
The Tissue Factor Pathway in Cancer: Overview and Role of Heparan Sulfate Proteoglycans






{kind=link}
{kind=link}
{kind=link}
{kind=link}
Simple Summary
Abstract
1. Introduction
2. Tissue Factor—Structure and Expression Profile
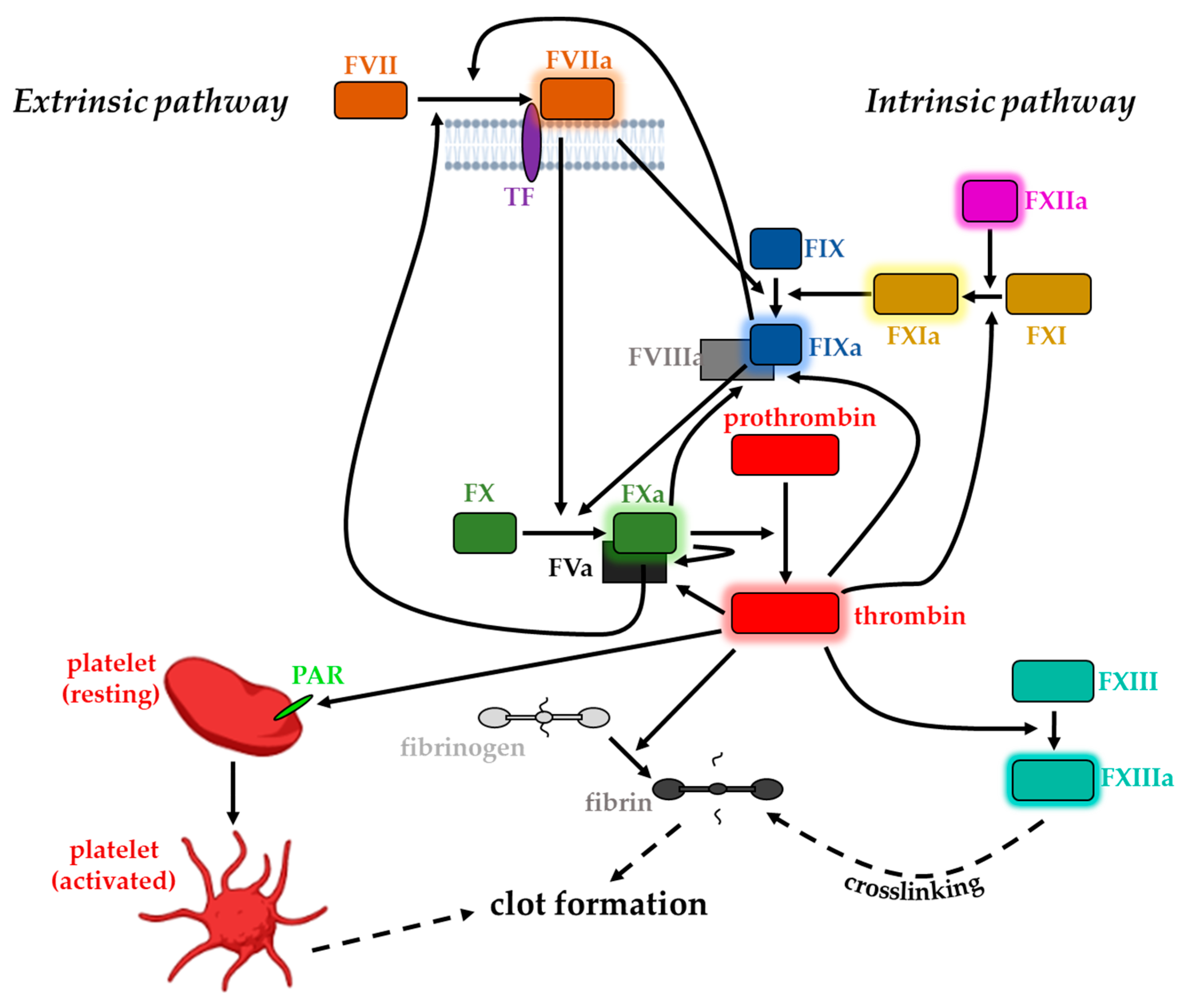
3. Coagulation Factors and Blood Clotting
4. Regulation of Tissue Factor and Blood Coagulation
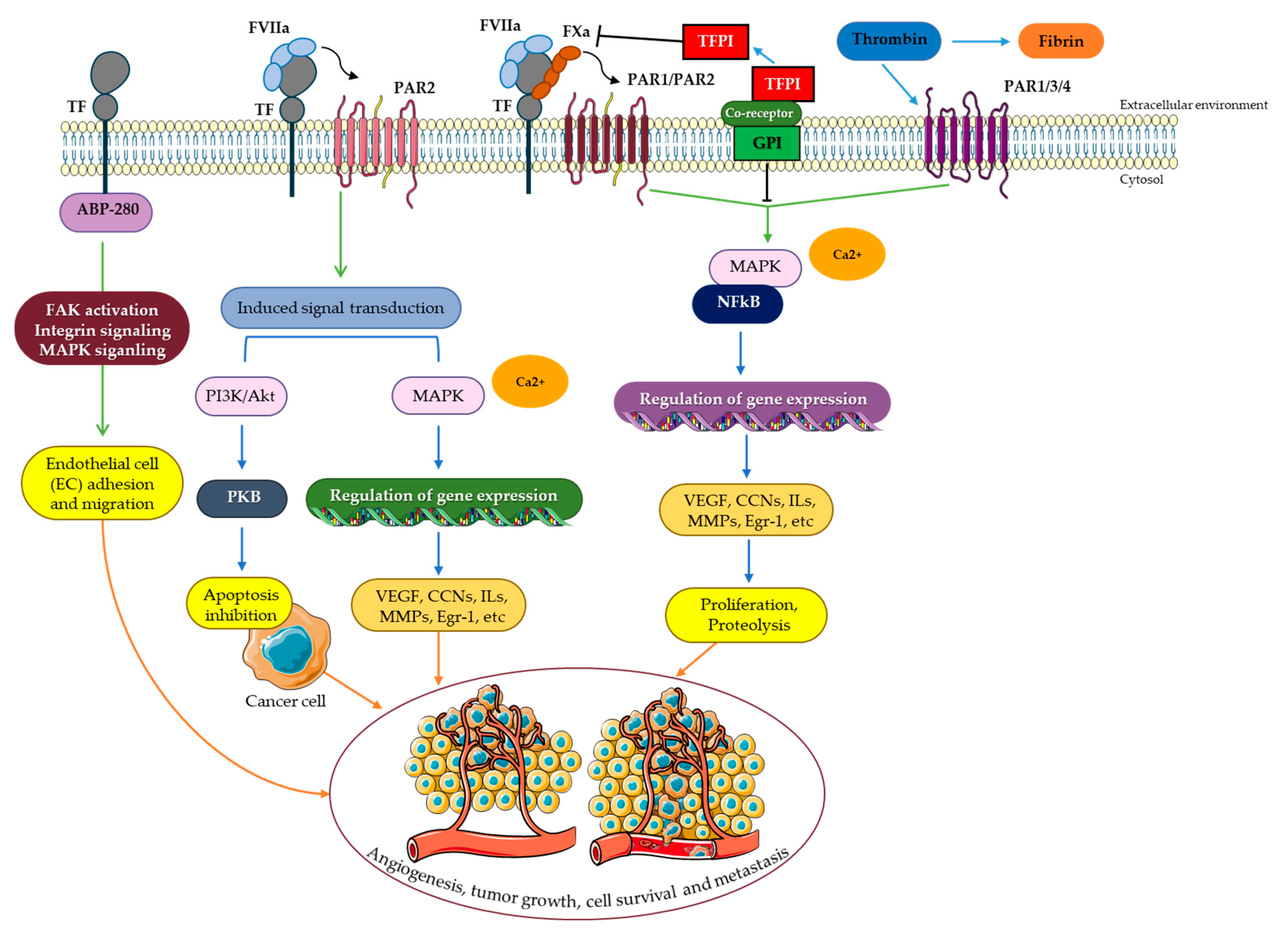
5. TF/FVIIa Intracellular Signaling
5.1. PAR Signaling
5.2. Receptor Tyrosine Kinase (RTK) Signaling
5.3. IGF-1R Signaling and the Tissue Factor Pathway
5.4. Integrin Signaling
5.5. MAPK Signaling
6. The Tissue Factor Pathway and Cancer
6.1. The Tissue Factor Pathway and Cancer-Induced Hypercoagulability
6.2. The Tissue Factor Pathway and Angiogenesis
6.3. The Tissue Factor Pathway and Cancer Cell Survival
6.4. The Tissue Factor Pathway and Metastasis
6.5. Impact of TF Domains and Complexity of TF and Its Effector FVIIa on Cancer Progression
7. Tissue Factor Pathway Inhibitor and Cancer
8. TF, TFPI, and Proteoglycans
9. Functional Enrichment Study of Tissue Factor Pathway Targets with HSPG-Related Proteins on the Cell Surface
10. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Camera, M.; Giesen, P.L.; Fallon, J.; Aufiero, B.M.; Taubman, M.; Tremoli, E.; Nemerson, Y. Cooperation between VEGF and TNF-alpha is necessary for exposure of active tissue factor on the surface of human endothelial cells. Arterioscler. Thromb. Vasc. Biol. 1999, 19, 531–537. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Carlsen, E.; Flatmark, A.; Prydz, H. Cytokine-induced procoagulant activity in monocytes and endothelial cells. Further enhancement by cyclosporine. Transplantation 1988, 46, 575–580. [Google Scholar] [CrossRef] [PubMed]
- Weiss, H.J.; Turitto, V.T.; Baumgartner, H.R.; Nemerson, Y.; Hoffmann, T. Evidence for the presence of tissue factor activity on subendothelium. Blood 1989, 73, 968–975. [Google Scholar] [CrossRef] [PubMed]
- Wilcox, J.N.; Smith, K.M.; Schwartz, S.M.; Gordon, D. Localization of tissue factor in the normal vessel wall and in the atherosclerotic plaque. Proc. Natl. Acad. Sci. USA 1989, 86, 2839–2843. [Google Scholar] [CrossRef]
- Yang, H.L.; Lu, F.J.; Wung, S.L.; Chiu, H.C. Humic acid induces expression of tissue factor by cultured endothelial cells: Regulation by cytosolic calcium and protein kinase C. Thromb. Haemost. 1994, 71, 325–330. [Google Scholar] [CrossRef]
- Dahlback, B. Blood coagulation. Lancet 2000, 355, 1627–1632. [Google Scholar] [CrossRef]
- Jackson, C.M.; Nemerson, Y. Blood coagulation. Annu. Rev. Biochem. 1980, 49, 765–811. [Google Scholar] [CrossRef]
- Mackman, N.; Tilley, R.E.; Key, N.S. Role of the extrinsic pathway of blood coagulation in hemostasis and thrombosis. Arterioscler. Thromb. Vasc. Biol. 2007, 27, 1687–1693. [Google Scholar] [CrossRef]
- Broze, G.J., Jr.; Leykam, J.E.; Schwartz, B.D.; Miletich, J.P. Purification of human brain tissue factor. J. Biol. Chem. 1985, 260, 10917–10920. [Google Scholar] [CrossRef]
- Cimmino, G.; Ciccarelli, G.; Golino, P. Role of Tissue Factor in the Coagulation Network. Semin. Thromb. Hemost. 2015, 41, 708–717. [Google Scholar] [CrossRef]
- Mackman, N. Role of tissue factor in hemostasis, thrombosis, and vascular development. Arterioscler. Thromb. Vasc. Biol. 2004, 24, 1015–1022. [Google Scholar] [CrossRef] [PubMed]
- Sharma, L.; Melis, E.; Hickey, M.J.; Clyne, C.D.; Erlich, J.; Khachigian, L.M.; Davenport, P.; Morand, E.; Carmeliet, P.; Tipping, P.G. The cytoplasmic domain of tissue factor contributes to leukocyte recruitment and death in endotoxemia. Am. J. Pathol. 2004, 165, 331–340. [Google Scholar] [CrossRef] [PubMed]
- Drake, T.A.; Morrissey, J.H.; Edgington, T.S. Selective cellular expression of tissue factor in human tissues. Implications for disorders of hemostasis and thrombosis. Am. J. Pathol. 1989, 134, 1087–1097. [Google Scholar] [PubMed]
- Erlich, J.; Parry, G.C.; Fearns, C.; Muller, M.; Carmeliet, P.; Luther, T.; Mackman, N. Tissue factor is required for uterine hemostasis and maintenance of the placental labyrinth during gestation. Proc. Natl. Acad. Sci. USA 1999, 96, 8138–8143. [Google Scholar] [CrossRef] [PubMed]
- Fleck, R.A.; Rao, L.V.; Rapaport, S.I.; Varki, N. Localization of human tissue factor antigen by immunostaining with monospecific, polyclonal anti-human tissue factor antibody. Thromb. Res. 1990, 59, 421–437. [Google Scholar] [CrossRef] [PubMed]
- Hartzell, S.; Ryder, K.; Lanahan, A.; Lau, L.F.; Nathan, D. A growth factor-responsive gene of murine BALB/c 3T3 cells encodes a protein homologous to human tissue factor. Mol. Cell. Biol. 1989, 9, 2567–2573. [Google Scholar] [CrossRef] [PubMed]
- Hoffman, M. The Tissue Factor Pathway and Wound Healing. Semin. Thromb. Hemost. 2018, 44, 142–150. [Google Scholar] [CrossRef] [PubMed]
- Mackman, N.; Sawdey, M.S.; Keeton, M.R.; Loskutoff, D.J. Murine tissue factor gene expression in vivo. Tissue and cell specificity and regulation by lipopolysaccharide. Am. J. Pathol. 1993, 143, 76–84. [Google Scholar] [PubMed]
- Giesen, P.L.; Rauch, U.; Bohrmann, B.; Kling, D.; Roque, M.; Fallon, J.T.; Badimon, J.J.; Himber, J.; Riederer, M.A.; Nemerson, Y. Blood-borne tissue factor: Another view of thrombosis. Proc. Natl. Acad. Sci. USA 1999, 96, 2311–2315. [Google Scholar] [CrossRef]
- Brambilla, M.; Camera, M.; Colnago, D.; Marenzi, G.; De Metrio, M.; Giesen, P.L.; Balduini, A.; Veglia, F.; Gertow, K.; Biglioli, P.; et al. Tissue factor in patients with acute coronary syndromes: Expression in platelets, leukocytes, and platelet-leukocyte aggregates. Arterioscler. Thromb. Vasc. Biol. 2008, 28, 947–953. [Google Scholar] [CrossRef]
- Osterud, B.; Bjorklid, E. Sources of tissue factor. Semin. Thromb. Hemost. 2006, 32, 11–23. [Google Scholar] [CrossRef]
- Lechner, D.; Weltermann, A. Circulating tissue factor-exposing microparticles. Thromb. Res. 2008, 122 (Suppl. 1), S47–S54. [Google Scholar] [CrossRef] [PubMed]
- Bogdanov, V.Y.; Balasubramanian, V.; Hathcock, J.; Vele, O.; Lieb, M.; Nemerson, Y. Alternatively spliced human tissue factor: A circulating, soluble, thrombogenic protein. Nat. Med. 2003, 9, 458–462. [Google Scholar] [CrossRef] [PubMed]
- Albrecht, S.; Kotzsch, M.; Siegert, G.; Luther, T.; Grossmann, H.; Grosser, M.; Muller, M. Detection of circulating tissue factor and factor VII in a normal population. Thromb. Haemost. 1996, 75, 772–777. [Google Scholar] [CrossRef] [PubMed]
- Koyama, T.; Nishida, K.; Ohdama, S.; Sawada, M.; Murakami, N.; Hirosawa, S.; Kuriyama, R.; Matsuzawa, K.; Hasegawa, R.; Aoki, N. Determination of plasma tissue factor antigen and its clinical significance. Br. J. Haematol. 1994, 87, 343–347. [Google Scholar] [CrossRef] [PubMed]
- Smith, S.A.; Travers, R.J.; Morrissey, J.H. How it all starts: Initiation of the clotting cascade. Crit. Rev. Biochem. Mol. Biol. 2015, 50, 326–336. [Google Scholar] [CrossRef]
- Cimmino, G.; Cirillo, P. Tissue factor: Newer concepts in thrombosis and its role beyond thrombosis and hemostasis. Cardiovasc. Diagn. Ther. 2018, 8, 581–593. [Google Scholar] [CrossRef]
- Butenas, S.; Dee, J.D.; Mann, K.G. The function of factor XI in tissue factor-initiated thrombin generation. J. Thromb. Haemost. 2003, 1, 2103–2111. [Google Scholar] [CrossRef] [PubMed]
- Eichinger, S.; Mannucci, P.M.; Tradati, F.; Arbini, A.A.; Rosenberg, R.D.; Bauer, K.A. Determinants of plasma factor VIIa levels in humans. Blood 1995, 86, 3021–3025. [Google Scholar] [CrossRef] [PubMed]
- Morrissey, J.H.; Macik, B.G.; Neuenschwander, P.F.; Comp, P.C. Quantitation of activated factor VII levels in plasma using a tissue factor mutant selectively deficient in promoting factor VII activation. Blood 1993, 81, 734–744. [Google Scholar] [CrossRef] [PubMed]
- Sabharwal, A.K.; Birktoft, J.J.; Gorka, J.; Wildgoose, P.; Petersen, L.C.; Bajaj, S.P. High affinity Ca2+-binding site in the serine protease domain of human factor VIIa and its role in tissue factor binding and development of catalytic activity. J. Biol. Chem. 1995, 270, 15523–15530. [Google Scholar] [CrossRef] [PubMed]
- Fair, D.S. Quantitation of factor VII in the plasma of normal and warfarin-treated individuals by radioimmunoassay. Blood 1983, 62, 784–791. [Google Scholar] [CrossRef] [PubMed]
- Nemerson, Y. The reaction between bovine brain tissue factor and factors VII and X. Biochemistry 1966, 5, 601–608. [Google Scholar] [CrossRef] [PubMed]
- Bom, V.J.; Bertina, R.M. The contributions of Ca2+, phospholipids and tissue-factor apoprotein to the activation of human blood-coagulation factor X by activated factor VII. Biochem. J. 1990, 265, 327–336. [Google Scholar] [CrossRef]
- Komiyama, Y.; Pedersen, A.H.; Kisiel, W. Proteolytic activation of human factors IX and X by recombinant human factor VIIa: Effects of calcium, phospholipids, and tissue factor. Biochemistry 1990, 29, 9418–9425. [Google Scholar] [CrossRef]
- Kalafatis, M.; Rand, M.D.; Mann, K.G. The mechanism of inactivation of human factor V and human factor Va by activated protein C. J. Biol. Chem. 1994, 269, 31869–31880. [Google Scholar] [CrossRef]
- Mann, K.G.; Nesheim, M.E.; Tracy, P.B.; Hibbard, L.S.; Bloom, J.W. Assembly of the prothrombinase complex. Biophys. J. 1982, 37, 106–107. [Google Scholar] [CrossRef][Green Version]
- Nesheim, M.E.; Taswell, J.B.; Mann, K.G. The contribution of bovine Factor V and Factor Va to the activity of prothrombinase. J. Biol. Chem. 1979, 254, 10952–10962. [Google Scholar] [CrossRef]
- Papahadjopoulos, D.; Yin, E.T.; Hanahan, D.J. Purification and Properties of Bovine Factor X: Molecular Changes during Activation. Biochemistry 1964, 3, 1931–1939. [Google Scholar] [CrossRef]
- Foster, W.B.; Nesheim, M.E.; Mann, K.G. The factor Xa-catalyzed activation of factor V. J. Biol. Chem. 1983, 258, 13970–13977. [Google Scholar] [CrossRef]
- Gailani, D.; Broze, G.J., Jr. Factor XI activation in a revised model of blood coagulation. Science 1991, 253, 909–912. [Google Scholar] [CrossRef] [PubMed]
- Lawson, J.H.; Kalafatis, M.; Stram, S.; Mann, K.G. A model for the tissue factor pathway to thrombin. I. An empirical study. J. Biol. Chem. 1994, 269, 23357–23366. [Google Scholar] [CrossRef] [PubMed]
- Mann, K.G.; Brummel, K.; Butenas, S. What is all that thrombin for? J. Thromb. Haemost. 2003, 1, 1504–1514. [Google Scholar] [CrossRef] [PubMed]
- Naito, K.; Fujikawa, K. Activation of human blood coagulation factor XI independent of factor XII. Factor XI is activated by thrombin and factor XIa in the presence of negatively charged surfaces. J. Biol. Chem. 1991, 266, 7353–7358. [Google Scholar] [CrossRef] [PubMed]
- Rak, J.; Milsom, C.; Magnus, N.; Yu, J. Tissue factor in tumour progression. Best Pract. Res. Clin. Haematol. 2009, 22, 71–83. [Google Scholar] [CrossRef] [PubMed]
- Furie, B.; Furie, B.C. The molecular basis of blood coagulation. Cell 1988, 53, 505–518. [Google Scholar] [CrossRef] [PubMed]
- Walsh, P.N. Roles of factor XI, platelets and tissue factor-initiated blood coagulation. J. Thromb. Haemost. 2003, 1, 2081–2086. [Google Scholar] [CrossRef]
- Davie, E.W. Biochemical and molecular aspects of the coagulation cascade. Thromb. Haemost. 1995, 74, 1–6. [Google Scholar] [CrossRef]
- Furie, B.; Furie, B.C. Molecular and cellular biology of blood coagulation. N. Engl. J. Med. 1992, 326, 800–806. [Google Scholar] [CrossRef]
- Di Cera, E.; Page, M.J.; Bah, A.; Bush-Pelc, L.A.; Garvey, L.C. Thrombin llostery. Phys. Chem. Chem. Phys. 2007, 9, 1291–1306. [Google Scholar] [CrossRef]
- Rawlings, N.D.; Tolle, D.P.; Barrett, A.J. MEROPS: The peptidase database. Nucleic Acids Res. 2004, 32, D160–D164. [Google Scholar] [CrossRef]
- Di Cera, E. Thrombin interactions. Chest 2003, 124, 11S–17S. [Google Scholar] [CrossRef]
- Dihanich, M.; Kaser, M.; Reinhard, E.; Cunningham, D.; Monard, D. Prothrombin mRNA is expressed by cells of the nervous system. Neuron 1991, 6, 575–581. [Google Scholar] [CrossRef]
- Wells, J.E.; Howlett, M.; Cole, C.H.; Kees, U.R. Deregulated expression of connective tissue growth factor (CTGF/CCN2) is linked to poor outcome in human cancer. Int. J. Cancer 2015, 137, 504–511. [Google Scholar] [CrossRef]
- Brass, L.F. Thrombin and platelet activation. Chest 2003, 124, 18S–25S. [Google Scholar] [CrossRef]
- Coughlin, S.R.; Vu, T.K.; Hung, D.T.; Wheaton, V.I. Characterization of a functional thrombin receptor. Issues and opportunities. J. Clin. Investig. 1992, 89, 351–355. [Google Scholar] [CrossRef] [PubMed]
- Lorand, L.; Downey, J.; Gotoh, T.; Jacobsen, A.; Tokura, S. The transpeptidase system which crosslinks fibrin by gamma-glutamyle-episilon-lysine bonds. Biochem. Biophys. Res. Commun. 1968, 31, 222–230. [Google Scholar] [CrossRef] [PubMed]
- Rao, L.V.; Pendurthi, U.R. Tissue factor-factor VIIa signaling. Arterioscler. Thromb. Vasc. Biol. 2005, 25, 47–56. [Google Scholar] [CrossRef] [PubMed]
- De Candia, E. Mechanisms of platelet activation by thrombin: A short history. Thromb. Res. 2012, 129, 250–256. [Google Scholar] [CrossRef] [PubMed]
- Kahn, M.L.; Zheng, Y.W.; Huang, W.; Bigornia, V.; Zeng, D.; Moff, S.; Farese, R.V., Jr.; Tam, C.; Coughlin, S.R. A dual thrombin receptor system for platelet activation. Nature 1998, 394, 690–694. [Google Scholar] [CrossRef] [PubMed]
- Esmon, C.T.; Xu, J.; Gu, J.M.; Qu, D.; Laszik, Z.; Ferrell, G.; Stearns-Kurosawa, D.J.; Kurosawa, S.; Taylor, F.B., Jr.; Esmon, N.L. Endothelial protein C receptor. Thromb. Haemost. 1999, 82, 251–258. [Google Scholar] [CrossRef] [PubMed]
- Taylor, F.B., Jr.; Peer, G.T.; Lockhart, M.S.; Ferrell, G.; Esmon, C.T. Endothelial cell protein C receptor plays an important role in protein C activation in vivo. Blood 2001, 97, 1685–1688. [Google Scholar] [CrossRef] [PubMed]
- Dahlback, B. The protein C anticoagulant system: Inherited defects as basis for venous thrombosis. Thromb. Res. 1995, 77, 1–43. [Google Scholar] [CrossRef] [PubMed]
- Esmon, C.T. The protein C pathway. Chest 2003, 124, 26S–32S. [Google Scholar] [CrossRef] [PubMed]
- Mann, K.G. Thrombin formation. Chest 2003, 124, 4S–10S. [Google Scholar] [CrossRef]
- Hjort, P.F. Intermediate reactions in the coagulation of blood with tissue thromboplastin; convertin, accelerin, prothrombinase. Scand. J. Clin. Lab. Investig. 1957, 9 (Suppl. 27), 1–183. [Google Scholar]
- Broze, G.J., Jr.; Miletich, J.P. Isolation of the tissue factor inhibitor produced by HepG2 hepatoma cells. Proc. Natl. Acad. Sci. USA 1987, 84, 1886–1890. [Google Scholar] [CrossRef]
- Hubbard, A.R.; Jennings, C.A. Inhibition of the tissue factor-factor VII complex: Involvement of factor Xa and lipoproteins. Thromb. Res. 1987, 46, 527–537. [Google Scholar] [CrossRef]
- Kondo, S.; Kisiel, W. Evidence that plasma lipoproteins inhibit the factor VIIa-tissue factor complex by a different mechanism that extrinsic pathway inhibitor. Blood 1987, 70, 1947–1954. [Google Scholar] [CrossRef]
- Warn-Cramer, B.J.; Maki, S.L.; Zivelin, A.; Rapaport, S.I. Partial purification and characterization of extrinsic pathway inhibitor (the factor Xa-dependent plasma inhibitor of factor VIIa/tissue factor). Thromb. Res. 1987, 48, 11–22. [Google Scholar] [CrossRef]
- Wun, T.C.; Kretzmer, K.K.; Girard, T.J.; Miletich, J.P.; Broze, G.J., Jr. Cloning and characterization of a cDNA coding for the lipoprotein-associated coagulation inhibitor shows that it consists of three tandem Kunitz-type inhibitory domains. J. Biol. Chem. 1988, 263, 6001–6004. [Google Scholar] [CrossRef] [PubMed]
- Baugh, R.J.; Broze, G.J., Jr.; Krishnaswamy, S. Regulation of extrinsic pathway factor Xa formation by tissue factor pathway inhibitor. J. Biol. Chem. 1998, 273, 4378–4386. [Google Scholar] [CrossRef] [PubMed]
- Broze, G.J., Jr.; Miletich, J.P. Characterization of the inhibition of tissue factor in serum. Blood 1987, 69, 150–155. [Google Scholar] [CrossRef]
- Broze, G.J., Jr. Tissue factor pathway inhibitor. Thromb. Haemost. 1995, 74, 90–93. [Google Scholar] [CrossRef] [PubMed]
- Girard, T.J.; Warren, L.A.; Novotny, W.F.; Likert, K.M.; Brown, S.G.; Miletich, J.P.; Broze, G.J., Jr. Functional significance of the Kunitz-type inhibitory domains of lipoprotein-associated coagulation inhibitor. Nature 1989, 338, 518–520. [Google Scholar] [CrossRef] [PubMed]
- Maroney, S.A.; Ellery, P.E.; Wood, J.P.; Ferrel, J.P.; Martinez, N.D.; Mast, A.E. Comparison of the inhibitory activities of human tissue factor pathway inhibitor (TFPI)alpha and TFPIbeta. J. Thromb. Haemost. 2013, 11, 911–918. [Google Scholar] [CrossRef]
- Rao, L.V.; Rapaport, S.I. Studies of a mechanism inhibiting the initiation of the extrinsic pathway of coagulation. Blood 1987, 69, 645–651. [Google Scholar] [CrossRef]
- Chang, J.Y.; Monroe, D.M.; Oliver, J.A.; Roberts, H.R. TFPIbeta, a second product from the mouse tissue factor pathway inhibitor (TFPI) gene. Thromb. Haemost. 1999, 81, 45–49. [Google Scholar]
- Zhang, Y.; Sun, L.G.; Shang, H.; Gao, H.; Yu, H.Y.; Gao, S.M.; Liu, N. Expression of aFGF in ovarian epithelial cancer and its signal transduction pathway. Zhonghua Yi Xue Za Zhi 2003, 83, 976–980. [Google Scholar]
- Bajaj, M.S.; Kuppuswamy, M.N.; Saito, H.; Spitzer, S.G.; Bajaj, S.P. Cultured normal human hepatocytes do not synthesize lipoprotein-associated coagulation inhibitor: Evidence that endothelium is the principal site of its synthesis. Proc. Natl. Acad. Sci. USA 1990, 87, 8869–8873. [Google Scholar] [CrossRef]
- Maroney, S.A.; Haberichter, S.L.; Friese, P.; Collins, M.L.; Ferrel, J.P.; Dale, G.L.; Mast, A.E. Active tissue factor pathway inhibitor is expressed on the surface of coated platelets. Blood 2007, 109, 1931–1937. [Google Scholar] [CrossRef] [PubMed]
- Novotny, W.F.; Girard, T.J.; Miletich, J.P.; Broze, G.J., Jr. Platelets secrete a coagulation inhibitor functionally and antigenically similar to the lipoprotein associated coagulation inhibitor. Blood 1988, 72, 2020–2025. [Google Scholar] [CrossRef] [PubMed]
- Lupu, C.; Goodwin, C.A.; Westmuckett, A.D.; Emeis, J.J.; Scully, M.F.; Kakkar, V.V.; Lupu, F. Tissue factor pathway inhibitor in endothelial cells colocalizes with glycolipid microdomains/caveolae. Regulatory mechanism(s) of the anticoagulant properties of the endothelium. Arterioscler. Thromb. Vasc. Biol. 1997, 17, 2964–2974. [Google Scholar] [CrossRef] [PubMed]
- Novotny, W.F.; Palmier, M.; Wun, T.C.; Broze, G.J., Jr.; Miletich, J.P. Purification and properties of heparin-releasable lipoprotein-associated coagulation inhibitor. Blood 1991, 78, 394–400. [Google Scholar] [CrossRef] [PubMed]
- Sandset, P.M.; Abildgaard, U.; Larsen, M.L. Heparin induces release of extrinsic coagulation pathway inhibitor (EPI). Thromb. Res. 1988, 50, 803–813. [Google Scholar] [CrossRef]
- Wood, J.P.; Bunce, M.W.; Maroney, S.A.; Tracy, P.B.; Camire, R.M.; Mast, A.E. Tissue factor pathway inhibitor-alpha inhibits prothrombinase during the initiation of blood coagulation. Proc. Natl. Acad. Sci. USA 2013, 110, 17838–17843. [Google Scholar] [CrossRef]
- Kojima, T.; Katsumi, A.; Yamazaki, T.; Muramatsu, T.; Nagasaka, T.; Ohsumi, K.; Saito, H. Human ryudocan from endothelium-like cells binds basic fibroblast growth factor, midkine, and tissue factor pathway inhibitor. J. Biol. Chem. 1996, 271, 5914–5920. [Google Scholar] [CrossRef]
- Mast, A.E.; Higuchi, D.A.; Huang, Z.F.; Warshawsky, I.; Schwartz, A.L.; Broze, G.J., Jr. Glypican-3 is a binding protein on the HepG2 cell surface for tissue factor pathway inhibitor. Biochem. J. 1997, 327 Pt 2, 577–583. [Google Scholar] [CrossRef]
- Zhang, J.; Piro, O.; Lu, L.; Broze, G.J., Jr. Glycosyl phosphatidylinositol anchorage of tissue factor pathway inhibitor. Circulation 2003, 108, 623–627. [Google Scholar] [CrossRef][Green Version]
- Ellery, P.E.; Maroney, S.A.; Martinez, N.D.; Wickens, M.P.; Mast, A.E. Translation of human tissue factor pathway inhibitor-beta mRNA is controlled by alternative splicing within the 5’ untranslated region. Arterioscler. Thromb. Vasc. Biol. 2014, 34, 187–195. [Google Scholar] [CrossRef]
- Olson, S.T.; Bjork, I.; Shore, J.D. Kinetic characterization of heparin-catalyzed and uncatalyzed inhibition of blood coagulation proteinases by antithrombin. Methods Enzymol. 1993, 222, 525–559. [Google Scholar] [CrossRef] [PubMed]
- Church, F.C.; Pratt, C.W.; Hoffman, M. Leukocyte chemoattractant peptides from the serpin heparin cofactor II. J. Biol. Chem. 1991, 266, 704–709. [Google Scholar] [CrossRef] [PubMed]
- Gettins, P.G. Serpin structure, mechanism, and function. Chem. Rev. 2002, 102, 4751–4804. [Google Scholar] [CrossRef]
- Lindahl, A.K.; Jacobsen, P.B.; Sandset, P.M.; Abildgaard, U. Tissue factor pathway inhibitor with high anticoagulant activity is increased in post-heparin plasma and in plasma from cancer patients. Blood Coagul. Fibrinolysis 1991, 2, 713–721. [Google Scholar] [CrossRef] [PubMed]
- Olson, S.T.; Chuang, Y.J. Heparin activates antithrombin anticoagulant function by generating new interaction sites (exosites) for blood clotting proteinases. Trends Cardiovasc. Med. 2002, 12, 331–338. [Google Scholar] [CrossRef] [PubMed]
- Tollefsen, D.M. Heparin cofactor II modulates the response to vascular injury. Arterioscler. Thromb. Vasc. Biol. 2007, 27, 454–460. [Google Scholar] [CrossRef] [PubMed]
- Tollefsen, D.M.; Majerus, D.W.; Blank, M.K. Heparin cofactor II. Purification and properties of a heparin-dependent inhibitor of thrombin in human plasma. J. Biol. Chem. 1982, 257, 2162–2169. [Google Scholar] [CrossRef]
- Sandset, P.M. Tissue factor pathway inhibitor (TFPI)--an update. Haemostasis 1996, 26 (Suppl. 4), 154–165. [Google Scholar] [CrossRef]
- Hansen, J.B.; Sandset, P.M.; Huseby, K.R.; Huseby, N.E.; Bendz, B.; Ostergaard, P.; Nordoy, A. Differential effect of unfractionated heparin and low molecular weight heparin on intravascular tissue factor pathway inhibitor: Evidence for a difference in antithrombotic action. Br. J. Haematol. 1998, 101, 638–646. [Google Scholar] [CrossRef]
- Maroney, S.A.; Mast, A.E. New insights into the biology of tissue factor pathway inhibitor. J. Thromb. Haemost. 2015, 13 (Suppl. 1), S200–S207. [Google Scholar] [CrossRef]
- Hockin, M.F.; Kalafatis, M.; Shatos, M.; Mann, K.G. Protein C activation and factor Va inactivation on human umbilical vein endothelial cells. Arterioscler. Thromb. Vasc. Biol. 1997, 17, 2765–2775. [Google Scholar] [CrossRef]
- Suzuki, K.; Stenflo, J.; Dahlback, B.; Teodorsson, B. Inactivation of human coagulation factor V by activated protein C. J. Biol. Chem. 1983, 258, 1914–1920. [Google Scholar] [CrossRef]
- Walker, F.J. Regulation of activated protein C by protein S. The role of phospholipid in factor Va inactivation. J. Biol. Chem. 1981, 256, 11128–11131. [Google Scholar] [CrossRef] [PubMed]
- Peraramelli, S.; Rosing, J.; Hackeng, T.M. TFPI-dependent activities of protein S. Thromb. Res. 2012, 129 (Suppl. 2), S23–S26. [Google Scholar] [CrossRef] [PubMed]
- Lindahl, U.; Kjellen, L. Heparin or heparan sulfate--what is the difference? Thromb. Haemost. 1991, 66, 44–48. [Google Scholar] [CrossRef] [PubMed]
- Mannucci, P.M.; Tripodi, A.; Bottasso, B.; Baudo, F.; Finazzi, G.; De Stefano, V.; Palareti, G.; Manotti, C.; Mazzucconi, M.G.; Castaman, G. Markers of procoagulant imbalance in patients with inherited thrombophilic syndromes. Thromb. Haemost. 1992, 67, 200–202. [Google Scholar] [CrossRef] [PubMed]
- Koster, T.; Blann, A.D.; Briet, E.; Vandenbroucke, J.P.; Rosendaal, F.R. Role of clotting factor VIII in effect of von Willebrand factor on occurrence of deep-vein thrombosis. Lancet 1995, 345, 152–155. [Google Scholar] [CrossRef]
- Poort, S.R.; Rosendaal, F.R.; Reitsma, P.H.; Bertina, R.M. A common genetic variation in the 3’-untranslated region of the prothrombin gene is associated with elevated plasma prothrombin levels and an increase in venous thrombosis. Blood 1996, 88, 3698–3703. [Google Scholar] [CrossRef]
- Eisenreich, A.; Leppert, U. The impact of microRNAs on the regulation of tissue factor biology. Trends Cardiovasc. Med. 2014, 24, 128–132. [Google Scholar] [CrossRef]
- Teruel, R.; Perez-Sanchez, C.; Corral, J.; Herranz, M.T.; Perez-Andreu, V.; Saiz, E.; Garcia-Barbera, N.; Martinez-Martinez, I.; Roldan, V.; Vicente, V.; et al. Identification of miRNAs as potential modulators of tissue factor expression in patients with systemic lupus erythematosus and antiphospholipid syndrome. J. Thromb. Haemost. 2011, 9, 1985–1992. [Google Scholar] [CrossRef]
- Witkowski, M.; Weithauser, A.; Tabaraie, T.; Steffens, D.; Krankel, N.; Witkowski, M.; Stratmann, B.; Tschoepe, D.; Landmesser, U.; Rauch-Kroehnert, U. Micro-RNA-126 Reduces the Blood Thrombogenicity in Diabetes Mellitus via Targeting of Tissue Factor. Arterioscler. Thromb. Vasc. Biol. 2016, 36, 1263–1271. [Google Scholar] [CrossRef] [PubMed]
- Bach, R.R.; Moldow, C.F. Mechanism of tissue factor activation on HL-60 cells. Blood 1997, 89, 3270–3276. [Google Scholar] [CrossRef] [PubMed]
- Carson, S.D. Manifestation of cryptic fibroblast tissue factor occurs at detergent concentrations which dissolve the plasma membrane. Blood Coagul. Fibrinolysis 1996, 7, 303–313. [Google Scholar] [CrossRef] [PubMed]
- Penn, M.S.; Cui, M.Z.; Winokur, A.L.; Bethea, J.; Hamilton, T.A.; DiCorleto, P.E.; Chisolm, G.M. Smooth muscle cell surface tissue factor pathway activation by oxidized low-density lipoprotein requires cellular lipid peroxidation. Blood 2000, 96, 3056–3063. [Google Scholar] [CrossRef]
- Bochkov, V.N.; Mechtcheriakova, D.; Lucerna, M.; Huber, J.; Malli, R.; Graier, W.F.; Hofer, E.; Binder, B.R.; Leitinger, N. Oxidized phospholipids stimulate tissue factor expression in human endothelial cells via activation of ERK/EGR-1 and Ca(++)/NFAT. Blood 2002, 99, 199–206. [Google Scholar] [CrossRef]
- Drake, T.A.; Ruf, W.; Morrissey, J.H.; Edgington, T.S. Functional tissue factor is entirely cell surface expressed on lipopolysaccharide-stimulated human blood monocytes and a constitutively tissue factor-producing neoplastic cell line. J. Cell Biol. 1989, 109, 389–395. [Google Scholar] [CrossRef] [PubMed]
- Rottingen, J.A.; Enden, T.; Camerer, E.; Iversen, J.G.; Prydz, H. Binding of human factor VIIa to tissue factor induces cytosolic Ca2+ signals in J82 cells, transfected COS-1 cells, Madin-Darby canine kidney cells and in human endothelial cells induced to synthesize tissue factor. J. Biol. Chem. 1995, 270, 4650–4660. [Google Scholar] [CrossRef]
- Aberg, M.; Siegbahn, A. Tissue factor non-coagulant signaling—Molecular mechanisms and biological consequences with a focus on cell migration and apoptosis. J. Thromb. Haemost. 2013, 11, 817–825. [Google Scholar] [CrossRef]
- Poulsen, L.K.; Jacobsen, N.; Sorensen, B.B.; Bergenhem, N.C.; Kelly, J.D.; Foster, D.C.; Thastrup, O.; Ezban, M.; Petersen, L.C. Signal transduction via the mitogen-activated protein kinase pathway induced by binding of coagulation factor VIIa to tissue factor. J. Biol. Chem. 1998, 273, 6228–6232. [Google Scholar] [CrossRef]
- Versteeg, H.H.; Hoedemaeker, I.; Diks, S.H.; Stam, J.C.; Spaargaren, M.; van Bergen En Henegouwen, P.M.; van Deventer, S.J.; Peppelenbosch, M.P. Factor VIIa/tissue factor-induced signaling via activation of Src-like kinases, phosphatidylinositol 3-kinase, and Rac. J. Biol. Chem. 2000, 275, 28750–28756. [Google Scholar] [CrossRef]
- Camerer, E.; Rottingen, J.A.; Gjernes, E.; Larsen, K.; Skartlien, A.H.; Iversen, J.G.; Prydz, H. Coagulation factors VIIa and Xa induce cell signaling leading to up-regulation of the egr-1 gene. J. Biol. Chem. 1999, 274, 32225–32233. [Google Scholar] [CrossRef]
- Wang, X.; Gjernes, E.; Prydz, H. Factor VIIa induces tissue factor-dependent up-regulation of interleukin-8 in a human keratinocyte line. J. Biol. Chem. 2002, 277, 23620–23626. [Google Scholar] [CrossRef] [PubMed]
- Versteeg, H.H.; Spek, C.A.; Peppelenbosch, M.P.; Richel, D.J. Tissue factor and cancer metastasis: The role of intracellular and extracellular signaling pathways. Mol. Med. 2004, 10, 6–11. [Google Scholar] [CrossRef] [PubMed]
- Ollivier, V.; Bentolila, S.; Chabbat, J.; Hakim, J.; de Prost, D. Tissue factor-dependent vascular endothelial growth factor production by human fibroblasts in response to activated factor VII. Blood 1998, 91, 2698–2703. [Google Scholar] [CrossRef] [PubMed]
- Shoji, M.; Hancock, W.W.; Abe, K.; Micko, C.; Casper, K.A.; Baine, R.M.; Wilcox, J.N.; Danave, I.; Dillehay, D.L.; Matthews, E.; et al. Activation of coagulation and angiogenesis in cancer: Immunohistochemical localization in situ of clotting proteins and vascular endothelial growth factor in human cancer. Am. J. Pathol. 1998, 152, 399–411. [Google Scholar] [PubMed]
- Siegbahn, A.; Johnell, M.; Rorsman, C.; Ezban, M.; Heldin, C.H.; Ronnstrand, L. Binding of factor VIIa to tissue factor on human fibroblasts leads to activation of phospholipase C and enhanced PDGF-BB-stimulated chemotaxis. Blood 2000, 96, 3452–3458. [Google Scholar] [CrossRef]
- Sorensen, B.B.; Rao, L.V.; Tornehave, D.; Gammeltoft, S.; Petersen, L.C. Antiapoptotic effect of coagulation factor VIIa. Blood 2003, 102, 1708–1715. [Google Scholar] [CrossRef]
- Vu, T.K.; Hung, D.T.; Wheaton, V.I.; Coughlin, S.R. Molecular cloning of a functional thrombin receptor reveals a novel proteolytic mechanism of receptor activation. Cell 1991, 64, 1057–1068. [Google Scholar] [CrossRef]
- Camerer, E.; Rottingen, J.A.; Iversen, J.G.; Prydz, H. Coagulation factors VII and X induce Ca2+ oscillations in Madin-Darby canine kidney cells only when proteolytically active. J. Biol. Chem. 1996, 271, 29034–29042. [Google Scholar] [CrossRef]
- Sorensen, B.B.; Freskgard, P.O.; Nielsen, L.S.; Rao, L.V.; Ezban, M.; Petersen, L.C. Factor VIIa-induced p44/42 mitogen-activated protein kinase activation requires the proteolytic activity of factor VIIa and is independent of the tissue factor cytoplasmic domain. J. Biol. Chem. 1999, 274, 21349–21354. [Google Scholar] [CrossRef]
- Camerer, E.; Huang, W.; Coughlin, S.R. Tissue factor- and factor X-dependent activation of protease-activated receptor 2 by factor VIIa. Proc. Natl. Acad. Sci. USA 2000, 97, 5255–5260. [Google Scholar] [CrossRef] [PubMed]
- Dorfleutner, A.; Ruf, W. Regulation of tissue factor cytoplasmic domain phosphorylation by palmitoylation. Blood 2003, 102, 3998–4005. [Google Scholar] [CrossRef] [PubMed]
- Aberg, M.; Eriksson, O.; Siegbahn, A. Tissue Factor Noncoagulant Signaling: Mechanisms and Implications for Cell Migration and Apoptosis. Semin. Thromb. Hemost. 2015, 41, 691–699. [Google Scholar] [CrossRef] [PubMed]
- Petersen, L.C.; Albrektsen, T.; Hjorto, G.M.; Kjalke, M.; Bjorn, S.E.; Sorensen, B.B. Factor VIIa/tissue factor-dependent gene regulation and pro-coagulant activity: Effect of factor VIIa concentration. Thromb. Haemost. 2007, 98, 909–911. [Google Scholar] [PubMed]
- Schaffner, F.; Versteeg, H.H.; Schillert, A.; Yokota, N.; Petersen, L.C.; Mueller, B.M.; Ruf, W. Cooperation of tissue factor cytoplasmic domain and PAR2 signaling in breast cancer development. Blood 2010, 116, 6106–6113. [Google Scholar] [CrossRef] [PubMed]
- Albrektsen, T.; Sorensen, B.B.; Hjorto, G.M.; Fleckner, J.; Rao, L.V.; Petersen, L.C. Transcriptional program induced by factor VIIa-tissue factor, PAR1 and PAR2 in MDA-MB-231 cells. J. Thromb. Haemost. 2007, 5, 1588–1597. [Google Scholar] [CrossRef] [PubMed]
- Samad, F.; Ruf, W. Inflammation, obesity, and thrombosis. Blood 2013, 122, 3415–3422. [Google Scholar] [CrossRef]
- Lemmon, M.A.; Schlessinger, J. Cell signaling by receptor tyrosine kinases. Cell 2010, 141, 1117–1134. [Google Scholar] [CrossRef]
- Wagner, M.J.; Stacey, M.M.; Liu, B.A.; Pawson, T. Molecular mechanisms of SH2- and PTB-domain-containing proteins in receptor tyrosine kinase signaling. Cold Spring Harb. Perspect. Biol. 2013, 5, a008987. [Google Scholar] [CrossRef]
- Katz, M.; Amit, I.; Yarden, Y. Regulation of MAPKs by growth factors and receptor tyrosine kinases. Biochim. Biophys. Acta 2007, 1773, 1161–1176. [Google Scholar] [CrossRef]
- Sever, R.; Brugge, J.S. Signal transduction in cancer. Cold Spring Harb. Perspect. Med. 2015, 5, a006098. [Google Scholar] [CrossRef] [PubMed]
- Hsu, J.L.; Hung, M.C. The role of HER2, EGFR, and other receptor tyrosine kinases in breast cancer. Cancer Metastasis Rev. 2016, 35, 575–588. [Google Scholar] [CrossRef] [PubMed]
- Eriksson, O.; Ramstrom, M.; Hornaeus, K.; Bergquist, J.; Mokhtari, D.; Siegbahn, A. The Eph tyrosine kinase receptors EphB2 and EphA2 are novel proteolytic substrates of tissue factor/coagulation factor VIIa. J. Biol. Chem. 2014, 289, 32379–32391. [Google Scholar] [CrossRef] [PubMed]
- Aberg, M.; Eriksson, O.; Mokhtari, D.; Siegbahn, A. Tissue factor/factor VIIa induces cell survival and gene transcription by transactivation of the insulin-like growth factor 1 receptor. Thromb. Haemost. 2014, 111, 748–760. [Google Scholar] [CrossRef] [PubMed]
- Siegbahn, A.; Johnell, M.; Nordin, A.; Aberg, M.; Velling, T. TF/FVIIa transactivate PDGFRbeta to regulate PDGF-BB-induced chemotaxis in different cell types: Involvement of Src and PLC. Arterioscler. Thromb. Vasc. Biol. 2008, 28, 135–141. [Google Scholar] [CrossRef]
- Wiiger, M.T.; Prydz, H. The epidermal growth factor receptor (EGFR) and proline rich tyrosine kinase 2 (PYK2) are involved in tissue factor dependent factor VIIa signalling in HaCaT cells. Thromb. Haemost. 2004, 92, 13–22. [Google Scholar] [CrossRef]
- Boucher, J.; Tseng, Y.H.; Kahn, C.R. Insulin and insulin-like growth factor-1 receptors act as ligand-specific amplitude modulators of a common pathway regulating gene transcription. J. Biol. Chem. 2010, 285, 17235–17245. [Google Scholar] [CrossRef]
- Laron, Z. Insulin-like growth factor 1 (IGF-1): A growth hormone. Mol. Pathol. 2001, 54, 311–316. [Google Scholar] [CrossRef]
- Spiliotaki, M.; Mavroudis, D.; Kokotsaki, M.; Vetsika, E.K.; Stoupis, I.; Matikas, A.; Kallergi, G.; Georgoulias, V.; Agelaki, S. Expression of insulin-like growth factor-1 receptor in circulating tumor cells of patients with breast cancer is associated with patient outcomes. Mol. Oncol. 2018, 12, 21–32. [Google Scholar] [CrossRef]
- De Meyts, P. The Insulin Receptor and Its Signal Transduction Network. In Endotext; Feingold, K.R., Anawalt, B., Boyce, A., Chrousos, G., de Herder, W.W., Dhatariya, K., Dungan, K., Hershman, J.M., Hofland, J., Kalra, S., et al., Eds.; MDText.com, Inc.: South Dartmouth, MA, USA, 2000. [Google Scholar]
- Hubbard, S.R. The insulin receptor: Both a prototypical and atypical receptor tyrosine kinase. Cold Spring Harb. Perspect. Biol. 2013, 5, a008946. [Google Scholar] [CrossRef]
- Larsson, O.; Girnita, A.; Girnita, L. Role of insulin-like growth factor 1 receptor signalling in cancer. Br. J. Cancer 2005, 92, 2097–2101. [Google Scholar] [CrossRef] [PubMed]
- Riedemann, J.; Macaulay, V.M. IGF1R signalling and its inhibition. Endocr. Relat. Cancer 2006, 13 (Suppl. 1), S33–S43. [Google Scholar] [CrossRef]
- Aberg, M.; Johnell, M.; Wickstrom, M.; Siegbahn, A. Tissue Factor/ FVIIa prevents the extrinsic pathway of apoptosis by regulation of the tumor suppressor Death-Associated Protein Kinase 1 (DAPK1). Thromb. Res. 2011, 127, 141–148. [Google Scholar] [CrossRef] [PubMed]
- Osher, E.; Macaulay, V.M. Therapeutic Targeting of the IGF Axis. Cells 2019, 8, 895. [Google Scholar] [CrossRef] [PubMed]
- Pan, G.; Ni, J.; Wei, Y.F.; Yu, G.; Gentz, R.; Dixit, V.M. An antagonist decoy receptor and a death domain-containing receptor for TRAIL. Science 1997, 277, 815–818. [Google Scholar] [CrossRef]
- Sato, K.; Niessner, A.; Kopecky, S.L.; Frye, R.L.; Goronzy, J.J.; Weyand, C.M. TRAIL-expressing T cells induce apoptosis of vascular smooth muscle cells in the atherosclerotic plaque. J. Exp. Med. 2006, 203, 239–250. [Google Scholar] [CrossRef]
- Yuan, X.; Gajan, A.; Chu, Q.; Xiong, H.; Wu, K.; Wu, G.S. Developing TRAIL/TRAIL death receptor-based cancer therapies. Cancer Metastasis Rev. 2018, 37, 733–748. [Google Scholar] [CrossRef]
- Zhao, D.; Bakirtzi, K.; Zhan, Y.; Zeng, H.; Koon, H.W.; Pothoulakis, C. Insulin-like growth factor-1 receptor transactivation modulates the inflammatory and proliferative responses of neurotensin in human colonic epithelial cells. J. Biol. Chem. 2011, 286, 6092–6099. [Google Scholar] [CrossRef]
- Awasthi, V.; Mandal, S.K.; Papanna, V.; Rao, L.V.; Pendurthi, U.R. Modulation of tissue factor-factor VIIa signaling by lipid rafts and caveolae. Arterioscler. Thromb. Vasc. Biol. 2007, 27, 1447–1455. [Google Scholar] [CrossRef]
- Huo, H.; Guo, X.; Hong, S.; Jiang, M.; Liu, X.; Liao, K. Lipid rafts/caveolae are essential for insulin-like growth factor-1 receptor signaling during 3T3-L1 preadipocyte differentiation induction. J. Biol. Chem. 2003, 278, 11561–11569. [Google Scholar] [CrossRef]
- Aberg, M.; Eden, D.; Siegbahn, A. Activation of beta1 integrins and caveolin-1 by TF/FVIIa promotes IGF-1R signaling and cell survival. Apoptosis 2020, 25, 519–534. [Google Scholar] [CrossRef] [PubMed]
- Giancotti, F.G.; Ruoslahti, E. Integrin signaling. Science 1999, 285, 1028–1032. [Google Scholar] [CrossRef] [PubMed]
- Goel, H.L.; Breen, M.; Zhang, J.; Das, I.; Aznavoorian-Cheshire, S.; Greenberg, N.M.; Elgavish, A.; Languino, L.R. beta1A integrin expression is required for type 1 insulin-like growth factor receptor mitogenic and transforming activities and localization to focal contacts. Cancer Res. 2005, 65, 6692–6700. [Google Scholar] [CrossRef]
- Bennett, J.S.; Berger, B.W.; Billings, P.C. The structure and function of platelet integrins. J. Thromb. Haemost. 2009, 7 (Suppl. 1), 200–205. [Google Scholar] [CrossRef] [PubMed]
- Rothmeier, A.S.; Liu, E.; Chakrabarty, S.; Disse, J.; Mueller, B.M.; Ostergaard, H.; Ruf, W. Identification of the integrin-binding site on coagulation factor VIIa required for proangiogenic PAR2 signaling. Blood 2018, 131, 674–685. [Google Scholar] [CrossRef] [PubMed]
- Versteeg, H.H.; Schaffner, F.; Kerver, M.; Petersen, H.H.; Ahamed, J.; Felding-Habermann, B.; Takada, Y.; Mueller, B.M.; Ruf, W. Inhibition of tissue factor signaling suppresses tumor growth. Blood 2008, 111, 190–199. [Google Scholar] [CrossRef] [PubMed]
- Tkach, M.; Rosemblit, C.; Rivas, M.A.; Proietti, C.J.; Diaz Flaque, M.C.; Mercogliano, M.F.; Beguelin, W.; Maronna, E.; Guzman, P.; Gercovich, F.G.; et al. p42/p44 MAPK-mediated Stat3Ser727 phosphorylation is required for progestin-induced full activation of Stat3 and breast cancer growth. Endocr. Relat. Cancer 2013, 20, 197–212. [Google Scholar] [CrossRef]
- Braicu, C.; Buse, M.; Busuioc, C.; Drula, R.; Gulei, D.; Raduly, L.; Rusu, A.; Irimie, A.; Atanasov, A.G.; Slaby, O.; et al. A Comprehensive Review on MAPK: A Promising Therapeutic Target in Cancer. Cancers 2019, 11, 1618. [Google Scholar] [CrossRef]
- Versteeg, H.H.; Bresser, H.L.; Spek, C.A.; Richel, D.J.; Van Deventer, S.J.; Peppelenbosch, M.P. Regulation of the p21Ras-MAP kinase pathway by factor VIIa. J. Thromb. Haemost. 2003, 1, 1012–1018. [Google Scholar] [CrossRef]
- Chen, J.K.; Capdevila, J.; Harris, R.C. Heparin-binding EGF-like growth factor mediates the biological effects of P450 arachidonate epoxygenase metabolites in epithelial cells. Proc. Natl. Acad. Sci. USA 2002, 99, 6029–6034. [Google Scholar] [CrossRef]
- Ott, I.; Weigand, B.; Michl, R.; Seitz, I.; Sabbari-Erfani, N.; Neumann, F.J.; Schomig, A. Tissue factor cytoplasmic domain stimulates migration by activation of the GTPase Rac1 and the mitogen-activated protein kinase p38. Circulation 2005, 111, 349–355. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Guba, M.; Yezhelyev, M.; Eichhorn, M.E.; Schmid, G.; Ischenko, I.; Papyan, A.; Graeb, C.; Seeliger, H.; Geissler, E.K.; Jauch, K.W.; et al. Rapamycin induces tumor-specific thrombosis via tissue factor in the presence of VEGF. Blood 2005, 105, 4463–4469. [Google Scholar] [CrossRef] [PubMed]
- Chu, A.J. Tissue factor, blood coagulation, and beyond: An overview. Int. J. Inflamm. 2011, 2011, 367284. [Google Scholar] [CrossRef] [PubMed]
- Cirillo, P.; Cali, G.; Golino, P.; Calabro, P.; Forte, L.; De Rosa, S.; Pacileo, M.; Ragni, M.; Scopacasa, F.; Nitsch, L.; et al. Tissue factor binding of activated factor VII triggers smooth muscle cell proliferation via extracellular signal-regulated kinase activation. Circulation 2004, 109, 2911–2916. [Google Scholar] [CrossRef] [PubMed]
- van den Berg, Y.W.; van den Hengel, L.G.; Myers, H.R.; Ayachi, O.; Jordanova, E.; Ruf, W.; Spek, C.A.; Reitsma, P.H.; Bogdanov, V.Y.; Versteeg, H.H. Alternatively spliced tissue factor induces angiogenesis through integrin ligation. Proc. Natl. Acad. Sci. USA 2009, 106, 19497–19502. [Google Scholar] [CrossRef] [PubMed]
- van den Berg, Y.W.; Versteeg, H.H. Alternatively spliced tissue factor. A crippled protein in coagulation or a key player in non-haemostatic processes? Hamostaseologie 2010, 30, 144–149. [Google Scholar] [CrossRef] [PubMed]
- Falanga, A. Thrombophilia in cancer. Semin. Thromb. Hemost. 2005, 31, 104–110. [Google Scholar] [CrossRef]
- Petralia, G.A.; Lemoine, N.R.; Kakkar, A.K. Mechanisms of disease: The impact of antithrombotic therapy in cancer patients. Nat. Clin. Pract. Oncol. 2005, 2, 356–363. [Google Scholar] [CrossRef]
- Rickles, F.R.; Patierno, S.; Fernandez, P.M. Tissue factor, thrombin, and cancer. Chest 2003, 124, 58S–68S. [Google Scholar] [CrossRef]
- ten Cate, H.; Falanga, A. Overview of the postulated mechanisms linking cancer and thrombosis. Pathophysiol. Haemost. Thromb. 2008, 36, 122–130. [Google Scholar] [CrossRef]
- Nitori, N.; Ino, Y.; Nakanishi, Y.; Yamada, T.; Honda, K.; Yanagihara, K.; Kosuge, T.; Kanai, Y.; Kitajima, M.; Hirohashi, S. Prognostic significance of tissue factor in pancreatic ductal adenocarcinoma. Clin. Cancer Res. 2005, 11, 2531–2539. [Google Scholar] [CrossRef]
- Forster, Y.; Meye, A.; Albrecht, S.; Kotzsch, M.; Fussel, S.; Wirth, M.P.; Schwenzer, B. Tissue specific expression and serum levels of human tissue factor in patients with urological cancer. Cancer Lett. 2003, 193, 65–73. [Google Scholar] [CrossRef] [PubMed]
- Lwaleed, B.A.; Francis, J.L.; Chisholm, M. Urinary tissue factor levels in patients with bladder and prostate cancer. Eur. J. Surg. Oncol. 2000, 26, 44–49. [Google Scholar] [CrossRef] [PubMed]
- Patry, G.; Hovington, H.; Larue, H.; Harel, F.; Fradet, Y.; Lacombe, L. Tissue factor expression correlates with disease-specific survival in patients with node-negative muscle-invasive bladder cancer. Int. J. Cancer 2008, 122, 1592–1597. [Google Scholar] [CrossRef] [PubMed]
- Brat, D.J.; Van Meir, E.G. Vaso-occlusive and prothrombotic mechanisms associated with tumor hypoxia, necrosis, and accelerated growth in glioblastoma. Lab. Investig. 2004, 84, 397–405. [Google Scholar] [CrossRef]
- Guan, M.; Jin, J.; Su, B.; Liu, W.W.; Lu, Y. Tissue factor expression and angiogenesis in human glioma. Clin. Biochem. 2002, 35, 321–325. [Google Scholar] [CrossRef]
- Hamada, K.; Kuratsu, J.; Saitoh, Y.; Takeshima, H.; Nishi, T.; Ushio, Y. Expression of tissue factor correlates with grade of malignancy in human glioma. Cancer 1996, 77, 1877–1883. [Google Scholar] [CrossRef]
- Takano, S.; Tsuboi, K.; Tomono, Y.; Mitsui, Y.; Nose, T. Tissue factor, osteopontin, alphavbeta3 integrin expression in microvasculature of gliomas associated with vascular endothelial growth factor expression. Br. J. Cancer 2000, 82, 1967–1973. [Google Scholar] [CrossRef]
- Takeshima, H.; Nishi, T.; Kuratsu, J.; Kamikubo, Y.; Kochi, M.; Ushio, Y. Suppression of the tissue factor-dependent coagulation cascade: A contributing factor for the development of intratumoral hemorrhage in glioblastoma. Int. J. Mol. Med. 2000, 6, 271–276. [Google Scholar] [CrossRef]
- Hron, G.; Kollars, M.; Weber, H.; Sagaster, V.; Quehenberger, P.; Eichinger, S.; Kyrle, P.A.; Weltermann, A. Tissue factor-positive microparticles: Cellular origin and association with coagulation activation in patients with colorectal cancer. Thromb. Haemost. 2007, 97, 119–123. [Google Scholar] [CrossRef]
- Kataoka, H.; Uchino, H.; Asada, Y.; Hatakeyama, K.; Nabeshima, K.; Sumiyoshi, A.; Koono, M. Analysis of tissue factor and tissue factor pathway inhibitor expression in human colorectal carcinoma cell lines and metastatic sublines to the liver. Int. J. Cancer 1997, 72, 878–884. [Google Scholar] [CrossRef]
- Nakasaki, T.; Wada, H.; Shigemori, C.; Miki, C.; Gabazza, E.C.; Nobori, T.; Nakamura, S.; Shiku, H. Expression of tissue factor and vascular endothelial growth factor is associated with angiogenesis in colorectal cancer. Am. J. Hematol. 2002, 69, 247–254. [Google Scholar] [CrossRef]
- Seto, S.; Onodera, H.; Kaido, T.; Yoshikawa, A.; Ishigami, S.; Arii, S.; Imamura, M. Tissue factor expression in human colorectal carcinoma: Correlation with hepatic metastasis and impact on prognosis. Cancer 2000, 88, 295–301. [Google Scholar] [CrossRef]
- Shigemori, C.; Wada, H.; Matsumoto, K.; Shiku, H.; Nakamura, S.; Suzuki, H. Tissue factor expression and metastatic potential of colorectal cancer. Thromb. Haemost. 1998, 80, 894–898. [Google Scholar]
- Belting, M.; Ahamed, J.; Ruf, W. Signaling of the tissue factor coagulation pathway in angiogenesis and cancer. Arterioscler. Thromb. Vasc. Biol. 2005, 25, 1545–1550. [Google Scholar] [CrossRef]
- Han, L.Y.; Landen, C.N., Jr.; Kamat, A.A.; Lopez, A.; Bender, D.P.; Mueller, P.; Schmandt, R.; Gershenson, D.M.; Sood, A.K. Preoperative serum tissue factor levels are an independent prognostic factor in patients with ovarian carcinoma. J. Clin. Oncol. 2006, 24, 755–761. [Google Scholar] [CrossRef] [PubMed]
- Uno, K.; Homma, S.; Satoh, T.; Nakanishi, K.; Abe, D.; Matsumoto, K.; Oki, A.; Tsunoda, H.; Yamaguchi, I.; Nagasawa, T.; et al. Tissue factor expression as a possible determinant of thromboembolism in ovarian cancer. Br. J. Cancer 2007, 96, 290–295. [Google Scholar] [CrossRef] [PubMed]
- Zacharski, L.R.; Memoli, V.A.; Ornstein, D.L.; Rousseau, S.M.; Kisiel, W.; Kudryk, B.J. Tumor cell procoagulant and urokinase expression in carcinoma of the ovary. J. Natl. Cancer Inst. 1993, 85, 1225–1230. [Google Scholar] [CrossRef] [PubMed]
- Del Conde, I.; Bharwani, L.D.; Dietzen, D.J.; Pendurthi, U.; Thiagarajan, P.; Lopez, J.A. Microvesicle-associated tissue factor and Trousseau’s syndrome. J. Thromb. Haemost. 2007, 5, 70–74. [Google Scholar] [CrossRef]
- Goldin-Lang, P.; Tran, Q.V.; Fichtner, I.; Eisenreich, A.; Antoniak, S.; Schulze, K.; Coupland, S.E.; Poller, W.; Schultheiss, H.P.; Rauch, U. Tissue factor expression pattern in human non-small cell lung cancer tissues indicate increased blood thrombogenicity and tumor metastasis. Oncol. Rep. 2008, 20, 123–128. [Google Scholar] [CrossRef]
- Haas, S.L.; Jesnowski, R.; Steiner, M.; Hummel, F.; Ringel, J.; Burstein, C.; Nizze, H.; Liebe, S.; Lohr, J.M. Expression of tissue factor in pancreatic adenocarcinoma is associated with activation of coagulation. World J. Gastroenterol. 2006, 12, 4843–4849. [Google Scholar] [CrossRef] [PubMed]
- Kakkar, A.K.; Lemoine, N.R.; Scully, M.F.; Tebbutt, S.; Williamson, R.C. Tissue factor expression correlates with histological grade in human pancreatic cancer. Br. J. Surg. 1995, 82, 1101–1104. [Google Scholar] [CrossRef] [PubMed]
- Khorana, A.A.; Ahrendt, S.A.; Ryan, C.K.; Francis, C.W.; Hruban, R.H.; Hu, Y.C.; Hostetter, G.; Harvey, J.; Taubman, M.B. Tissue factor expression, angiogenesis, and thrombosis in pancreatic cancer. Clin. Cancer Res. 2007, 13, 2870–2875. [Google Scholar] [CrossRef] [PubMed]
- Langer, F.; Spath, B.; Haubold, K.; Holstein, K.; Marx, G.; Wierecky, J.; Brummendorf, T.H.; Dierlamm, J.; Bokemeyer, C.; Eifrig, B. Tissue factor procoagulant activity of plasma microparticles in patients with cancer-associated disseminated intravascular coagulation. Ann. Hematol. 2008, 87, 451–457. [Google Scholar] [CrossRef] [PubMed]
- Rauch, U.; Antoniak, S.; Boots, M.; Schulze, K.; Goldin-Lang, P.; Stein, H.; Schultheiss, H.P.; Coupland, S.E. Association of tissue-factor upregulation in squamous-cell carcinoma of the lung with increased tissue factor in circulating blood. Lancet Oncol. 2005, 6, 254. [Google Scholar] [CrossRef] [PubMed]
- Akashi, T.; Furuya, Y.; Ohta, S.; Fuse, H. Tissue factor expression and prognosis in patients with metastatic prostate cancer. Urology 2003, 62, 1078–1082. [Google Scholar] [CrossRef] [PubMed]
- Kaido, T.; Oe, H.; Yoshikawa, A.; Mori, A.; Arii, S.; Imamura, M. Tissue factor is a useful prognostic factor of recurrence in hepatocellular carcinoma in 5-year survivors. Hepatogastroenterology 2005, 52, 1383–1387. [Google Scholar]
- Milsom, C.; Rak, J. Tissue factor and cancer. Pathophysiol. Haemost. Thromb. 2008, 36, 160–176. [Google Scholar] [CrossRef]
- Ueno, T.; Toi, M.; Koike, M.; Nakamura, S.; Tominaga, T. Tissue factor expression in breast cancer tissues: Its correlation with prognosis and plasma concentration. Br. J. Cancer 2000, 83, 164–170. [Google Scholar] [CrossRef]
- Yamashita, H.; Kitayama, J.; Ishikawa, M.; Nagawa, H. Tissue factor expression is a clinical indicator of lymphatic metastasis and poor prognosis in gastric cancer with intestinal phenotype. J. Surg. Oncol. 2007, 95, 324–331. [Google Scholar] [CrossRef]
- Yu, J.L.; May, L.; Lhotak, V.; Shahrzad, S.; Shirasawa, S.; Weitz, J.I.; Coomber, B.L.; Mackman, N.; Rak, J.W. Oncogenic events regulate tissue factor expression in colorectal cancer cells: Implications for tumor progression and angiogenesis. Blood 2005, 105, 1734–1741. [Google Scholar] [CrossRef]
- Rong, Y.; Belozerov, V.E.; Tucker-Burden, C.; Chen, G.; Durden, D.L.; Olson, J.J.; Van Meir, E.G.; Mackman, N.; Brat, D.J. Epidermal growth factor receptor and PTEN modulate tissue factor expression in glioblastoma through JunD/activator protein-1 transcriptional activity. Cancer Res. 2009, 69, 2540–2549. [Google Scholar] [CrossRef] [PubMed]
- Tallman, M.S.; Lefebvre, P.; Baine, R.M.; Shoji, M.; Cohen, I.; Green, D.; Kwaan, H.C.; Paietta, E.; Rickles, F.R. Effects of all-trans retinoic acid or chemotherapy on the molecular regulation of systemic blood coagulation and fibrinolysis in patients with acute promyelocytic leukemia. J. Thromb. Haemost. 2004, 2, 1341–1350. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.L.; May, L.; Klement, P.; Weitz, J.I.; Rak, J. Oncogenes as regulators of tissue factor expression in cancer: Implications for tumor angiogenesis and anti-cancer therapy. Semin. Thromb. Hemost. 2004, 30, 21–30. [Google Scholar] [CrossRef] [PubMed]
- Kato, S.; Pinto, M.; Carvajal, A.; Espinoza, N.; Monso, C.; Bravo, L.; Villalon, M.; Cuello, M.; Quest, A.F.; Suenaga, A.; et al. Tissue factor is regulated by epidermal growth factor in normal and malignant human endometrial epithelial cells. Thromb. Haemost. 2005, 94, 444–453. [Google Scholar] [CrossRef][Green Version]
- Milsom, C.C.; Yu, J.L.; Mackman, N.; Micallef, J.; Anderson, G.M.; Guha, A.; Rak, J.W. Tissue factor regulation by epidermal growth factor receptor and epithelial-to-mesenchymal transitions: Effect on tumor initiation and angiogenesis. Cancer Res. 2008, 68, 10068–10076. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.L.; Xing, R.; Milsom, C.; Rak, J. Modulation of the oncogene-dependent tissue factor expression by kinase suppressor of ras 1. Thromb. Res. 2010, 126, e6–e10. [Google Scholar] [CrossRef] [PubMed]
- Rong, Y.; Post, D.E.; Pieper, R.O.; Durden, D.L.; Van Meir, E.G.; Brat, D.J. PTEN and hypoxia regulate tissue factor expression and plasma coagulation by glioblastoma. Cancer Res. 2005, 65, 1406–1413. [Google Scholar] [CrossRef]
- Rao, B.; Gao, Y.; Huang, J.; Gao, X.; Fu, X.; Huang, M.; Yao, J.; Wang, J.; Li, W.; Zhang, J.; et al. Mutations of p53 and K-ras correlate TF expression in human colorectal carcinomas: TF downregulation as a marker of poor prognosis. Int. J. Color. Dis. 2011, 26, 593–601. [Google Scholar] [CrossRef]
- Regina, S.; Rollin, J.; Blechet, C.; Iochmann, S.; Reverdiau, P.; Gruel, Y. Tissue factor expression in non-small cell lung cancer: Relationship with vascular endothelial growth factor expression, microvascular density, and K-ras mutation. J. Thorac. Oncol. 2008, 3, 689–697. [Google Scholar] [CrossRef]
- Sun, L.; Liu, Y.; Lin, S.; Shang, J.; Liu, J.; Li, J.; Yuan, S.; Zhang, L. Early growth response gene-1 and hypoxia-inducible factor-1alpha affect tumor metastasis via regulation of tissue factor. Acta Oncol. 2013, 52, 842–851. [Google Scholar] [CrossRef] [PubMed]
- Boccaccio, C.; Sabatino, G.; Medico, E.; Girolami, F.; Follenzi, A.; Reato, G.; Sottile, A.; Naldini, L.; Comoglio, P.M. The MET oncogene drives a genetic programme linking cancer to haemostasis. Nature 2005, 434, 396–400. [Google Scholar] [CrossRef] [PubMed]
- Pennacchietti, S.; Michieli, P.; Galluzzo, M.; Mazzone, M.; Giordano, S.; Comoglio, P.M. Hypoxia promotes invasive growth by transcriptional activation of the met protooncogene. Cancer Cell 2003, 3, 347–361. [Google Scholar] [CrossRef] [PubMed]
- Bourcy, M.; Suarez-Carmona, M.; Lambert, J.; Francart, M.E.; Schroeder, H.; Delierneux, C.; Skrypek, N.; Thompson, E.W.; Jerusalem, G.; Berx, G.; et al. Tissue Factor Induced by Epithelial-Mesenchymal Transition Triggers a Procoagulant State That Drives Metastasis of Circulating Tumor Cells. Cancer Res. 2016, 76, 4270–4282. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.N.; Ljungdahl, S.; Shoshan, M.C.; Swedenborg, J.; Linder, S. Activation of tissue-factor gene expression in breast carcinoma cells by stimulation of the RAF-ERK signaling pathway. Mol. Carcinog. 1998, 21, 234–243. [Google Scholar] [CrossRef]
- Hamilton, J.R.; Trejo, J. Challenges and Opportunities in Protease-Activated Receptor Drug Development. Annu. Rev. Pharmacol. Toxicol. 2017, 57, 349–373. [Google Scholar] [CrossRef] [PubMed]
- Ruf, W.; Yokota, N.; Schaffner, F. Tissue factor in cancer progression and angiogenesis. Thromb. Res. 2010, 125 (Suppl. 2), S36–S38. [Google Scholar] [CrossRef]
- Zelaya, H.; Rothmeier, A.S.; Ruf, W. Tissue factor at the crossroad of coagulation and cell signaling. J. Thromb. Haemost. 2018, 16, 1941–1952. [Google Scholar] [CrossRef]
- Austin, K.M.; Covic, L.; Kuliopulos, A. Matrix metalloproteases and PAR1 activation. Blood 2013, 121, 431–439. [Google Scholar] [CrossRef]
- Camerer, E.; Kataoka, H.; Kahn, M.; Lease, K.; Coughlin, S.R. Genetic evidence that protease-activated receptors mediate factor Xa signaling in endothelial cells. J. Biol. Chem. 2002, 277, 16081–16087. [Google Scholar] [CrossRef]
- Schaffner, F.; Ruf, W. Tissue factor and PAR2 signaling in the tumor microenvironment. Arterioscler. Thromb. Vasc. Biol. 2009, 29, 1999–2004. [Google Scholar] [CrossRef] [PubMed]
- Ge, L.; Ly, Y.; Hollenberg, M.; DeFea, K. A beta-arrestin-dependent scaffold is associated with prolonged MAPK activation in pseudopodia during protease-activated receptor-2-induced chemotaxis. J. Biol. Chem. 2003, 278, 34418–34426. [Google Scholar] [CrossRef] [PubMed]
- Roy, A.; Ansari, S.A.; Das, K.; Prasad, R.; Bhattacharya, A.; Mallik, S.; Mukherjee, A.; Sen, P. Coagulation factor VIIa-mediated protease-activated receptor 2 activation leads to beta-catenin accumulation via the AKT/GSK3beta pathway and contributes to breast cancer progression. J. Biol. Chem. 2017, 292, 13688–13701. [Google Scholar] [CrossRef] [PubMed]
- Jiang, X.; Bailly, M.A.; Panetti, T.S.; Cappello, M.; Konigsberg, W.H.; Bromberg, M.E. Formation of tissue factor-factor VIIa-factor Xa complex promotes cellular signaling and migration of human breast cancer cells. J. Thromb. Haemost. 2004, 2, 93–101. [Google Scholar] [CrossRef]
- Hjortoe, G.M.; Petersen, L.C.; Albrektsen, T.; Sorensen, B.B.; Norby, P.L.; Mandal, S.K.; Pendurthi, U.R.; Rao, L.V. Tissue factor-factor VIIa-specific up-regulation of IL-8 expression in MDA-MB-231 cells is mediated by PAR-2 and results in increased cell migration. Blood 2004, 103, 3029–3037. [Google Scholar] [CrossRef]
- Holmes, W.E.; Lee, J.; Kuang, W.J.; Rice, G.C.; Wood, W.I. Structure and functional expression of a human interleukin-8 receptor. Science 1991, 253, 1278–1280. [Google Scholar] [CrossRef]
- Murphy, P.M.; Tiffany, H.L. Cloning of complementary DNA encoding a functional human interleukin-8 receptor. Science 1991, 253, 1280–1283. [Google Scholar] [CrossRef]
- Schraufstatter, I.U.; Chung, J.; Burger, M. IL-8 activates endothelial cell CXCR1 and CXCR2 through Rho and Rac signaling pathways. Am. J. Physiol. Lung Cell. Mol. Physiol. 2001, 280, L1094–L1103. [Google Scholar] [CrossRef]
- Waugh, D.J.; Wilson, C. The interleukin-8 pathway in cancer. Clin. Cancer Res. 2008, 14, 6735–6741. [Google Scholar] [CrossRef]
- DeFea, K.A.; Zalevsky, J.; Thoma, M.S.; Dery, O.; Mullins, R.D.; Bunnett, N.W. beta-arrestin-dependent endocytosis of proteinase-activated receptor 2 is required for intracellular targeting of activated ERK1/2. J. Cell Biol. 2000, 148, 1267–1281. [Google Scholar] [CrossRef]
- Ge, L.; Shenoy, S.K.; Lefkowitz, R.J.; DeFea, K. Constitutive protease-activated receptor-2-mediated migration of MDA MB-231 breast cancer cells requires both beta-arrestin-1 and -2. J. Biol. Chem. 2004, 279, 55419–55424. [Google Scholar] [CrossRef]
- Zoudilova, M.; Kumar, P.; Ge, L.; Wang, P.; Bokoch, G.M.; DeFea, K.A. Beta-arrestin-dependent regulation of the cofilin pathway downstream of protease-activated receptor-2. J. Biol. Chem. 2007, 282, 20634–20646. [Google Scholar] [CrossRef] [PubMed]
- Ahamed, J.; Versteeg, H.H.; Kerver, M.; Chen, V.M.; Mueller, B.M.; Hogg, P.J.; Ruf, W. Disulfide isomerization switches tissue factor from coagulation to cell signaling. Proc. Natl. Acad. Sci. USA 2006, 103, 13932–13937. [Google Scholar] [CrossRef] [PubMed]
- Abe, K.; Shoji, M.; Chen, J.; Bierhaus, A.; Danave, I.; Micko, C.; Casper, K.; Dillehay, D.L.; Nawroth, P.P.; Rickles, F.R. Regulation of vascular endothelial growth factor production and angiogenesis by the cytoplasmic tail of tissue factor. Proc. Natl. Acad. Sci. USA 1999, 96, 8663–8668. [Google Scholar] [CrossRef] [PubMed]
- Versteeg, H.H.; Schaffner, F.; Kerver, M.; Ellies, L.G.; Andrade-Gordon, P.; Mueller, B.M.; Ruf, W. Protease-activated receptor (PAR) 2, but not PAR1, signaling promotes the development of mammary adenocarcinoma in polyoma middle T mice. Cancer Res. 2008, 68, 7219–7227. [Google Scholar] [CrossRef]
- Prandoni, P.; Lensing, A.W.; Piccioli, A.; Bernardi, E.; Simioni, P.; Girolami, B.; Marchiori, A.; Sabbion, P.; Prins, M.H.; Noventa, F.; et al. Recurrent venous thromboembolism and bleeding complications during anticoagulant treatment in patients with cancer and venous thrombosis. Blood 2002, 100, 3484–3488. [Google Scholar] [CrossRef]
- Hu, T.; Bach, R.R.; Horton, R.; Konigsberg, W.H.; Todd, M.B. Procoagulant activity in cancer cells is dependent on tissue factor expression. Oncol. Res. 1994, 6, 321–327. [Google Scholar]
- Kobayashi, Y.; Yoshimura, N.; Nakamura, K.; Yamagishi, H.; Oka, T. Expression of tissue factor in hepatic ischemic-reperfusion injury of the rat. Transplantation 1998, 66, 708–716. [Google Scholar] [CrossRef]
- Tesselaar, M.E.; Romijn, F.P.; Van Der Linden, I.K.; Prins, F.A.; Bertina, R.M.; Osanto, S. Microparticle-associated tissue factor activity: A link between cancer and thrombosis? J. Thromb. Haemost. 2007, 5, 520–527. [Google Scholar] [CrossRef]
- Goldhaber, S.Z. Tamoxifen: Preventing breast cancer and placing the risk of deep vein thrombosis in perspective. Circulation 2005, 111, 539–541. [Google Scholar] [CrossRef]
- Elice, F.; Rodeghiero, F.; Falanga, A.; Rickles, F.R. Thrombosis associated with angiogenesis inhibitors. Best Pract. Res. Clin. Haematol. 2009, 22, 115–128. [Google Scholar] [CrossRef]
- Duvvuri, B.; Lood, C. Cell-Free DNA as a Biomarker in Autoimmune Rheumatic Diseases. Front. Immunol. 2019, 10, 502. [Google Scholar] [CrossRef]
- Holroyd, E.W.; Delacroix, S.; Larsen, K.; Harbuzariu, A.; Psaltis, P.J.; Wang, L.; Pan, S.; White, T.A.; Witt, T.A.; Kleppe, L.S.; et al. Tissue factor pathway inhibitor blocks angiogenesis via its carboxyl terminus. Arterioscler. Thromb. Vasc. Biol. 2012, 32, 704–711. [Google Scholar] [CrossRef]
- Xu, Z.; Maiti, D.; Kisiel, W.; Duh, E.J. Tissue factor pathway inhibitor-2 is upregulated by vascular endothelial growth factor and suppresses growth factor-induced proliferation of endothelial cells. Arterioscler. Thromb. Vasc. Biol. 2006, 26, 2819–2825. [Google Scholar] [CrossRef]
- Hembrough, T.A.; Swartz, G.M.; Papathanassiu, A.; Vlasuk, G.P.; Rote, W.E.; Green, S.J.; Pribluda, V.S. Tissue factor/factor VIIa inhibitors block angiogenesis and tumor growth through a nonhemostatic mechanism. Cancer Res. 2003, 63, 2997–3000. [Google Scholar]
- Rak, J.; Milsom, C.; May, L.; Klement, P.; Yu, J. Tissue factor in cancer and angiogenesis: The molecular link between genetic tumor progression, tumor neovascularization, and cancer coagulopathy. Semin. Thromb. Hemost. 2006, 32, 54–70. [Google Scholar] [CrossRef]
- Zhang, Y.; Deng, Y.; Luther, T.; Muller, M.; Ziegler, R.; Waldherr, R.; Stern, D.M.; Nawroth, P.P. Tissue factor controls the balance of angiogenic and antiangiogenic properties of tumor cells in mice. J. Clin. Investig. 1994, 94, 1320–1327. [Google Scholar] [CrossRef]
- Griffin, C.T.; Srinivasan, Y.; Zheng, Y.W.; Huang, W.; Coughlin, S.R. A role for thrombin receptor signaling in endothelial cells during embryonic development. Science 2001, 293, 1666–1670. [Google Scholar] [CrossRef]
- Rak, J.; Klement, G. Impact of oncogenes and tumor suppressor genes on deregulation of hemostasis and angiogenesis in cancer. Cancer Metastasis Rev. 2000, 19, 93–96. [Google Scholar] [CrossRef]
- Kerbel, R.S. Tumor angiogenesis. N. Engl. J. Med. 2008, 358, 2039–2049. [Google Scholar] [CrossRef]
- Rak, J.; Yu, J.L. Oncogenes and tumor angiogenesis: The question of vascular “supply” and vascular “demand”. Semin. Cancer Biol. 2004, 14, 93–104. [Google Scholar] [CrossRef] [PubMed]
- Tehrani, M.; Friedman, T.M.; Olson, J.J.; Brat, D.J. Intravascular thrombosis in central nervous system malignancies: A potential role in astrocytoma progression to glioblastoma. Brain Pathol. 2008, 18, 164–171. [Google Scholar] [CrossRef]
- Liu, Y.; Mueller, B.M. Protease-activated receptor-2 regulates vascular endothelial growth factor expression in MDA-MB-231 cells via MAPK pathways. Biochem. Biophys. Res. Commun. 2006, 344, 1263–1270. [Google Scholar] [CrossRef] [PubMed]
- Belting, M.; Dorrell, M.I.; Sandgren, S.; Aguilar, E.; Ahamed, J.; Dorfleutner, A.; Carmeliet, P.; Mueller, B.M.; Friedlander, M.; Ruf, W. Regulation of angiogenesis by tissue factor cytoplasmic domain signaling. Nat. Med. 2004, 10, 502–509. [Google Scholar] [CrossRef] [PubMed]
- Bar-Shavit, R.; Kahn, A.; Fenton, J.W., 2nd; Wilner, G.D. Chemotactic response of monocytes to thrombin. J. Cell Biol. 1983, 96, 282–285. [Google Scholar] [CrossRef] [PubMed]
- Norfleet, A.M.; Bergmann, J.S.; Carney, D.H. Thrombin peptide, TP508, stimulates angiogenic responses in animal models of dermal wound healing, in chick chorioallantoic membranes, and in cultured human aortic and microvascular endothelial cells. Gen. Pharmacol. 2000, 35, 249–254. [Google Scholar] [CrossRef] [PubMed]
- Pilcher, B.K.; Kim, D.W.; Carney, D.H.; Tomasek, J.J. Thrombin stimulates fibroblast-mediated collagen lattice contraction by its proteolytically activated receptor. Exp. Cell Res. 1994, 211, 368–373. [Google Scholar] [CrossRef]
- Tsopanoglou, N.E.; Maragoudakis, M.E. On the mechanism of thrombin-induced angiogenesis. Potentiation of vascular endothelial growth factor activity on endothelial cells by up-regulation of its receptors. J. Biol. Chem. 1999, 274, 23969–23976. [Google Scholar] [CrossRef]
- Jiang, X.; Zhu, S.; Panetti, T.S.; Bromberg, M.E. Formation of tissue factor-factor VIIa-factor Xa complex induces activation of the mTOR pathway which regulates migration of human breast cancer cells. Thromb. Haemost. 2008, 100, 127–133. [Google Scholar] [CrossRef]
- Wong, R.S. Apoptosis in cancer: From pathogenesis to treatment. J. Exp. Clin. Cancer Res. 2011, 30, 87. [Google Scholar] [CrossRef]
- Versteeg, H.H.; Spek, C.A.; Slofstra, S.H.; Diks, S.H.; Richel, D.J.; Peppelenbosch, M.P. FVIIa:TF induces cell survival via G12/G13-dependent Jak/STAT activation and BclXL production. Circ. Res. 2004, 94, 1032–1040. [Google Scholar] [CrossRef]
- Bluff, J.E.; Brown, N.J.; Reed, M.W.; Staton, C.A. Tissue factor, angiogenesis and tumour progression. Breast Cancer Res. 2008, 10, 204. [Google Scholar] [CrossRef]
- Unruh, D.; Horbinski, C. Beyond thrombosis: The impact of tissue factor signaling in cancer. J. Hematol. Oncol. 2020, 13, 93. [Google Scholar] [CrossRef]
- Palumbo, J.S.; Talmage, K.E.; Massari, J.V.; La Jeunesse, C.M.; Flick, M.J.; Kombrinck, K.W.; Hu, Z.; Barney, K.A.; Degen, J.L. Tumor cell-associated tissue factor and circulating hemostatic factors cooperate to increase metastatic potential through natural killer cell-dependent and-independent mechanisms. Blood 2007, 110, 133–141. [Google Scholar] [CrossRef] [PubMed]
- Ott, I.; Fischer, E.G.; Miyagi, Y.; Mueller, B.M.; Ruf, W. A role for tissue factor in cell adhesion and migration mediated by interaction with actin-binding protein 280. J. Cell Biol. 1998, 140, 1241–1253. [Google Scholar] [CrossRef] [PubMed]
- Bromberg, M.E.; Konigsberg, W.H.; Madison, J.F.; Pawashe, A.; Garen, A. Tissue factor promotes melanoma metastasis by a pathway independent of blood coagulation. Proc. Natl. Acad. Sci. USA 1995, 92, 8205–8209. [Google Scholar] [CrossRef]
- Ricono, J.M.; Huang, M.; Barnes, L.A.; Lau, S.K.; Weis, S.M.; Schlaepfer, D.D.; Hanks, S.K.; Cheresh, D.A. Specific cross-talk between epidermal growth factor receptor and integrin alphavbeta5 promotes carcinoma cell invasion and metastasis. Cancer Res. 2009, 69, 1383–1391. [Google Scholar] [CrossRef] [PubMed]
- Boire, A.; Covic, L.; Agarwal, A.; Jacques, S.; Sherifi, S.; Kuliopulos, A. PAR1 is a matrix metalloprotease-1 receptor that promotes invasion and tumorigenesis of breast cancer cells. Cell 2005, 120, 303–313. [Google Scholar] [CrossRef] [PubMed]
- Liu, F.; Zhang, Y.; Men, T.; Jiang, X.; Yang, C.; Li, H.; Wei, X.; Yan, D.; Feng, G.; Yang, J.; et al. Quantitative proteomic analysis of gastric cancer tissue reveals novel proteins in platelet-derived growth factor b signaling pathway. Oncotarget 2017, 8, 22059–22075. [Google Scholar] [CrossRef]
- Zhang, X.; Yu, H.; Lou, J.R.; Zheng, J.; Zhu, H.; Popescu, N.I.; Lupu, F.; Lind, S.E.; Ding, W.Q. MicroRNA-19 (miR-19) regulates tissue factor expression in breast cancer cells. J. Biol. Chem. 2011, 286, 1429–1435. [Google Scholar] [CrossRef]
- Yu, G.; Li, H.; Wang, X.; Wu, T.; Zhu, J.; Huang, S.; Wan, Y.; Tang, J. MicroRNA-19a targets tissue factor to inhibit colon cancer cells migration and invasion. Mol. Cell. Biochem. 2013, 380, 239–247. [Google Scholar] [CrossRef] [PubMed]
- Amarzguioui, M.; Peng, Q.; Wiiger, M.T.; Vasovic, V.; Babaie, E.; Holen, T.; Nesland, J.M.; Prydz, H. Ex vivo and in vivo delivery of anti-tissue factor short interfering RNA inhibits mouse pulmonary metastasis of B16 melanoma cells. Clin. Cancer Res. 2006, 12, 4055–4061. [Google Scholar] [CrossRef] [PubMed]
- Mueller, B.M.; Reisfeld, R.A.; Edgington, T.S.; Ruf, W. Expression of tissue factor by melanoma cells promotes efficient hematogenous metastasis. Proc. Natl. Acad. Sci. USA 1992, 89, 11832–11836. [Google Scholar] [CrossRef] [PubMed]
- Ngo, C.V.; Picha, K.; McCabe, F.; Millar, H.; Tawadros, R.; Tam, S.H.; Nakada, M.T.; Anderson, G.M. CNTO 859, a humanized anti-tissue factor monoclonal antibody, is a potent inhibitor of breast cancer metastasis and tumor growth in xenograft models. Int. J. Cancer 2007, 120, 1261–1267. [Google Scholar] [CrossRef]
- Ahamed, J.; Ruf, W. Protease-activated receptor 2-dependent phosphorylation of the tissue factor cytoplasmic domain. J. Biol. Chem. 2004, 279, 23038–23044. [Google Scholar] [CrossRef]
- Muller, M.; Albrecht, S.; Golfert, F.; Hofer, A.; Funk, R.H.; Magdolen, V.; Flossel, C.; Luther, T. Localization of tissue factor in actin-filament-rich membrane areas of epithelial cells. Exp. Cell Res. 1999, 248, 136–147. [Google Scholar] [CrossRef]
- Ettelaie, C.; Elkeeb, A.M.; Maraveyas, A.; Collier, M.E. p38alpha phosphorylates serine 258 within the cytoplasmic domain of tissue factor and prevents its incorporation into cell-derived microparticles. Biochim. Biophys. Acta 2013, 1833, 613–621. [Google Scholar] [CrossRef]
- Kurakula, K.; Koenis, D.S.; Herzik, M.A., Jr.; Liu, Y.; Craft, J.W., Jr.; van Loenen, P.B.; Vos, M.; Tran, M.K.; Versteeg, H.H.; Goumans, M.T.H.; et al. Structural and cellular mechanisms of peptidyl-prolyl isomerase Pin1-mediated enhancement of Tissue Factor gene expression, protein half-life, and pro-coagulant activity. Haematologica 2018, 103, 1073–1082. [Google Scholar] [CrossRef]
- Sen, M.; Herzik, M.; Craft, J.W.; Creath, A.L.; Agrawal, S.; Ruf, W.; Legge, G.B. Spectroscopic Characterization of Successive Phosphorylation of the Tissue Factor Cytoplasmic Region. Open Spectrosc. J. 2009, 3, 58–64. [Google Scholar] [CrossRef]
- Koizume, S.; Ito, S.; Yoshioka, Y.; Kanayama, T.; Nakamura, Y.; Yoshihara, M.; Yamada, R.; Ochiya, T.; Ruf, W.; Miyagi, E.; et al. High-level secretion of tissue factor-rich extracellular vesicles from ovarian cancer cells mediated by filamin-A and protease-activated receptors. Thromb. Haemost. 2016, 115, 299–310. [Google Scholar] [CrossRef]
- Savoy, R.M.; Ghosh, P.M. The dual role of filamin A in cancer: Can’t live with (too much of) it, can’t live without it. Endocr. Relat. Cancer 2013, 20, R341–R356. [Google Scholar] [CrossRef] [PubMed]
- Shao, Q.Q.; Zhang, T.P.; Zhao, W.J.; Liu, Z.W.; You, L.; Zhou, L.; Guo, J.C.; Zhao, Y.P. Filamin A: Insights into its Exact Role in Cancers. Pathol. Oncol. Res. 2016, 22, 245–252. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.H.; Hall, P.; Milenkovski, G.; Sharma, L.; Hutchinson, P.; Melis, E.; Carmeliet, P.; Tipping, P.; Morand, E. Reduction in arthritis severity and modulation of immune function in tissue factor cytoplasmic domain mutant mice. Am. J. Pathol. 2004, 164, 109–117. [Google Scholar] [CrossRef] [PubMed]
- Yin, Y.J.; Salah, Z.; Grisaru-Granovsky, S.; Cohen, I.; Even-Ram, S.C.; Maoz, M.; Uziely, B.; Peretz, T.; Bar-Shavit, R. Human protease-activated receptor 1 expression in malignant epithelia: A role in invasiveness. Arterioscler. Thromb. Vasc. Biol. 2003, 23, 940–944. [Google Scholar] [CrossRef] [PubMed]
- Ryden, L.; Grabau, D.; Schaffner, F.; Jonsson, P.E.; Ruf, W.; Belting, M. Evidence for tissue factor phosphorylation and its correlation with protease-activated receptor expression and the prognosis of primary breast cancer. Int. J. Cancer 2010, 126, 2330–2340. [Google Scholar] [CrossRef]
- Zhao, J.; Guan, J.L. Signal transduction by focal adhesion kinase in cancer. Cancer Metastasis Rev. 2009, 28, 35–49. [Google Scholar] [CrossRef] [PubMed]
- Hisada, Y.; Mackman, N. Tissue Factor and Cancer: Regulation, Tumor Growth, and Metastasis. Semin. Thromb. Hemost. 2019, 45, 385–395. [Google Scholar] [CrossRef]
- Yu, J.; May, L.; Milsom, C.; Anderson, G.M.; Weitz, J.I.; Luyendyk, J.P.; Broze, G.; Mackman, N.; Rak, J. Contribution of host-derived tissue factor to tumor neovascularization. Arterioscler. Thromb. Vasc. Biol. 2008, 28, 1975–1981. [Google Scholar] [CrossRef]
- Al-Nedawi, K.; Meehan, B.; Micallef, J.; Lhotak, V.; May, L.; Guha, A.; Rak, J. Intercellular transfer of the oncogenic receptor EGFRvIII by microvesicles derived from tumour cells. Nat. Cell Biol. 2008, 10, 619–624. [Google Scholar] [CrossRef]
- Chand, H.S.; Ness, S.A.; Kisiel, W. Identification of a novel human tissue factor splice variant that is upregulated in tumor cells. Int. J. Cancer 2006, 118, 1713–1720. [Google Scholar] [CrossRef]
- Hobbs, J.E.; Zakarija, A.; Cundiff, D.L.; Doll, J.A.; Hymen, E.; Cornwell, M.; Crawford, S.E.; Liu, N.; Signaevsky, M.; Soff, G.A. Alternatively spliced human tissue factor promotes tumor growth and angiogenesis in a pancreatic cancer tumor model. Thromb. Res. 2007, 120 (Suppl. 2), S13–S21. [Google Scholar] [CrossRef] [PubMed]
- Szotowski, B.; Antoniak, S.; Rauch, U. Alternatively spliced tissue factor: A previously unknown piece in the puzzle of hemostasis. Trends Cardiovasc. Med. 2006, 16, 177–182. [Google Scholar] [CrossRef] [PubMed]
- Kocaturk, B.; Tieken, C.; Vreeken, D.; Unlu, B.; Engels, C.C.; de Kruijf, E.M.; Kuppen, P.J.; Reitsma, P.H.; Bogdanov, V.Y.; Versteeg, H.H. Alternatively spliced tissue factor synergizes with the estrogen receptor pathway in promoting breast cancer progression. J. Thromb. Haemost. 2015, 13, 1683–1693. [Google Scholar] [CrossRef] [PubMed]
- Kocaturk, B.; Versteeg, H.H. Tissue factor-integrin interactions in cancer and thrombosis: Every Jack has his Jill. J. Thromb. Haemost. 2013, 11 (Suppl. 1), 285–293. [Google Scholar] [CrossRef]
- Signaevsky, M.; Hobbs, J.; Doll, J.; Liu, N.; Soff, G.A. Role of alternatively spliced tissue factor in pancreatic cancer growth and angiogenesis. Semin. Thromb. Hemost. 2008, 34, 161–169. [Google Scholar] [CrossRef]
- Xu, C.; Wang, H.; He, H.; Zheng, F.; Chen, Y.; Zhang, J.; Lin, X.; Ma, D.; Zhang, H. Low expression of TFPI-2 associated with poor survival outcome in patients with breast cancer. BMC Cancer 2013, 13, 118. [Google Scholar] [CrossRef]
- Stavik, B.; Skretting, G.; Sletten, M.; Sandset, P.M.; Iversen, N. Overexpression of both TFPIalpha and TFPIbeta induces apoptosis and expression of genes involved in the death receptor pathway in breast cancer cells. Mol. Carcinog. 2010, 49, 951–963. [Google Scholar] [CrossRef]
- Stavik, B.; Skretting, G.; Aasheim, H.C.; Tinholt, M.; Zernichow, L.; Sletten, M.; Sandset, P.M.; Iversen, N. Downregulation of TFPI in breast cancer cells induces tyrosine phosphorylation signaling and increases metastatic growth by stimulating cell motility. BMC Cancer 2011, 11, 357. [Google Scholar] [CrossRef]
- McCall-Culbreath, K.D.; Zutter, M.M. Collagen receptor integrins: Rising to the challenge. Curr. Drug Targets 2008, 9, 139–149. [Google Scholar] [CrossRef]
- Rao, C.N.; Lakka, S.S.; Kin, Y.; Konduri, S.D.; Fuller, G.N.; Mohanam, S.; Rao, J.S. Expression of tissue factor pathway inhibitor 2 inversely correlates during the progression of human gliomas. Clin. Cancer Res. 2001, 7, 570–576. [Google Scholar]
- Guo, H.; Lin, Y.; Zhang, H.; Liu, J.; Zhang, N.; Li, Y.; Kong, D.; Tang, Q.; Ma, D. Tissue factor pathway inhibitor-2 was repressed by CpG hypermethylation through inhibition of KLF6 binding in highly invasive breast cancer cells. BMC Mol. Biol. 2007, 8, 110. [Google Scholar] [CrossRef] [PubMed]
- Nobeyama, Y.; Okochi-Takada, E.; Furuta, J.; Miyagi, Y.; Kikuchi, K.; Yamamoto, A.; Nakanishi, Y.; Nakagawa, H.; Ushijima, T. Silencing of tissue factor pathway inhibitor-2 gene in malignant melanomas. Int. J. Cancer 2007, 121, 301–307. [Google Scholar] [CrossRef] [PubMed]
- Hibi, K.; Goto, T.; Kitamura, Y.H.; Yokomizo, K.; Sakuraba, K.; Shirahata, A.; Mizukami, H.; Saito, M.; Ishibashi, K.; Kigawa, G.; et al. Methylation of TFPI2 gene is frequently detected in advanced well-differentiated colorectal cancer. Anticancer Res. 2010, 30, 1205–1207. [Google Scholar] [PubMed]
- Sato, N.; Parker, A.R.; Fukushima, N.; Miyagi, Y.; Iacobuzio-Donahue, C.A.; Eshleman, J.R.; Goggins, M. Epigenetic inactivation of TFPI-2 as a common mechanism associated with growth and invasion of pancreatic ductal adenocarcinoma. Oncogene 2005, 24, 850–858. [Google Scholar] [CrossRef] [PubMed]
- Hube, F.; Reverdiau, P.; Iochmann, S.; Cherpi-Antar, C.; Gruel, Y. Characterization and functional analysis of TFPI-2 gene promoter in a human choriocarcinoma cell line. Thromb. Res. 2003, 109, 207–215. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Jia, Y.; Yang, Y.; Brock, M.V.; Cao, B.; Zhan, Q.; Li, Y.; Yu, Y.; Herman, J.G.; Guo, M. Methylation of TFPI-2 is an early event of esophageal carcinogenesis. Epigenomics 2012, 4, 135–146. [Google Scholar] [CrossRef]
- Takada, H.; Wakabayashi, N.; Dohi, O.; Yasui, K.; Sakakura, C.; Mitsufuji, S.; Taniwaki, M.; Yoshikawa, T. Tissue factor pathway inhibitor 2 (TFPI2) is frequently silenced by aberrant promoter hypermethylation in gastric cancer. Cancer Genet. Cytogenet. 2010, 197, 16–24. [Google Scholar] [CrossRef]
- Geng, G.; Liu, X.; Xu, A.; Lu, Z.; Chen, K.; He, J.; Qi, D.; Yuan, X. Low abundance of TFPI-2 by both promoter methylation and miR-27a-3p regulation is linked with poor clinical outcome in gastric cancer. J. Gene Med. 2020, 22, e3166. [Google Scholar] [CrossRef]
- Amirkhosravi, A.; Meyer, T.; Chang, J.Y.; Amaya, M.; Siddiqui, F.; Desai, H.; Francis, J.L. Tissue factor pathway inhibitor reduces experimental lung metastasis of B16 melanoma. Thromb. Haemost. 2002, 87, 930–936. [Google Scholar]
- Gessler, F.; Voss, V.; Seifert, V.; Gerlach, R.; Kogel, D. Knockdown of TFPI-2 promotes migration and invasion of glioma cells. Neurosci. Lett. 2011, 497, 49–54. [Google Scholar] [CrossRef]
- Wang, S.; Xiao, X.; Zhou, X.; Huang, T.; Du, C.; Yu, N.; Mo, Y.; Lin, L.; Zhang, J.; Ma, N.; et al. TFPI-2 is a putative tumor suppressor gene frequently inactivated by promoter hypermethylation in nasopharyngeal carcinoma. BMC Cancer 2010, 10, 617. [Google Scholar] [CrossRef] [PubMed]
- Kondraganti, S.; Gondi, C.S.; Gujrati, M.; McCutcheon, I.; Dinh, D.H.; Rao, J.S.; Olivero, W.C. Restoration of tissue factor pathway inhibitor inhibits invasion and tumor growth in vitro and in vivo in a malignant meningioma cell line. Int. J. Oncol. 2006, 29, 25–32. [Google Scholar] [CrossRef] [PubMed]
- Konduri, S.D.; Tasiou, A.; Chandrasekar, N.; Rao, J.S. Overexpression of tissue factor pathway inhibitor-2 (TFPI-2), decreases the invasiveness of prostate cancer cells in vitro. Int. J. Oncol. 2001, 18, 127–131. [Google Scholar] [CrossRef] [PubMed]
- George, J.; Gondi, C.S.; Dinh, D.H.; Gujrati, M.; Rao, J.S. Restoration of tissue factor pathway inhibitor-2 in a human glioblastoma cell line triggers caspase-mediated pathway and apoptosis. Clin. Cancer Res. 2007, 13, 3507–3517. [Google Scholar] [CrossRef] [PubMed]
- Tang, Z.; Geng, G.; Huang, Q.; Xu, G.; Hu, H.; Chen, J.; Li, J. Expression of tissue factor pathway inhibitor 2 in human pancreatic carcinoma and its effect on tumor growth, invasion, and migration in vitro and in vivo. J. Surg. Res. 2011, 167, 62–69. [Google Scholar] [CrossRef]
- Lavergne, M.; Jourdan, M.L.; Blechet, C.; Guyetant, S.; Pape, A.L.; Heuze-Vourc’h, N.; Courty, Y.; Lerondel, S.; Sobilo, J.; Iochmann, S.; et al. Beneficial role of overexpression of TFPI-2 on tumour progression in human small cell lung cancer. FEBS Open Bio 2013, 3, 291–301. [Google Scholar] [CrossRef]
- Fischer, E.G.; Riewald, M.; Huang, H.Y.; Miyagi, Y.; Kubota, Y.; Mueller, B.M.; Ruf, W. Tumor cell adhesion and migration supported by interaction of a receptor-protease complex with its inhibitor. J. Clin. Investig. 1999, 104, 1213–1221. [Google Scholar] [CrossRef]
- Lorenzet, R.; Donati, M.B. Blood clotting activation, angiogenesis and tumor metastasis: Any role for TFPI? Thromb. Haemost. 2002, 87, 928–929. [Google Scholar]
- Espinoza-Sanchez, N.A.; Gotte, M. Role of cell surface proteoglycans in cancer immunotherapy. Semin. Cancer Biol. 2020, 62, 48–67. [Google Scholar] [CrossRef]
- Wight, T.N. A role for proteoglycans in vascular disease. Matrix Biol. 2018, 71–72, 396–420. [Google Scholar] [CrossRef]
- Peterson, J.A.; Maroney, S.A.; Martinez, N.D.; Mast, A.E. Major Reservoir for Heparin-Releasable TFPIalpha (Tissue Factor Pathway Inhibitor alpha) Is Extracellular Matrix. Arterioscler. Thromb. Vasc. Biol. 2021, 41, 1942–1955. [Google Scholar] [CrossRef] [PubMed]
- Chowdary, P. Inhibition of Tissue Factor Pathway Inhibitor (TFPI) as a Treatment for Haemophilia: Rationale with Focus on Concizumab. Drugs 2018, 78, 881–890. [Google Scholar] [CrossRef] [PubMed]
- Reitsma, S.; Slaaf, D.W.; Vink, H.; van Zandvoort, M.A.; oude Egbrink, M.G. The endothelial glycocalyx: Composition, functions, and visualization. Pflug. Arch. 2007, 454, 345–359. [Google Scholar] [CrossRef] [PubMed]
- Wesselschmidt, R.; Likert, K.; Girard, T.; Wun, T.C.; Broze, G.J., Jr. Tissue factor pathway inhibitor: The carboxy-terminus is required for optimal inhibition of factor Xa. Blood 1992, 79, 2004–2010. [Google Scholar] [CrossRef] [PubMed]
- Wun, T.C. Lipoprotein-associated coagulation inhibitor (LACI) is a cofactor for heparin: Synergistic anticoagulant action between LACI and sulfated polysaccharides. Blood 1992, 79, 430–438. [Google Scholar] [CrossRef] [PubMed]
- Warshawsky, I.; Herz, J.; Broze, G.J., Jr.; Schwartz, A.L. The low density lipoprotein receptor-related protein can function independently from heparan sulfate proteoglycans in tissue factor pathway inhibitor endocytosis. J. Biol. Chem. 1996, 271, 25873–25879. [Google Scholar] [CrossRef] [PubMed]
- Swieringa, F.; Spronk, H.M.H.; Heemskerk, J.W.M.; van der Meijden, P.E.J. Integrating platelet and coagulation activation in fibrin clot formation. Res. Pract. Thromb. Haemost. 2018, 2, 450–460. [Google Scholar] [CrossRef]
- Bode, M.; Mackman, N. Regulation of tissue factor gene expression in monocytes and endothelial cells: Thromboxane A2 as a new player. Vasc. Pharmacol. 2014, 62, 57–62. [Google Scholar] [CrossRef]
- Petit, L.; Lesnik, P.; Dachet, C.; Moreau, M.; Chapman, M.J. Tissue factor pathway inhibitor is expressed by human monocyte-derived macrophages: Relationship to tissue factor induction by cholesterol and oxidized LDL. Arterioscler. Thromb. Vasc. Biol. 1999, 19, 309–315. [Google Scholar] [CrossRef]
- Ott, I.; Miyagi, Y.; Miyazaki, K.; Heeb, M.J.; Mueller, B.M.; Rao, L.V.; Ruf, W. Reversible regulation of tissue factor-induced coagulation by glycosyl phosphatidylinositol-anchored tissue factor pathway inhibitor. Arterioscler. Thromb. Vasc. Biol. 2000, 20, 874–882. [Google Scholar] [CrossRef]
- Piro, O.; Broze, G.J., Jr. Role for the Kunitz-3 domain of tissue factor pathway inhibitor-alpha in cell surface binding. Circulation 2004, 110, 3567–3572. [Google Scholar] [CrossRef] [PubMed]
- Warshawsky, I.; Bu, G.; Mast, A.; Saffitz, J.E.; Broze, G.J., Jr.; Schwartz, A.L. The carboxy terminus of tissue factor pathway inhibitor is required for interacting with hepatoma cells in vitro and in vivo. J. Clin. Investig. 1995, 95, 1773–1781. [Google Scholar] [CrossRef] [PubMed]
- Novotny, W.F.; Brown, S.G.; Miletich, J.P.; Rader, D.J.; Broze, G.J., Jr. Plasma antigen levels of the lipoprotein-associated coagulation inhibitor in patient samples. Blood 1991, 78, 387–393. [Google Scholar] [CrossRef] [PubMed]
- Haynes, A., 3rd; Ruda, F.; Oliver, J.; Hamood, A.N.; Griswold, J.A.; Park, P.W.; Rumbaugh, K.P. Syndecan 1 shedding contributes to Pseudomonas aeruginosa sepsis. Infect. Immun. 2005, 73, 7914–7921. [Google Scholar] [CrossRef] [PubMed]
- Osuka, A.; Kusuki, H.; Yoneda, K.; Matsuura, H.; Matsumoto, H.; Ogura, H.; Ueyama, M. Glycocalyx Shedding is Enhanced by Age and Correlates with Increased Fluid Requirement in Patients with Major Burns. Shock 2018, 50, 60–65. [Google Scholar] [CrossRef] [PubMed]
- Vigiola Cruz, M.; Carney, B.C.; Luker, J.N.; Monger, K.W.; Vazquez, J.S.; Moffatt, L.T.; Johnson, L.S.; Shupp, J.W. Plasma Ameliorates Endothelial Dysfunction in Burn Injury. J. Surg. Res. 2019, 233, 459–466. [Google Scholar] [CrossRef]
- Keyloun, J.W.; Le, T.D.; Pusateri, A.E.; Ball, R.L.; Carney, B.C.; Orfeo, T.; Brummel-Ziedins, K.E.; Bravo, M.C.; McLawhorn, M.M.; Moffatt, L.T.; et al. Circulating Syndecan-1 and Tissue Factor Pathway Inhibitor, Biomarkers of Endothelial Dysfunction, Predict Mortality in Burn Patients. Shock 2021, 56, 237–244. [Google Scholar] [CrossRef]
- Ho, G.; Broze, G.J., Jr.; Schwartz, A.L. Role of heparan sulfate proteoglycans in the uptake and degradation of tissue factor pathway inhibitor-coagulation factor Xa complexes. J. Biol. Chem. 1997, 272, 16838–16844. [Google Scholar] [CrossRef]
- Nassar, E.; Hassan, N.; El-Ghonaimy, E.A.; Hassan, H.; Abdullah, M.S.; Rottke, T.V.; Kiesel, L.; Greve, B.; Ibrahim, S.A.; Gotte, M. Syndecan-1 Promotes Angiogenesis in Triple-Negative Breast Cancer through the Prognostically Relevant Tissue Factor Pathway and Additional Angiogenic Routes. Cancers 2021, 13, 2318. [Google Scholar] [CrossRef]
- Hassan, N.; Greve, B.; Espinoza-Sanchez, N.A.; Gotte, M. Cell-surface heparan sulfate proteoglycans as multifunctional integrators of signaling in cancer. Cell. Signal. 2021, 77, 109822. [Google Scholar] [CrossRef]
- Szklarczyk, D.; Franceschini, A.; Wyder, S.; Forslund, K.; Heller, D.; Huerta-Cepas, J.; Simonovic, M.; Roth, A.; Santos, A.; Tsafou, K.P.; et al. STRING v10: Protein-protein interaction networks, integrated over the tree of life. Nucleic Acids Res. 2015, 43, D447–D452. [Google Scholar] [CrossRef] [PubMed]
- Tinholt, M.; Stavik, B.; Louch, W.; Carlson, C.R.; Sletten, M.; Ruf, W.; Skretting, G.; Sandset, P.M.; Iversen, N. Syndecan-3 and TFPI colocalize on the surface of endothelial-, smooth muscle-, and cancer cells. PLoS ONE 2015, 10, e0117404. [Google Scholar] [CrossRef] [PubMed]
- Girard, T.J.; Tuley, E.; Broze, G.J., Jr. TFPIbeta is the GPI-anchored TFPI isoform on human endothelial cells and placental microsomes. Blood 2012, 119, 1256–1262. [Google Scholar] [CrossRef] [PubMed]
- Saikali, Z.; Sinnett, D. Expression of glypican 3 (GPC3) in embryonal tumors. Int. J. Cancer 2000, 89, 418–422. [Google Scholar] [CrossRef] [PubMed]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hassan, N.; Efing, J.; Kiesel, L.; Bendas, G.; Götte, M. The Tissue Factor Pathway in Cancer: Overview and Role of Heparan Sulfate Proteoglycans. Cancers 2023, 15, 1524. https://doi.org/10.3390/cancers15051524
Hassan N, Efing J, Kiesel L, Bendas G, Götte M. The Tissue Factor Pathway in Cancer: Overview and Role of Heparan Sulfate Proteoglycans. Cancers. 2023; 15(5):1524. https://doi.org/10.3390/cancers15051524
Chicago/Turabian StyleHassan, Nourhan, Janes Efing, Ludwig Kiesel, Gerd Bendas, and Martin Götte. 2023. "The Tissue Factor Pathway in Cancer: Overview and Role of Heparan Sulfate Proteoglycans" Cancers 15, no. 5: 1524. https://doi.org/10.3390/cancers15051524
APA StyleHassan, N., Efing, J., Kiesel, L., Bendas, G., & Götte, M. (2023). The Tissue Factor Pathway in Cancer: Overview and Role of Heparan Sulfate Proteoglycans. Cancers, 15(5), 1524. https://doi.org/10.3390/cancers15051524