
The Classification of Myeloproliferative Neoplasms: Rationale, Historical Background and Future Perspectives with Focus on Unclassifiable Cases

 ,
,  , , and
, , and
Abstract
:Simple Summary
Abstract
1. Introduction
2. History and Rationale of MPN Classification

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| A. EARLY CLINICALLY-ORIENTED CLASSIFICATIONS | ||
| Damashek’s Classification [5] | 1976 WHO Classification [9] | |
|
| |
| B. HISTOLOGICALLY-ORIENTED CLASSIFICATIONS | ||
| Working Classification [10] | Hannover Classification [11] | |
a. Typical CMPDs
| a. Primary diseases
| |
| C. INTEGRATED CLINICAL-PATHOLOGICAL CLASSIFICATIONS | ||
| 2001 WHO Classification [15] | 2008 WHO Classification [20] | 2016 WHO Classification [1] |
|
|
|
3. Evolution of the Diagnostic Criteria for MPNs
3.1. Evolution of the Diagnostic Criteria for PV
3.2. Evolution of the Diagnostic Criteria for ET
3.3. Evolution of the Diagnostic Criteria for PMF
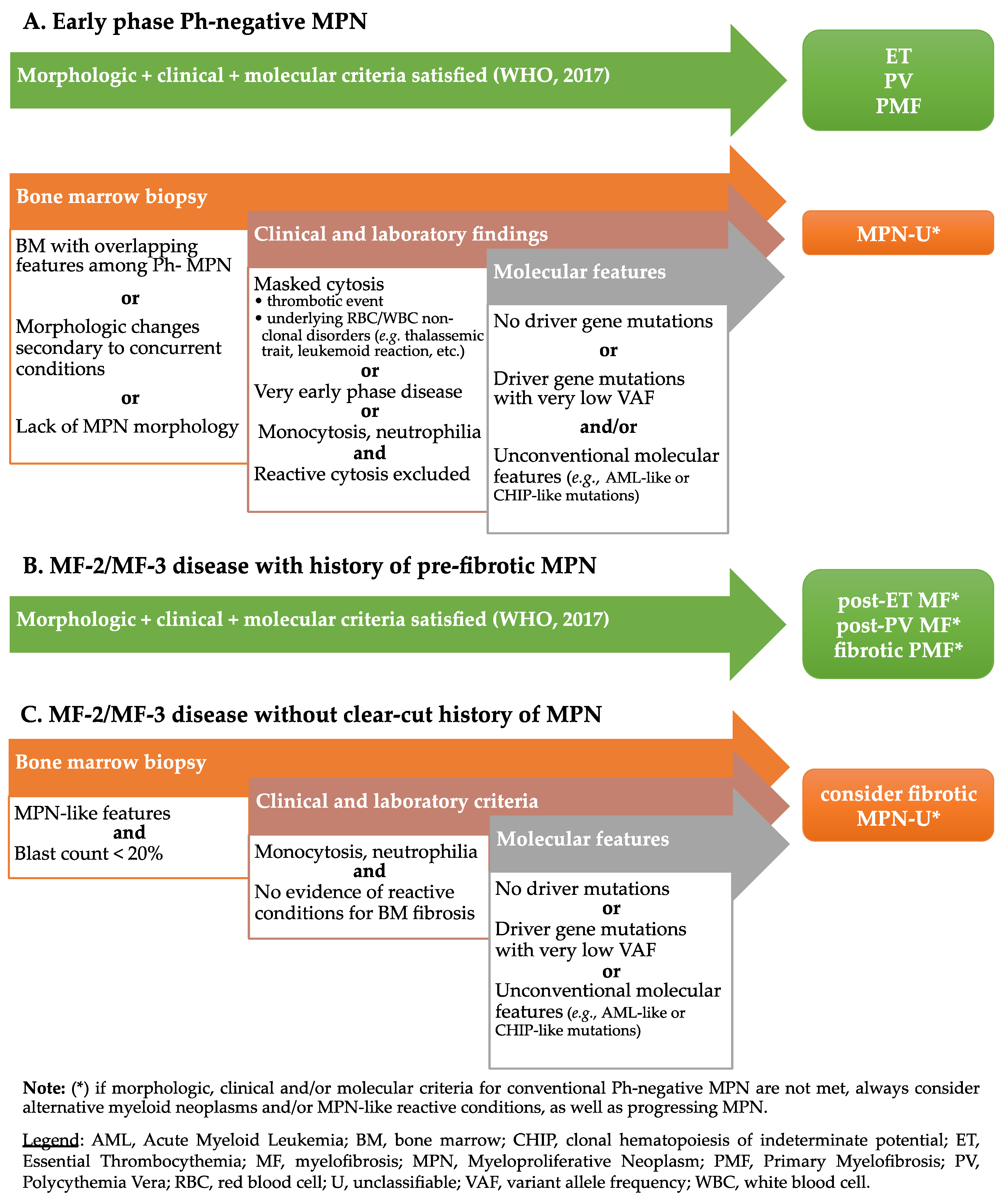
4. Myeloproliferative Neoplasms, Unclassifiable (MPN-U)
5. Challenges and Perspectives in the Classification of MPN-U
5.1. MPN with Clinical-Morphological Mismatch
5.1.1. Cases with MPN-like Clinical Findings, Lacking MPN Morphologic Criteria
Case 1—Synopsis and Discussion
5.1.2. Cases with MPN-like Morphology, Lacking MPN Clinical Criteria
Case 2—Synopsis and Discussion
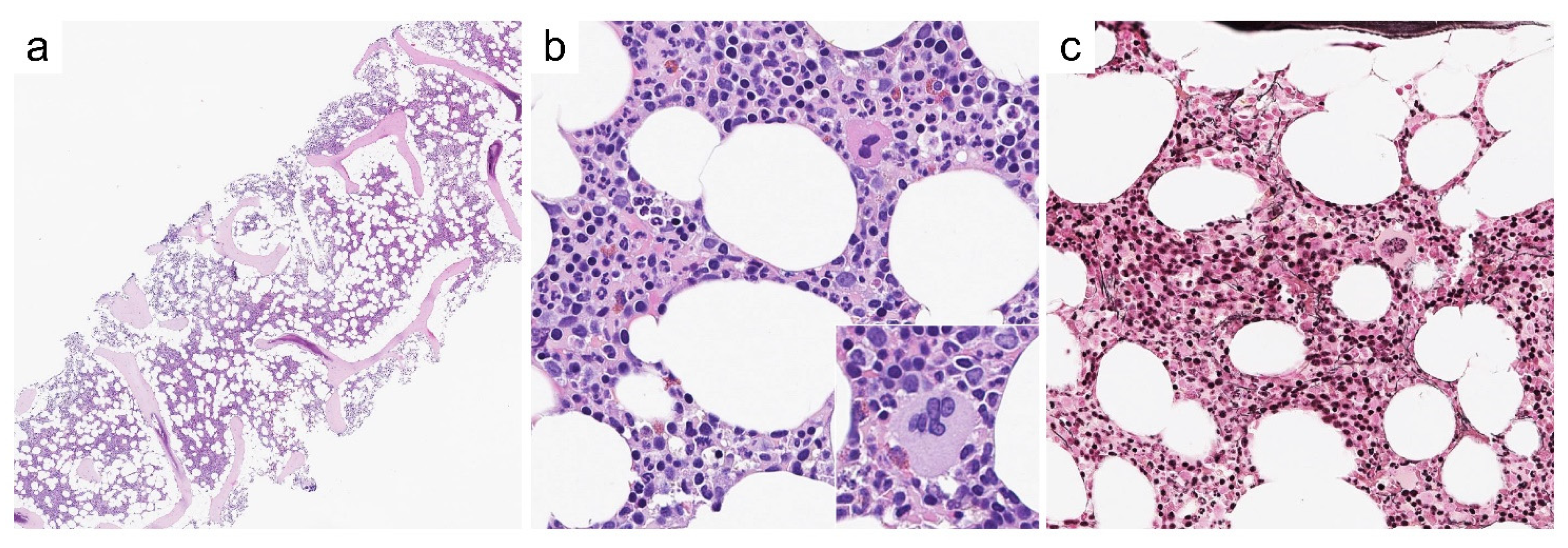
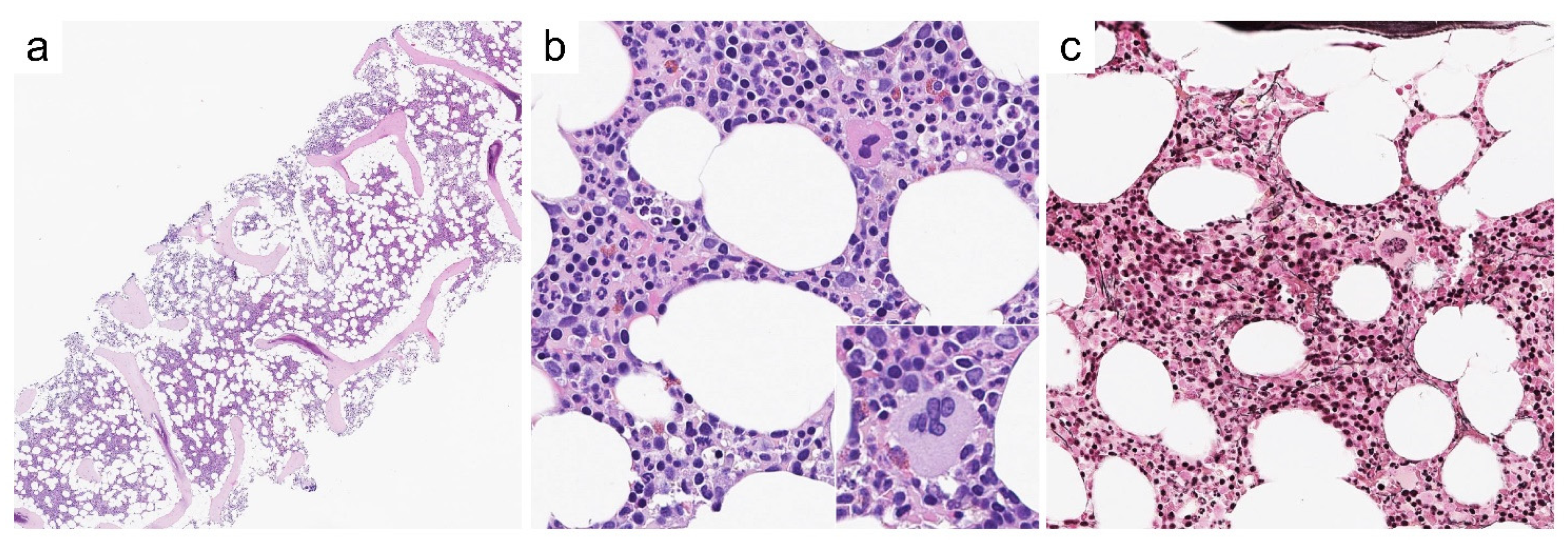
Case 3—Synopsis and Discussion
Case 4—Synopsis and Discussion
5.1.3. MPN with Unusual Morphology
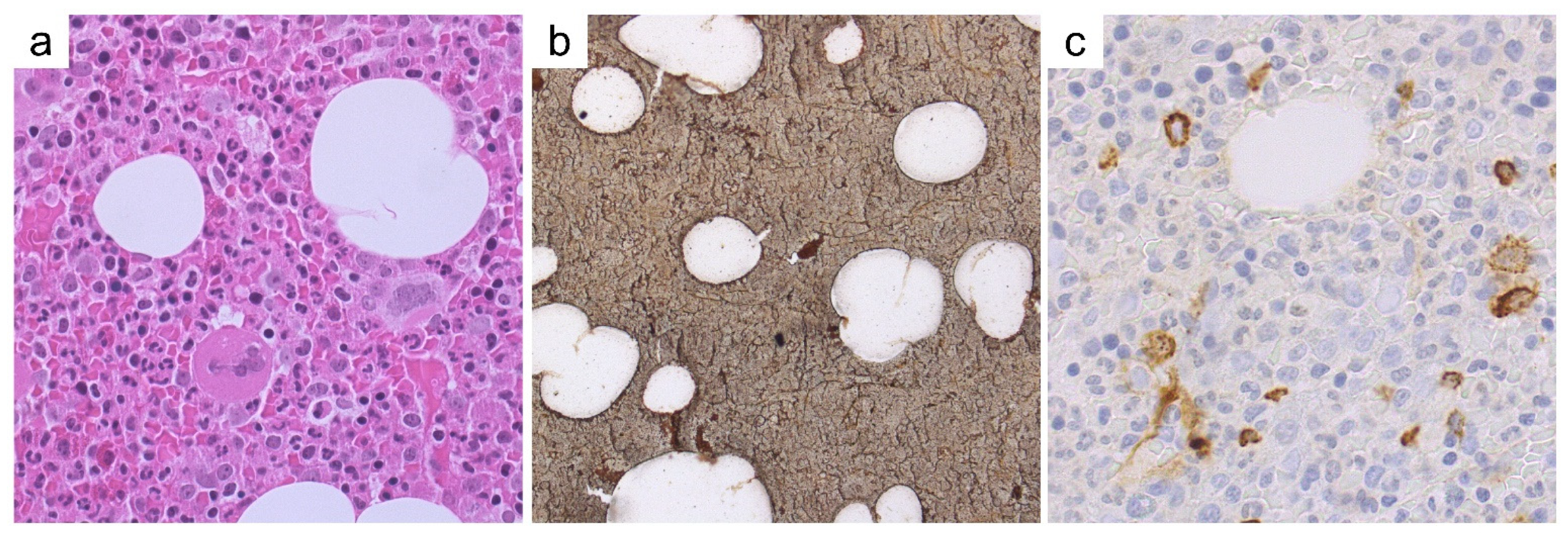
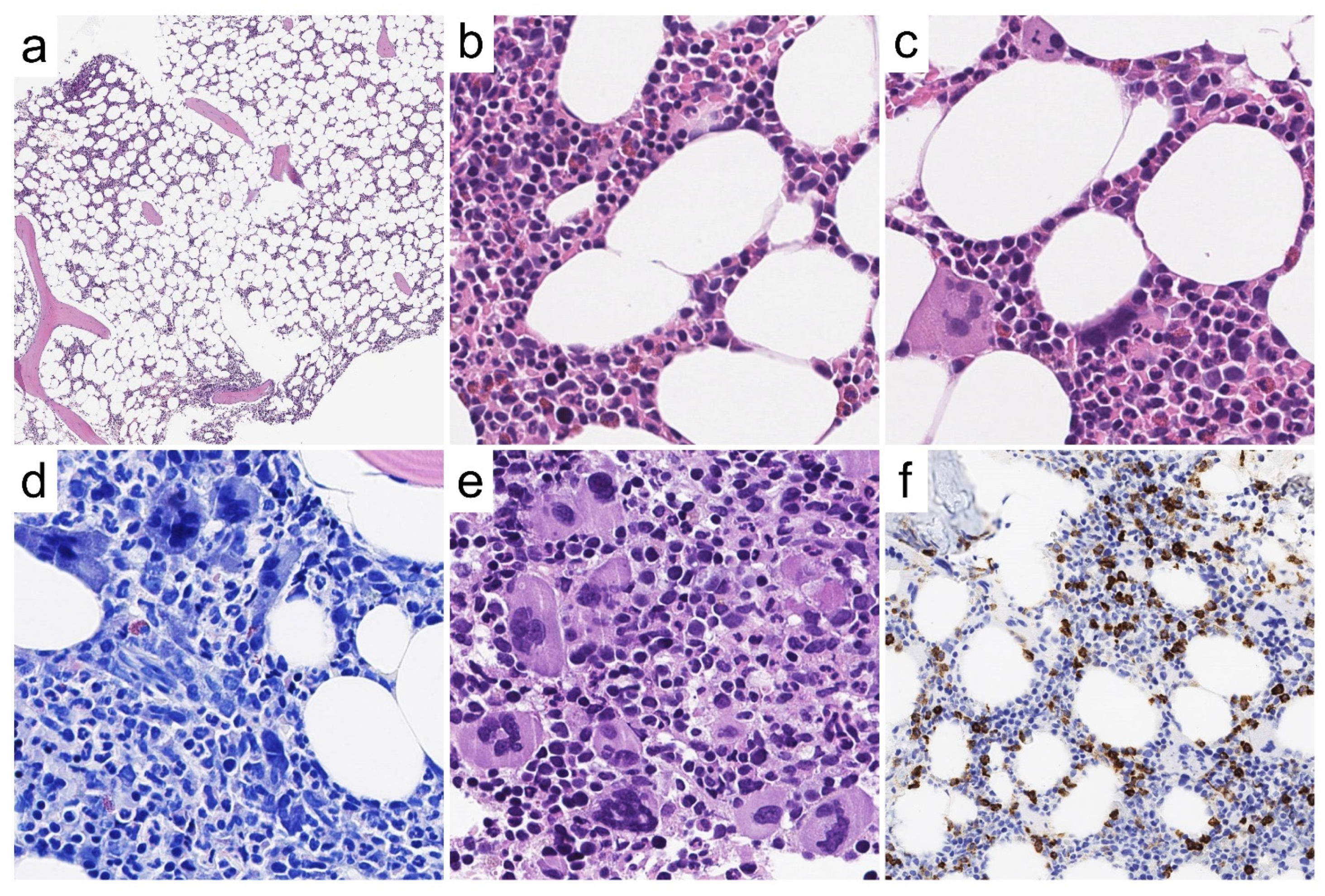
Case 5—Synopsis and Discussion
Case 6—Synopsis and Discussion
5.2. MPN with Unconventional Molecular Features
5.2.1. JAK2-Negative Erythrocytosis
5.2.2. MPN with “High Risk” Molecular Features
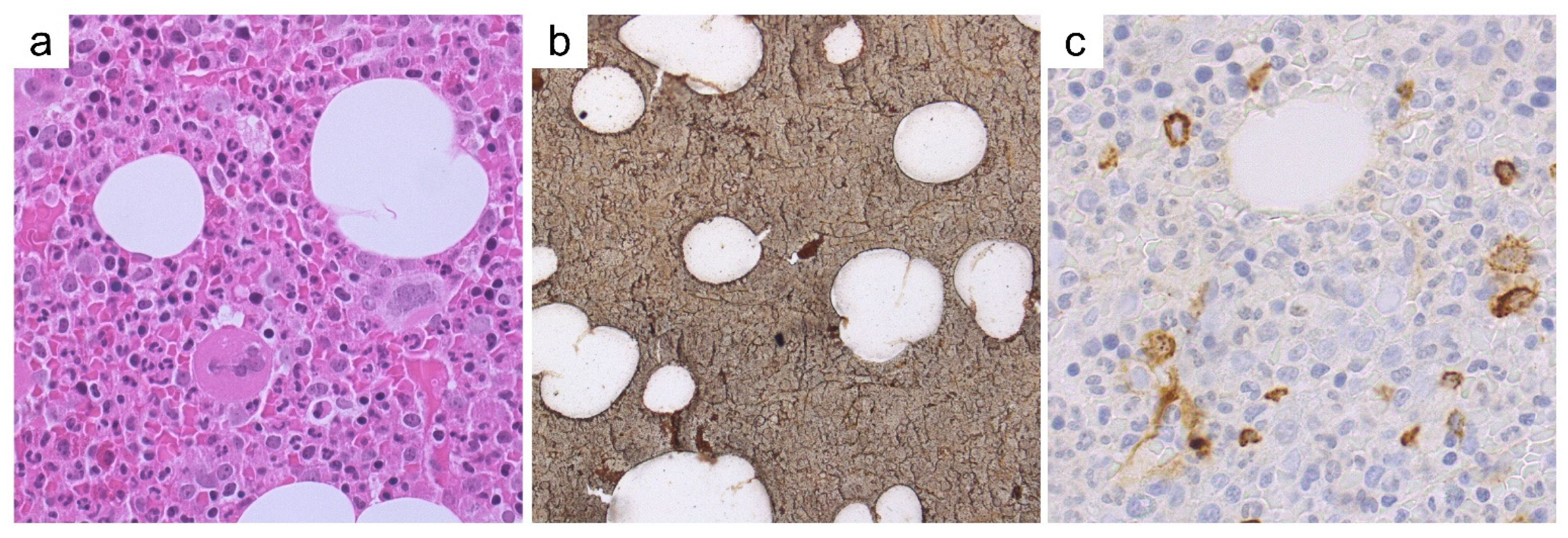
Case 7—Synopsis and Discussion
5.2.3. MPN with CHIP-like Molecular Features
6. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Swerdlow, S.H.; Campo, E.; Harris, N.L.; Jaffe, E.S.; Pileri, S.A.; Stein, H.; Thiele, J. WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues; IARC Press: Lyon, France, 2017; ISBN 978-92-832-4494-3. [Google Scholar]
- Anderson, L.A.; McMullin, M.F. Epidemiology of MPN: What Do We Know? Curr. Hematol. Malign. Rep. 2014, 9, 340–349. [Google Scholar] [CrossRef]
- Szuber, N.; Mudireddy, M.; Nicolosi, M.; Penna, D.; Vallapureddy, R.R.; Lasho, T.L.; Finke, C.; Begna, K.H.; Elliott, M.A.; Hook, C.C.; et al. 3023 Mayo Clinic Patients with Myeloproliferative Neoplasms: Risk-Stratified Comparison of Survival and Outcomes Data Among Disease Subgroups. Mayo Clin. Proc. 2019, 94, 599–610. [Google Scholar] [CrossRef]
- Tefferi, A. The history of myeloproliferative disorders: Before and after Dameshek. Leukemia 2007, 22, 3–13. [Google Scholar] [CrossRef]
- Dameshek, W. Some speculations on the myeloproliferative syndromes. Blood 1951, 6, 372–375. [Google Scholar] [CrossRef] [Green Version]
- Ward, H.P.; Block, M.H. The natural history of agnogenic myeloid metaplasia (AMM) and a critical evaluation of its relationship with the myeloproliferative syndrome. Medicine 1971, 50, 357–420. [Google Scholar] [CrossRef] [PubMed]
- Arber, D.A. The 2016 WHO classification of acute myeloid leukemia: What the practicing clinician needs to know. Semin. Hematol. 2019, 56, 90–95. [Google Scholar] [CrossRef] [PubMed]
- Mughal, T.I.; Abdel-Wahab, O.; Rampal, R.; Mesa, R.; Koschmieder, S.; Levine, R.; Hehlmann, R.; Saglio, G.; Barbui, T.; Van Etten, R.A. Contemporary insights into the pathogenesis and treatment of chronic myeloproliferative neoplasms. Leuk. Lymphoma 2016, 57, 1517–1526. [Google Scholar] [CrossRef] [PubMed]
- Mathé, G.; Rappaport, H. Histological and Cytological Typing of Neoplastic Diseases of Haematopoietic and Lymphoid Tissues; International Histological Classification of Tumours; World Health Organization: Geneva, Switzerland, 1976; ISBN 92-4-176014-1. [Google Scholar]
- Burkhardt, R.; Bartl, R.; Jäger, K.; Frisch, B.; Kettner, G.; Mahl, G.; Sund, M. Chronic Myeloproliferative Disorders (CMPD). Pathol. Res. Pr. 1984, 179, 131–186. [Google Scholar] [CrossRef]
- Georgii, A.; Vykoupil, K.-F.; Buhr, T.; Choritz, H.; Döhler, U.; Kaloutsi, V.; Werner, M. Chronic Myeloproliferative Disorders in Bone Marrow Biopsies. Pathol. Res. Pr. 1990, 186, 3–27. [Google Scholar] [CrossRef]
- Thiele, J.; Kvasnicka, H.-M.; Werden, C.; Zankovich, R.; Diehl, V.; Fischer, R. Idiopathic Primary Osteo-myelofibrosis: A Clinico-Pathological Study on 208 Patients with Special Emphasis on Evolution of Disease Features, Differentiation from Essential Thrombocythemia and Variables of Prognostic Impact. Leuk. Lymphoma 1996, 22, 303–317. [Google Scholar] [CrossRef]
- Szuber, N.; Tefferi, A. Chronic neutrophilic leukemia: New science and new diagnostic criteria. Blood Cancer J. 2018, 8, 1–19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cassi, E.; De Paoli, A.; Fava, S.; Luoni, M.; Tosi, A.; Turri, C.; Grimi, E. Idiopathic hypereosinophilic syndrome and “eosinophilic leukemia”. Haematologica 1992, 77, 430–432. [Google Scholar]
- Jaffe, E.S.; Harris, N.L.; Stein, H.; Vardiman, J.W. Pathology and Genetics of Tumours of Haematopoietic and Lymphoid Tissues; IARC Press: Lyon, France, 2001. [Google Scholar]
- Harris, N.L.; Jaffe, E.S.; Stein, H.; Banks, P.M.; Chan, J.K.; Cleary, M.L.; Delsol, G.; De Wolf-Peeters, C.; Falini, B.; Gatter, K.C. A revised European-American classification of lymphoid neoplasms: A proposal from the International Lymphoma Study Group. Blood 1994, 84. [Google Scholar] [CrossRef] [Green Version]
- Spivak, J.L.; Silver, R.T. The revised World Health Organization diagnostic criteria for polycythemia vera, essential thrombocytosis, and primary myelofibrosis: An alternative proposal. Blood 2008, 112, 231–239. [Google Scholar] [CrossRef] [Green Version]
- Vainchenker, W.; Kralovics, R. Genetic basis and molecular pathophysiology of classical myeloproliferative neoplasms. Blood 2017, 129, 667–679. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Longley, B.J.; Tyrrell, L.; Lu, S.-Z.; Ma, Y.-S.; Langley, K.; Ding, T.-G.; Duffy, T.; Jacobs, P.; Tang, L.H.; Modlin, I. Somatic c-KIT activating mutation in urticaria pigmentosa and aggressive mastocytosis: Establishment of clonality in a human mast cell neoplasm. Nat. Genet. 1996, 12, 312–314. [Google Scholar] [CrossRef] [PubMed]
- Reiter, A.; Gotlib, J. Myeloid neoplasms with eosinophilia. Blood 2017, 129, 704–714. [Google Scholar] [CrossRef] [Green Version]
- Swerdlow, S.H.; Campo, E.; Harris, N.L.; Jaffe, E.S.; Pileri, S.; Stein, H.; Thiele, J.; Vardiman, J.W. WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues; IARC Press: Lyon, France, 2008. [Google Scholar]
- Tefferi, A.; Thiele, J.; Orazi, A.; Kvasnicka, H.M.; Barbui, T.; Hanson, C.A.; Barosi, G.; Verstovsek, S.; Birgegard, G.; Mesa, R.; et al. Proposals and rationale for revision of the World Health Organization diagnostic criteria for polycythemia vera, essential thrombocythemia, and primary myelofibrosis: Recommendations from an ad hoc international expert panel. Blood 2007, 110, 1092–1097. [Google Scholar] [CrossRef]
- Arber, D.A.; Orazi, A.; Hasserjian, R.; Thiele, J.; Borowitz, M.J.; Le Beau, M.M.; Bloomfield, C.D.; Cazzola, M.; Vardiman, J.W. The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood 2016, 127, 2391–2405. [Google Scholar] [CrossRef]
- Wasserman, L.R. THE MANAGEMENT OF POLYCYTHAEMIA VERA. Br. J. Haematol. 1971, 21, 371–376. [Google Scholar] [CrossRef]
- Murphy, S.; Iland, H.; Rosenthal, D.; Laszlo, J. Essential thrombocythemia: An interim report from the Polycythemia Vera Study Group. Semin. Hematol. 1986, 23. [Google Scholar]
- Michiels, J. Diagnostic criteria of the myeloproliferative disorders (MPD): Essential thrombocythaemia, polycythaemia vera and chronic megakaryocytic granulocytic metaplasia. Neth. J. Med. 1997, 51, 57–64. [Google Scholar] [CrossRef]
- Barbui, T.; Thiele, J.; Carobbio, A.; Guglielmelli, P.; Rambaldi, A.; Vannucchi, A.M.; Tefferi, A. Discriminating between essential thrombocythemia and masked polycythemia vera inJAK2mutated patients. Am. J. Hematol. 2014, 89, 588–590. [Google Scholar] [CrossRef] [PubMed]
- Barbui, T.; Thiele, J.; Gisslinger, H.; Kvasnicka, H.M.; Vannucchi, A.M.; Guglielmelli, P.; Orazi, A.; Tefferi, A. The 2016 WHO classification and diagnostic criteria for myeloproliferative neoplasms: Document summary and in-depth discussion. Blood Cancer J. 2018, 8, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Gisslinger, B.; Jeryczynski, G.; Wolfler, A.; Burgstaller, S.; Buxhoferausch, V.; Schalling, M.; Krauth, M.-T.; Schiefer, A.-I.; Kornauth, C.; Simonitschklupp, I.; et al. Clinical impact of bone marrow morphology for the diagnosis of essential thrombocythemia: Comparison between the BCSH and the WHO criteria. Leukemia 2015, 30, 1126–1132. [Google Scholar] [CrossRef] [PubMed]
- Thiele, J.; Kvasnicka, H.M. Chronic myeloproliferative disorders with thrombocythemia: A comparative study of two classification systems (PVSG, WHO) on 839 patients. Ann. Hematol. 2003, 82, 148–152. [Google Scholar] [CrossRef]
- Florena, A.M.; Tripodo, C.; Iannitto, E.; Porcasi, R.; Ingrao, S.; Franco, V. Value of bone marrow biopsy in the diagnosis of essential thrombocythemia. Haematologica 2004, 89, 911–919. [Google Scholar]
- Thiele, J.; Kvasnicka, H.M.; Diehl, V.; Fischer, R.; Michiels, J.J. Clinicopathological Diagnosis and Differential Criteria of Thrombocythemias in Various Myeloproliferative Disorders by Histopathology, Histochemistry and Immunostaining from Bone Marrow Biopsies. Leuk. Lymphoma 1999, 33, 207–218. [Google Scholar] [CrossRef]
- Gianelli, U.; Cattaneo, D.; Bossi, A.; Cortinovis, I.; Boiocchi, L.; Liu, Y.-C.; Augello, C.; Bonometti, A.; Fiori, S.; Orofino, N.; et al. Erratum: The myeloproliferative neoplasms, unclassifiable: Clinical and pathological considerations. Mod. Pathol. 2017, 30, 1430. [Google Scholar] [CrossRef] [Green Version]
- Primignani, M.; Barosi, G.; Bergamaschi, G.; Gianelli, U.; Fabris, F.; Reati, R.; Dell’Era, A.; Bucciarelli, P.; Mannucci, P.M. Role of theJAK2 mutation in the diagnosis of chronic myeloproliferative disorders in splanchnic vein thrombosis. Hepatology 2006, 44, 1528–1534. [Google Scholar] [CrossRef]
- Iurlo, A.; Gianelli, U.; Cattaneo, D.; Thiele, J.; Orazi, A. Impact of the 2016 revised WHO criteria for myeloproliferative neoplasms, unclassifiable: Comparison with the 2008 version. Am. J. Hematol. 2017, 92, E48–E51. [Google Scholar] [CrossRef] [Green Version]
- Gianelli, U.; Iurlo, A.; Cattaneo, D.; Bossi, A.; Cortinovis, I.; Augello, C.; Moro, A.; Savi, F.; Castelli, R.; Brambilla, C.; et al. Discrepancies between bone marrow histopathology and clinical phenotype in BCR-ABL1-negative myeloproliferative neoplasms associated with splanchnic vein thrombosis. Leuk. Res. 2015, 39, 525–529. [Google Scholar] [CrossRef] [PubMed]
- Deschamps, P.; Moonim, M.; Radia, D.; Curto-Garcia, N.; Woodley, C.; Bassiony, S.; O’Sullivan, J.; Harrington, P.; Raj, K.; Francis, Y.; et al. Clinicopathological characterisation of myeloproliferative neoplasm-unclassifiable (MPN-U): A retrospective analysis from a large UK tertiary referral centre. Br. J. Haematol. 2021, 193, 792–797. [Google Scholar] [CrossRef]
- Gianelli, U.; Bossi, A.; Cortinovis, I.; Sabattini, E.; Tripodo, C.; Boveri, E.; Moro, A.; Valli, R.; Ponzoni, M.; Florena, A.M.; et al. Reproducibility of the WHO histological criteria for the diagnosis of Philadelphia chromosome-negative myeloproliferative neoplasms. Mod. Pathol. 2013, 27, 814–822. [Google Scholar] [CrossRef] [Green Version]
- Kvasnicka, H.M.; Orazi, A.; Thiele, J.; Barosi, G.; Bueso-Ramos, C.E.; Vannucchi, A.M.; Hasserjian, R.P.; Kiladjian, J.-J.; Gianelli, U.; Silver, R.; et al. European LeukemiaNet study on the reproducibility of bone marrow features in masked polycythemia vera and differentiation from essential thrombocythemia. Am. J. Hematol. 2017, 92, 1062–1067. [Google Scholar] [CrossRef]
- Thiele, J.; Orazi, A.; Kvasnicka, H.M.; Franco, V.; Boveri, E.; Gianelli, U.; Gisslinger, H.; Passamonti, F.; Tefferi, A.; Barbui, T. European Bone Marrow Working Group trial on reproducibility of World Health Organization criteria to discriminate essential thrombocythemia from prefibrotic primary myelofibrosis. Haematologica 2012, 97, e5–e6. [Google Scholar] [CrossRef]
- Perricone, M.; Polverelli, N.; Martinelli, G.; Catani, L.; Ottaviani, E.; Zuffa, E.; Franchini, E.; Dizdari, A.; Forte, D.; Sabattini, E.; et al. The relevance of a low JAK2V617F allele burden in clinical practice: A monocentric study. Oncotarget 2017, 8, 37239–37249. [Google Scholar] [CrossRef] [Green Version]
- Jovanovic, J.V.; Ivey, A.; Vannucchi, A.M.; Lippert, E.; Leibundgut, E.O.; Cassinat, B.; Pallisgaard, N.; Maroc, N.; Hermouet, S.; Nickless, G.; et al. Establishing optimal quantitative-polymerase chain reaction assays for routine diagnosis and tracking of minimal residual disease in JAK2-V617F-associated myeloproliferative neoplasms: A joint European LeukemiaNet/MPN&MPNr-EuroNet (COST action BM0902) study. Leukemia 2013, 27, 2032–2039. [Google Scholar] [CrossRef] [Green Version]
- Carobbio, A.; Ferrari, A.; Masciulli, A.; Ghirardi, A.; Barosi, G.; Barbui, T. Leukocytosis and thrombosis in essential thrombocythemia and polycythemia vera: A systematic review and meta-analysis. Blood Adv. 2019, 3, 1729–1737. [Google Scholar] [CrossRef] [PubMed]
- Tefferi, A.; Barbui, T. Polycythemia vera and essential thrombocythemia: 2021 update on diagnosis, risk-stratification and management. Am. J. Hematol. 2020, 95, 1599–1613. [Google Scholar] [CrossRef] [PubMed]
- Debureaux, P.-E.; Cassinat, B.; Soret-Dulphy, J.; Mora, B.; Verger, E.; Maslah, N.; Plessier, A.; Rautou, P.-E.; Ollivier-Hourman, I.; De Ledinghen, V.; et al. Molecular profiling and risk classification of patients with myeloproliferative neoplasms and splanchnic vein thromboses. Blood Adv. 2020, 4, 3708–3715. [Google Scholar] [CrossRef]
- Kottas, K.; Marathonitis, A.; Nodarou, A.; Kanellis, G.; Christopoulos, K. Polycythemia Vera in a Patient With Heterozygous Beta-Thalassemia: Coincidence or Causal Relationship? Cureus 2020, 12. [Google Scholar] [CrossRef]
- Delhommeau, F.; Dupont, S.; Della Valle, V.; James, C.; Trannoy, S.; Massé, A.; Kosmider, O.; Le Couedic, J.-P.; Robert, F.; Alberdi, A.; et al. Mutation inTET2in Myeloid Cancers. N. Engl. J. Med. 2009, 360, 2289–2301. [Google Scholar] [CrossRef]
- Steensma, D.P.; Bejar, R.; Jaiswal, S.; Lindsley, R.C.; Sekeres, M.; Hasserjian, R.P.; Ebert, B.L. Clonal hematopoiesis of indeterminate potential and its distinction from myelodysplastic syndromes. Blood 2015, 126, 9–16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kang, M.-G.; Choi, H.-W.; Lee, J.H.; Choi, Y.J.; Shin, J.-H.; Suh, S.-P.; Szardenings, M.; Kim, H.-R.; Shin, M.-G.; Choi, H.-J. Coexistence of JAK2 and CALR mutations and their clinical implications in patients with essential thrombocythemia. Oncotarget 2016, 7, 57036–57049. [Google Scholar] [CrossRef]
- Palomo, L.; Meggendorfer, M.; Hutter, S.; Twardziok, S.; Ademà, V.; Fuhrmann, I.; Fuster-Tormo, F.; Xicoy, B.; Zamora, L.; Acha, P.; et al. Molecular landscape and clonal architecture of adult myelodysplastic/myeloproliferative neoplasms. Blood 2020, 136, 1851–1862. [Google Scholar] [CrossRef] [PubMed]
- Chia, Y.; Islam, A.; Hider, P.; Woon, P.; Johan, M.; Hassan, R.; Ramli, M. The Prevalence of TET2 Gene Mutations in Patients with BCR-ABL-Negative Myeloproliferative Neoplasms (MPN): A Systematic Review and Meta-Analysis. Cancers 2021, 13, 3078. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, R.Z.; Rashid, M.; Ahmed, N.; Nadeem, M.; Shamsi, T.S. Coexisting JAK2V617F and CALR Exon 9 Mutations in Myeloproliferative Neoplasms—Do They Designate a New Subtype? Asian Pac. J. Cancer Prev. 2016, 17, 923–926. [Google Scholar] [CrossRef] [Green Version]
- Boiocchi, L.; Espinal-Witter, R.; Geyer, J.T.; Steinhilber, J.; Bonzheim, I.; Knowles, D.M.; Fend, F.; Orazi, A. Development of monocytosis in patients with primary myelofibrosis indicates an accelerated phase of the disease. Mod. Pathol. 2012, 26, 204–212. [Google Scholar] [CrossRef] [PubMed]
- Mangaonkar, A.A.; Tande, A.J.; Bekele, D.I. Differential Diagnosis and Workup of Monocytosis: A Systematic Approach to a Common Hematologic Finding. Curr. Hematol. Malign- Rep. 2021, 16, 267–275. [Google Scholar] [CrossRef]
- Cattaneo, D.; Croci, G.A.; Bucelli, C.; Tabano, S.; Cannone, M.G.; Gaudioso, G.; Barbanti, M.C.; Barbullushi, K.; Bianchi, P.; Fermo, E.; et al. Triple-Negative Essential Thrombocythemia: Clinical-Pathological and Molecular Features. A Single-Center Cohort Study. Front. Oncol. 2021, 11. [Google Scholar] [CrossRef]
- Acha, P.; Xandri, M.; Fuster-Tormo, F.; Palomo, L.; Xicoy, B.; Cabezón, M.; Marcé, S.; Granada, I.; Vela, D.; Sagüés, M.; et al. Diagnostic and prognostic contribution of targeted NGS in patients with triple-negative myeloproliferative neoplasms. Am. J. Hematol. 2019, 94, E264–E267. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feenstra, J.D.M.; Nivarthi, H.; Gisslinger, H.; Leroy, E.; Rumi, E.; Chachoua, I.; Bagienski, K.; Kubesova, B.; Pietra, D.; Gisslinger, B.; et al. Whole-exome sequencing identifies novel MPL and JAK2 mutations in triple-negative myeloproliferative neoplasms. Blood 2016, 127, 325–332. [Google Scholar] [CrossRef]
- Grinfeld, J.; Nangalia, J.; Baxter, E.J.; Wedge, D.; Angelopoulos, N.; Cantrill, R.; Godfrey, A.L.; Papaemmanuil, E.; Gundem, G.; MacLean, C.; et al. Classification and Personalized Prognosis in Myeloproliferative Neoplasms. N. Engl. J. Med. 2018, 379, 1416–1430. [Google Scholar] [CrossRef]
- Cazzola, M.; Kralovics, R. From Janus kinase 2 to calreticulin: The clinically relevant genomic landscape of myeloproliferative neoplasms. Blood 2014, 123, 3714–3719. [Google Scholar] [CrossRef]
- Loscocco, G.; Coltro, G.; Guglielmelli, P.; Vannucchi, A. Integration of Molecular Information in Risk Assessment of Patients with Myeloproliferative Neoplasms. Cells 2021, 10, 1962. [Google Scholar] [CrossRef]
- Iurlo, A.; Palandri, F.; Elli, E.M.; Cattaneo, D.; Bucelli, C.; Sciumè, M.; Vincelli, D.; Brioschi, F.; Auteri, G.; Croci, G.A.; et al. Cytogenetic study in primary myelofibrosis at diagnosis: Clinical and histological association and impact on survival according to WHO 2017 classification in an Italian multicenter series. Hematol. Oncol. 2020, 39, 123–128. [Google Scholar] [CrossRef]
- Gangat, N.; Szuber, N.; Pardanani, A.; Tefferi, A. JAK2 unmutated erythrocytosis: Current diagnostic approach and therapeutic views. Leukemia 2021, 35, 2166–2181. [Google Scholar] [CrossRef] [PubMed]
- Broséus, J.; Park, J.-H.; Carillo, S.; Hermouet, S.; Girodon, F. Presence of calreticulin mutations in JAK2-negative polycythemia vera. Blood 2014, 124, 3964–3966. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quattrocchi, A.; Maiorca, C.; Billi, M.; Tomassini, S.; De Marinis, E.; Cenfra, N.; Equitani, F.; Gentile, M.; Ceccherelli, A.; Banella, C.; et al. Genetic lesions disrupting calreticulin 3′-untranslated region in JAK2 mutation-negative polycythemia vera. Am. J. Hematol. 2020, 95, E263–E267. [Google Scholar] [CrossRef]
- Maslah, N.; Cassinat, B.; Verger, E.; Kiladjian, J.-J.; Velazquez, L. The role of LNK/SH2B3 genetic alterations in myeloproliferative neoplasms and other hematological disorders. Leukemia 2017, 31, 1661–1670. [Google Scholar] [CrossRef]
- Pardanani, A.; Lasho, T.L.; Finke, C.M.; Oh, S.T.; Gotlib, J.; Tefferi, A. LNK mutation studies in blast-phase myeloproliferative neoplasms, and in chronic-phase disease with TET2, IDH, JAK2 or MPL mutations. Leukemia 2010, 24, 1713–1718. [Google Scholar] [CrossRef]
- Skov, V. Next Generation Sequencing in MPNs. Lessons from the Past and Prospects for Use as Predictors of Prognosis and Treatment Responses. Cancers 2020, 12, 2194. [Google Scholar] [CrossRef] [PubMed]
- Brown, A.L.; Arts, P.; Carmichael, C.; Babic, M.; Dobbins, J.; Chong, C.-E.; Schreiber, A.W.; Feng, J.; Phillips, K.; Wang, P.P.S.; et al. RUNX1-mutated families show phenotype heterogeneity and a somatic mutation profile unique to germline predisposed AML. Blood Adv. 2020, 4, 1131–1144. [Google Scholar] [CrossRef] [Green Version]
- Young, A.L.; Challen, G.A.; Birmann, B.; Druley, T.E. Clonal haematopoiesis harbouring AML-associated mutations is ubiquitous in healthy adults. Nat. Commun. 2016, 7, 12484. [Google Scholar] [CrossRef] [PubMed]
- Bejar, R.; Stevenson, K.; Abdel-Wahab, O.; Galili, N.; Nilsson, B.; Garcia-Manero, G.; Kantarjian, H.; Raza, A.; Levine, R.L.; Neuberg, D.; et al. Clinical Effect of Point Mutations in Myelodysplastic Syndromes. N. Engl. J. Med. 2011, 364, 2496–2506. [Google Scholar] [CrossRef] [Green Version]
- Schischlik, F.; Jäger, R.; Rosebrock, F.; Hug, E.; Schuster, M.; Holly, R.; Fuchs, E.; Feenstra, J.D.M.; Bogner, E.; Gisslinger, B.; et al. Mutational landscape of the transcriptome offers putative targets for immunotherapy of myeloproliferative neoplasms. Blood 2019, 134, 199–210. [Google Scholar] [CrossRef] [Green Version]
- Kjær, L. Clonal Hematopoiesis and Mutations of Myeloproliferative Neoplasms. Cancers 2020, 12, 2100. [Google Scholar] [CrossRef]
- Jaiswal, S.; Fontanillas, P.; Flannick, J.; Manning, A.; Grauman, P.V.; Mar, B.; Lindsley, C.; Mermel, C.; Burtt, N.; Chavez, A.; et al. Age-Related Clonal Hematopoiesis Associated with Adverse Outcomes. N. Engl. J. Med. 2014, 371, 2488–2498. [Google Scholar] [CrossRef] [Green Version]
- Veninga, A.; De Simone, I.; Heemskerk, J.W.; Cate, H.T.; Van Der Meijden, P.E. Clonal hematopoietic mutations linked to platelet traits and the risk of thrombosis or bleeding. Haematologica 2020, 105, 2020–2031. [Google Scholar] [CrossRef] [PubMed]
- Buscarlet, M.; Provost, S.; Zada, Y.F.; Barhdadi, A.; Bourgoin, V.; Lépine, G.; Mollica, L.; Szuber, N.; Dubé, M.-P.; Busque, L. DNMT3A and TET2 dominate clonal hematopoiesis and demonstrate benign phenotypes and different genetic predispositions. Blood 2017, 130, 753–762. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zink, F.; Stacey, S.N.; Norddahl, G.L.; Frigge, M.L.; Magnusson, O.T.; Jonsdottir, I.; Thorgeirsson, T.E.; Sigurdsson, A.; Gudjonsson, S.A.; Gudmundsson, J.; et al. Clonal hematopoiesis, with and without candidate driver mutations, is common in the elderly. Blood 2017, 130, 742–752. [Google Scholar] [CrossRef] [PubMed] [Green Version]

| PVSG Criteria | WHO Criteria 2001 | WHO Criteria 2008 | WHO Criteria 2016 |
|---|---|---|---|
Major criteria
| Major criteria
| Major criteria
| Major criteria
|
| Diagnosis posed if A1 + A2 + A3 or A1 + A2 and 2 B criteria are fulfilled | Diagnosis posed if A1 + A2 and any other A or A1 + A2 and 2 B criteria are fulfilled | Diagnosis posed if both the major and one minor criteria or the first major and 2 minor criteria are fulfilled | Diagnosis posed if all major criteria or the first 2 major and the minor criteria are fulfilled |
| PVSG Criteria | WHO Criteria 2001 | WHO Criteria 2008 | WHO Criteria 2016 |
|---|---|---|---|
| Positive criteria
|
| Major criteria
|
| Diagnosis posed if all criteria are fulfilled | Diagnosis posed if all criteria are fulfilled | Diagnosis posed if all criteria are fulfilled | Diagnosis posed if all major criteria or the first 3 major and the minor criteria are fulfilled |
| WHO Criteria 2001 | WHO Criteria 2008 | WHO Criteria 2016 |
|---|---|---|
| Prefibrotic phase | Prefibrotic phase | Prefibrotic phase |
Clinical findings
| Major criteria
| Major criteria
|
| Fibrotic phase | Fibrotic phase | Fibrotic phase |
Clinical findings
| Major criteria
| Major criteria
|
| Minimal criteria for diagnosis not formally provided | Diagnosis posed if all major and 2 minor criteria are fulfilled | Diagnosis posed if all major and ≥1 minor criterion are fulfilled |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pizzi, M.; Croci, G.A.; Ruggeri, M.; Tabano, S.; Dei Tos, A.P.; Sabattini, E.; Gianelli, U. The Classification of Myeloproliferative Neoplasms: Rationale, Historical Background and Future Perspectives with Focus on Unclassifiable Cases. Cancers 2021, 13, 5666. https://doi.org/10.3390/cancers13225666
Pizzi M, Croci GA, Ruggeri M, Tabano S, Dei Tos AP, Sabattini E, Gianelli U. The Classification of Myeloproliferative Neoplasms: Rationale, Historical Background and Future Perspectives with Focus on Unclassifiable Cases. Cancers. 2021; 13(22):5666. https://doi.org/10.3390/cancers13225666
Chicago/Turabian StylePizzi, Marco, Giorgio Alberto Croci, Marco Ruggeri, Silvia Tabano, Angelo Paolo Dei Tos, Elena Sabattini, and Umberto Gianelli. 2021. "The Classification of Myeloproliferative Neoplasms: Rationale, Historical Background and Future Perspectives with Focus on Unclassifiable Cases" Cancers 13, no. 22: 5666. https://doi.org/10.3390/cancers13225666
APA StylePizzi, M., Croci, G. A., Ruggeri, M., Tabano, S., Dei Tos, A. P., Sabattini, E., & Gianelli, U. (2021). The Classification of Myeloproliferative Neoplasms: Rationale, Historical Background and Future Perspectives with Focus on Unclassifiable Cases. Cancers, 13(22), 5666. https://doi.org/10.3390/cancers13225666