [Google Scholar] [CrossRef]
- Hurvitz, S.A.; Bachelot, T.; Bianchini, G.; Harbeck, N.; Loi, S.; Park, Y.H.; Prat, A.; Gilham, L.; Boulet, T.; Gochitashvili, N.; et al. ASTEFANIA: Adjuvant Ado-Trastuzumab Emtansine and Atezolizumab for High-Risk, HER2-Positive Breast Cancer. Future Oncol. 2022, 18, 3563–3572. [Google Scholar] [CrossRef]
- Chia, S.; Bedard, P.L.; Hilton, J.; Amir, E.; Gelmon, K.; Goodwin, R.; Villa, D.; Cabanero, M.; Tu, D.; Tsao, M.; et al. A Phase Ib Trial of Durvalumab in Combination with Trastuzumab in HER2-Positive Metastatic Breast Cancer (CCTG IND.229). Oncologist 2019, 24, 1439–1445. [Google Scholar] [CrossRef]
- Dirix, L.Y.; Takacs, I.; Jerusalem, G.; Nikolinakos, P.; Arkenau, H.-T.; Forero-Torres, A.; Boccia, R.; Lippman, M.E.; Somer, R.; Smakal, M.; et al. Avelumab, an Anti-PD-L1 Antibody, in Patients with Locally Advanced or Metastatic Breast Cancer: A Phase 1b JAVELIN Solid Tumor Study. Breast Cancer Res. Treat. 2018, 167, 671–686. [Google Scholar] [CrossRef] [PubMed]
- André, P.; Denis, C.; Soulas, C.; Bourbon-Caillet, C.; Lopez, J.; Arnoux, T.; Bléry, M.; Bonnafous, C.; Gauthier, L.; Morel, A.; et al. Anti-NKG2A mAb Is a Checkpoint Inhibitor That Promotes Anti-Tumor Immunity by Unleashing Both T and NK Cells. Cell 2018, 175, 1731–1743.e13. [Google Scholar] [CrossRef] [PubMed]
- Kwok, G.; Yau, T.C.C.; Chiu, J.W.; Tse, E.; Kwong, Y.-L. Pembrolizumab (Keytruda). Hum. Vaccin. Immunother. 2016, 12, 2777–2789. [Google Scholar] [CrossRef]
- Herbst, R.S.; Soria, J.-C.; Kowanetz, M.; Fine, G.D.; Hamid, O.; Gordon, M.S.; Sosman, J.A.; McDermott, D.F.; Powderly, J.D.; Gettinger, S.N.; et al. Predictive Correlates of Response to the Anti-PD-L1 Antibody MPDL3280A in Cancer Patients. Nature 2014, 515, 563–567. [Google Scholar] [CrossRef]
- Stewart, R.; Morrow, M.; Hammond, S.A.; Mulgrew, K.; Marcus, D.; Poon, E.; Watkins, A.; Mullins, S.; Chodorge, M.; Andrews, J.; et al. Identification and Characterization of MEDI4736, an Antagonistic Anti-PD-L1 Monoclonal Antibody. Cancer Immunol. Res. 2015, 3, 1052–1062. [Google Scholar] [CrossRef] [PubMed]
- Mittal, D.; Vijayan, D.; Neijssen, J.; Kreijtz, J.; Habraken, M.M.J.M.; Van Eenennaam, H.; Van Elsas, A.; Smyth, M.J. Blockade of ErbB2 and PD-L1 Using a Bispecific Antibody to Improve Targeted Anti-ErbB2 Therapy. Oncoimmunology 2019, 8, e1648171. [Google Scholar] [CrossRef]
- Gu, C.-L.; Zhu, H.-X.; Deng, L.; Meng, X.-Q.; Li, K.; Xu, W.; Zhao, L.; Liu, Y.-Q.; Zhu, Z.-P.; Huang, H.-M. Bispecific Antibody Simultaneously Targeting PD1 and HER2 Inhibits Tumor Growth via Direct Tumor Cell Killing in Combination with PD1/PDL1 Blockade and HER2 Inhibition. Acta Pharmacol. Sin. 2022, 43, 672–680. [Google Scholar] [CrossRef]
- Chen, Y.-L.; Cui, Y.; Liu, X.; Liu, G.; Dong, X.; Tang, L.; Hung, Y.; Wang, C.; Feng, M.-Q. A Bispecific Antibody Targeting HER2 and PD-L1 Inhibits Tumor Growth with Superior Efficacy. J. Biol. Chem. 2021, 297, 101420. [Google Scholar] [CrossRef]
- Biologics, I. Innovent Releases Preliminary Results of the Phase Ia Dose-Escalation Study of IBI315 (Anti-Her2/PD-1 Bispecific Antibody) in Patients with Advanced Solid Tumors at CSCO Annual Meeting. 2021. Available online: https://www.prnewswire.com/news-releases/innovent-releases-preliminary-results-of-the-phase-ia-dose-escalation-study-of-ibi315-anti-her2pd-1-bispecific-antibody-in-patients-with-advanced-solid-tumors-at-csco-annual-meeting-2021-301386697.html (accessed on 15 February 2023).
- Sun, C.; Xu, J.; Huang, Q.; Huang, M.; Wen, H.; Zhang, C.; Wang, J.; Song, J.; Zheng, M.; Sun, H.; et al. High NKG2A Expression Contributes to NK Cell Exhaustion and Predicts a Poor Prognosis of Patients with Liver Cancer. Oncoimmunology 2017, 6, e1264562. [Google Scholar] [CrossRef]
- Borst, L.; van der Burg, S.H.; van Hall, T. The NKG2A-HLA-E Axis as a Novel Checkpoint in the Tumor Microenvironment. Clin. Cancer Res. 2020, 26, 5549–5556. [Google Scholar] [CrossRef]
- Mantovani, A.; Longo, D.L. Macrophage Checkpoint Blockade in Cancer—Back to the Future. N. Engl. J. Med. 2018, 379, 1777–1779. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Ma, Y.; Gao, P.; Yao, Z. Targeting CD47: The Achievements and Concerns of Current Studies on Cancer Immunotherapy. J. Thorac. Dis. 2017, 9, E168–E174. [Google Scholar] [CrossRef] [PubMed]
- Tsao, L.-C.; Crosby, E.J.; Trotter, T.N.; Agarwal, P.; Hwang, B.-J.; Acharya, C.; Shuptrine, C.W.; Wang, T.; Wei, J.; Yang, X.; et al. CD47 Blockade Augmentation of Trastuzumab Antitumor Efficacy Dependent on Antibody-Dependent Cellular Phagocytosis. JCI Insight 2019, 4, e131882. [Google Scholar] [CrossRef]
- Kryczek, I.; Zou, L.; Rodriguez, P.; Zhu, G.; Wei, S.; Mottram, P.; Brumlik, M.; Cheng, P.; Curiel, T.; Myers, L.; et al. B7-H4 Expression Identifies a Novel Suppressive Macrophage Population in Human Ovarian Carcinoma. J. Exp. Med. 2006, 203, 871–881. [Google Scholar] [CrossRef] [PubMed]
- Hu, X.; Liu, Y.; Zhang, X.; Kong, D.; Kong, J.; Zhao, D.; Guo, Y.; Sun, L.; Chu, L.; Liu, S.; et al. The Anti-B7-H4 Checkpoint Synergizes Trastuzumab Treatment to Promote Phagocytosis and Eradicate Breast Cancer. Neoplasia 2020, 22, 539–553. [Google Scholar] [CrossRef] [PubMed]
- Wortzman, M.E.; Clouthier, D.L.; McPherson, A.J.; Lin, G.H.Y.; Watts, T.H. The Contextual Role of TNFR Family Members in CD8(+) T-Cell Control of Viral Infections. Immunol. Rev. 2013, 255, 125–148. [Google Scholar] [CrossRef]
- Hinner, M.J.; Aiba, R.-S.B.; Schlosser, C.; Wiedenmann, A.; Allersdorfer, A.; Matschiner, G.; Berger, S.; Moebius, U.; Rothe, C.; Olwill, S.A. Abstract 556: Costimulatory T-Cell Engagement by the HER2/CD137 Bispecific PRS-343 Leads to Strong Antitumor Effect in Humanized Mouse Model. Cancer Res. 2016, 76, 556. [Google Scholar] [CrossRef]
- Hinner, M.J.; Aiba, R.S.B.; Jaquin, T.J.; Berger, S.; Dürr, M.C.; Schlosser, C.; Allersdorfer, A.; Wiedenmann, A.; Matschiner, G.; Schüler, J.; et al. Tumor-Localized Costimulatory T-Cell Engagement by the 4-1BB/HER2 Bispecific Antibody-Anticalin Fusion PRS-343. Clin. Cancer Res. 2019, 25, 5878–5889. [Google Scholar] [CrossRef]
- Ku, G.; Bendell, J.C.; Tolcher, A.W.; Hurvitz, S.A.; Krishnamurthy, A.; El-Khoueiry, A.B.; Patnaik, A.; Shroff, R.T.; Noonan, A.; Hahn, N.M.; et al. 525O A Phase I Dose Escalation Study of PRS-343, a HER2/4-1BB Bispecific Molecule, in Patients with HER2-Positive Malignancies. Ann. Oncol. 2020, 31, S462–S463. [Google Scholar] [CrossRef]
- Rizzo, A.; Ricci, A.D. Biomarkers for Breast Cancer Immunotherapy: PD-L1, TILs, and beyond. Expert Opin. Investig. Drugs 2022, 31, 549–555. [Google Scholar] [CrossRef]
- Park, S.E.; Park, K.; Lee, E.; Kim, J.-Y.; Ahn, J.S.; Im, Y.-H.; Lee, C.; Jung, H.; Cho, S.Y.; Park, W.-Y.; et al. Clinical Implication of Tumor Mutational Burden in Patients with HER2-Positive Refractory Metastatic Breast Cancer. Oncoimmunology 2018, 7, e1466768. [Google Scholar] [CrossRef] [PubMed]
- Salgado, R.; Denkert, C.; Demaria, S.; Sirtaine, N.; Klauschen, F.; Pruneri, G.; Wienert, S.; Van den Eynden, G.; Baehner, F.L.; Penault-Llorca, F.; et al. The Evaluation of Tumor-Infiltrating Lymphocytes (TILs) in Breast Cancer: Recommendations by an International TILs Working Group 2014. Ann. Oncol. 2015, 26, 259–271. [Google Scholar] [CrossRef]
- Perez, E.A.; Ballman, K.V.; Tenner, K.S.; Thompson, E.A.; Badve, S.S.; Bailey, H.; Baehner, F.L. Association of Stromal Tumor-Infiltrating Lymphocytes With Recurrence-Free Survival in the N9831 Adjuvant Trial in Patients With Early-Stage HER2-Positive Breast Cancer. JAMA Oncol. 2016, 2, 56–64. [Google Scholar] [CrossRef] [PubMed]
- Triulzi, T.; Forte, L.; Regondi, V.; Di Modica, M.; Ghirelli, C.; Carcangiu, M.L.; Sfondrini, L.; Balsari, A.; Tagliabue, E. HER2 Signaling Regulates the Tumor Immune Microenvironment and Trastuzumab Efficacy. Oncoimmunology 2019, 8, e1512942. [Google Scholar] [CrossRef] [PubMed]
- Luen, S.J.; Salgado, R.; Fox, S.; Savas, P.; Eng-Wong, J.; Clark, E.; Kiermaier, A.; Swain, S.M.; Baselga, J.; Michiels, S.; et al. Tumour-Infiltrating Lymphocytes in Advanced HER2-Positive Breast Cancer Treated with Pertuzumab or Placebo in Addition to Trastuzumab and Docetaxel: A Retrospective Analysis of the CLEOPATRA Study. Lancet Oncol. 2017, 18, 52–62. [Google Scholar] [CrossRef]
- Dieci, M.V.; Conte, P.; Bisagni, G.; Brandes, A.A.; Frassoldati, A.; Cavanna, L.; Musolino, A.; Giotta, F.; Rimanti, A.; Garrone, O.; et al. Association of Tumor-Infiltrating Lymphocytes with Distant Disease-Free Survival in the ShortHER Randomized Adjuvant Trial for Patients with Early HER2+ Breast Cancer. Ann. Oncol. 2019, 30, 418–423. [Google Scholar] [CrossRef]
- Force, J.; Howie, L.J.; Abbott, S.E.; Bentley, R.; Marcom, P.K.; Kimmick, G.; Westbrook, K.; Sammons, S.L.; Parks, M.; Topping, D.L.; et al. Early Stage HER2-Positive Breast Cancers Not Achieving a pCR From Neoadjuvant Trastuzumab- or Pertuzumab-Based Regimens Have an Immunosuppressive Phenotype. Clin. Breast Cancer 2018, 18, 410–417. [Google Scholar] [CrossRef]
- Ochi, T.; Bianchini, G.; Ando, M.; Nozaki, F.; Kobayashi, D.; Criscitiello, C.; Curigliano, G.; Iwamoto, T.; Niikura, N.; Takei, H.; et al. Predictive and Prognostic Value of Stromal Tumour-Infiltrating Lymphocytes before and after Neoadjuvant Therapy in Triple Negative and HER2-Positive Breast Cancer. Eur. J. Cancer 2019, 118, 41–48. [Google Scholar] [CrossRef]
- Kim, A.; Lee, S.J.; Kim, Y.K.; Park, W.Y.; Park, D.Y.; Kim, J.Y.; Lee, C.H.; Gong, G.; Huh, G.Y.; Choi, K.U. Programmed Death-Ligand 1 (PD-L1) Expression in Tumour Cell and Tumour Infiltrating Lymphocytes of HER2-Positive Breast Cancer and Its Prognostic Value. Sci. Rep. 2017, 7, 11671. [Google Scholar] [CrossRef]
- Bertucci, F.; Gonçalves, A. Immunotherapy in Breast Cancer: The Emerging Role of PD-1 and PD-L1. Curr. Oncol. Rep. 2017, 19, 64. [Google Scholar] [CrossRef]
- Kurozumi, S.; Inoue, K.; Matsumoto, H.; Fujii, T.; Horiguchi, J.; Oyama, T.; Kurosumi, M.; Shirabe, K. Clinicopathological Values of PD-L1 Expression in HER2-Positive Breast Cancer. Sci. Rep. 2019, 9, 16662. [Google Scholar] [CrossRef]
- Shang, M.; Chi, Y.; Zhang, J.; Chang, J.; Yang, H.; Yin, S.; Tan, Q.; Man, X.; Li, H. The Therapeutic Effectiveness of Neoadjuvant Trastuzumab Plus Chemotherapy for HER2-Positive Breast Cancer Can Be Predicted by Tumor-Infiltrating Lymphocytes and PD-L1 Expression. Front. Oncol. 2021, 11, 706606. [Google Scholar] [CrossRef] [PubMed]
- Phelan, K.W.; Advani, A.S. Novel Therapies in Acute Lymphoblastic Leukemia. Curr. Hematol. Malig. Rep. 2018, 13, 289–299. [Google Scholar] [CrossRef] [PubMed]
- Frey, N. The What, When and How of CAR T Cell Therapy for ALL. Best Pract. Res. Clin. Haematol. 2017, 30, 275–281. [Google Scholar] [CrossRef]
- Dai, H.; Wang, Y.; Lu, X.; Han, W. Chimeric Antigen Receptors Modified T-Cells for Cancer Therapy. J. Natl. Cancer Inst. 2016, 108, djv439. [Google Scholar] [CrossRef] [PubMed]
- Grupp, S.A.; Kalos, M.; Barrett, D.; Aplenc, R.; Porter, D.L.; Rheingold, S.R.; Teachey, D.T.; Chew, A.; Hauck, B.; Wright, J.F.; et al. Chimeric Antigen Receptor-Modified T Cells for Acute Lymphoid Leukemia. N. Engl. J. Med. 2013, 368, 1509–1518. [Google Scholar] [CrossRef]
- Raponi, S.; De Propris, M.S.; Intoppa, S.; Milani, M.L.; Vitale, A.; Elia, L.; Perbellini, O.; Pizzolo, G.; Foá, R.; Guarini, A. Flow Cytometric Study of Potential Target Antigens (CD19, CD20, CD22, CD33) for Antibody-Based Immunotherapy in Acute Lymphoblastic Leukemia: Analysis of 552 Cases. Leuk. Lymphoma 2011, 52, 1098–1107. [Google Scholar] [CrossRef]
- Scheuermann, R.H.; Racila, E. CD19 Antigen in Leukemia and Lymphoma Diagnosis and Immunotherapy. Leuk. Lymphoma 1995, 18, 385–397. [Google Scholar] [CrossRef]
- Elia, A.R.; Grioni, M.; Basso, V.; Curnis, F.; Freschi, M.; Corti, A.; Mondino, A.; Bellone, M. Targeting Tumor Vasculature with TNF Leads Effector T Cells to the Tumor and Enhances Therapeutic Efficacy of Immune Checkpoint Blockers in Combination with Adoptive Cell Therapy. Clin. Cancer Res. 2018, 24, 2171–2181. [Google Scholar] [CrossRef]
- Piali, L.; Fichtel, A.; Terpe, H.J.; Imhof, B.A.; Gisler, R.H. Endothelial Vascular Cell Adhesion Molecule 1 Expression Is Suppressed by Melanoma and Carcinoma. J. Exp. Med. 1995, 181, 811–816. [Google Scholar] [CrossRef]
- Bellone, M.; Calcinotto, A. Ways to Enhance Lymphocyte Trafficking into Tumors and Fitness of Tumor Infiltrating Lymphocytes. Front. Oncol. 2013, 3, 231. [Google Scholar] [CrossRef]
- Lindo, L.; Wilkinson, L.H.; Hay, K.A. Befriending the Hostile Tumor Microenvironment in CAR T-Cell Therapy. Front. Immunol. 2020, 11, 618387. [Google Scholar] [CrossRef] [PubMed]
- Gajewski, T.F.; Schreiber, H.; Fu, Y.-X. Innate and Adaptive Immune Cells in the Tumor Microenvironment. Nat. Immunol. 2013, 14, 1014–1022. [Google Scholar] [CrossRef] [PubMed]
- Silzle, T.; Randolph, G.J.; Kreutz, M.; Kunz-Schughart, L.A. The Fibroblast: Sentinel Cell and Local Immune Modulator in Tumor Tissue. Int. J. Cancer 2004, 108, 173–180. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.-C.S.; Lo, A.; Scholler, J.; Sun, J.; Majumdar, R.S.; Kapoor, V.; Antzis, M.; Cotner, C.E.; Johnson, L.A.; Durham, A.C.; et al. Targeting Fibroblast Activation Protein in Tumor Stroma with Chimeric Antigen Receptor T Cells Can Inhibit Tumor Growth and Augment Host Immunity without Severe Toxicity. Cancer Immunol. Res. 2014, 2, 154–166. [Google Scholar] [CrossRef]
- Caruana, I.; Savoldo, B.; Hoyos, V.; Weber, G.; Liu, H.; Kim, E.S.; Ittmann, M.M.; Marchetti, D.; Dotti, G. Heparanase Promotes Tumor Infiltration and Antitumor Activity of CAR-Redirected T Lymphocytes. Nat. Med. 2015, 21, 524–529. [Google Scholar] [CrossRef]
- Maude, S.L.; Barrett, D.; Teachey, D.T.; Grupp, S.A. Managing Cytokine Release Syndrome Associated with Novel T Cell-Engaging Therapies. Cancer J. 2014, 20, 119–122. [Google Scholar] [CrossRef]
- Brentjens, R.J.; Davila, M.L.; Riviere, I.; Park, J.; Wang, X.; Cowell, L.G.; Bartido, S.; Stefanski, J.; Taylor, C.; Olszewska, M.; et al. CD19-Targeted T Cells Rapidly Induce Molecular Remissions in Adults with Chemotherapy-Refractory Acute Lymphoblastic Leukemia. Sci. Transl. Med. 2013, 5, 177ra38. [Google Scholar] [CrossRef]
- Frey, N.; Porter, D. Cytokine Release Syndrome with Chimeric Antigen Receptor T Cell Therapy. Biol. Blood Marrow Transplant. 2019, 25, e123–e127. [Google Scholar] [CrossRef]
- Freyer, C.W.; Porter, D.L. Cytokine Release Syndrome and Neurotoxicity Following CAR T-Cell Therapy for Hematologic Malignancies. J. Allergy Clin. Immunol. 2020, 146, 940–948. [Google Scholar] [CrossRef]
- Vora, S.B.; Waghmare, A.; Englund, J.A.; Qu, P.; Gardner, R.A.; Hill, J.A. Infectious Complications Following CD19 Chimeric Antigen Receptor T-Cell Therapy for Children, Adolescents, and Young Adults. Open Forum Infect. Dis. 2020, 7, ofaa121. [Google Scholar] [CrossRef] [PubMed]
- Boulch, M.; Cazaux, M.; Loe-Mie, Y.; Thibaut, R.; Corre, B.; Lemaître, F.; Grandjean, C.L.; Garcia, Z.; Bousso, P. A Cross-Talk between CAR T Cell Subsets and the Tumor Microenvironment Is Essential for Sustained Cytotoxic Activity. Sci. Immunol. 2021, 6, eabd4344. [Google Scholar] [CrossRef]
- Maher, J.; Brentjens, R.J.; Gunset, G.; Rivière, I.; Sadelain, M. Human T-Lymphocyte Cytotoxicity and Proliferation Directed by a Single Chimeric TCRzeta /CD28 Receptor. Nat. Biotechnol. 2002, 20, 70–75. [Google Scholar] [CrossRef] [PubMed]
- Mullard, A. FDA Approves First CAR T Therapy. Nat. Rev. Drug Discov. 2017, 16, 669. [Google Scholar] [CrossRef]
- Melenhorst, J.J.; Chen, G.M.; Wang, M.; Porter, D.L.; Chen, C.; Collins, M.A.; Gao, P.; Bandyopadhyay, S.; Sun, H.; Zhao, Z.; et al. Decade-Long Leukaemia Remissions with Persistence of CD4+ CAR T Cells. Nature 2022, 602, 503–509. [Google Scholar] [CrossRef] [PubMed]
- Rafiq, S.; Hackett, C.S.; Brentjens, R.J. Engineering Strategies to Overcome the Current Roadblocks in CAR T Cell Therapy. Nat. Rev. Clin. Oncol. 2019, 17, 147–167. [Google Scholar] [CrossRef]
- Dwivedi, A.; Karulkar, A.; Ghosh, S.; Rafiq, A.; Purwar, R. Lymphocytes in Cellular Therapy: Functional Regulation of CAR T Cells. Front. Immunol. 2018, 9, 3180. [Google Scholar] [CrossRef]
- Poorebrahim, M.; Mohammadkhani, N.; Mahmoudi, R.; Gholizadeh, M.; Fakhr, E.; Cid-Arregui, A. TCR-like CARs and TCR-CARs Targeting Neoepitopes: An Emerging Potential. Cancer Gene Ther. 2021, 28, 581–589. [Google Scholar] [CrossRef]
- Mo, F.; Duan, S.; Jiang, X.; Yang, X.; Hou, X.; Shi, W.; Carlos, C.J.J.; Liu, A.; Yin, S.; Wang, W.; et al. Nanobody-Based Chimeric Antigen Receptor T Cells Designed by CRISPR/Cas9 Technology for Solid Tumor Immunotherapy. Signal Transduct. Target. Ther. 2021, 6, 80. [Google Scholar] [CrossRef]
- Inaguma, Y.; Akahori, Y.; Murayama, Y.; Shiraishi, K.; Tsuzuki-Iba, S.; Endoh, A.; Tsujikawa, J.; Demachi-Okamura, A.; Hiramatsu, K.; Saji, H.; et al. Construction and Molecular Characterization of a T-Cell Receptor-like Antibody and CAR-T Cells Specific for Minor Histocompatibility Antigen HA-1H. Gene Ther. 2014, 21, 575–584. [Google Scholar] [CrossRef]
- Srivastava, S.; Riddell, S.R. Engineering CAR-T Cells: Design Concepts. Trends Immunol. 2015, 36, 494–502. [Google Scholar] [CrossRef] [PubMed]
- Bridgeman, J.S.; Hawkins, R.E.; Bagley, S.; Blaylock, M.; Holland, M.; Gilham, D.E. The Optimal Antigen Response of Chimeric Antigen Receptors Harboring the CD3zeta Transmembrane Domain Is Dependent upon Incorporation of the Receptor into the Endogenous TCR/CD3 Complex. J. Immunol. 2010, 184, 6938–6949. [Google Scholar] [CrossRef] [PubMed]
- Hong, M.; Clubb, J.D.; Chen, Y.Y. Engineering CAR-T Cells for Next-Generation Cancer Therapy. Cancer Cell 2020, 38, 473–488. [Google Scholar] [CrossRef] [PubMed]
- Guedan, S.; Calderon, H.; Posey, A.D., Jr.; Maus, M.V. Engineering and Design of Chimeric Antigen Receptors. Mol. Ther. Methods Clin. Dev. 2019, 12, 145–156. [Google Scholar] [CrossRef]
- Levine, B.L.; Miskin, J.; Wonnacott, K.; Keir, C. Global Manufacturing of CAR T Cell Therapy. Mol. Ther. Methods Clin. Dev. 2017, 4, 92–101. [Google Scholar] [CrossRef]
- Luangwattananun, P.; Junking, M.; Sujjitjoon, J.; Wutti-In, Y.; Poungvarin, N.; Thuwajit, C.; Yenchitsomanus, P.-T. Fourth-Generation Chimeric Antigen Receptor T Cells Targeting Folate Receptor Alpha Antigen Expressed on Breast Cancer Cells for Adoptive T Cell Therapy. Breast Cancer Res. Treat. 2021, 186, 25–36. [Google Scholar] [CrossRef] [PubMed]
- Chmielewski, M.; Abken, H. TRUCKS, the Fourth-generation CAR T Cells: Current Developments and Clinical Translation. Adv. Cell Gene Ther. 2020, 3, e84. [Google Scholar] [CrossRef]
- Kagoya, Y.; Tanaka, S.; Guo, T.; Anczurowski, M.; Wang, C.-H.; Saso, K.; Butler, M.O.; Minden, M.D.; Hirano, N. A Novel Chimeric Antigen Receptor Containing a JAK–STAT Signaling Domain Mediates Superior Antitumor Effects. Nat. Med. 2018, 24, 352–359. [Google Scholar] [CrossRef]
- Mehrabadi, A.Z.; Ranjbar, R.; Farzanehpour, M.; Shahriary, A.; Dorostkar, R.; Hamidinejad, M.A.; Ghaleh, H.E.G. Therapeutic Potential of CAR T Cell in Malignancies: A Scoping Review. Biomed. Pharmacother. 2022, 146, 112512. [Google Scholar] [CrossRef]
- Zah, E.; Lin, M.-Y.; Silva-Benedict, A.; Jensen, M.C.; Chen, Y.Y. T Cells Expressing CD19/CD20 Bispecific Chimeric Antigen Receptors Prevent Antigen Escape by Malignant B Cells. Cancer Immunol. Res. 2016, 4, 498–508. [Google Scholar] [CrossRef]
- Yang, M.; Tang, X.; Zhang, Z.; Gu, L.; Wei, H.; Zhao, S.; Zhong, K.; Mu, M.; Huang, C.; Jiang, C.; et al. Tandem CAR-T Cells Targeting CD70 and B7-H3 Exhibit Potent Preclinical Activity against Multiple Solid Tumors. Theranostics 2020, 10, 7622–7634. [Google Scholar] [CrossRef]
- Pan, K.; Farrukh, H.; Chittepu, V.C.S.R.; Xu, H.; Pan, C.-X.; Zhu, Z. CAR Race to Cancer Immunotherapy: From CAR T, CAR NK to CAR Macrophage Therapy. J. Exp. Clin. Cancer Res. 2022, 41, 119. [Google Scholar] [CrossRef] [PubMed]
- Ma, S.; Li, X.; Wang, X.; Cheng, L.; Li, Z.; Zhang, C.; Ye, Z.; Qian, Q. Current Progress in CAR-T Cell Therapy for Solid Tumors. Int. J. Biol. Sci. 2019, 15, 2548–2560. [Google Scholar] [CrossRef] [PubMed]
- Abramson, J.S. Anti-CD19 CAR T-Cell Therapy for B-Cell Non-Hodgkin Lymphoma. Transfus. Med. Rev. 2020, 34, 29–33. [Google Scholar] [CrossRef] [PubMed]
- Weisel, K.; Martin, T.; Krishnan, A.; Jagannath, S.; Londhe, A.; Nair, S.; Diels, J.; Vogel, M.; Schecter, J.M.; Banerjee, A.; et al. Comparative Efficacy of Ciltacabtagene Autoleucel in CARTITUDE-1 vs Physician’s Choice of Therapy in the Long-Term Follow-Up of POLLUX, CASTOR, and EQUULEUS Clinical Trials for the Treatment of Patients with Relapsed or Refractory Multiple Myeloma. Clin. Drug Investig. 2022, 42, 29–41. [Google Scholar] [CrossRef]
- Mullard, A. FDA Approves First BCMA-Targeted CAR-T Cell Therapy. Nat. Rev. Drug Discov. 2021, 20, 332. [Google Scholar] [CrossRef]
- Maggs, L.; Cattaneo, G.; Dal, A.E.; Moghaddam, A.S.; Ferrone, S. CAR T Cell-Based Immunotherapy for the Treatment of Glioblastoma. Front. Neurosci. 2021, 15, 662064. [Google Scholar] [CrossRef] [PubMed]
- Morgan, R.A.; Yang, J.C.; Kitano, M.; Dudley, M.E.; Laurencot, C.M.; Rosenberg, S.A. Case Report of a Serious Adverse Event Following the Administration of T Cells Transduced with a Chimeric Antigen Receptor Recognizing ERBB2. Mol. Ther. 2010, 18, 843–851. [Google Scholar] [CrossRef]
- Ahmed, N.; Salsman, V.S.; Kew, Y.; Shaffer, D.; Powell, S.; Zhang, Y.J.; Grossman, R.G.; Heslop, H.E.; Gottschalk, S. HER2-Specific T Cells Target Primary Glioblastoma Stem Cells and Induce Regression of Autologous Experimental Tumors. Clin. Cancer Res. 2010, 16, 474–485. [Google Scholar] [CrossRef]
- Ahmed, N.; Brawley, V.; Hegde, M.; Bielamowicz, K.; Kalra, M.; Landi, D.; Robertson, C.; Gray, T.L.; Diouf, O.; Wakefield, A.; et al. HER2-Specific Chimeric Antigen Receptor-Modified Virus-Specific T Cells for Progressive Glioblastoma: A Phase 1 Dose-Escalation Trial. JAMA Oncol. 2017, 3, 1094–1101. [Google Scholar] [CrossRef]
- Priceman, S.J.; Tilakawardane, D.; Jeang, B.; Aguilar, B.; Murad, J.P.; Park, A.K.; Chang, W.-C.; Ostberg, J.R.; Neman, J.; Jandial, R.; et al. Regional Delivery of Chimeric Antigen Receptor-Engineered T Cells Effectively Targets HER2+ Breast Cancer Metastasis to the Brain. Clin. Cancer Res. 2018, 24, 95–105. [Google Scholar] [CrossRef]
- Memory-Enriched T Cells in Treating Patients with Recurrent or Refractory Grade III-IV Glioma. Available online: https://clinicaltrials.gov/ct2/show/NCT03389230 (accessed on 13 February 2023).
- HER2-CAR T Cells in Treating Patients with Recurrent Brain or Leptomeningeal Metastases. Available online: https://clinicaltrials.gov/ct2/show/NCT03696030 (accessed on 13 February 2023).
- HER2-Specific CAR T Cell Locoregional Immunotherapy for HER2-Positive Recurrent/Refractory Pediatric CNS Tumors. Available online: https://clinicaltrials.gov/ct2/show/NCT03500991 (accessed on 13 February 2023).
- T Cells Expressing HER2-Specific Chimeric Antigen Receptors(CAR) for Patients With HER2-Positive CNS Tumors—Full Text View—ClinicalTrials.Gov. Available online: https://clinicaltrials.gov/ct2/show/NCT02442297 (accessed on 13 February 2023).
- HER2-Specific Chimeric Antigen Receptor (CAR) T Cells for Children with Ependymoma. Available online: https://clinicaltrials.gov/ct2/show/NCT04903080 (accessed on 13 February 2023).
- Tóth, G.; Szöllősi, J.; Abken, H.; Vereb, G.; Szöőr, Á. A Small Number of HER2 Redirected CAR T Cells Significantly Improves Immune Response of Adoptively Transferred Mouse Lymphocytes against Human Breast Cancer Xenografts. Int. J. Mol. Sci. 2020, 21, 1039. [Google Scholar] [CrossRef] [PubMed]
- Chen, A.C.; Migliaccio, I.; Rimawi, M.; Lopez-Tarruella, S.; Creighton, C.J.; Massarweh, S.; Huang, C.; Wang, Y.-C.; Batra, S.K.; Gutierrez, M.C.; et al. Upregulation of mucin4 in ER-positive/HER2-Overexpressing Breast Cancer Xenografts with Acquired Resistance to Endocrine and HER2-Targeted Therapies. Breast Cancer Res. Treat. 2012, 134, 583–593. [Google Scholar] [CrossRef] [PubMed]
- Singha, N.C.; Nekoroski, T.; Zhao, C.; Symons, R.; Jiang, P.; Frost, G.I.; Huang, Z.; Shepard, H.M. Tumor-Associated Hyaluronan Limits Efficacy of Monoclonal Antibody Therapy. Mol. Cancer Ther. 2015, 14, 523–532. [Google Scholar] [CrossRef] [PubMed]
- Globerson-Levin, A.; Waks, T.; Eshhar, Z. Elimination of Progressive Mammary Cancer by Repeated Administrations of Chimeric Antigen Receptor-Modified T Cells. Mol. Ther. 2014, 22, 1029–1038. [Google Scholar] [CrossRef] [PubMed]
- Wei, J.; Sun, H.; Zhang, A.; Wu, X.; Li, Y.; Liu, J.; Duan, Y.; Xiao, F.; Wang, H.; Lv, M.; et al. A Novel AXL Chimeric Antigen Receptor Endows T Cells with Anti-Tumor Effects against Triple Negative Breast Cancers. Cell. Immunol. 2018, 331, 49–58. [Google Scholar] [CrossRef]
- Szöőr, Á.; Tóth, G.; Zsebik, B.; Szabó, V.; Eshhar, Z.; Abken, H.; Vereb, G. Trastuzumab Derived HER2-Specific CARs for the Treatment of Trastuzumab-Resistant Breast Cancer: CAR T Cells Penetrate and Eradicate Tumors That Are Not Accessible to Antibodies. Cancer Lett. 2020, 484, 1–8. [Google Scholar] [CrossRef]
- Azim, H.A.; Azim, H.A., Jr. Systemic Treatment of Brain Metastases in HER2-Positive Breast Cancer: Current Status and Future Directions. Future Oncol. 2012, 8, 135–144. [Google Scholar] [CrossRef]
- Leone, J.P.; Leone, B.A. Breast Cancer Brain Metastases: The Last Frontier. Exp. Hematol. Oncol. 2015, 4, 33. [Google Scholar] [CrossRef]
- Binary Oncolytic Adenovirus in Combination with HER2-Specific Autologous CAR VST, Advanced HER2 Positive Solid Tumors—Full Text View—ClinicalTrials.Gov. Available online: https://clinicaltrials.gov/ct2/show/NCT03740256?term=CAR+T&recrs=abde&cond=Breast+Cancer&draw=2&rank=12 (accessed on 13 February 2023).
- Autologous huMNC2-CAR44 T Cells for Breast Cancer Targeting Cleaved Form of MUC1 (MUC1*). Available online: https://clinicaltrials.gov/ct2/show/study/NCT04020575 (accessed on 13 February 2023).
- A Phase I Trial of CCT303-406 in Patients with Relapsed or Refractory HER2 Positive Solid Tumors. Available online: https://clinicaltrials.gov/ct2/show/NCT04511871?term=CAR+T&recrs=abde&cond=Breast+Cancer&draw=2&rank=15 (accessed on 13 February 2023).
- Safety and Activity Study of HER2-Targeted Dual Switch CAR-T Cells (BPX-603) in Subjects With HER2-Positive Solid Tumors. Available online: https://clinicaltrials.gov/ct2/show/study/NCT04650451 (accessed on 13 February 2023).
- Cao, Y.J.; Wang, X.; Wang, Z.; Zhao, L.; Li, S.; Zhang, Z.; Wei, X.; Yun, H.; Choi, S.-H.; Liu, Z.; et al. Switchable CAR-T Cells Outperformed Traditional Antibody-Redirected Therapeutics Targeting Breast Cancers. ACS Synth. Biol. 2021, 10, 1176–1183. [Google Scholar] [CrossRef]
- NCI. Drug Dictionary. Available online: https://www.cancer.gov/publications/dictionaries/cancer-drug/def/autologous-her2-targeted-dual-switch-car-t-cells-bpx-603 (accessed on 13 February 2023).
- DeRose, R.; Miyamoto, T.; Inoue, T. Manipulating Signaling at Will: Chemically-Inducible Dimerization (CID) Techniques Resolve Problems in Cell Biology. Pflug. Arch. 2013, 465, 409–417. [Google Scholar] [CrossRef] [PubMed]
- Gargett, T.; Brown, M.P. The Inducible Caspase-9 Suicide Gene System as a “Safety Switch” to Limit on-Target, off-Tumor Toxicities of Chimeric Antigen Receptor T Cells. Front. Pharmacol. 2014, 5, 235. [Google Scholar] [CrossRef] [PubMed]
- Di Stasi, A.; Tey, S.-K.; Dotti, G.; Fujita, Y.; Kennedy-Nasser, A.; Martinez, C.; Straathof, K.; Liu, E.; Durett, A.G.; Grilley, B.; et al. Inducible Apoptosis as a Safety Switch for Adoptive Cell Therapy. N. Engl. J. Med. 2011, 365, 1673–1683. [Google Scholar] [CrossRef] [PubMed]
- Zhang, P.-F.; Huang, Y.; Liang, X.; Li, D.; Jiang, L.; Yang, X.; Zhu, M.; Gou, H.-F.; Gong, Y.-L.; Wei, Y.-Q.; et al. Enhancement of the Antitumor Effect of HER2-Directed CAR-T Cells through Blocking Epithelial-Mesenchymal Transition in Tumor Cells. FASEB J. 2020, 34, 11185–11199. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Yuan, W.; Bin, S.; Wu, G.; Li, P.; Liu, M.; Yang, J.; Li, X.; Yang, K.; Gu, H. Overcome Trastuzumab Resistance of Breast Cancer Using Anti-HER2 Chimeric Antigen Receptor T Cells and PD1 Blockade. Am. J. Cancer Res. 2020, 10, 688–703. [Google Scholar] [PubMed]
- Li, P.; Yang, L.; Li, T.; Bin, S.; Sun, B.; Huang, Y.; Yang, K.; Shan, D.; Gu, H.; Li, H. The Third Generation Anti-HER2 Chimeric Antigen Receptor Mouse T Cells Alone or Together With Anti-PD1 Antibody Inhibits the Growth of Mouse Breast Tumor Cells Expressing HER2 In Vitro and in Immune Competent Mice. Front. Oncol. 2020, 10, 1143. [Google Scholar] [CrossRef] [PubMed]
- Thakur, A.; Scholler, J.; Kubicka, E.; Bliemeister, E.T.; Schalk, D.L.; June, C.H.; Lum, L.G. Bispecific Antibody Armed Metabolically Enhanced Headless CAR T Cells. Front. Immunol. 2021, 12, 690437. [Google Scholar] [CrossRef]
- Trastuzumab Deruxtecan for Metastatic HER2-Low Breast Cancer. Available online: https://www.cancer.gov/news-events/cancer-currents-blog/2022/enhertu-her2-low-breast-cancer (accessed on 14 February 2023).
- Eiger, D.; Agostinetto, E.; Saúde-Conde, R.; de Azambuja, E. The Exciting New Field of HER2-Low Breast Cancer Treatment. Cancers 2021, 13, 1015. [Google Scholar] [CrossRef]
- Siddiqui, T.; Rani, P.; Ashraf, T.; Ellahi, A. Enhertu (Fam-Trastuzumab-Deruxtecan-Nxki)—Revolutionizing Treatment Paradigm for HER2-Low Breast Cancer. Ann. Med. Surg. 2022, 82, 104665. [Google Scholar] [CrossRef]
- Yang, Y.-H.; Liu, J.-W.; Lu, C.; Wei, J.-F. CAR-T Cell Therapy for Breast Cancer: From Basic Research to Clinical Application. Int. J. Biol. Sci. 2022, 18, 2609–2626. [Google Scholar] [CrossRef]
- Mamessier, E.; Sylvain, A.; Thibult, M.-L.; Houvenaeghel, G.; Jacquemier, J.; Castellano, R.; Gonçalves, A.; André, P.; Romagné, F.; Thibault, G.; et al. Human Breast Cancer Cells Enhance Self Tolerance by Promoting Evasion from NK Cell Antitumor Immunity. J. Clin. Investig. 2011, 121, 3609–3622. [Google Scholar] [CrossRef] [PubMed]
- Viel, S.; Marçais, A.; Guimaraes, F.S.-F.; Loftus, R.; Rabilloud, J.; Grau, M.; Degouve, S.; Djebali, S.; Sanlaville, A.; Charrier, E.; et al. TGF-β Inhibits the Activation and Functions of NK Cells by Repressing the mTOR Pathway. Sci. Signal. 2016, 9, ra19. [Google Scholar] [CrossRef] [PubMed]
- Loftus, R.M.; Assmann, N.; Kedia-Mehta, N.; O’Brien, K.L.; Garcia, A.; Gillespie, C.; Hukelmann, J.L.; Oefner, P.J.; Lamond, A.I.; Gardiner, C.M.; et al. Amino Acid-Dependent cMyc Expression Is Essential for NK Cell Metabolic and Functional Responses in Mice. Nat. Commun. 2018, 9, 1–15. [Google Scholar] [CrossRef] [PubMed]
- O’Brien, K.L.; Finlay, D.K. Immunometabolism and Natural Killer Cell Responses. Nat. Rev. Immunol. 2019, 19, 282–290. [Google Scholar] [CrossRef]
- Marofi, F.; Al-Awad, A.S.; Sulaiman Rahman, H.; Markov, A.; Abdelbasset, W.K.; Ivanovna Enina, Y.; Mahmoodi, M.; Hassanzadeh, A.; Yazdanifar, M.; Stanley Chartrand, M.; et al. CAR-NK Cell: A New Paradigm in Tumor Immunotherapy. Front. Oncol. 2021, 11, 673276. [Google Scholar] [CrossRef]
- Vaněk, O.; Kalousková, B.; Abreu, C.; Nejadebrahim, S.; Skořepa, O. Natural Killer Cell-Based Strategies for Immunotherapy of Cancer. Adv. Protein Chem. Struct. Biol. 2022, 129, 91–133. [Google Scholar] [CrossRef]
- Basar, R.; Daher, M.; Rezvani, K. Next-Generation Cell Therapies: The Emerging Role of CAR-NK Cells. Blood Adv. 2020, 4, 5868–5876. [Google Scholar] [CrossRef]
- Ao, X.; Yang, Y.; Li, W.; Tan, Y.; Guo, W.; Ao, L.; He, X.; Wu, X.; Xia, J.; Xu, X.; et al. Anti-αFR CAR-Engineered NK-92 Cells Display Potent Cytotoxicity Against αFR-Positive Ovarian Cancer. J. Immunother. 2019, 42, 284–296. [Google Scholar] [CrossRef]
- Ingegnere, T.; Mariotti, F.R.; Pelosi, A.; Quintarelli, C.; De Angelis, B.; Tumino, N.; Besi, F.; Cantoni, C.; Locatelli, F.; Vacca, P.; et al. Human CAR NK Cells: A New Non-Viral Method Allowing High Efficient Transfection and Strong Tumor Cell Killing. Front. Immunol. 2019, 10, 957. [Google Scholar] [CrossRef]
- Jamali, A.; Hadjati, J.; Madjd, Z.; Mirzaei, H.R.; Thalheimer, F.B.; Agarwal, S.; Bonig, H.; Ullrich, E.; Hartmann, J. Highly Efficient Generation of Transgenically Augmented CAR NK Cells Overexpressing CXCR4. Front. Immunol. 2020, 11, 2028. [Google Scholar] [CrossRef]
- Zhang, Q.; Zhang, H.; Ding, J.; Liu, H.; Li, H.; Li, H.; Lu, M.; Miao, Y.; Li, L.; Zheng, J. Combination Therapy with EpCAM-CAR-NK-92 Cells and Regorafenib against Human Colorectal Cancer Models. J. Immunol. Res. 2018, 2018, 4263520. [Google Scholar] [CrossRef]
- Müller, S.; Bexte, T.; Gebel, V.; Kalensee, F.; Stolzenberg, E.; Hartmann, J.; Koehl, U.; Schambach, A.; Wels, W.S.; Modlich, U.; et al. High Cytotoxic Efficiency of Lentivirally and Alpharetrovirally Engineered CD19-Specific Chimeric Antigen Receptor Natural Killer Cells Against Acute Lymphoblastic Leukemia. Front. Immunol. 2020, 10, 3123. [Google Scholar] [CrossRef]
- Lee, M.Y.; Robbins, Y.; Sievers, C.; Friedman, J.; Abdul Sater, H.; Clavijo, P.E.; Judd, N.; Tsong, E.; Silvin, C.; Soon-Shiong, P.; et al. Chimeric Antigen Receptor Engineered NK Cellular Immunotherapy Overcomes the Selection of T-Cell Escape Variant Cancer Cells. J. Immunother. Cancer 2021, 9, e002128. [Google Scholar] [CrossRef] [PubMed]
- Daher, M.; Rezvani, K. Outlook for New CAR-Based Therapies with a Focus on CAR NK Cells: What Lies Beyond CAR-Engineered T Cells in the Race against Cancer. Cancer Discov. 2021, 11, 45–58. [Google Scholar] [CrossRef]
- Töpfer, K.; Cartellieri, M.; Michen, S.; Wiedemuth, R.; Müller, N.; Lindemann, D.; Bachmann, M.; Füssel, M.; Schackert, G.; Temme, A. DAP12-Based Activating Chimeric Antigen Receptor for NK Cell Tumor Immunotherapy. J. Immunol. 2015, 194, 3201–3212. [Google Scholar] [CrossRef] [PubMed]
- Upshaw, J.L.; Arneson, L.N.; Schoon, R.A.; Dick, C.J.; Billadeau, D.D.; Leibson, P.J. NKG2D-Mediated Signaling Requires a DAP10-Bound Grb2-Vav1 Intermediate and Phosphatidylinositol-3-Kinase in Human Natural Killer Cells. Nat. Immunol. 2006, 7, 524–532. [Google Scholar] [CrossRef]
- Xu, Y.; Liu, Q.; Zhong, M.; Wang, Z.; Chen, Z.; Zhang, Y.; Xing, H.; Tian, Z.; Tang, K.; Liao, X.; et al. 2B4 Costimulatory Domain Enhancing Cytotoxic Ability of Anti-CD5 Chimeric Antigen Receptor Engineered Natural Killer Cells against T Cell Malignancies. J. Hematol. Oncol. 2019, 12, 49. [Google Scholar] [CrossRef] [PubMed]
- Ebrahimiyan, H.; Tamimi, A.; Shokoohian, B.; Minaei, N.; Memarnejadian, A.; Hossein-Khannazer, N.; Hassan, M.; Vosough, M. Novel Insights in CAR-NK Cells beyond CAR-T Cell Technology; Promising Advantages. Int. Immunopharmacol. 2022, 106, 108587. [Google Scholar] [CrossRef] [PubMed]
- Cao, B.; Liu, M.; Wang, L.; Liang, B.; Feng, Y.; Chen, X.; Shi, Y.; Zhang, J.; Ye, X.; Tian, Y.; et al. Use of Chimeric Antigen Receptor NK-92 Cells to Target Mesothelin in Ovarian Cancer. Biochem. Biophys. Res. Commun. 2020, 524, 96–102. [Google Scholar] [CrossRef]
- Cienfuegos-Jimenez, O.; Vazquez-Garza, E.; Rojas-Martinez, A. CAR-NK Cells for Cancer Therapy: Molecular Redesign of the Innate Antineoplastic Response. Curr. Gene Ther. 2022, 22, 303–318. [Google Scholar] [CrossRef]
- Klingemann, H. Are Natural Killer Cells Superior CAR Drivers? Oncoimmunology 2014, 3, e28147. [Google Scholar] [CrossRef] [PubMed]
- Hunter, B.D.; Jacobson, C.A. CAR T-Cell Associated Neurotoxicity: Mechanisms, Clinicopathologic Correlates, and Future Directions. J. Natl. Cancer Inst. 2019, 111, 646–654. [Google Scholar] [CrossRef] [PubMed]
- Fehniger, T.A.; Cooper, M.A. Harnessing NK Cell Memory for Cancer Immunotherapy. Trends Immunol. 2016, 37, 877–888. [Google Scholar] [CrossRef] [PubMed]
- Gang, M.; Marin, N.D.; Wong, P.; Neal, C.C.; Marsala, L.; Foster, M.; Schappe, T.; Meng, W.; Tran, J.; Schaettler, M.; et al. CAR-Modified Memory-like NK Cells Exhibit Potent Responses to NK-Resistant Lymphomas. Blood 2020, 136, 2308–2318. [Google Scholar] [CrossRef]
- Paust, S.; von Andrian, U.H. Natural Killer Cell Memory. Nat. Immunol. 2011, 12, 500–508. [Google Scholar] [CrossRef]
- Shoae-Hassani, A.; Behfar, M.; Mortazavi-Tabatabaei, S.A.; Ai, J.; Mohseni, R.; Hamidieh, A.A. Natural Killer Cells from the Subcutaneous Adipose Tissue Underexpress the NKp30 and NKp44 in Obese Persons and Are Less Active against Major Histocompatibility Complex Class I Non-Expressing Neoplastic Cells. Front. Immunol. 2017, 8, 1486. [Google Scholar] [CrossRef]
- Martín-Antonio, B.; Suñe, G.; Perez-Amill, L.; Castella, M.; Urbano-Ispizua, A. Natural Killer Cells: Angels and Devils for Immunotherapy. Int. J. Mol. Sci. 2017, 18, 1868. [Google Scholar] [CrossRef]
- Bae, D.S.; Lee, J.K. Development of NK Cell Expansion Methods Using Feeder Cells from Human Myelogenous Leukemia Cell Line. Blood Res. 2014, 49, 154–161. [Google Scholar] [CrossRef]
- Oran, B.; Shpall, E. Umbilical Cord Blood Transplantation: A Maturing Technology. Hematol. Am. Soc. Hematol. Educ. Program 2012, 2012, 215–222. [Google Scholar] [CrossRef]
- Nomura, A.; Takada, H.; Jin, C.H.; Tanaka, T.; Ohga, S.; Hara, T. Functional Analyses of Cord Blood Natural Killer Cells and T Cells: A Distinctive Interleukin-18 Response. Exp. Hematol. 2001, 29, 1169–1176. [Google Scholar] [CrossRef]
- Luevano, M.; Daryouzeh, M.; Alnabhan, R.; Querol, S.; Khakoo, S.; Madrigal, A.; Saudemont, A. The Unique Profile of Cord Blood Natural Killer Cells Balances Incomplete Maturation and Effective Killing Function upon Activation. Hum. Immunol. 2012, 73, 248–257. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, H.; Kai, S.; Yamaguchi, M.; Misawa, M.; Fujimori, Y.; Yamamoto, M.; Hara, H. Analysis of Natural Killer (NK) Cell Activity and Adhesion Molecules on NK Cells from Umbilical Cord Blood. Eur. J. Haematol. 2003, 71, 29–38. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.; Tian, Z.; Zhang, C. Natural Killer Cell-Based Immunotherapy for Cancer: Advances and Prospects. Proc. Est. Acad. Sci. Eng. 2019, 5, 106–114. [Google Scholar] [CrossRef]
- Zhu, H.; Kaufman, D.S. An Improved Method to Produce Clinical-Scale Natural Killer Cells from Human Pluripotent Stem Cells. Methods Mol. Biol. 2019, 2048, 107–119. [Google Scholar] [CrossRef]
- Bernareggi, D.; Pouyanfard, S.; Kaufman, D.S. Development of Innate Immune Cells from Human Pluripotent Stem Cells. Exp. Hematol. 2019, 71, 13–23. [Google Scholar] [CrossRef]
- Saetersmoen, M.L.; Hammer, Q.; Valamehr, B.; Kaufman, D.S.; Malmberg, K.-J. Off-the-Shelf Cell Therapy with Induced Pluripotent Stem Cell-Derived Natural Killer Cells. Semin. Immunopathol. 2019, 41, 59–68. [Google Scholar] [CrossRef]
- Yagita, M.; Huang, C.L.; Umehara, H.; Matsuo, Y.; Tabata, R.; Miyake, M.; Konaka, Y.; Takatsuki, K. A Novel Natural Killer Cell Line (KHYG-1) from a Patient with Aggressive Natural Killer Cell Leukemia Carrying a p53 Point Mutation. Leukemia 2000, 14, 922–930. [Google Scholar] [CrossRef]
- Klingemann, H.; Boissel, L.; Toneguzzo, F. Natural Killer Cells for Immunotherapy—Advantages of the NK-92 Cell Line over Blood NK Cells. Front. Immunol. 2016, 7, 91. [Google Scholar] [CrossRef]
- Zhang, C.; Oberoi, P.; Oelsner, S.; Waldmann, A.; Lindner, A.; Tonn, T.; Wels, W.S. Chimeric Antigen Receptor-Engineered NK-92 Cells: An Off-the-Shelf Cellular Therapeutic for Targeted Elimination of Cancer Cells and Induction of Protective Antitumor Immunity. Front. Immunol. 2017, 8, 533. [Google Scholar] [CrossRef] [PubMed]
- Hermanson, D.L.; Kaufman, D.S. Utilizing Chimeric Antigen Receptors to Direct Natural Killer Cell Activity. Front. Immunol. 2015, 6, 195. [Google Scholar] [CrossRef]
- Klingemann, H.G.; Wong, E.; Maki, G. A Cytotoxic NK-Cell Line (NK-92) for Ex Vivo Purging of Leukemia from Blood. Biol. Blood Marrow Transplant. 1996, 2, 68–75. [Google Scholar] [PubMed]
- Karagiannis, P.; Kim, S.-I. iPSC-Derived Natural Killer Cells for Cancer Immunotherapy. Mol. Cells 2021, 44, 541–548. [Google Scholar] [CrossRef] [PubMed]
- Klöß, S.; Oberschmidt, O.; Morgan, M.; Dahlke, J.; Arseniev, L.; Huppert, V.; Granzin, M.; Gardlowski, T.; Matthies, N.; Soltenborn, S.; et al. Optimization of Human NK Cell Manufacturing: Fully Automated Separation, Improved Ex Vivo Expansion Using IL-21 with Autologous Feeder Cells, and Generation of Anti-CD123-CAR-Expressing Effector Cells. Hum. Gene Ther. 2017, 28, 897–913. [Google Scholar] [CrossRef]
- Tanaka, J.; Miller, J.S. Recent Progress in and Challenges in Cellular Therapy Using NK Cells for Hematological Malignancies. Blood Rev. 2020, 44, 100678. [Google Scholar] [CrossRef] [PubMed]
- Pedroza-Pacheco, I.; Madrigal, A.; Saudemont, A. Interaction between Natural Killer Cells and Regulatory T Cells: Perspectives for Immunotherapy. Cell. Mol. Immunol. 2013, 10, 222–229. [Google Scholar] [CrossRef]
- Waldmann, T.A.; Lugli, E.; Roederer, M.; Perera, L.P.; Smedley, J.V.; Macallister, R.P.; Goldman, C.K.; Bryant, B.R.; Decker, J.M.; Fleisher, T.A.; et al. Safety (toxicity), Pharmacokinetics, Immunogenicity, and Impact on Elements of the Normal Immune System of Recombinant Human IL-15 in Rhesus Macaques. Blood 2011, 117, 4787–4795. [Google Scholar] [CrossRef]
- Liu, E.; Tong, Y.; Dotti, G.; Savoldo, B.; Muftuoglu, M.; Kondo, K.; Mukherjee, M.; Orange, J.S.; Sobieski, C.; Alsuliman, A.; et al. Cord Blood Derived Natural Killer Cells Engineered with a Chimeric Antigen Receptor Targeting CD19 and Expressing IL-15 Have Long Term Persistence and Exert Potent Anti-Leukemia Activity. Blood 2015, 126, 3091. [Google Scholar] [CrossRef]
- Liu, E.; Marin, D.; Banerjee, P.; Macapinlac, H.A.; Thompson, P.; Basar, R.; Nassif Kerbauy, L.; Overman, B.; Thall, P.; Kaplan, M.; et al. Use of CAR-Transduced Natural Killer Cells in CD19-Positive Lymphoid Tumors. N. Engl. J. Med. 2020, 382, 545–553. [Google Scholar] [CrossRef]
- Zhang, L.; Meng, Y.; Feng, X.; Han, Z. CAR-NK Cells for Cancer Immunotherapy: From Bench to Bedside. Biomark. Res. 2022, 10, 12. [Google Scholar] [CrossRef]
- Luanpitpong, S.; Poohadsuan, J.; Klaihmon, P.; Issaragrisil, S. Selective Cytotoxicity of Single and Dual Anti-CD19 and Anti-CD138 Chimeric Antigen Receptor-Natural Killer Cells against Hematologic Malignancies. J. Immunol. Res. 2021, 2021, 5562630. [Google Scholar] [CrossRef] [PubMed]
- Daldrup-Link, H.E.; Meier, R.; Rudelius, M.; Piontek, G.; Piert, M.; Metz, S.; Settles, M.; Uherek, C.; Wels, W.; Schlegel, J.; et al. In Vivo Tracking of Genetically Engineered, Anti-HER2/neu Directed Natural Killer Cells to HER2/neu Positive Mammary Tumors with Magnetic Resonance Imaging. Eur. Radiol. 2005, 15, 4–13. [Google Scholar] [CrossRef]
- Schönfeld, K.; Sahm, C.; Zhang, C.; Naundorf, S.; Brendel, C.; Odendahl, M.; Nowakowska, P.; Bönig, H.; Köhl, U.; Kloess, S.; et al. Selective Inhibition of Tumor Growth by Clonal NK Cells Expressing an ErbB2/HER2-Specific Chimeric Antigen Receptor. Mol. Ther. 2015, 23, 330–338. [Google Scholar] [CrossRef]
- Eitler, J.; Wotschel, N.; Miller, N.; Boissel, L.; Klingemann, H.G.; Wels, W.; Tonn, T. Inability of Granule Polarization by NK Cells Defines Tumor Resistance and Can Be Overcome by CAR or ADCC Mediated Targeting. J. Immunother. Cancer 2021, 9, e001334. [Google Scholar] [CrossRef]
- Zhang, C.; Röder, J.; Scherer, A.; Bodden, M.; Pfeifer Serrahima, J.; Bhatti, A.; Waldmann, A.; Müller, N.; Oberoi, P.; Wels, W.S. Bispecific Antibody-Mediated Redirection of NKG2D-CAR Natural Killer Cells Facilitates Dual Targeting and Enhances Antitumor Activity. J. Immunother. Cancer 2021, 9, e002980. [Google Scholar] [CrossRef]
- Intracranial Injection of NK-92/5.28.Z Cells in Combination with Intravenous Ezabenlimab in Patients with Recurrent HER2-Positive Glioblastoma—No Study Results Posted—ClinicalTrials.Gov. Available online: https://clinicaltrials.gov/ct2/show/results/NCT03383978?term=CAR+NK+cells&draw=2&rank=62 (accessed on 14 February 2023).
- NKG2D-CAR-NK92 Cells Immunotherapy for Solid Tumors. Available online: https://clinicaltrials.gov/ct2/show/NCT05528341?term=chimeric+antigen+receptor+NK+cells&draw=2&rank=22 (accessed on 14 February 2023).
- Study of Anti-5T4 CAR-raNK Cell Therapy in Locally Advanced or Metastatic Solid Tumors. Available online: https://clinicaltrials.gov/ct2/show/NCT05137275?term=CAR+NK+cells&recrs=abde&draw=2&rank=48 (accessed on 14 February 2023).
- Single-Arm. Open-Label Clinical Study of SZ011 in the Treatment of Advanced Triple Negative Breast Cancer. Available online: https://clinicaltrials.gov/ct2/show/NCT05686720?term=CAR+NK&draw=3&rank=37 (accessed on 14 February 2023).
- Moreno, C.; Haynie, C.; Johnson, A.; Weber, K.S. Alternative CAR Therapies: Recent Approaches in Engineering Chimeric Antigen Receptor Immune Cells to Combat Cancer. Biomedicines 2022, 10, 1493. [Google Scholar] [CrossRef]
- Fujiwara, N.; Kobayashi, K. Macrophages in Inflammation. Curr. Drug Targets Inflamm. Allergy 2005, 4, 281–286. [Google Scholar] [CrossRef]
- Shapouri-Moghaddam, A.; Mohammadian, S.; Vazini, H.; Taghadosi, M.; Esmaeili, S.-A.; Mardani, F.; Seifi, B.; Mohammadi, A.; Afshari, J.T.; Sahebkar, A. Macrophage Plasticity, Polarization, and Function in Health and Disease. J. Cell. Physiol. 2018, 233, 6425–6440. [Google Scholar] [CrossRef]
- Jaguin, M.; Houlbert, N.; Fardel, O.; Lecureur, V. Polarization Profiles of Human M-CSF-Generated Macrophages and Comparison of M1-Markers in Classically Activated Macrophages from GM-CSF and M-CSF Origin. Cell. Immunol. 2013, 281, 51–61. [Google Scholar] [CrossRef]
- Fleetwood, A.J.; Lawrence, T.; Hamilton, J.A.; Cook, A.D. Granulocyte-Macrophage Colony-Stimulating Factor (CSF) and Macrophage CSF-Dependent Macrophage Phenotypes Display Differences in Cytokine Profiles and Transcription Factor Activities: Implications for CSF Blockade in Inflammation. J. Immunol. 2007, 178, 5245–5252. [Google Scholar] [CrossRef]
- Anderson, N.R.; Minutolo, N.G.; Gill, S.; Klichinsky, M. Macrophage-Based Approaches for Cancer Immunotherapy. Cancer Res. 2021, 81, 1201–1208. [Google Scholar] [CrossRef]
- Stein, M.; Keshav, S.; Harris, N.; Gordon, S. Interleukin 4 Potently Enhances Murine Macrophage Mannose Receptor Activity: A Marker of Alternative Immunologic Macrophage Activation. J. Exp. Med. 1992, 176, 287–292. [Google Scholar] [CrossRef]
- Gordon, S.; Martinez, F.O. Alternative Activation of Macrophages: Mechanism and Functions. Immunity 2010, 32, 593–604. [Google Scholar] [CrossRef]
- Doherty, T.M.; Kastelein, R.; Menon, S.; Andrade, S.; Coffman, R.L. Modulation of Murine Macrophage Function by IL-13. J. Immunol. 1993, 151, 7151–7160. [Google Scholar] [CrossRef]
- Zhang, F.; Wang, H.; Wang, X.; Jiang, G.; Liu, H.; Zhang, G.; Wang, H.; Fang, R.; Bu, X.; Cai, S.; et al. TGF-β Induces M2-like Macrophage Polarization via SNAIL-Mediated Suppression of a pro-Inflammatory Phenotype. Oncotarget 2016, 7, 52294–52306. [Google Scholar] [CrossRef]
- Mantovani, A.; Sozzani, S.; Locati, M.; Allavena, P.; Sica, A. Macrophage Polarization: Tumor-Associated Macrophages as a Paradigm for Polarized M2 Mononuclear Phagocytes. Trends Immunol. 2002, 23, 549–555. [Google Scholar] [CrossRef]
- Yang, L.; Zhang, Y. Tumor-Associated Macrophages: From Basic Research to Clinical Application. J. Hematol. Oncol. 2017, 10, 58. [Google Scholar] [CrossRef]
- Qian, B.-Z.; Li, J.; Zhang, H.; Kitamura, T.; Zhang, J.; Campion, L.R.; Kaiser, E.A.; Snyder, L.A.; Pollard, J.W. CCL2 Recruits Inflammatory Monocytes to Facilitate Breast-Tumour Metastasis. Nature 2011, 475, 222–225. [Google Scholar] [CrossRef]
- Sanford, D.E.; Belt, B.A.; Panni, R.Z.; Mayer, A.; Deshpande, A.D.; Carpenter, D.; Mitchem, J.B.; Plambeck-Suess, S.M.; Worley, L.A.; Goetz, B.D.; et al. Inflammatory Monocyte Mobilization Decreases Patient Survival in Pancreatic Cancer: A Role for Targeting the CCL2/CCR2 Axis. Clin. Cancer Res. 2013, 19, 3404–3415. [Google Scholar] [CrossRef]
- Zhao, L.; Lim, S.Y.; Gordon-Weeks, A.N.; Tapmeier, T.T.; Im, J.H.; Cao, Y.; Beech, J.; Allen, D.; Smart, S.; Muschel, R.J. Recruitment of a Myeloid Cell Subset (CD11b/Gr1 Mid) via CCL2/CCR2 Promotes the Development of Colorectal Cancer Liver Metastasis. Hepatology 2013, 57, 829–839. [Google Scholar] [CrossRef]
- Peña, C.G.; Nakada, Y.; Saatcioglu, H.D.; Aloisio, G.M.; Cuevas, I.; Zhang, S.; Miller, D.S.; Lea, J.S.; Wong, K.-K.; DeBerardinis, R.J.; et al. LKB1 Loss Promotes Endometrial Cancer Progression via CCL2-Dependent Macrophage Recruitment. J. Clin. Investig. 2015, 125, 4063–4076. [Google Scholar] [CrossRef]
- Jahchan, N.S.; Mujal, A.M.; Pollack, J.L.; Binnewies, M.; Sriram, V.; Reyno, L.; Krummel, M.F. Tuning the Tumor Myeloid Microenvironment to Fight Cancer. Front. Immunol. 2019, 10, 1611. [Google Scholar] [CrossRef]
- De Henau, O.; Rausch, M.; Winkler, D.; Campesato, L.F.; Liu, C.; Cymerman, D.H.; Budhu, S.; Ghosh, A.; Pink, M.; Tchaicha, J.; et al. Overcoming Resistance to Checkpoint Blockade Therapy by Targeting PI3Kγ in Myeloid Cells. Nature 2016, 539, 443–447. [Google Scholar] [CrossRef]
- Kaneda, M.M.; Messer, K.S.; Ralainirina, N.; Li, H.; Leem, C.J.; Gorjestani, S.; Woo, G.; Nguyen, A.V.; Figueiredo, C.C.; Foubert, P.; et al. PI3Kγ Is a Molecular Switch That Controls Immune Suppression. Nature 2016, 539, 437–442. [Google Scholar] [CrossRef]
- Georgoudaki, A.-M.; Prokopec, K.E.; Boura, V.F.; Hellqvist, E.; Sohn, S.; Östling, J.; Dahan, R.; Harris, R.A.; Rantalainen, M.; Klevebring, D.; et al. Reprogramming Tumor-Associated Macrophages by Antibody Targeting Inhibits Cancer Progression and Metastasis. Cell Rep. 2016, 15, 2000–2011. [Google Scholar] [CrossRef]
- Guerriero, J.L.; Sotayo, A.; Ponichtera, H.E.; Castrillon, J.A.; Pourzia, A.L.; Schad, S.; Johnson, S.F.; Carrasco, R.D.; Lazo, S.; Bronson, R.T.; et al. Class IIa HDAC Inhibition Reduces Breast Tumours and Metastases through Anti-Tumour Macrophages. Nature 2017, 543, 428–432. [Google Scholar] [CrossRef]
- Schmid, M.C.; Khan, S.Q.; Kaneda, M.M.; Pathria, P.; Shepard, R.; Louis, T.L.; Anand, S.; Woo, G.; Leem, C.; Faridi, M.H.; et al. Integrin CD11b Activation Drives Anti-Tumor Innate Immunity. Nat. Commun. 2018, 9, 5379. [Google Scholar] [CrossRef]
- Morrissey, M.A.; Williamson, A.P.; Steinbach, A.M.; Roberts, E.W.; Kern, N.; Headley, M.B.; Vale, R.D. Chimeric Antigen Receptors That Trigger Phagocytosis. elife 2018, 7, e36688. [Google Scholar] [CrossRef]
- Klichinsky, M.; Ruella, M.; Shestova, O.; Lu, X.M.; Best, A.; Zeeman, M.; Schmierer, M.; Gabrusiewicz, K.; Anderson, N.R.; Petty, N.E.; et al. Human Chimeric Antigen Receptor Macrophages for Cancer Immunotherapy. Nat. Biotechnol. 2020, 38, 947–953. [Google Scholar] [CrossRef]
- Pierini, S.; Gabbasov, R.; Gabitova, L.; Ohtani, Y.; Shestova, O.; Gill, S.; Abramson, S.; Condamine, T.; Klichinsky, M. Abstract 63: Chimeric Antigen Receptor Macrophages (CAR-M) Induce Anti-Tumor Immunity and Synergize with T Cell Checkpoint Inhibitors in Pre-Clinical Solid Tumor Models. Cancer Res. 2021, 81, 63. [Google Scholar] [CrossRef]
- CAR-Macrophages for the Treatment of HER2 Overexpressing Solid Tumors. Available online: https://clinicaltrials.gov/ct2/show/NCT04660929 (accessed on 14 February 2023).
- Cohort Study to Determine the Antitumor Activity of New CAR-Macrophages in Breast Cancer Patients’ Derived Organoids—Full Text View—ClinicalTrials.Gov. Available online: https://clinicaltrials.gov/ct2/show/NCT05007379 (accessed on 14 February 2023).
- Wynn, T.A.; Vannella, K.M. Macrophages in Tissue Repair, Regeneration, and Fibrosis. Immunity 2016, 44, 450–462. [Google Scholar] [CrossRef]
- Zhang, W.; Liu, L.; Su, H.; Liu, Q.; Shen, J.; Dai, H.; Zheng, W.; Lu, Y.; Zhang, W.; Bei, Y.; et al. Chimeric Antigen Receptor Macrophage Therapy for Breast Tumours Mediated by Targeting the Tumour Extracellular Matrix. Br. J. Cancer 2019, 121, 837–845. [Google Scholar] [CrossRef] [PubMed]
- Handy, C.E.; Antonarakis, E.S. Sipuleucel-T for the Treatment of Prostate Cancer: Novel Insights and Future Directions. Future Oncol. 2018, 14, 907–917. [Google Scholar] [CrossRef] [PubMed]
- Al-Awadhi, A.; Lee Murray, J.; Ibrahim, N.K. Developing Anti-HER2 Vaccines: Breast Cancer Experience. Int. J. Cancer 2018, 143, 2126–2132. [Google Scholar] [CrossRef]
- Lollini, P.-L.; Cavallo, F.; Nanni, P.; Forni, G. Vaccines for Tumour Prevention. Nat. Rev. Cancer 2006, 6, 204–216. [Google Scholar] [CrossRef]
- Pallerla, S.; Abdul, A.U.R.M.; Comeau, J.; Jois, S. Cancer Vaccines, Treatment of the Future: With Emphasis on HER2-Positive Breast Cancer. Int. J. Mol. Sci. 2021, 22, 779. [Google Scholar] [CrossRef] [PubMed]
- Gajria, D.; Chandarlapaty, S. HER2-Amplified Breast Cancer: Mechanisms of Trastuzumab Resistance and Novel Targeted Therapies. Expert Rev. Anticancer Ther. 2011, 11, 263–275. [Google Scholar] [CrossRef]
- Crosby, E.J.; Gwin, W.; Blackwell, K.; Marcom, P.K.; Chang, S.; Maecker, H.T.; Broadwater, G.; Hyslop, T.; Kim, S.; Rogatko, A.; et al. Vaccine-Induced Memory CD8 T Cells Provide Clinical Benefit in HER2 Expressing Breast Cancer: A Mouse to Human Translational Study. Clin. Cancer Res. 2019, 25, 2725–2736. [Google Scholar] [CrossRef] [PubMed]
- Koski, G.K.; Cohen, P.A.; Roses, R.E.; Xu, S.; Czerniecki, B.J. Reengineering Dendritic Cell-Based Anti-Cancer Vaccines. Immunol. Rev. 2008, 222, 256–276. [Google Scholar] [CrossRef]
- Rosenberg, S.A.; Yang, J.C.; Restifo, N.P. Cancer Immunotherapy: Moving beyond Current Vaccines. Nat. Med. 2004, 10, 909–915. [Google Scholar] [CrossRef] [PubMed]
- Badve, S.S.; Gökmen-Polar, Y. Ductal Carcinoma In Situ of Breast: Update 2019. Pathology 2019, 51, 563–569. [Google Scholar] [CrossRef]
- Xu, S.; Koldovsky, U.; Xu, M.; Wang, D.; Fitzpatrick, E.; Son, G.; Koski, G.; Czerniecki, B.J. High-Avidity Antitumor T-Cell Generation by Toll Receptor 8-Primed, Myeloid- Derived Dendritic Cells Is Mediated by IL-12 Production. Surgery 2006, 140, 170–178. [Google Scholar] [CrossRef]
- Xu, S.; Koski, G.K.; Faries, M.; Bedrosian, I.; Mick, R.; Maeurer, M.; Cheever, M.A.; Cohen, P.A.; Czerniecki, B.J. Rapid High Efficiency Sensitization of CD8+ T Cells to Tumor Antigens by Dendritic Cells Leads to Enhanced Functional Avidity and Direct Tumor Recognition through an IL-12-Dependent Mechanism. J. Immunol. 2003, 171, 2251–2261. [Google Scholar] [CrossRef]
- Schmidt, C.S.; Mescher, M.F. Peptide Antigen Priming of Naive, But Not Memory, CD8 T Cells Requires a Third Signal That Can Be Provided by IL-12. J. Immunol. 2002, 168, 5521–5529. [Google Scholar] [CrossRef] [PubMed]
- Czerniecki, B.J.; Koski, G.K.; Koldovsky, U.; Xu, S.; Cohen, P.A.; Mick, R.; Nisenbaum, H.; Pasha, T.; Xu, M.; Fox, K.R.; et al. Targeting HER-2/neu in Early Breast Cancer Development Using Dendritic Cells with Staged Interleukin-12 Burst Secretion. Cancer Res. 2007, 67, 1842–1852. [Google Scholar] [CrossRef]
- Perez, S.A.; Sotiropoulou, P.A.; Sotiriadou, N.N.; Mamalaki, A.; Gritzapis, A.D.; Echner, H.; Voelter, W.; Pawelec, G.; Papamichail, M.; Baxevanis, C.N. HER-2/neu-Derived Peptide 884–899 Is Expressed by Human Breast, Colorectal and Pancreatic Adenocarcinomas and Is Recognized by in-Vitro-Induced Specific CD4 T Cell Clones. Cancer Immunol. Immunother. 2002, 50, 615–624. [Google Scholar] [CrossRef]
- Kristensen, V.N.; Vaske, C.J.; Ursini-Siegel, J.; Van Loo, P.; Nordgard, S.H.; Sachidanandam, R.; Sørlie, T.; Wärnberg, F.; Haakensen, V.D.; Helland, Å.; et al. Integrated Molecular Profiles of Invasive Breast Tumors and Ductal Carcinoma In Situ (DCIS) Reveal Differential Vascular and Interleukin Signaling. Proc. Natl. Acad. Sci. USA 2012, 109, 2802–2807. [Google Scholar] [CrossRef]
- Koski, G.K.; Koldovsky, U.; Xu, S.; Mick, R.; Sharma, A.; Fitzpatrick, E.; Weinstein, S.; Nisenbaum, H.; Levine, B.L.; Fox, K.; et al. A Novel Dendritic Cell-Based Immunization Approach for the Induction of Durable Th1-Polarized Anti-HER-2/neu Responses in Women with Early Breast Cancer. J. Immunother. 2012, 35, 54–65. [Google Scholar] [CrossRef]
- Sharma, A.; Koldovsky, U.; Xu, S.; Mick, R.; Roses, R.; Fitzpatrick, E.; Weinstein, S.; Nisenbaum, H.; Levine, B.L.; Fox, K.; et al. HER-2 Pulsed Dendritic Cell Vaccine Can Eliminate HER-2 Expression and Impact Ductal Carcinoma in Situ. Cancer 2012, 118, 4354–4362. [Google Scholar] [CrossRef] [PubMed]
- Disis, M.L.N.; Guthrie, K.A.; Liu, Y.; Coveler, A.L.; Higgins, D.M.; Childs, J.S.; Dang, Y.; Salazar, L.G. Safety and Outcomes of a Plasmid DNA Vaccine Encoding the ERBB2 Intracellular Domain in Patients With Advanced-Stage ERBB2-Positive Breast Cancer: A Phase 1 Nonrandomized Clinical Trial. JAMA Oncol. 2023, 9, 71–78. [Google Scholar] [CrossRef] [PubMed]
- Knutson, K.L.; Schiffman, K.; Cheever, M.A.; Disis, M.L. Immunization of Cancer Patients with a HER-2/neu, HLA-A2 Peptide, p369-377, Results in Short-Lived Peptide-Specific Immunity. Clin. Cancer Res. 2002, 8, 1014–1018. [Google Scholar]
- Disis, M.L.; Schiffman, K.; Guthrie, K.; Salazar, L.G.; Knutson, K.L.; Goodell, V.; dela Rosa, C.; Cheever, M.A. Effect of Dose on Immune Response in Patients Vaccinated with an Her-2/neu Intracellular Domain Protein--Based Vaccine. J. Clin. Oncol. 2004, 22, 1916–1925. [Google Scholar] [CrossRef] [PubMed]
- Disis, M.L.; Scholler, N.; Dahlin, A.; Pullman, J.; Knutson, K.L.; Hellström, K.-E.; Hellström, I. Plasmid-Based Vaccines Encoding Rat Neu and Immune Stimulatory Molecules Can Elicit Rat Neu-Specific Immunity. Mol. Cancer Ther. 2003, 2, 995–1002. [Google Scholar] [PubMed]
- Gall, V.A.; Philips, A.V.; Qiao, N.; Clise-Dwyer, K.; Perakis, A.A.; Zhang, M.; Clifton, G.T.; Sukhumalchandra, P.; Ma, Q.; Reddy, S.M.; et al. Trastuzumab Increases HER2 Uptake and Cross-Presentation by Dendritic Cells. Cancer Res. 2017, 77, 5374–5383. [Google Scholar] [CrossRef] [PubMed]
- Mittendorf, E.A.; Storrer, C.; Shriver, C.; Ponniah, S.; Peoples, G. Evaluation of the HER2/neu-Derived Peptide GP2 for Use in a Peptide-Based Breast Cancer Vaccine Trial. J. Am. Coll. Surg. 2005, 201, S79. [Google Scholar] [CrossRef]
- Ferris, R.L.; Jaffee, E.M.; Ferrone, S. Tumor Antigen-Targeted, Monoclonal Antibody-Based Immunotherapy: Clinical Response, Cellular Immunity, and Immunoescape. J. Clin. Oncol. 2010, 28, 4390–4399. [Google Scholar] [CrossRef]
- Clifton, G.T.; Hale, D.; Vreeland, T.J.; Hickerson, A.T.; Litton, J.K.; Alatrash, G.; Murthy, R.K.; Qiao, N.; Philips, A.V.; Lukas, J.J.; et al. Results of a Randomized Phase IIb Trial of Nelipepimut-S + Trastuzumab versus Trastuzumab to Prevent Recurrences in Patients with High-Risk HER2 Low-Expressing Breast Cancer. Clin. Cancer Res. 2020, 26, 2515–2523. [Google Scholar] [CrossRef]
- Zhu, S.-Y.; Yu, K.-D. Breast Cancer Vaccines: Disappointing or Promising? Front. Immunol. 2022, 13, 190. [Google Scholar] [CrossRef]
- Holmes, J.P.; Clifton, G.T.; Patil, R.; Benavides, L.C.; Gates, J.D.; Stojadinovic, A.; Mittendorf, E.A.; Ponniah, S.; Peoples, G.E. Use of Booster Inoculations to Sustain the Clinical Effect of an Adjuvant Breast Cancer Vaccine: From US Military Cancer Institute Clinical Trials Group Study I-01 and I-02. Cancer 2011, 117, 463–471. [Google Scholar] [CrossRef]
- Gasparri, M.L.; Ruscito, I.; Taghavi, K.; Farooqi, A.A.; Papadia, A.; Focaccetti, C.; Barnaba, V.; Panici, P.B.; Mueller, M.D. The Immunobiology of Cancer: From Tumor Escape to Cancer Immunoediting Towards Immunotherapy in Gynecologic Oncology. In Molecular Oncology: Underlying Mechanisms and Translational Advancements; Springer: Cham, Switzerland, 2017; pp. 193–204. [Google Scholar]
- Azmi, A.S.; Bao, B.; Sarkar, F.H. Exosomes in Cancer Development, Metastasis, and Drug Resistance: A Comprehensive Review. Cancer Metast. Rev. 2013, 32, 623–642. [Google Scholar] [CrossRef]
- Petersen, K.E.; Manangon, E.; Hood, J.L.; Wickline, S.A.; Fernandez, D.P.; Johnson, W.P.; Gale, B.K. A Review of Exosome Separation Techniques and Characterization of B16-F10 Mouse Melanoma Exosomes with AF4-UV-MALS-DLS-TEM. Anal. Bioanal. Chem. 2014, 406, 7855–7866. [Google Scholar] [CrossRef]
- Phinney, D.G.; Pittenger, M.F. Concise Review: MSC-Derived Exosomes for Cell-Free Therapy. Stem Cells 2017, 35, 851–858. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, K.A.; Munegowda, M.A.; Xie, Y.; Xiang, J. Intercellular Trogocytosis Plays an Important Role in Modulation of Immune Responses. Cell. Mol. Immunol. 2008, 5, 261–269. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Xie, Y.; Ahmed, K.A.; Ahmed, S.; Sami, A.; Chibbar, R.; Xu, Q.; Kane, S.E.; Hao, S.; Mulligan, S.J.; et al. Exosomal pMHC-I Complex Targets T Cell-Based Vaccine to Directly Stimulate CTL Responses Leading to Antitumor Immunity in Transgenic FVBneuN and HLA-A2/HER2 Mice and Eradicating Trastuzumab-Resistant Tumor in Athymic Nude Mice. Breast Cancer Res. Treat. 2013, 140, 273–284. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.; Xu, A.; Zhang, X.; Wu, J.; Freywald, A.; Xu, J.; Xiang, J. Novel Exosome-Targeted T-Cell-Based Vaccine Counteracts T-Cell Anergy and Converts CTL Exhaustion in Chronic Infection via CD40L Signaling through the mTORC1 Pathway. Cell. Mol. Immunol. 2017, 14, 529–545. [Google Scholar] [CrossRef]
- Sabzevary-Ghahfarokhi, M.; Shirzad, H.; Rafieian-Kopaei, M.; Ghatreh-Samani, M.; Shohan, M. The Role of Inflammatory Cytokines in Creating T Cell Exhaustion in Cancer. Cancer Biother. Radiopharm. 2018, 33, 267–273. [Google Scholar] [CrossRef]
- Li, R.; Chibbar, R.; Xiang, J. Novel EXO-T Vaccine Using Polyclonal CD4 T Cells Armed with HER2-Specific Exosomes for HER2-Positive Breast Cancer. Onco. Targets Ther. 2018, 11, 7089–7093. [Google Scholar] [CrossRef]
- Degenhardt, K.; Mathew, R.; Beaudoin, B.; Bray, K.; Anderson, D.; Chen, G.; Mukherjee, C.; Shi, Y.; Gélinas, C.; Fan, Y.; et al. Autophagy Promotes Tumor Cell Survival and Restricts Necrosis, Inflammation, and Tumorigenesis. Cancer Cell 2006, 10, 51–64. [Google Scholar] [CrossRef]
- Bars, R.L.; Le Bars, R.; Marion, J.; Le Borgne, R.; Satiat-Jeunemaitre, B.; Bianchi, M.W. ATG5 Defines a Phagophore Domain Connected to the Endoplasmic Reticulum during Autophagosome Formation in Plants. Nat. Commun. 2014, 5, 4121. [Google Scholar] [CrossRef]
- Han, M.; Hu, J.; Lu, P.; Cao, H.; Yu, C.; Li, X.; Qian, X.; Yang, X.; Yang, Y.; Han, N.; et al. Exosome-Transmitted miR-567 Reverses Trastuzumab Resistance by Inhibiting ATG5 in Breast Cancer. Cell Death Dis. 2020, 11, 43. [Google Scholar] [CrossRef]
- Tan, A.; De La Peña, H.; Seifalian, A.M. The Application of Exosomes as a Nanoscale Cancer Vaccine. Int. J. Nanomed. 2010, 5, 889–900. [Google Scholar] [CrossRef]
- Wahlgren, J.; Karlson, T.D.L.; Brisslert, M.; Vaziri Sani, F.; Telemo, E.; Sunnerhagen, P.; Valadi, H. Plasma Exosomes Can Deliver Exogenous Short Interfering RNA to Monocytes and Lymphocytes. Nucleic Acids Res. 2012, 40, e130. [Google Scholar] [CrossRef] [PubMed]
- Limoni, S.K.; Moghadam, M.F.; Moazzeni, S.M.; Gomari, H.; Salimi, F. Engineered Exosomes for Targeted Transfer of siRNA to HER2 Positive Breast Cancer Cells. Appl. Biochem. Biotechnol. 2019, 187, 352–364. [Google Scholar] [CrossRef]
- Tang, X.; Huang, Y.; Lei, J.; Luo, H.; Zhu, X. The Single-Cell Sequencing: New Developments and Medical Applications. Cell Biosci. 2019, 9, 53. [Google Scholar] [CrossRef] [PubMed]
- Okamoto, S.; Shiga, T.; Tamaki, N. Clinical Perspectives of Theranostics. Molecules 2021, 26, 2232. [Google Scholar] [CrossRef] [PubMed]
- Williams, C.G.; Lee, H.J.; Asatsuma, T.; Vento-Tormo, R.; Haque, A. An Introduction to Spatial Transcriptomics for Biomedical Research. Genome Med. 2022, 14, 68. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, R.; Zaman, T.; Chowdhury, F.; Mraiche, F.; Tariq, M.; Ahmad, I.S.; Hasan, A. Single-Cell RNA Sequencing with Spatial Transcriptomics of Cancer Tissues. Int. J. Mol. Sci. 2022, 23, 3042. [Google Scholar] [CrossRef]
- Lundberg, E.; Borner, G.H.H. Spatial Proteomics: A Powerful Discovery Tool for Cell Biology. Nat. Rev. Mol. Cell Biol. 2019, 20, 285–302. [Google Scholar] [CrossRef]

| ID | Type of Study | Status | No. Patients | Population | Treatment |
|---|---|---|---|---|---|
| Pembrolizumab (anti PD-1 antibody) | |||||
| NCT02129556 PANACEA | Phase 1/2 Single arm | Completed | 58 | Metastatic HER2+ breast cancer, trastuzumab-resistant | Pembrolizumab with trastuzumab [205] |
| NCT03747120 | Phase 2 open-label, randomized | Recruiting | 174 | Naive patients with invasive human HER2+ breast cancer whose primary tumors are > 2 cm and/or clinically lymph node-positive | Neoadjuvant trastuzumab, pertuzumab, and paclitaxel Arm A: trastuzumab + pertuzumab + paclitaxel, Arm B: trastuzumab + pertuzumab + paclitaxel+ pembrolizumab or Arm C: trastuzumab + pembrolizumab + paclitaxel [206]. |
| NCT03032107 | Phase 1b | Active, not recruiting | 27 | Metastatic HER2+ breast cancer | Pembrolizumab + T-DM1 |
| NCT04789096 TUGETHER | Two arms, phase 2 | Not yet recruiting | 50 | Women or men with HER2+, metastatic breast cancer, who have progressed since previous treatment | Pembrolizumab + tucatinib + trastuzumab (PD-L1+) or Pembrolizumab + tucatinib + trastuzumab + capecitabine (PD-L1-) |
| NCT04660929 | Phase 1, open label | Recruiting | 48 | HER2+ recurrent or metastatic solid tumors | Anti-HER2 CAR macrophages + pembrolizumab |
| NCT05020860 I-SPY trial | Phase 2, open label | Not yet recruiting | 185 | Early HER2+ breast cancer | Neoadjuvant paclitaxel + trastuzumab + pertuzumab in combination with pembrolizumab |
| NCT03272334 Breast-47 | Phase 1/2 | Recruiting | 33 | Metastatic HER2+ breast cancer | Pembrolizumab administered in combination with HER2 and CD3 bispecific antibody armed activated T cell (BATs) infusions |
| Atezolizumab (anti-PD-L1 antibody) | |||||
| NCT02924883 KATE2 | Phase 2, double blind | Completed | 133 | Locally advaced or metastatic HER2+ breast cancer | Atezolizumab and trastuzumab-emtansine (T-DM1) Arm 1: T-DM1 + atezolizumab, Arm 2: T-DM1 + placebo [207] |
| NCT04740918 KATE3 | Phase 3, doble blind | Recruiting | 320 | Locally advanced or metastatic HER2+ and PD-L1+ breast cancer who have received prior trastuzumab- (+/− pertuzumab) and taxane-based therapies | Atezolizumab and T-DM1 Arm A: T-DM1 + placebo, Arm B: T-DM1 + atezolizumab |
| NCT03726879 IMpassion050 | Phase 3, doble blind | Active, not recruiting | 454 | High-risk early HER2+ breast cancer | Atezolizumab or placebo in combination with neoadjuvant doxorubicin + cyclophosphamide followed by paclitaxel + trastuzumab + pertuzumab (ddAC-PacHP) Arm 1: Atezolizumab + ddAC-PacHP. Arm 2: placebo + ddAC-PacHP [208] |
| NCT04873362 Astefania | Phase 3, doble blind | Recruiting | 1700 | High risk HER2+ breast cancer following preoperative therapy | Adjuvant atezolizumab or placebo and T-DM1. Arm A: placebo + T-DM1. Arm B: Atezolizumab + T-DM1 [209] |
| NCT02605915 | Phase 1, open label | Completed | 98 | HER2+ and HER2− breast cancer | Atezolizumab + T-DM1 or with trastuzumab and pertuzumab (with and without docetaxel) in patients with HER2+ breast cancer and atezolizumab + doxorubicin and cyclophosphamide in HER2− breast cancer |
| NCT03417544 | Phase 2 | Active, not recruiting | 33 | Central nervous system metastases in patients with HER2+ breast cancer | Atezolizumab + pertuzumab + high-dose trastuzumab |
| NCT03199885 | Phase 3, doble blind | Active, not recruiting | 600 | First-line metastatic HER2+ breast cancer | Arm I: pertuzumab + trastuzumab + taxane therapy + atezolizumab. Arm II: pertuzumab + trastuzumab + taxane therapy + placebo |
| NCT04759248 ATREZZO | Phase 2, open label | Recruiting | 110 | Advanced/metastatic HER2+ breast cancer | Atezolizumab + trastuzumab + vinorelbine |
| NCT03595592 APTneo | Phase 3, open label | Active, not recruiting | 650 | Early high-risk and locally advanced HER2+ breast cancer | Arm 1:Trastuzumab + pertuzumab + carboplatin + paclitaxel (HPCT). Arm 2: Doxorubicin + cyclophosphamide (AC) followed by HPCT + atezolizumab, Arm 3: HPCT + atezolizumab |
| Durvalumab (anti PD-L1 antibody) | |||||
| NCT02649686 CCTG IND.229 | Phase 1, open label | Completed | 15 | Metastatic HER2+ breast cancer receiving trastuzumab | Durvalumab + trastuzumab [210] |
| NCT04538742 DB-07 | Phase 1b/2, open label | Recruiting | 450 | Metastatic HER2+ breast cancer | Trastuzumab Deruxtecan (T-DXd) in Combination With Other Anti-cancer Agents |
| Avelumab (anti PD-L1 antibody) | |||||
| NCT01772004 JAVELIN solid tumor | Phase 1, open label | Completed | 1756 | Metastatic or locally advaced solid tumors | Avelumab monotherapy to 26 HER2+ breast cancer [211] |
| NCT03414658 AVIATOR | Phase 2, open label | Recruiting | 100 | Advanced HER2+ breast cancer | Trastuzumab + vinorelbine with avelumab or avelumab + utomilumab (anti CD137) |
| Monalizumab (anti-NKG2A antibody) | |||||
| NCT04307329 MIMOSA | Phase 2, open label | Active, not recruiting | 38 | Metastatic HER2+ breast cancer | Molalizumab + trastuzumab in cohort of low TILS (<5%) or cohort of high TILS (≥5%) [212] |
| IMM2902 (HER2/SIRPα Bispecific mAb-Trap Antibody-receptor Fusion Protein) | |||||
| NCT05076591 | Phase 1, open label | Recruiting | 135 | Advanced solid tumors HER2+ | IMM2902, dose escalation |
| Utomilumab (anti-CD137 antibody) | |||||
| NCT03414658 AVIATOR | Phase 2, open label | Recruiting | 100 | Advanced HER2+ breast cancer | Trastuzumab + vinorelbine with avelumab or avelumab + utomilumab (anti CD137) |
| NCT03364348 | Phase 1, open label | Completed | 18 | Advanced HER2+ breast cancer | Utomilumab + T-DM1 or trastuzumab |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mercogliano, M.F.; Bruni, S.; Mauro, F.L.; Schillaci, R. Emerging Targeted Therapies for HER2-Positive Breast Cancer. Cancers 2023, 15, 1987. https://doi.org/10.3390/cancers15071987
Mercogliano MF, Bruni S, Mauro FL, Schillaci R. Emerging Targeted Therapies for HER2-Positive Breast Cancer. Cancers. 2023; 15(7):1987. https://doi.org/10.3390/cancers15071987
Chicago/Turabian StyleMercogliano, María Florencia, Sofía Bruni, Florencia Luciana Mauro, and Roxana Schillaci. 2023. "Emerging Targeted Therapies for HER2-Positive Breast Cancer" Cancers 15, no. 7: 1987. https://doi.org/10.3390/cancers15071987
APA StyleMercogliano, M. F., Bruni, S., Mauro, F. L., & Schillaci, R. (2023). Emerging Targeted Therapies for HER2-Positive Breast Cancer. Cancers, 15(7), 1987. https://doi.org/10.3390/cancers15071987