
Dysregulated TET Family Genes and Aberrant 5mC Oxidation in Breast Cancer: Causes and Consequences


{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
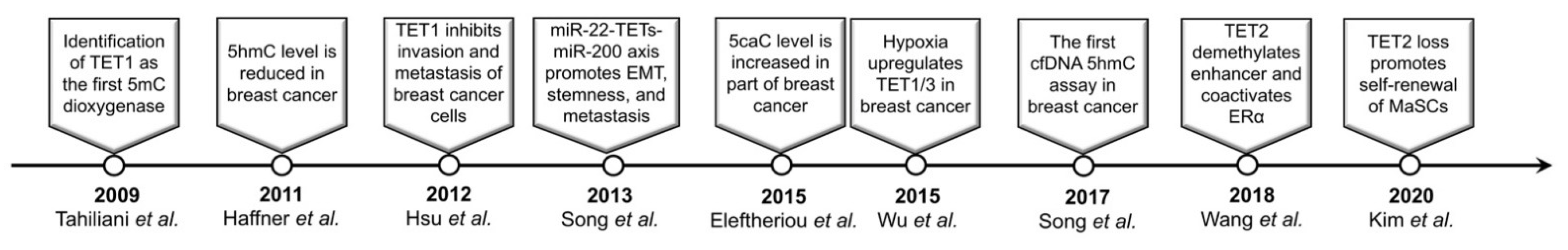
1. Introduction
2. Aberrant 5mC Oxidation in Breast Cancer
3. Dysregulation of TET Family Genes in Breast Cancer
3.1. Genetic Alterations of TET Family Genes in Breast Cancer
3.2. Aberrant Expression of TET Family Genes in Breast Cancer
3.3. The Rewired Catalytic Activity of TET Proteins in Breast Cancer
3.4. Aberrant Genomic Targeting of TET Proteins in Breast Cancer
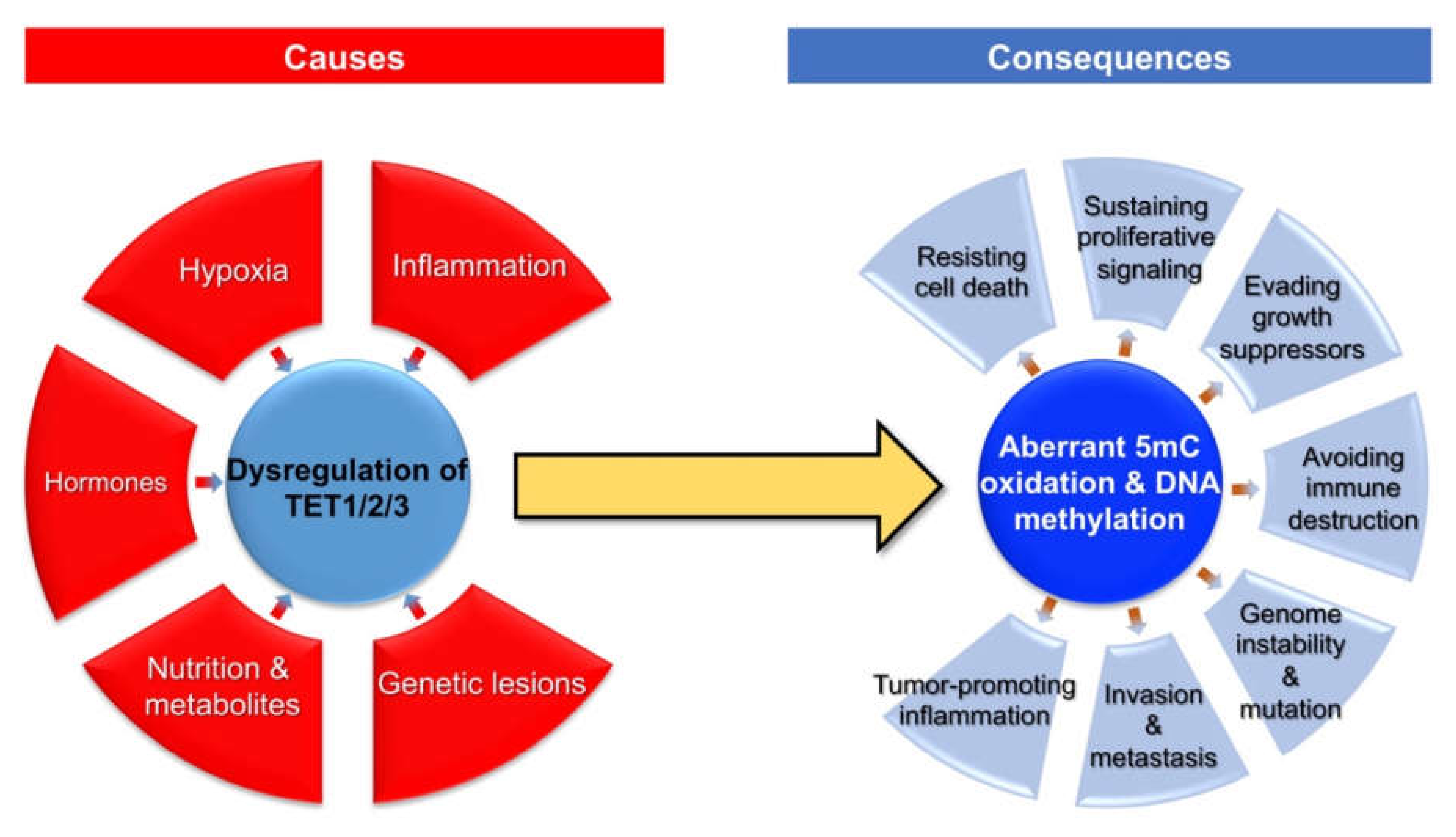
4. Consequences of Dysregulated TET Family Genes and Aberrant 5mC Oxidation in Breast Cancer
4.1. Genomic Instability and Mutation
4.2. Stemness
4.3. Cell Proliferation
4.4. Invasion and Metastasis
4.5. Cell Death
4.6. Inflammation and Tumor Immunity
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Łukasiewicz, S.; Czeczelewski, M.; Forma, A.; Baj, J.; Sitarz, R.; Stanisławek, A. Breast Cancer—Epidemiology, Risk Factors, Classification, Prognostic Markers, and Current Treatment Strategies—An Updated Review. Cancers 2021, 13, 4287. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Martinez, L.; Zhang, Y.; Nakata, Y.; Chan, H.L.; Morey, L. Epigenetic mechanisms in breast cancer therapy and resistance. Nat. Commun. 2021, 12, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Tahiliani, M.; Koh, K.P.; Shen, Y.; Pastor, W.A.; Bandukwala, H.; Brudno, Y.; Agarwal, S.; Iyer, L.M.; Liu, D.R.; Aravind, L.; et al. Conversion of 5-Methylcytosine to 5-Hydroxymethylcytosine in Mammalian DNA by MLL Partner TET1. Science 2009, 324, 930–935. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, Y.-F.; Li, B.-Z.; Li, Z.; Liu, P.; Wang, Y.; Tang, Q.; Ding, J.; Jia, Y.; Chen, Z.; Li, L.; et al. Tet-Mediated Formation of 5-Carboxylcytosine and Its Excision by TDG in Mammalian DNA. Science 2011, 333, 1303–1307. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ito, S.; Shen, L.; Dai, Q.; Wu, S.C.; Collins, L.B.; Swenberg, J.A.; He, C.; Zhang, Y. Tet Proteins Can Convert 5-Methylcytosine to 5-Formylcytosine and 5-Carboxylcytosine. Science 2011, 333, 1300–1303. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lorsbach, R.B.; Moore, J.; Mathew, S.; Raimondi, S.C.; Mukatira, S.T.; Downing, J.R. TET1, a member of a novel protein family, is fused to MLL in acute myeloid leukemia containing the t(10;11)(q22;q23). Leukemia 2003, 17, 637–641. [Google Scholar] [CrossRef] [Green Version]
- Tan, L.; Shi, Y.G. Tet family proteins and 5-hydroxymethylcytosine in development and disease. Development 2012, 139, 1895–1902. [Google Scholar] [CrossRef] [Green Version]
- Haffner, M.C.; Chaux, A.; Meeker, A.K.; Esopi, D.M.; Gerber, J.; Pellakuru, L.G.; Toubaji, A.; Argani, P.; Iacobuzio-Donahue, C.; Nelson, W.G.; et al. Global 5-hydroxymethylcytosine content is significantly reduced in tissue stem/progenitor cell compartments and in human cancers. Oncotarget 2011, 2, 627–637. [Google Scholar] [CrossRef] [Green Version]
- Nestor, C.E.; Ottaviano, R.; Reddington, J.; Sproul, D.; Reinhardt, D.; Dunican, D.; Katz, E.; Dixon, J.M.; Harrison, D.J.; Meehan, R.R. Tissue type is a major modifier of the 5-hydroxymethylcytosine content of human genes. Genome Res. 2011, 22, 467–477. [Google Scholar] [CrossRef] [Green Version]
- Nestor, C.E.; Ottaviano, R.; Reinhardt, D.; Cruickshanks, H.A.; Mjoseng, H.K.; McPherson, R.C.; Lentini, A.; Thomson, J.P.; Dunican, D.S.; Pennings, S.; et al. Rapid reprogramming of epigenetic and transcriptional profiles in mammalian culture systems. Genome Biol. 2015, 16, 1–17. [Google Scholar] [CrossRef] [Green Version]
- Bachman, M.; Uribe-Lewis, S.; Yang, X.; Williams, M.; Murrell, A.; Balasubramanian, S. 5-Hydroxymethylcytosine is a predominantly stable DNA modification. Nat. Chem. 2014, 6, 1049–1055. [Google Scholar] [CrossRef] [Green Version]
- Tsai, K.-W.; Li, G.-C.; Chen, C.-H.; Yeh, M.-H.; Huang, J.-S.; Tseng, H.-H.; Fu, T.-Y.; Liou, H.-H.; Pan, H.-W.; Huang, S.-F.; et al. Reduction of global 5-hydroxymethylcytosine is a poor prognostic factor in breast cancer patients, especially for an ER/PR-negative subtype. Breast Cancer Res. Treat. 2015, 153, 219–234. [Google Scholar] [CrossRef]
- Wu, M.Z.; Chen, S.F.; Nieh, S.; Benner, C.; Ger, L.P.; Jan, C.I.; Ma, L.; Chen, C.H.; Hishida, T.; Chang, H.T.; et al. Hypoxia Drives Breast Tumor Malignancy through a TET-TNFalpha-p38-MAPK Signaling Axis. Cancer Res. 2015, 75, 3912–3924. [Google Scholar] [CrossRef] [Green Version]
- Eleftheriou, M.M.; Pascual, A.J.A.; Wheldon, L.L.; Perry, C.C.; Abakir, A.H.; Arora, A.A.; Johnson, A.D.A.; Auer, D.D.; Ellis, I.O.; Madhusudan, S.S.; et al. 5-Carboxylcytosine levels are elevated in human breast cancers and gliomas. Clin. Epigenet. 2015, 7, 1–6. [Google Scholar] [CrossRef]
- Matthias, W.; Liou, W.; Pulverer, W.; Singer, C.F.; Rappaport-Fuerhauser, C.; Kandioler, D.; Egger, G.; Weinhäusel, A. Cytosine 5-Hydroxymethylation of the LZTS1 Gene Is Reduced in Breast Cancer. Transl. Oncol. 2013, 6, 715–721. [Google Scholar] [CrossRef] [Green Version]
- Zhang, D.; An, X.; Li, Z.; Zhang, S. Role of gene promoter methylation regulated by TETs and DNMTs in the overexpression of HLA-G in MCF-7 cells. Exp. Ther. Med. 2019, 17, 4709–4714. [Google Scholar] [CrossRef]
- Collignon, E.; Canale, A.; Al Wardi, C.; Bizet, M.; Calonne, E.; Dedeurwaerder, S.; Garaud, S.; Naveaux, C.; Barham, W.; Wilson, A.; et al. Immunity drives TET1 regulation in cancer through NF-kappaB. Sci. Adv. 2018, 4, eaap7309. [Google Scholar] [CrossRef] [Green Version]
- Song, C.-X.; Yin, S.; Ma, L.; Wheeler, A.; Chen, Y.; Zhang, Y.; Liu, B.; Xiong, J.; Zhang, W.; Hu, J.; et al. 5-Hydroxymethylcytosine signatures in cell-free DNA provide information about tumor types and stages. Cell Res. 2017, 27, 1231–1242. [Google Scholar] [CrossRef] [Green Version]
- Cai, J.; Chen, L.; Zhang, Z.; Zhang, X.; Lu, X.; Liu, W.; Shi, G.; Ge, Y.; Gao, P.; Yang, Y.; et al. Genome-wide mapping of 5-hydroxymethylcytosines in circulating cell-free DNA as a non-invasive approach for early detection of hepatocellular carcinoma. Gut 2019, 68, 2195–2205. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Han, X.; Gao, C.; Xing, Y.; Qi, Z.; Liu, R.; Wang, Y.; Zhang, X.; Yang, Y.-G.; Li, X.; et al. 5-Hydroxymethylome in Circulating Cell-free DNA as A Potential Biomarker for Non-small-cell Lung Cancer. Genom. Proteom. Bioinform. 2018, 16, 187–199. [Google Scholar] [CrossRef]
- Li, W.; Zhang, X.; Lu, X.; You, L.; Song, Y.; Luo, Z.; Zhang, J.; Nie, J.; Zheng, W.; Xu, D.; et al. 5-Hydroxymethylcytosine signatures in circulating cell-free DNA as diagnostic biomarkers for human cancers. Cell Res. 2017, 27, 1243–1257. [Google Scholar] [CrossRef] [Green Version]
- Zeng, C.; Stroup, E.K.; Zhang, Z.; Chiu, B.C.-H.; Zhang, W. Towards precision medicine: Advances in 5-hydroxymethylcytosine cancer biomarker discovery in liquid biopsy. Cancer Commun. 2019, 39, 12–19. [Google Scholar] [CrossRef] [Green Version]
- Li, N.; Rowley, S.M.; Thompson, E.R.; McInerny, S.; Devereux, L.; Amarasinghe, K.C.; Zethoven, M.; Lupat, R.; Goode, D.; Li, J.; et al. Evaluating the breast cancer predisposition role of rare variants in genes associated with low-penetrance breast cancer risk SNPs. Breast Cancer Res. 2018, 20, 3. [Google Scholar] [CrossRef] [Green Version]
- Jaiswal, S.; Fontanillas, P.; Flannick, J.; Manning, A.; Grauman, P.V.; Mar, B.; Lindsley, C.; Mermel, C.; Burtt, N.; Chavez, A.; et al. Age-Related Clonal Hematopoiesis Associated with Adverse Outcomes. N. Engl. J. Med. 2014, 371, 2488–2498. [Google Scholar] [CrossRef] [Green Version]
- Xie, M.; Lu, C.; Wang, J.; McLellan, M.D.; Johnson, K.J.; Wendl, M.C.; McMichael, J.F.; Schmidt, H.K.; Yellapantula, V.; Miller, C.A.; et al. Age-related mutations associated with clonal hematopoietic expansion and malignancies. Nat. Med. 2014, 20, 1472–1478. [Google Scholar] [CrossRef]
- Busque, L.; Buscarlet, M.; Mollica, L.; Levine, R.L. Concise Review: Age-Related Clonal Hematopoiesis: Stem Cells Tempting the Devil. Stem Cells 2018, 36, 1287–1294. [Google Scholar] [CrossRef] [Green Version]
- Yang, H.; Liu, Y.; Bai, F.; Zhang, J.-Y.; Ma, S.-H.; Liu, J.; Xu, Z.-D.; Zhu, H.-G.; Ling, Z.-Q.; Ye, D.; et al. Tumor development is associated with decrease of TET gene expression and 5-methylcytosine hydroxylation. Oncogene 2013, 32, 663–669. [Google Scholar] [CrossRef] [Green Version]
- Yang, L.; Yu, S.-J.; Hong, Q.; Yang, Y.; Shao, Z.-M. Reduced Expression of TET1, TET2, TET3 and TDG mRNAs Are Associated with Poor Prognosis of Patients with Early Breast Cancer. PLoS ONE 2015, 10, e0133896. [Google Scholar] [CrossRef]
- Sklias, A.; Halaburkova, A.; Vanzan, L.; Jimenez, N.F.; Cuenin, C.; Bouaoun, L.; Cahais, V.; Ythier, V.; Sallé, A.; Renard, C.; et al. Epigenetic remodelling of enhancers in response to estrogen deprivation and re-stimulation. Nucleic Acids Res. 2021, 49, 9738–9754. [Google Scholar] [CrossRef]
- Wang, L.; Ozark, P.A.; Smith, E.R.; Zhao, Z.; Marshall, S.A.; Rendleman, E.J.; Piunti, A.; Ryan, C.; Whelan, A.L.; Helmin, K.A.; et al. TET2 coactivates gene expression through demethylation of enhancers. Sci. Adv. 2018, 4, eaau6986. [Google Scholar] [CrossRef] [Green Version]
- Li, L.; Li, C.; Mao, H.; Du, Z.; Chan, W.Y.; Murray, P.; Luo, B.; Chan, A.T.C.; Mok, T.; Chan, F.K.; et al. Epigenetic inactivation of the CpG demethylase TET1 as a DNA methylation feedback loop in human cancers. Sci. Rep. 2016, 6, 26591. [Google Scholar] [CrossRef] [PubMed]
- Good, C.R.; Madzo, J.; Patel, B.; Maegawa, S.; Engel, N.; Jelinek, J.; Issa, J.-P.J. A novel isoform of TET1 that lacks a CXXC domain is overexpressed in cancer. Nucleic Acids Res. 2017, 45, 8269–8281. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lou, H.; Li, H.; Ho, K.J.; Cai, L.; Huang, A.S.; Shank, T.R.; Verneris, M.R.; Nickerson, M.L.; Dean, M.; Anderson, S.K. The Human TET2 Gene Contains Three Distinct Promoter Regions with Differing Tissue and Developmental Specificities. Front. Cell Dev. Biol. 2019, 7, 99. [Google Scholar] [CrossRef] [PubMed]
- Song, S.J.; Poliseno, L.; Song, M.S.; Ala, U.; Webster, K.; Ng, C.; Beringer, G.; Brikbak, N.J.; Yuan, X.; Cantley, L.C.; et al. MicroRNA-antagonism regulates breast cancer stemness and metastasis via TET-family-dependent chromatin remodeling. Cell 2013, 154, 311–324. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.W.; Wang, Z.; Xie, W.; Cai, Y.; Xia, L.; Easwaran, H.; Luo, J.; Yen, R.-W.C.; Li, Y.; Baylin, S.B. Acetylation Enhances TET2 Function in Protecting against Abnormal DNA Methylation during Oxidative Stress. Mol. Cell 2017, 65, 323–335. [Google Scholar] [CrossRef] [Green Version]
- Wu, D.; Hu, D.; Chen, H.; Shi, G.; Fetahu, I.; Wu, F.; Rabidou, K.; Fang, R.; Tan, L.; Xu, S.; et al. Glucose-regulated phosphorylation of TET2 by AMPK reveals a pathway linking diabetes to cancer. Nat. Cell Biol. 2018, 559, 637–641. [Google Scholar] [CrossRef]
- Chen, H.; Yu, D.; Fang, R.; Rabidou, K.; Wu, D.; Hu, D.; Jia, P.; Zhao, Z.; Wu, Z.; Peng, J.; et al. TET2 stabilization by 14-3-3 binding to the phosphorylated Serine 99 is deregulated by mutations in cancer. Cell Res. 2019, 29, 248–250. [Google Scholar] [CrossRef] [Green Version]
- Kundu, A.; Shelar, S.; Ghosh, A.P.; Ballestas, M.; Kirkman, R.; Nam, H.; Brinkley, G.; Karki, S.; Mobley, J.A.; Bae, S.; et al. 14-3-3 proteins protect AMPK-phosphorylated ten-eleven translocation-2 (TET2) from PP2A-mediated dephosphorylation. J. Biol. Chem. 2020, 295, 1754–1766. [Google Scholar] [CrossRef]
- Jeong, J.J.; Gu, X.; Nie, J.; Sundaravel, S.; Liu, H.; Kuo, W.L.; Bhagat, T.D.; Pradhan, K.; Cao, J.; Nischal, S.; et al. Cytokine-Regulated Phosphorylation and Activation of TET2 by JAK2 in Hematopoiesis. Cancer Discov. 2019, 9, 778–795. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Tan, P.; Guo, L.; Gong, J.; Ma, J.; Li, J.; Lee, M.; Fang, S.; Jing, J.; Johnson, G.; et al. p53-dependent autophagic degradation of TET2 modulates cancer therapeutic resistance. Oncogene 2019, 38, 1905–1919. [Google Scholar] [CrossRef]
- Xu, W.; Yang, H.; Liu, Y.; Yang, Y.; Wang, P.; Kim, S.-H.; Ito, S.; Yang, C.; Wang, P.; Xiao, M.-T.; et al. Oncometabolite 2-Hydroxyglutarate Is a Competitive Inhibitor of α-Ketoglutarate-Dependent Dioxygenases. Cancer Cell 2011, 19, 17–30. [Google Scholar] [CrossRef] [Green Version]
- Hoekstra, A.S.; De Graaff, M.A.; Bruijn, I.H.B.-D.; Ras, C.; Seifar, R.M.; Van Minderhout, I.; Cornelisse, C.J.; Hogendoorn, P.; Breuning, M.H.; Suijker, J.; et al. Inactivation of SDH and FH cause loss of 5hmC and increased H3K9me3 in paraganglioma/pheochromocytoma and smooth muscle tumors. Oncotarget 2015, 6, 38777–38788. [Google Scholar] [CrossRef] [Green Version]
- Yang, H.; Lin, H.; Xu, H.; Zhang, L.; Cheng, L.; Wen, B.; Shou, J.; Guan, K.; Xiong, Y.; Ye, D. TET-catalyzed 5-methylcytosine hydroxylation is dynamically regulated by metabolites. Cell Res. 2014, 24, 1017–1020. [Google Scholar] [CrossRef] [Green Version]
- Yin, R.; Mao, S.-Q.; Zhao, B.; Chong, Z.; Yang, Y.; Zhao, C.; Zhang, D.; Huang, H.; Gao, J.; Li, Z.; et al. Ascorbic Acid Enhances Tet-Mediated 5-Methylcytosine Oxidation and Promotes DNA Demethylation in Mammals. J. Am. Chem. Soc. 2013, 135, 10396–10403. [Google Scholar] [CrossRef]
- Agathocleous, M.; Meacham, C.E.; Burgess, R.J.; Piskounova, E.; Zhao, Z.; Crane, G.; Cowin, B.L.; Bruner, E.; Murphy, M.M.; Chen, W.; et al. Ascorbate regulates haematopoietic stem cell function and leukaemogenesis. Nat. Cell Biol. 2017, 549, 476–481. [Google Scholar] [CrossRef]
- Sant, D.W.; Mustafi, S.; Gustafson, C.B.; Chen, J.; Slingerland, J.M.; Wang, G. Vitamin C promotes apoptosis in breast cancer cells by increasing TRAIL expression. Sci. Rep. 2018, 8, 1–11. [Google Scholar] [CrossRef]
- Thienpont, B.; Steinbacher, J.; Zhao, H.; D’Anna, F.; Kuchnio, A.; Ploumakis, A.; Ghesquière, B.; Van Dyck, L.; Boeckx, B.; Schoonjans, L.; et al. Tumour hypoxia causes DNA hypermethylation by reducing TET activity. Nat. Cell Biol. 2016, 537, 63–68. [Google Scholar] [CrossRef]
- Zhang, H.; Zhang, X.; Clark, E.; Mulcahey, M.; Huang, S.; Shi, Y.G. TET1 is a DNA-binding protein that modulates DNA methylation and gene transcription via hydroxylation of 5-methylcytosine. Cell Res. 2010, 20, 1390–1393. [Google Scholar] [CrossRef] [Green Version]
- Xu, Y.; Wu, F.; Tan, L.; Kong, L.; Xiong, L.; Deng, J.; Barbera, A.J.; Zheng, L.; Zhang, H.; Huang, S.; et al. Genome-wide Regulation of 5hmC, 5mC, and Gene Expression by Tet1 Hydroxylase in Mouse Embryonic Stem Cells. Mol. Cell 2011, 42, 451–464. [Google Scholar] [CrossRef] [Green Version]
- Xu, Y.; Xu, C.; Kato, A.; Tempel, W.; Abreu, J.G.; Bian, C.; Hu, Y.; Hu, D.; Zhao, B.; Cerovina, T.; et al. Tet3 CXXC Domain and Dioxygenase Activity Cooperatively Regulate Key Genes for Xenopus Eye and Neural Development. Cell 2012, 151, 1200–1213. [Google Scholar] [CrossRef] [Green Version]
- Ko, M.; An, J.; Bandukwala, H.S.; Chavez, L.; Äijö, T.; Pastor, W.A.; Segal, M.F.; Li, H.; Koh, K.P.; Lähdesmäki, H.; et al. Modulation of TET2 expression and 5-methylcytosine oxidation by the CXXC domain protein IDAX. Nat. Cell Biol. 2013, 497, 122–126. [Google Scholar] [CrossRef]
- Rasmussen, K.D.; Jia, G.; Johansen, J.V.; Pedersen, M.T.; Rapin, N.; Bagger, F.O.; Porse, B.T.; Bernard, O.A.; Christensen, J.; Helin, K. Loss of TET2 in hematopoietic cells leads to DNA hypermethylation of active enhancers and induction of leukemogenesis. Genes Dev. 2015, 29, 910–922. [Google Scholar] [CrossRef] [Green Version]
- Sardina, J.L.; Collombet, S.; Tian, T.; Gómez, A.; Di Stefano, B.; Berenguer, C.; Brumbaugh, J.; Stadhouders, R.; Segura-Morales, C.; Gut, M.; et al. Transcription Factors Drive Tet2-Mediated Enhancer Demethylation to Reprogram Cell Fate. Cell Stem Cell 2018, 23, 905–906. [Google Scholar] [CrossRef] [Green Version]
- Yang, Y.A.; Zhao, J.C.; Fong, K.-W.; Kim, J.; Li, S.; Song, C.-X.; Song, B.; Zheng, B.; He, C.; Yu, J. FOXA1 potentiates lineage-specific enhancer activation through modulating TET1 expression and function. Nucleic Acids Res. 2016, 44, 8153–8164. [Google Scholar] [CrossRef]
- Broome, R.; Chernukhin, I.; Jamieson, S.; Kishore, K.; Papachristou, E.K.; Mao, S.-Q.; Tejedo, C.G.; Mahtey, A.; Theodorou, V.; Groen, A.J.; et al. TET2 is a component of the estrogen receptor complex and controls 5mC to 5hmC conversion at estrogen receptor cis-regulatory regions. Cell Rep. 2021, 34, 108776. [Google Scholar] [CrossRef]
- Lyu, R.; Zhu, X.; Shen, Y.; Xiong, L.; Liu, L.; Liu, H.; Wu, F.; Argueta, C.; Tan, L. Tumour suppressor TET2 safeguards enhancers from aberrant DNA methylation and epigenetic reprogramming in ERalpha-positive breast cancer cells. Epigenetics 2021, 2021, 1–15. [Google Scholar] [CrossRef]
- Sheaffer, K.L.; Elliott, E.N.; Kaestner, K.H. DNA Hypomethylation Contributes to Genomic Instability and Intestinal Cancer Initiation. Cancer Prev. Res. 2016, 9, 534–546. [Google Scholar] [CrossRef] [Green Version]
- Guo, J.U.; Su, Y.; Zhong, C.; Ming, G.-L.; Song, H. Hydroxylation of 5-Methylcytosine by TET1 Promotes Active DNA Demethylation in the Adult Brain. Cell 2011, 145, 423–434. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ficz, G.; Branco, M.; Seisenberger, S.; Santos, F.; Krueger, F.; Hore, T.; Marques, J.; Andrews, S.; Reik, W. Dynamic regulation of 5-hydroxymethylcytosine in mouse ES cells and during differentiation. Nat. Cell Biol. 2011, 473, 398–402. [Google Scholar] [CrossRef] [PubMed]
- Williams, K.; Christensen, J.; Pedersen, M.T.; Johansen, J.V.; Cloos, P.; Rappsilber, J.; Helin, K. TET1 and hydroxymethylcytosine in transcription and DNA methylation fidelity. Nat. Cell Biol. 2011, 473, 343–348. [Google Scholar] [CrossRef]
- Guallar, D.; Bi, X.; Pardavila, J.A.; Huang, X.; Saenz, C.; Shi, X.; Zhou, H.; Faiola, F.; Ding, J.; Haruehanroengra, P.; et al. RNA-dependent chromatin targeting of TET2 for endogenous retrovirus control in pluripotent stem cells. Nat. Genet. 2018, 50, 443–451. [Google Scholar] [CrossRef] [PubMed]
- Zhang, P.; Ludwig, A.K.; Hastert, F.D.; Rausch, C.; Lehmkuhl, A.; Hellmann, I.; Smets, M.; Leonhardt, H.; Cardoso, M.C. L1 retrotransposition is activated by Ten-eleven-translocation protein 1 and repressed by methyl-CpG binding proteins. Nucleus 2017, 8, 548–562. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De La Rica, L.; Deniz, Ö.; Cheng, K.C.L.; Todd, C.; Cruz, C.; Houseley, J.; Branco, M.R. TET-dependent regulation of retrotransposable elements in mouse embryonic stem cells. Genome Biol. 2016, 17, 1–14. [Google Scholar] [CrossRef] [Green Version]
- López-Moyado, I.F.; Tsagaratou, A.; Yuita, H.; Seo, H.; Delatte, B.; Heinz, S.; Benner, C.; Rao, A. Paradoxical association of TET loss of function with genome-wide DNA hypomethylation. Proc. Natl. Acad. Sci. USA 2019, 116, 16933–16942. [Google Scholar] [CrossRef] [Green Version]
- Wu, M.J.; Kim, M.R.; Chen, Y.S.; Yang, J.Y.; Chang, C.J. Retinoic acid directs breast cancer cell state changes through regulation of TET2-PKCzeta pathway. Oncogene 2017, 36, 3193–3206. [Google Scholar] [CrossRef]
- Kim, M.R.; Wu, M.-J.; Zhang, Y.; Yang, J.-Y.; Chang, C.J. TET2 directs mammary luminal cell differentiation and endocrine response. Nat. Commun. 2020, 11, 1–13. [Google Scholar] [CrossRef]
- Puig, I.; Tenbaum, S.P.; Chicote, I.; Arqués, O.; Martínez-Quintanilla, J.; Cuesta-Borrás, E.; Ramírez, L.; Gonzalo, P.; Soto, A.; Aguilar, S.; et al. TET2 controls chemoresistant slow-cycling cancer cell survival and tumor recurrence. J. Clin. Investig. 2018, 128, 3887–3905. [Google Scholar] [CrossRef]
- Hsu, C.-H.; Peng, K.-L.; Kang, M.-L.; Chen, Y.-R.; Yang, Y.-C.; Tsai, C.-H.; Chu, C.-S.; Jeng, Y.-M.; Chen, Y.-T.; Lin, F.-M.; et al. TET1 Suppresses Cancer Invasion by Activating the Tissue Inhibitors of Metalloproteinases. Cell Rep. 2012, 2, 568–579. [Google Scholar] [CrossRef] [Green Version]
- Sun, M.; Song, C.X.; Huang, H.; Frankenberger, C.A.; Sankarasharma, D.; Gomes, S.; Chen, P.; Chen, J.; Chada, K.K.; He, C.; et al. HMGA2/TET1/HOXA9 signaling pathway regulates breast cancer growth and metastasis. Proc. Natl. Acad. Sci. USA 2013, 110, 9920–9925. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.-Y.; Luo, C.-W.; Lai, Y.-S.; Wu, C.-C.; Hung, W.-C. Lysine demethylase KDM2A inhibits TET2 to promote DNA methylation and silencing of tumor suppressor genes in breast cancer. Oncogenesis 2017, 6, e369. [Google Scholar] [CrossRef] [Green Version]
- Song, C.; Wang, L.; Wu, X.; Wang, K.; Xie, D.; Xiao, Q.; Li, S.; Jiang, K.; Liao, L.; Yates, J.R., 3rd; et al. PML Recruits TET2 to Regulate DNA Modification and Cell Proliferation in Response to Chemotherapeutic Agent. Cancer Res 2018, 78, 2475–2489. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blaschke, K.; Ebata, K.; Karimi, M.M.; Zepeda-Martínez, J.A.; Goyal, P.; Mahapatra, S.; Tam, A.; Laird, D.J.; Hirst, M.; Rao, A.; et al. Vitamin C induces Tet-dependent DNA demethylation and a blastocyst-like state in ES cells. Nat. Cell Biol. 2013, 500, 222–226. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.-P.; Lv, L.; Liu, Y.; Smith, M.D.; Li, W.-C.; Tan, X.-M.; Cheng, M.; Li, Z.; Bovino, M.; Aubé, J.; et al. Tumor suppressor TET2 promotes cancer immunity and immunotherapy efficacy. J. Clin. Investig. 2019, 129, 4316–4331. [Google Scholar] [CrossRef] [PubMed]
- Shen, Y.; Liu, L.; Wang, M.; Xu, B.; Lyu, R.; Shi, Y.; Tan, L. TET2 Inhibits PD-L1 Gene Expression in Breast Cancer Cells through Histone Deacetylation. Cancers 2021, 13, 2207. [Google Scholar] [CrossRef]
- Li, S.; Feng, J.; Wu, F.; Cai, J.; Zhang, X.; Wang, H.; Fetahu, I.S.; Iwanicki, I.; Ma, D.; Hu, T.; et al. TET2 promotes anti-tumor immunity by governing G-MDSCs and CD8(+) T-cell numbers. EMBO Rep. 2020, 21, e49425. [Google Scholar] [CrossRef]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xu, B.; Wang, H.; Tan, L. Dysregulated TET Family Genes and Aberrant 5mC Oxidation in Breast Cancer: Causes and Consequences. Cancers 2021, 13, 6039. https://doi.org/10.3390/cancers13236039
Xu B, Wang H, Tan L. Dysregulated TET Family Genes and Aberrant 5mC Oxidation in Breast Cancer: Causes and Consequences. Cancers. 2021; 13(23):6039. https://doi.org/10.3390/cancers13236039
Chicago/Turabian StyleXu, Bo, Hao Wang, and Li Tan. 2021. "Dysregulated TET Family Genes and Aberrant 5mC Oxidation in Breast Cancer: Causes and Consequences" Cancers 13, no. 23: 6039. https://doi.org/10.3390/cancers13236039
APA StyleXu, B., Wang, H., & Tan, L. (2021). Dysregulated TET Family Genes and Aberrant 5mC Oxidation in Breast Cancer: Causes and Consequences. Cancers, 13(23), 6039. https://doi.org/10.3390/cancers13236039