
Enhancing T Cell Chemotaxis and Infiltration in Glioblastoma

 , , and
, , and
Abstract
Simple Summary
Abstract
1. Introduction
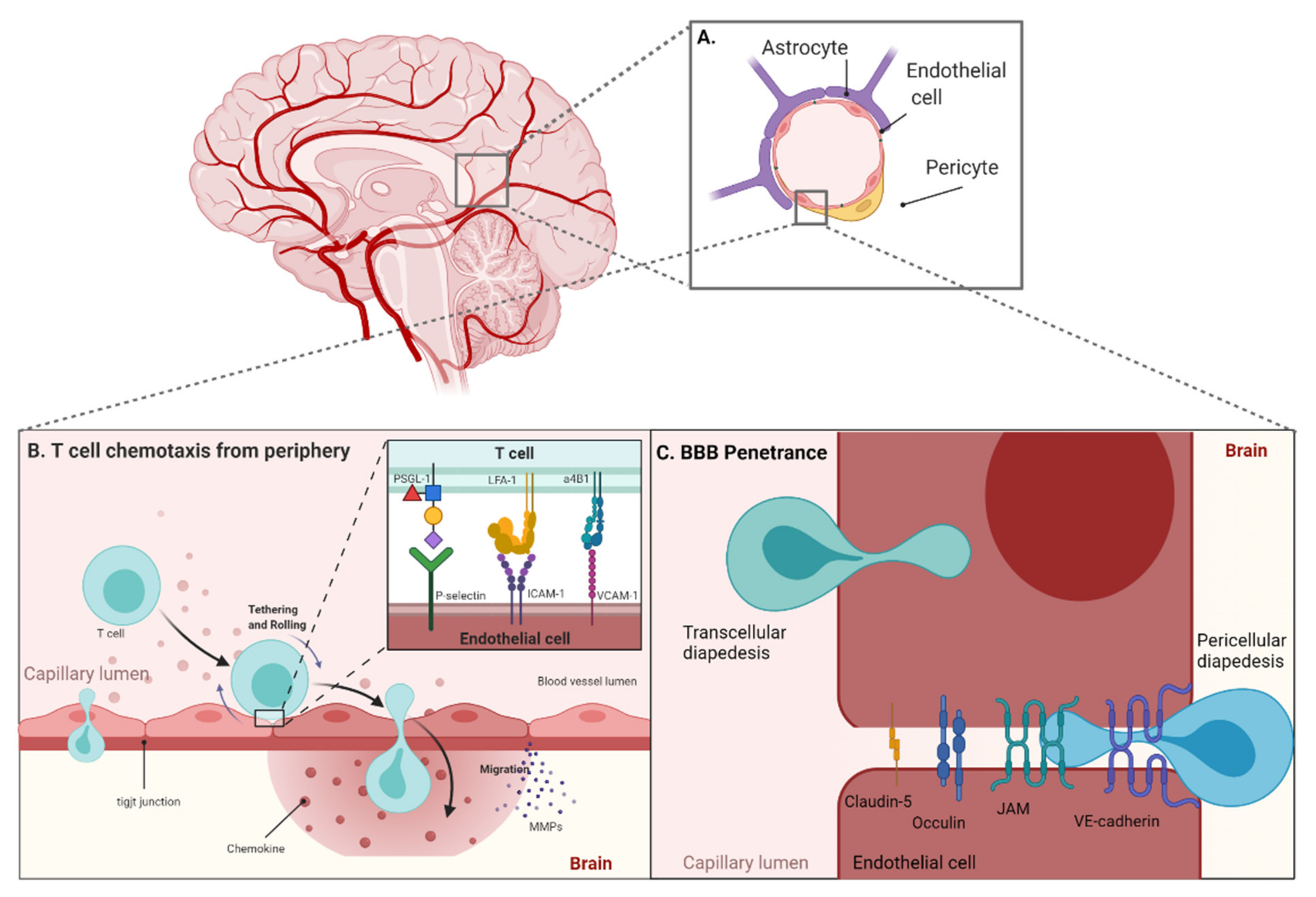
2. T Cell Trafficking from the Periphery to the Blood–Brain Barrier
3. Blood–Brain Barrier Specific Targets
4. The Glia Limitans—Accessing the Parenchyma
5. T Cell Trafficking through the Parenchyma
6. The Tumor Microenvironment
7. Modeling the BBB
8. Safety
9. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Klein, R.S.; Hunter, C.A. Protective and Pathological Immunity during Central Nervous System Infections. Immunity 2017, 46, 891–909. [Google Scholar] [CrossRef]
- Daneman, R.; Prat, A. The blood-brain barrier. Cold Spring Harb. Perspect. Biol. 2015, 7, a020412. [Google Scholar] [CrossRef] [PubMed]
- Engelhardt, B.; Sorokin, L. The blood-brain and the blood-cerebrospinal fluid barriers: Function and dysfunction. Semin. Immunopathol. 2009, 31, 497–511. [Google Scholar] [CrossRef] [PubMed]
- Khasraw, M.; Reardon, D.A.; Weller, M.; Sampson, J.H. PD-1 Inhibitors: Do they have a Future in the Treatment of Glioblastoma? Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2020, 26, 5287–5296. [Google Scholar] [CrossRef]
- Quail, D.F.; Joyce, J.A. Microenvironmental regulation of tumor progression and metastasis. Nat. Med. 2013, 19, 1423–1437. [Google Scholar] [CrossRef]
- Quail, D.F.; Joyce, J.A. The Microenvironmental Landscape of Brain Tumors. Cancer Cell 2017, 31, 326–341. [Google Scholar] [CrossRef] [PubMed]
- Yeung, J.T.; Hamilton, R.L.; Ohnishi, K.; Ikeura, M.; Potter, D.M.; Nikiforova, M.N.; Ferrone, S.; Jakacki, R.I.; Pollack, I.F.; Okada, H. LOH in the HLA Class I Region at 6p21 Is Associated with Shorter Survival in Newly Diagnosed Adult Glioblastoma. Clin. Cancer Res. 2013, 19, 1816–1826. [Google Scholar] [CrossRef] [PubMed]
- Johanns, T.M.; Bowman-Kirigin, J.A.; Liu, C.; Dunn, G.P. Targeting Neoantigens in Glioblastoma: An Overview of Cancer Immunogenomics and Translational Implications. Neurosurgery 2017, 64, 165–176. [Google Scholar] [CrossRef]
- Reardon, D.A.; Brandes, A.A.; Omuro, A.; Mulholland, P.; Lim, M.; Wick, A.; Baehring, J.; Ahluwalia, M.S.; Roth, P.; Bähr, O.; et al. Effect of Nivolumab vs Bevacizumab in Patients With Recurrent Glioblastoma: The CheckMate 143 Phase 3 Randomized Clinical Trial. JAMA Oncol. 2020, 6, 1003–1010. [Google Scholar] [CrossRef] [PubMed]
- Garzon-Muvdi, T.; Theodros, D.; Luksik, A.S.; Maxwell, R.; Kim, E.; Jackson, C.M.; Belcaid, Z.; Ganguly, S.; Tyler, B.; Brem, H.; et al. Dendritic cell activation enhances anti-PD-1 mediated immunotherapy against glioblastoma. Oncotarget 2018, 9, 20681–20697. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.E.; Patel, M.A.; Mangraviti, A.; Kim, E.S.; Theodros, D.; Velarde, E.; Liu, A.; Sankey, E.W.; Tam, A.; Xu, H.; et al. Combination Therapy with Anti-PD-1, Anti-TIM-3, and Focal Radiation Results in Regression of Murine Gliomas. Clin. Cancer Res. 2017, 23, 124–136. [Google Scholar] [CrossRef] [PubMed]
- Ellwardt, E.; Walsh, J.T.; Kipnis, J.; Zipp, F. Understanding the Role of T Cells in CNS Homeostasis. Trends Immunol. 2016, 37, 154–165. [Google Scholar] [CrossRef] [PubMed]
- Ziv, Y.; Ron, N.; Butovsky, O.; Landa, G.; Sudai, E.; Greenberg, N.; Cohen, H.; Kipnis, J.; Schwartz, M. Immune cells contribute to the maintenance of neurogenesis and spatial learning abilities in adulthood. Nat. Neurosci. 2006, 9, 268–275. [Google Scholar] [CrossRef]
- Kipnis, J.; Cohen, H.; Cardon, M.; Ziv, Y.; Schwartz, M. T cell deficiency leads to cognitive dysfunction: Implications for therapeutic vaccination for schizophrenia and other psychiatric conditions. Proc. Natl. Acad. Sci. USA 2004, 101, 8180–8185. [Google Scholar] [CrossRef] [PubMed]
- Engelhardt, B.; Vajkoczy, P.; Weller, R.O. The movers and shapers in immune privilege of the CNS. Nat. Immunol. 2017, 18, 123–131. [Google Scholar] [CrossRef] [PubMed]
- Baruch, K.; Schwartz, M. CNS-specific T cells shape brain function via the choroid plexus. Brain Behav. Immun. 2013, 34, 11–16. [Google Scholar] [CrossRef] [PubMed]
- Korin, B.; Ben-Shaanan, T.L.; Schiller, M.; Dubovik, T.; Azulay-Debby, H.; Boshnak, N.T.; Koren, T.; Rolls, A. High-dimensional, single-cell characterization of the brain’s immune compartment. Nat. Neurosci. 2017, 20, 1300–1309. [Google Scholar] [CrossRef] [PubMed]
- Murphy, J.B.; Sturm, E. Conditions determining the transplantability of tissues in the brain. J. Exp. Med. 1923, 38, 183–197. [Google Scholar] [CrossRef] [PubMed]
- Medawar, P.B. Immunity to homologous grafted skin; the fate of skin homografts transplanted to the brain, to subcutaneous tissue, and to the anterior chamber of the eye. Br. J. Exp. Pathol. 1948, 29, 58–69. [Google Scholar] [PubMed]
- Schläger, C.; Körner, H.; Krueger, M.; Vidoli, S.; Haberl, M.; Mielke, D.; Brylla, E.; Issekutz, T.; Cabañas, C.; Nelson, P.J.; et al. Effector T-cell trafficking between the leptomeninges and the cerebrospinal fluid. Nature 2016, 530, 349–353. [Google Scholar] [CrossRef] [PubMed]
- Hu, X.; Deng, Q.; Ma, L.; Li, Q.; Chen, Y.; Liao, Y.; Zhou, F.; Zhang, C.; Shao, L.; Feng, J.; et al. Meningeal lymphatic vessels regulate brain tumor drainage and immunity. Cell Res. 2020, 30, 229–243. [Google Scholar] [CrossRef] [PubMed]
- Song, E.; Mao, T.; Dong, H.; Boisserand, L.S.B.; Antila, S.; Bosenberg, M.; Alitalo, K.; Thomas, J.-L.; Iwasaki, A. VEGF-C-driven lymphatic drainage enables immunosurveillance of brain tumours. Nature 2020, 577, 689–694. [Google Scholar] [CrossRef] [PubMed]
- Tomaszewski, W.; Sanchez-Perez, L.; Gajewski, T.F.; Sampson, J.H. Brain Tumor Microenvironment and Host State: Implications for Immunotherapy. Clin. Cancer Res. 2019, 25, 4202–4210. [Google Scholar] [CrossRef] [PubMed]
- González, H.; Pacheco, R. T-cell-mediated regulation of neuroinflammation involved in neurodegenerative diseases. J. Neuroinflamm. 2014, 11, 201. [Google Scholar] [CrossRef] [PubMed]
- Sommer, A.; Winner, B.; Prots, I. The Trojan horse—Neuroinflammatory impact of T cells in neurodegenerative diseases. Mol. Neurodegener. 2017, 12, 78. [Google Scholar] [CrossRef] [PubMed]
- Muller, W.A. Getting leukocytes to the site of inflammation. Vet. Pathol. 2013, 50, 7–22. [Google Scholar] [CrossRef] [PubMed]
- Lawrence, M.B.; Springer, T.A. Leukocytes roll on a selectin at physiologic flow rates: Distinction from and prerequisite for adhesion through integrins. Cell 1991, 65, 859–873. [Google Scholar] [CrossRef]
- Von Andrian, U.H.; Chambers, J.D.; McEvoy, L.M.; Bargatze, R.F.; Arfors, K.E.; Butcher, E.C. Two-step model of leukocyte-endothelial cell interaction in inflammation: Distinct roles for LECAM-1 and the leukocyte beta 2 integrins in vivo. Proc. Natl. Acad. Sci. USA 1991, 88, 7538–7542. [Google Scholar] [CrossRef] [PubMed]
- Ley, K.; Laudanna, C.; Cybulsky, M.I.; Nourshargh, S. Getting to the site of inflammation: The leukocyte adhesion cascade updated. Nat. Rev. Immunol. 2007, 7, 678–689. [Google Scholar] [CrossRef] [PubMed]
- Nourshargh, S.; Alon, R. Leukocyte Migration into Inflamed Tissues. Immunity 2014, 41, 694–707. [Google Scholar] [CrossRef]
- Montresor, A.; Toffali, L.; Constantin, G.; Laudanna, C. Chemokines and the signaling modules regulating integrin affinity. Front. Immunol. 2012, 3, 127. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.H.J.; Hammer, D.A. Integrin cross-talk modulates stiffness-independent motility of CD4+ T lymphocytes. Mol. Biol. Cell 2021, 32, 1749–1757. [Google Scholar] [CrossRef]
- Marchetti, L.; Engelhardt, B. Immune cell trafficking across the blood-brain barrier in the absence and presence of neuroinflammation. Vasc. Biol. 2020, 2, H1–H18. [Google Scholar] [CrossRef]
- Nourshargh, S.; Hordijk, P.L.; Sixt, M. Breaching multiple barriers: Leukocyte motility through venular walls and the interstitium. Nat. Rev. Mol. Cell Biol. 2010, 11, 366–378. [Google Scholar] [CrossRef] [PubMed]
- Engelhardt, B.; Ransohoff, R.M. Capture, crawl, cross: The T cell code to breach the blood–brain barriers. Trends Immunol. 2012, 33, 579–589. [Google Scholar] [CrossRef] [PubMed]
- Vajkoczy, P.; Laschinger, M.; Engelhardt, B. α4-integrin-VCAM-1 binding mediates G protein–independent capture of encephalitogenic T cell blasts to CNS white matter microvessels. J. Clin. Investig. 2001, 108, 557–565. [Google Scholar] [CrossRef] [PubMed]
- Sporici, R.; Issekutz, T.B. CXCR3 blockade inhibits T-cell migration into the CNS during EAE and prevents development of adoptively transferred, but not actively induced, disease. Eur. J. Immunol. 2010, 40, 2751–2761. [Google Scholar] [CrossRef]
- Murphy, C.A.; Hoek, R.M.; Wiekowski, M.T.; Lira, S.A.; Sedgwick, J.D. Interactions between hemopoietically derived TNF and central nervous system-resident glial chemokines underlie initiation of autoimmune inflammation in the brain. J. Immunol. 2002, 169, 7054–7062. [Google Scholar] [CrossRef] [PubMed]
- Battistini, L.; Piccio, L.; Rossi, B.; Bach, S.; Galgani, S.; Gasperini, C.; Ottoboni, L.; Ciabini, D.; Caramia, M.D.; Bernardi, G.; et al. CD8+ T cells from patients with acute multiple sclerosis display selective increase of adhesiveness in brain venules: A critical role for P-selectin glycoprotein ligand-1. Blood 2003, 101, 4775–4782. [Google Scholar] [CrossRef] [PubMed]
- Steiner, O.; Coisne, C.; Cecchelli, R.; Boscacci, R.; Deutsch, U.; Engelhardt, B.; Lyck, R. Differential roles for endothelial ICAM-1, ICAM-2, and VCAM-1 in shear-resistant T cell arrest, polarization, and directed crawling on blood-brain barrier endothelium. J. Immunol. 2010, 185, 4846–4855. [Google Scholar] [CrossRef] [PubMed]
- Martin-Blondel, G.; Pignolet, B.; Tietz, S.; Yshii, L.; Gebauer, C.; Perinat, T.; Van Weddingen, I.; Blatti, C.; Engelhardt, B.; Liblau, R. Migration of encephalitogenic CD8 T cells into the central nervous system is dependent on the α4β1-integrin. Eur. J. Immunol. 2015, 45, 3302–3312. [Google Scholar] [CrossRef]
- Alvarez, J.I.; Kébir, H.; Cheslow, L.; Charabati, M.; Chabarati, M.; Larochelle, C.; Prat, A. JAML mediates monocyte and CD8 T cell migration across the brain endothelium. Ann. Clin. Transl. Neurol. 2015, 2, 1032–1037. [Google Scholar] [CrossRef]
- Minten, C.; Alt, C.; Gentner, M.; Frei, E.; Deutsch, U.; Lyck, R.; Schaeren-Wiemers, N.; Rot, A.; Engelhardt, B. DARC shuttles inflammatory chemokines across the blood-brain barrier during autoimmune central nervous system inflammation. Brain 2014, 137, 1454–1469. [Google Scholar] [CrossRef] [PubMed]
- Hauptmann, J.; Johann, L.; Marini, F.; Kitic, M.; Colombo, E.; Mufazalov, I.A.; Krueger, M.; Karram, K.; Moos, S.; Wanke, F.; et al. Interleukin-1 promotes autoimmune neuroinflammation by suppressing endothelial heme oxygenase-1 at the blood-brain barrier. Acta Neuropathol. 2020, 140, 549–567. [Google Scholar] [CrossRef]
- Frewert, S.; Stockhammer, F.; Warschewske, G.; Zenclussen, A.C.; Rupprecht, S.; Volk, H.D.; Woiciechowsky, C. Intratumoral infusion of interleukin-1beta and interferon-gamma induces tumor invasion with macrophages and lymphocytes in a rat glioma model. Neurosci. Lett. 2004, 364, 145–148. [Google Scholar] [CrossRef] [PubMed]
- Armulik, A.; Genové, G.; Mäe, M.; Nisancioglu, M.H.; Wallgard, E.; Niaudet, C.; He, L.; Norlin, J.; Lindblom, P.; Strittmatter, K.; et al. Pericytes regulate the blood-brain barrier. Nature 2010, 468, 557–561. [Google Scholar] [CrossRef]
- Daneman, R.; Zhou, L.; Kebede, A.A.; Barres, B.A. Pericytes are required for blood-brain barrier integrity during embryogenesis. Nature 2010, 468, 562–566. [Google Scholar] [CrossRef] [PubMed]
- Nikolakopoulou, A.M.; Montagne, A.; Kisler, K.; Dai, Z.; Wang, Y.; Huuskonen, M.T.; Sagare, A.P.; Lazic, D.; Sweeney, M.D.; Kong, P.; et al. Pericyte loss leads to circulatory failure and pleiotrophin depletion causing neuron loss. Nat. Neurosci. 2019, 22, 1089–1098. [Google Scholar] [CrossRef]
- Ben-Zvi, A.; Lacoste, B.; Kur, E.; Andreone, B.J.; Mayshar, Y.; Yan, H.; Gu, C. Mfsd2a is critical for the formation and function of the blood–brain barrier. Nature 2014, 509, 507–511. [Google Scholar] [CrossRef]
- Shulman, Z.; Cohen, S.J.; Roediger, B.; Kalchenko, V.; Jain, R.; Grabovsky, V.; Klein, E.; Shinder, V.; Stoler-Barak, L.; Feigelson, S.W.; et al. Transendothelial migration of lymphocytes mediated by intraendothelial vesicle stores rather than by extracellular chemokine depots. Nat. Immunol. 2012, 13, 67–76. [Google Scholar] [CrossRef]
- Török, O.; Schreiner, B.; Schaffenrath, J.; Tsai, H.-C.; Maheshwari, U.; Stifter, S.A.; Welsh, C.; Amorim, A.; Sridhar, S.; Utz, S.G.; et al. Pericytes regulate vascular immune homeostasis in the CNS. Proc. Natl. Acad. Sci. USA 2021, 118, e2016587118. [Google Scholar] [CrossRef]
- Park, J.S.; Kim, I.K.; Han, S.; Park, I.; Kim, C.; Bae, J.; Oh, S.J.; Lee, S.; Kim, J.H.; Woo, D.C.; et al. Normalization of Tumor Vessels by Tie2 Activation and Ang2 Inhibition Enhances Drug Delivery and Produces a Favorable Tumor Microenvironment. Cancer Cell 2016, 30, 953–967. [Google Scholar] [CrossRef]
- Zhou, W.; Chen, C.; Shi, Y.; Wu, Q.; Gimple, R.C.; Fang, X.; Huang, Z.; Zhai, K.; Ke, S.Q.; Ping, Y.F.; et al. Targeting Glioma Stem Cell-Derived Pericytes Disrupts the Blood-Tumor Barrier and Improves Chemotherapeutic Efficacy. Cell Stem Cell 2017, 21, 591–603.e594. [Google Scholar] [CrossRef]
- Zhang, X.-N.; Yang, K.-D.; Chen, C.; He, Z.-C.; Wang, Q.-H.; Feng, H.; Lv, S.-Q.; Wang, Y.; Mao, M.; Liu, Q.; et al. Pericytes augment glioblastoma cell resistance to temozolomide through CCL5-CCR5 paracrine signaling. Cell Res. 2021, 31, 1072–1087. [Google Scholar] [CrossRef] [PubMed]
- Berthiaume, A.A.; Grant, R.I.; McDowell, K.P.; Underly, R.G.; Hartmann, D.A.; Levy, M.; Bhat, N.R.; Shih, A.Y. Dynamic Remodeling of Pericytes In Vivo Maintains Capillary Coverage in the Adult Mouse Brain. Cell Rep. 2018, 22, 8–16. [Google Scholar] [CrossRef] [PubMed]
- Schlecker, E.; Stojanovic, A.; Eisen, C.; Quack, C.; Falk, C.S.; Umansky, V.; Cerwenka, A. Tumor-infiltrating monocytic myeloid-derived suppressor cells mediate CCR5-dependent recruitment of regulatory T cells favoring tumor growth. J. Immunol. 2012, 189, 5602–5611. [Google Scholar] [CrossRef]
- Pozo-Balado, M.M.; Martínez-Bonet, M.; Rosado, I.; Ruiz-Mateos, E.; Méndez-Lagares, G.; Rodríguez-Méndez, M.M.; Vidal, F.; Muñoz-Fernández, M.A.; Pacheco, Y.M.; Leal, M. Maraviroc Reduces the Regulatory T-Cell Frequency in Antiretroviral-Naive HIV-Infected Subjects. J. Infect. Dis. 2014, 210, 890–898. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Moreno-Fernandez, M.E.; Zapata, W.; Blackard, J.T.; Franchini, G.; Chougnet, C.A. Human regulatory T cells are targets for human immunodeficiency Virus (HIV) infection, and their susceptibility differs depending on the HIV type 1 strain. J. Virol. 2009, 83, 12925–12933. [Google Scholar] [CrossRef]
- Halama, N.; Zoernig, I.; Berthel, A.; Kahlert, C.; Klupp, F.; Suarez-Carmona, M.; Suetterlin, T.; Brand, K.; Krauss, J.; Lasitschka, F.; et al. Tumoral Immune Cell Exploitation in Colorectal Cancer Metastases Can Be Targeted Effectively by Anti-CCR5 Therapy in Cancer Patients. Cancer Cell 2016, 29, 587–601. [Google Scholar] [CrossRef]
- Joy, M.T.; Ben Assayag, E.; Shabashov-Stone, D.; Liraz-Zaltsman, S.; Mazzitelli, J.; Arenas, M.; Abduljawad, N.; Kliper, E.; Korczyn, A.D.; Thareja, N.S.; et al. CCR5 Is a Therapeutic Target for Recovery after Stroke and Traumatic Brain Injury. Cell 2019, 176, 1143–1157.e1113. [Google Scholar] [CrossRef] [PubMed]
- Struyf, S.; Menten, P.; Lenaerts, J.P.; Put, W.; D’Haese, A.; De Clercq, E.; Schols, D.; Proost, P.; Van Damme, J. Diverging binding capacities of natural LD78beta isoforms of macrophage inflammatory protein-1alpha to the CC chemokine receptors 1, 3 and 5 affect their anti-HIV-1 activity and chemotactic potencies for neutrophils and eosinophils. Eur. J. Immunol. 2001, 31, 2170–2178. [Google Scholar] [CrossRef]
- Liu, J.Y.; Li, F.; Wang, L.P.; Chen, X.F.; Wang, D.; Cao, L.; Ping, Y.; Zhao, S.; Li, B.; Thorne, S.H.; et al. CTL- vs Treg lymphocyte-attracting chemokines, CCL4 and CCL20, are strong reciprocal predictive markers for survival of patients with oesophageal squamous cell carcinoma. Br. J. Cancer 2015, 113, 747–755. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Liu, T.; Yang, N.; Xu, S.; Li, X.; Wang, D. Hypoxia and macrophages promote glioblastoma invasion by the CCL4-CCR5 axis. Oncol. Rep. 2016, 36, 3522–3528. [Google Scholar] [CrossRef] [PubMed]
- Sallusto, F.; Lenig, D.; Förster, R.; Lipp, M.; Lanzavecchia, A. Two subsets of memory T lymphocytes with distinct homing potentials and effector functions. Nature 1999, 401, 708–712. [Google Scholar] [CrossRef]
- Joshi, N.S.; Cui, W.; Chandele, A.; Lee, H.K.; Urso, D.R.; Hagman, J.; Gapin, L.; Kaech, S.M. Inflammation directs memory precursor and short-lived effector CD8(+) T cell fates via the graded expression of T-bet transcription factor. Immunity 2007, 27, 281–295. [Google Scholar] [CrossRef]
- Intlekofer, A.M.; Takemoto, N.; Wherry, E.J.; Longworth, S.A.; Northrup, J.T.; Palanivel, V.R.; Mullen, A.C.; Gasink, C.R.; Kaech, S.M.; Miller, J.D.; et al. Effector and memory CD8+ T cell fate coupled by T-bet and eomesodermin. Nat. Immunol. 2005, 6, 1236–1244. [Google Scholar] [CrossRef]
- Rutishauser, R.L.; Martins, G.A.; Kalachikov, S.; Chandele, A.; Parish, I.A.; Meffre, E.; Jacob, J.; Calame, K.; Kaech, S.M. Transcriptional repressor Blimp-1 promotes CD8(+) T cell terminal differentiation and represses the acquisition of central memory T cell properties. Immunity 2009, 31, 296–308. [Google Scholar] [CrossRef]
- Cannarile, M.A.; Lind, N.A.; Rivera, R.; Sheridan, A.D.; Camfield, K.A.; Wu, B.B.; Cheung, K.P.; Ding, Z.; Goldrath, A.W. Transcriptional regulator Id2 mediates CD8+ T cell immunity. Nat. Immunol. 2006, 7, 1317–1325. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.Y.; Best, J.A.; Knell, J.; Yang, E.; Sheridan, A.D.; Jesionek, A.K.; Li, H.S.; Rivera, R.R.; Lind, K.C.; D’Cruz, L.M.; et al. The transcriptional regulators Id2 and Id3 control the formation of distinct memory CD8+ T cell subsets. Nat. Immunol. 2011, 12, 1221–1229. [Google Scholar] [CrossRef] [PubMed]
- Soeda, A.; Hara, A.; Kunisada, T.; Yoshimura, S.-i.; Iwama, T.; Park, D.M. The Evidence of Glioblastoma Heterogeneity. Sci. Rep. 2015, 5, 1–7. [Google Scholar] [CrossRef]
- Hickey, W.F.; Hsu, B.L.; Kimura, H. T-lymphocyte entry into the central nervous system. J. Neurosci. Res. 1991, 28, 254–260. [Google Scholar] [CrossRef] [PubMed]
- Calzascia, T.; Masson, F.; Di Berardino-Besson, W.; Contassot, E.; Wilmotte, R.; Aurrand-Lions, M.; Rüegg, C.; Dietrich, P.Y.; Walker, P.R. Homing phenotypes of tumor-specific CD8 T cells are predetermined at the tumor site by crosspresenting APCs. Immunity 2005, 22, 175–184. [Google Scholar] [CrossRef]
- Ogawa, M.; Tsutsui, T.; Zou, J.P.; Mu, J.; Wijesuriya, R.; Yu, W.G.; Herrmann, S.; Kubo, T.; Fujiwara, H.; Hamaoka, T. Enhanced induction of very late antigen 4/lymphocyte function-associated antigen 1-dependent T-cell migration to tumor sites following administration of interleukin 12. Cancer Res. 1997, 57, 2216–2222. [Google Scholar] [PubMed]
- Odoardi, F.; Sie, C.; Streyl, K.; Ulaganathan, V.K.; Schläger, C.; Lodygin, D.; Heckelsmiller, K.; Nietfeld, W.; Ellwart, J.; Klinkert, W.E.; et al. T cells become licensed in the lung to enter the central nervous system. Nature 2012, 488, 675–679. [Google Scholar] [CrossRef] [PubMed]
- Karin, N. CXCR3 Ligands in Cancer and Autoimmunity, Chemoattraction of Effector T Cells, and Beyond. Front. Immunol. 2020, 11, 976. [Google Scholar] [CrossRef] [PubMed]
- Korn, T.; Kallies, A. T cell responses in the central nervous system. Nat. Rev. Immunol. 2017, 17, 179–194. [Google Scholar] [CrossRef]
- Smith, J.S.; Alagesan, P.; Desai, N.K.; Pack, T.F.; Wu, J.-H.; Inoue, A.; Freedman, N.J.; Rajagopal, S. CXC motif chemokine receptor 3 splice variants differentially activate beta-arrestins to regulate downstream signaling pathways. Mol. Pharmacol. 2017, 92, 136–150. [Google Scholar] [CrossRef]
- Nishimura, F.; Dusak, J.E.; Eguchi, J.; Zhu, X.; Gambotto, A.; Storkus, W.J.; Okada, H. Adoptive transfer of type 1 CTL mediates effective anti–central nervous system tumor response: Critical roles of IFN-inducible protein-10. Cancer Res. 2006, 66, 4478–4487. [Google Scholar] [CrossRef]
- Maru, S.V.; Holloway, K.A.; Flynn, G.; Lancashire, C.L.; Loughlin, A.J.; Male, D.K.; Romero, I.A. Chemokine production and chemokine receptor expression by human glioma cells: Role of CXCL10 in tumour cell proliferation. J. Neuroimmunol. 2008, 199, 35–45. [Google Scholar] [CrossRef]
- Barreira da Silva, R.; Laird, M.E.; Yatim, N.; Fiette, L.; Ingersoll, M.A.; Albert, M.L. Dipeptidylpeptidase 4 inhibition enhances lymphocyte trafficking, improving both naturally occurring tumor immunity and immunotherapy. Nat. Immunol. 2015, 16, 850–858. [Google Scholar] [CrossRef] [PubMed]
- Busek, P.; Stremenova, J.; Sromova, L.; Hilser, M.; Balaziova, E.; Kosek, D.; Trylcova, J.; Strnad, H.; Krepela, E.; Sedo, A. Dipeptidyl peptidase-IV inhibits glioma cell growth independent of its enzymatic activity. Int. J. Biochem. Cell Biol. 2012, 44, 738–747. [Google Scholar] [CrossRef]
- Zhu, X.; Fallert-Junecko, B.A.; Fujita, M.; Ueda, R.; Kohanbash, G.; Kastenhuber, E.R.; McDonald, H.A.; Liu, Y.; Kalinski, P.; Reinhart, T.A.; et al. Poly-ICLC promotes the infiltration of effector T cells into intracranial gliomas via induction of CXCL10 in IFN-alpha and IFN-gamma dependent manners. Cancer Immunol. Immunother. 2010, 59, 1401–1409. [Google Scholar] [CrossRef]
- Sarkaria, J.N.; Hu, L.S.; Parney, I.F.; Pafundi, D.H.; Brinkmann, D.H.; Laack, N.N.; Giannini, C.; Burns, T.C.; Kizilbash, S.H.; Laramy, J.K.; et al. Is the blood-brain barrier really disrupted in all glioblastomas? A critical assessment of existing clinical data. Neuro-Oncology 2018, 20, 184–191. [Google Scholar] [CrossRef] [PubMed]
- Kotrotsou, A.; Elakkad, A.; Sun, J.; Thomas, G.A.; Yang, D.; Abrol, S.; Wei, W.; Weinberg, J.S.; Bakhtiari, A.S.; Kircher, M.F.; et al. Multi-center study finds postoperative residual non-enhancing component of glioblastoma as a new determinant of patient outcome. J. Neuro-Oncol. 2018, 139, 125–133. [Google Scholar] [CrossRef] [PubMed]
- Turner, N.; Grose, R. Fibroblast growth factor signalling: From development to cancer. Nat. Rev. Cancer 2010, 10, 116–129. [Google Scholar] [CrossRef] [PubMed]
- Bastola, S.; Pavlyukov, M.S.; Yamashita, D.; Ghosh, S.; Cho, H.; Kagaya, N.; Zhang, Z.; Minata, M.; Lee, Y.; Sadahiro, H.; et al. Glioma-initiating cells at tumor edge gain signals from tumor core cells to promote their malignancy. Nat. Commun. 2020, 11, 4660. [Google Scholar] [CrossRef] [PubMed]
- Darmanis, S.; Sloan, S.A.; Croote, D.; Mignardi, M.; Chernikova, S.; Samghababi, P.; Zhang, Y.; Neff, N.; Kowarsky, M.; Caneda, C.; et al. Single-Cell RNA-Seq Analysis of Infiltrating Neoplastic Cells at the Migrating Front of Human Glioblastoma. Cell Rep. 2017, 21, 1399–1410. [Google Scholar] [CrossRef]
- Platten, M.; Wick, W.; Weller, M. Malignant glioma biology: Role for TGF-β in growth, motility, angiogenesis, and immune escape. Microsc. Res. Tech. 2001, 52, 401–410. [Google Scholar] [CrossRef]
- Li, C.; Jiang, P.; Wei, S.; Xu, X.; Wang, J. Regulatory T cells in tumor microenvironment: New mechanisms, potential therapeutic strategies and future prospects. Mol. Cancer 2020, 19, 116. [Google Scholar] [CrossRef] [PubMed]
- Chang, A.L.; Miska, J.; Wainwright, D.A.; Dey, M.; Rivetta, C.V.; Yu, D.; Kanojia, D.; Pituch, K.C.; Qiao, J.; Pytel, P.; et al. CCL2 Produced by the Glioma Microenvironment Is Essential for the Recruitment of Regulatory T Cells and Myeloid-Derived Suppressor Cells. Cancer Res. 2016, 76, 5671–5682. [Google Scholar] [CrossRef] [PubMed]
- Pan, Y.; Smithson, L.J.; Ma, Y.; Hambardzumyan, D.; Gutmann, D.H. Ccl5 establishes an autocrine high-grade glioma growth regulatory circuit critical for mesenchymal glioblastoma survival. Oncotarget 2017, 8, 32977–32989. [Google Scholar] [CrossRef]
- Vanlandewijck, M.; He, L.; Mäe, M.A.; Andrae, J.; Ando, K.; Del Gaudio, F.; Nahar, K.; Lebouvier, T.; Laviña, B.; Gouveia, L.; et al. A molecular atlas of cell types and zonation in the brain vasculature. Nature 2018, 554, 475–480. [Google Scholar] [CrossRef] [PubMed]
- Wolburg, H.; Wolburg-Buchholz, K.; Engelhardt, B. Diapedesis of mononuclear cells across cerebral venules during experimental autoimmune encephalomyelitis leaves tight junctions intact. Acta Neuropathol. 2005, 109, 181–190. [Google Scholar] [CrossRef]
- Wälchli, T.; Ghobrial, M.; Schwab, M.; Takada, S.; Zhong, H.; Suntharalingham, S.; Vetiska, S.; Rodrigues Rodrigues, D.; Rehrauer, H.; Wu, R.; et al. Molecular atlas of the human brain vasculature at the single-cell level. bioRxiv 2021. [Google Scholar] [CrossRef]
- Castro Dias, M.; Odriozola Quesada, A.; Soldati, S.; Bösch, F.; Gruber, I.; Hildbrand, T.; Sönmez, D.; Khire, T.; Witz, G.; McGrath, J.L.; et al. Brain endothelial tricellular junctions as novel sites for T cell diapedesis across the blood-brain barrier. J. Cell Sci. 2021, 134, jcs253880. [Google Scholar] [CrossRef] [PubMed]
- Ikenouchi, J.; Furuse, M.; Furuse, K.; Sasaki, H.; Tsukita, S.; Tsukita, S. Tricellulin constitutes a novel barrier at tricellular contacts of epithelial cells. J. Cell Biol. 2005, 171, 939–945. [Google Scholar] [CrossRef]
- Fujita, K.; Katahira, J.; Horiguchi, Y.; Sonoda, N.; Furuse, M.; Tsukita, S. Clostridium Perfringens enterotoxin binds to the second extracellular loop of claudin-3, a tight junction integral membrane protein. FEBS Lett. 2000, 476, 258–261. [Google Scholar] [CrossRef]
- Veshnyakova, A.; Piontek, J.; Protze, J.; Waziri, N.; Heise, I.; Krause, G. Mechanism of Clostridium Perfringens enterotoxin interaction with claudin-3/-4 protein suggests structural modifications of the toxin to target specific claudins. J. Biol. Chem. 2012, 287, 1698–1708. [Google Scholar] [CrossRef] [PubMed]
- Taddei, A.; Giampietro, C.; Conti, A.; Orsenigo, F.; Breviario, F.; Pirazzoli, V.; Potente, M.; Daly, C.; Dimmeler, S.; Dejana, E. Endothelial adherens junctions control tight junctions by VE-cadherin-mediated upregulation of claudin-5. Nat. Cell Biol. 2008, 10, 923–934. [Google Scholar] [CrossRef]
- Nitta, T.; Hata, M.; Gotoh, S.; Seo, Y.; Sasaki, H.; Hashimoto, N.; Furuse, M.; Tsukita, S. Size-selective loosening of the blood-brain barrier in claudin-5–deficient mice. J. Cell Biol. 2003, 161, 653–660. [Google Scholar] [CrossRef] [PubMed]
- Greene, C.; Hanley, N.; Campbell, M. Claudin-5: Gatekeeper of neurological function. Fluids Barriers CNS 2019, 16, 3. [Google Scholar] [CrossRef]
- Neuhaus, W.; Piontek, A.; Protze, J.; Eichner, M.; Mahringer, A.; Subileau, E.A.; Lee, I.M.; Schulzke, J.D.; Krause, G.; Piontek, J. Reversible opening of the blood-brain barrier by claudin-5-binding variants of Clostridium Perfringens enterotoxin’s claudin-binding domain. Biomaterials 2018, 161, 129–143. [Google Scholar] [CrossRef]
- Paul, D.; Baena, V.; Ge, S.; Jiang, X.; Jellison, E.R.; Kiprono, T.; Agalliu, D.; Pachter, J.S. Appearance of claudin-5(+) leukocytes in the central nervous system during neuroinflammation: A novel role for endothelial-derived extracellular vesicles. J. Neuroinflamm. 2016, 13, 292. [Google Scholar] [CrossRef] [PubMed]
- Wimmer, I.; Tietz, S.; Nishihara, H.; Deutsch, U.; Sallusto, F.; Gosselet, F.; Lyck, R.; Muller, W.A.; Lassmann, H.; Engelhardt, B. PECAM-1 Stabilizes Blood-Brain Barrier Integrity and Favors Paracellular T-Cell Diapedesis Across the Blood-Brain Barrier During Neuroinflammation. Front. Immunol. 2019, 10, 711. [Google Scholar] [CrossRef]
- Nag, S. Ultracytochemical studies of the compromised blood-brain barrier. Methods Mol. Med. 2003, 89, 145–160. [Google Scholar] [CrossRef] [PubMed]
- Claudio, L.; Brosnan, C.F. Effects of prazosin on the blood-brain barrier during experimental autoimmune encephalomyelitis. Brain Res. 1992, 594, 233–243. [Google Scholar] [CrossRef]
- Millán, J.; Hewlett, L.; Glyn, M.; Toomre, D.; Clark, P.; Ridley, A.J. Lymphocyte transcellular migration occurs through recruitment of endothelial ICAM-1 to caveola- and F-actin-rich domains. Nat. Cell Biol. 2006, 8, 113–123. [Google Scholar] [CrossRef]
- Abadier, M.; Haghayegh Jahromi, N.; Cardoso Alves, L.; Boscacci, R.; Vestweber, D.; Barnum, S.; Deutsch, U.; Engelhardt, B.; Lyck, R. Cell surface levels of endothelial ICAM-1 influence the transcellular or paracellular T-cell diapedesis across the blood–brain barrier. Eur. J. Immunol. 2015, 45, 1043–1058. [Google Scholar] [CrossRef] [PubMed]
- Lutz, S.E.; Smith, J.R.; Kim, D.H.; Olson, C.V.L.; Ellefsen, K.; Bates, J.M.; Gandhi, S.P.; Agalliu, D. Caveolin1 Is Required for Th1 Cell Infiltration, but Not Tight Junction Remodeling, at the Blood-Brain Barrier in Autoimmune Neuroinflammation. Cell Rep. 2017, 21, 2104–2117. [Google Scholar] [CrossRef]
- Engelhardt, B.; Carare, R.O.; Bechmann, I.; Flügel, A.; Laman, J.D.; Weller, R.O. Vascular, glial, and lymphatic immune gateways of the central nervous system. Acta Neuropathol. 2016, 132, 317–338. [Google Scholar] [CrossRef]
- Reboldi, A.; Coisne, C.; Baumjohann, D.; Benvenuto, F.; Bottinelli, D.; Lira, S.; Uccelli, A.; Lanzavecchia, A.; Engelhardt, B.; Sallusto, F. C-C chemokine receptor 6-regulated entry of TH-17 cells into the CNS through the choroid plexus is required for the initiation of EAE. Nat. Immunol. 2009, 10, 514–523. [Google Scholar] [CrossRef] [PubMed]
- Kondo, T.; Takata, H.; Takiguchi, M. Functional expression of chemokine receptor CCR6 on human effector memory CD8+ T cells. Eur. J. Immunol. 2007, 37, 54–65. [Google Scholar] [CrossRef] [PubMed]
- Liao, L.-S.; Zhang, M.-W.; Gu, Y.-J.; Sun, X.-C. Targeting CCL20 inhibits subarachnoid hemorrhage-related neuroinflammation in mice. Aging 2020, 12, 14849–14862. [Google Scholar] [CrossRef] [PubMed]
- Hippe, A.; Braun, S.A.; Oláh, P.; Gerber, P.A.; Schorr, A.; Seeliger, S.; Holtz, S.; Jannasch, K.; Pivarcsi, A.; Buhren, B.; et al. EGFR/Ras-induced CCL20 production modulates the tumour microenvironment. Br. J. Cancer 2020, 123, 942–954. [Google Scholar] [CrossRef]
- Fantini, M.C.; Becker, C.; Monteleone, G.; Pallone, F.; Galle, P.R.; Neurath, M.F. Cutting edge: TGF-beta induces a regulatory phenotype in CD4+CD25− T cells through Foxp3 induction and down-regulation of Smad7. J. Immunol. 2004, 172, 5149–5153. [Google Scholar] [CrossRef]
- Rivino, L.; Gruarin, P.; Häringer, B.; Steinfelder, S.; Lozza, L.; Steckel, B.; Weick, A.; Sugliano, E.; Jarrossay, D.; Kühl, A.A.; et al. CCR6 is expressed on an IL-10–producing, autoreactive memory T cell population with context-dependent regulatory function. J. Exp. Med. 2010, 207, 565–577. [Google Scholar] [CrossRef]
- Krumbholz, M.; Theil, D.; Steinmeyer, F.; Cepok, S.; Hemmer, B.; Hofbauer, M.; Farina, C.; Derfuss, T.; Junker, A.; Arzberger, T.; et al. CCL19 is constitutively expressed in the CNS, up-regulated in neuroinflammation, active and also inactive multiple sclerosis lesions. J. Neuroimmunol. 2007, 190, 72–79. [Google Scholar] [CrossRef]
- Yan, Y.; Zhao, W.; Liu, W.; Li, Y.; Wang, X.; Xun, J.; Davgadorj, C. CCL19 enhances CD8(+) T-cell responses and accelerates HBV clearance. J. Gastroenterol. 2021, 56, 769–785. [Google Scholar] [CrossRef]
- Ponda, M.P.; Breslow, J.L. Serum stimulation of CCR7 chemotaxis due to coagulation factor XIIa-dependent production of high-molecular-weight kininogen domain 5. Proc. Natl. Acad. Sci. USA 2016, 113, E7059–E7068. [Google Scholar] [CrossRef]
- Schneider, M.A.; Meingassner, J.G.; Lipp, M.; Moore, H.D.; Rot, A. CCR7 is required for the in vivo function of CD4+ CD25+ regulatory T cells. J. Exp. Med. 2007, 204, 735–745. [Google Scholar] [CrossRef]
- Greter, M.; Heppner, F.L.; Lemos, M.P.; Odermatt, B.M.; Goebels, N.; Laufer, T.; Noelle, R.J.; Becher, B. Dendritic cells permit immune invasion of the CNS in an animal model of multiple sclerosis. Nat. Med. 2005, 11, 328–334. [Google Scholar] [CrossRef]
- Owens, T.; Bechmann, I.; Engelhardt, B. Perivascular Spaces and the Two Steps to Neuroinflammation. J. Neuropathol. Exp. Neurol. 2008, 67, 1113–1121. [Google Scholar] [CrossRef]
- Mundt, S.; Mrdjen, D.; Utz, S.G.; Greter, M.; Schreiner, B.; Becher, B. Conventional DCs sample and present myelin antigens in the healthy CNS and allow parenchymal T cell entry to initiate neuroinflammation. Sci. Immunol. 2019, 4. [Google Scholar] [CrossRef]
- Agrawal, S.; Anderson, P.; Durbeej, M.; van Rooijen, N.; Ivars, F.; Opdenakker, G.; Sorokin, L.M. Dystroglycan is selectively cleaved at the parenchymal basement membrane at sites of leukocyte extravasation in experimental autoimmune encephalomyelitis. J. Exp. Med. 2006, 203, 1007–1019. [Google Scholar] [CrossRef]
- Tzartos, J.S.; Friese, M.A.; Craner, M.J.; Palace, J.; Newcombe, J.; Esiri, M.M.; Fugger, L. Interleukin-17 production in central nervous system-infiltrating T cells and glial cells is associated with active disease in multiple sclerosis. Am. J. Pathol. 2008, 172, 146–155. [Google Scholar] [CrossRef]
- Zhang, X.; Wang, Y.; Song, J.; Gerwien, H.; Chuquisana, O.; Chashchina, A.; Denz, C.; Sorokin, L. The endothelial basement membrane acts as a checkpoint for entry of pathogenic T cells into the brain. J. Exp. Med. 2020, 217, e20191339. [Google Scholar] [CrossRef]
- Sixt, M.; Engelhardt, B.; Pausch, F.; Hallmann, R.; Wendler, O.; Sorokin, L.M. Endothelial cell laminin isoforms, laminins 8 and 10, play decisive roles in T cell recruitment across the blood-brain barrier in experimental autoimmune encephalomyelitis. J. Cell Biol. 2001, 153, 933–946. [Google Scholar] [CrossRef] [PubMed]
- Pullen, N.A.; Anand, M.; Cooper, P.S.; Fillmore, H.L. Matrix metalloproteinase-1 expression enhances tumorigenicity as well as tumor-related angiogenesis and is inversely associated with TIMP-4 expression in a model of glioblastoma. J. Neuro-Oncol. 2012, 106, 461–471. [Google Scholar] [CrossRef]
- Steward, W.P.; Thomas, A.L. Marimastat: The clinical development of a matrix metalloproteinase inhibitor. Expert Opin. Investig. Drugs 2000, 9, 2913–2922. [Google Scholar] [CrossRef] [PubMed]
- Butler, G.S.; Overall, C.M. Updated biological roles for matrix metalloproteinases and new “intracellular” substrates revealed by degradomics. Biochemistry 2009, 48, 10830–10845. [Google Scholar] [CrossRef]
- McCandless, E.E.; Wang, Q.; Woerner, B.M.; Harper, J.M.; Klein, R.S. CXCL12 limits inflammation by localizing mononuclear infiltrates to the perivascular space during experimental autoimmune encephalomyelitis. J. Immunol. 2006, 177, 8053–8064. [Google Scholar] [CrossRef]
- Cruz-Orengo, L.; Holman, D.W.; Dorsey, D.; Zhou, L.; Zhang, P.; Wright, M.; McCandless, E.E.; Patel, J.R.; Luker, G.D.; Littman, D.R.; et al. CXCR7 influences leukocyte entry into the CNS parenchyma by controlling abluminal CXCL12 abundance during autoimmunity. J. Exp. Med. 2011, 208, 327–339. [Google Scholar] [CrossRef]
- Wiatr, M.; Stump-Guthier, C.; Latorre, D.; Uhlig, S.; Weiss, C.; Ilonen, J.; Engelhardt, B.; Ishikawa, H.; Schwerk, C.; Schroten, H.; et al. Distinct migratory pattern of naive and effector T cells through the blood-CSF barrier following Echovirus 30 infection. J. Neuroinflamm. 2019, 16, 232. [Google Scholar] [CrossRef] [PubMed]
- Zohar, Y.; Wildbaum, G.; Novak, R.; Salzman, A.L.; Thelen, M.; Alon, R.; Barsheshet, Y.; Karp, C.L.; Karin, N. CXCL11-dependent induction of FOXP3-negative regulatory T cells suppresses autoimmune encephalomyelitis. J. Clin. Investig. 2014, 124, 2009–2022. [Google Scholar] [CrossRef]
- Haynes, A.B.; Weiser, T.G.; Berry, W.R.; Lipsitz, S.R.; Breizat, A.H.; Dellinger, E.P.; Herbosa, T.; Joseph, S.; Kibatala, P.L.; Lapitan, M.C.; et al. A surgical safety checklist to reduce morbidity and mortality in a global population. New Engl. J. Med. 2009, 360, 491–499. [Google Scholar] [CrossRef]
- Jordan, J.T.; Sun, W.; Hussain, S.F.; DeAngulo, G.; Prabhu, S.S.; Heimberger, A.B. Preferential migration of regulatory T cells mediated by glioma-secreted chemokines can be blocked with chemotherapy. Cancer Immunol. Immunother. 2008, 57, 123–131. [Google Scholar] [CrossRef] [PubMed]
- Gotsch, U.; Jäger, U.; Dominis, M.; Vestweber, D. Expression of P-selectin on endothelial cells is upregulated by LPS and TNF-alpha in vivo. Cell Adhes. Commun. 1994, 2, 7–14. [Google Scholar] [CrossRef] [PubMed]
- Cloughesy, T.F.; Mochizuki, A.Y.; Orpilla, J.R.; Hugo, W.; Lee, A.H.; Davidson, T.B.; Wang, A.C.; Ellingson, B.M.; Rytlewski, J.A.; Sanders, C.M.; et al. Neoadjuvant anti-PD-1 immunotherapy promotes a survival benefit with intratumoral and systemic immune responses in recurrent glioblastoma. Nat. Med. 2019, 25, 477–486. [Google Scholar] [CrossRef] [PubMed]
- Strickland, M.; Stoll, E.A. Metabolic Reprogramming in Glioma. Front. Cell Dev. Biol. 2017, 5, 43. [Google Scholar] [CrossRef] [PubMed]
- Poff, A.; Koutnik, A.P.; Egan, K.M.; Sahebjam, S.; D’Agostino, D.; Kumar, N.B. Targeting the Warburg effect for cancer treatment: Ketogenic diets for management of glioma. Semin. Cancer Biol. 2019, 56, 135–148. [Google Scholar] [CrossRef]
- Macintyre, A.N.; Gerriets, V.A.; Nichols, A.G.; Michalek, R.D.; Rudolph, M.C.; Deoliveira, D.; Anderson, S.M.; Abel, E.D.; Chen, B.J.; Hale, L.P.; et al. The glucose transporter Glut1 is selectively essential for CD4 T cell activation and effector function. Cell Metab. 2014, 20, 61–72. [Google Scholar] [CrossRef] [PubMed]
- Mauro, C.; Fu, H.; Marelli-Berg, F.M. T cell trafficking and metabolism: Novel mechanisms and targets for immunomodulation. Curr. Opin. Pharm. 2012, 12, 452–457. [Google Scholar] [CrossRef] [PubMed]
- Marelli-Berg, F.M.; Jangani, M. Metabolic regulation of leukocyte motility and migration. J. Leukoc. Biol. 2018, 104, 285–293. [Google Scholar] [CrossRef]
- Chapman, N.M.; Boothby, M.R.; Chi, H. Metabolic coordination of T cell quiescence and activation. Nat. Rev. Immunol. 2020, 20, 55–70. [Google Scholar] [CrossRef] [PubMed]
- Lim, A.R.; Rathmell, W.K.; Rathmell, J.C. The tumor microenvironment as a metabolic barrier to effector T cells and immunotherapy. Elife 2020, 9, e55185. [Google Scholar] [CrossRef] [PubMed]
- Haas, R.; Smith, J.; Rocher-Ros, V.; Nadkarni, S.; Montero-Melendez, T.; D’Acquisto, F.; Bland, E.J.; Bombardieri, M.; Pitzalis, C.; Perretti, M.; et al. Lactate Regulates Metabolic and Pro-inflammatory Circuits in Control of T Cell Migration and Effector Functions. PLoS Biol. 2015, 13, e1002202. [Google Scholar] [CrossRef]
- Fischer, K.; Hoffmann, P.; Voelkl, S.; Meidenbauer, N.; Ammer, J.; Edinger, M.; Gottfried, E.; Schwarz, S.; Rothe, G.; Hoves, S.; et al. Inhibitory effect of tumor cell-derived lactic acid on human T cells. Blood 2007, 109, 3812–3819. [Google Scholar] [CrossRef] [PubMed]
- Hu, J.; Mao, Y.; Li, M.; Lu, Y. The profile of Th17 subset in glioma. Int. Immunopharmacol. 2011, 11, 1173–1179. [Google Scholar] [CrossRef] [PubMed]
- Kramer, P.A.; Chacko, B.K.; George, D.J.; Zhi, D.; Wei, C.C.; Dell’Italia, L.J.; Melby, S.J.; George, J.F.; Darley-Usmar, V.M. Decreased Bioenergetic Health Index in monocytes isolated from the pericardial fluid and blood of post-operative cardiac surgery patients. Biosci. Rep. 2015, 35, e00237. [Google Scholar] [CrossRef]
- Oelkrug, C.; Ramage, J.M. Enhancement of T cell recruitment and infiltration into tumours. Clin. Exp. Immunol. 2014, 178, 1–8. [Google Scholar] [CrossRef]
- You, R.; Artichoker, J.; Fries, A.; Edwards, A.W.; Combes, A.J.; Reeder, G.C.; Samad, B.; Krummel, M.F. Active surveillance characterizes human intratumoral T cell exhaustion. J. Clin. Investig. 2021, 131. [Google Scholar] [CrossRef]
- Kloog, Y.; Mor, A. Cytotoxic-T-lymphocyte antigen 4 receptor signaling for lymphocyte adhesion is mediated by C3G and Rap1. Mol. Cell. Biol. 2014, 34, 978–988. [Google Scholar] [CrossRef]
- Finlay, D.; Cantrell, D.A. Metabolism, migration and memory in cytotoxic T cells. Nat. Rev. Immunol. 2011, 11, 109–117. [Google Scholar] [CrossRef]
- Garris, C.S.; Blaho, V.A.; Hla, T.; Han, M.H. Sphingosine-1-phosphate receptor 1 signalling in T cells: Trafficking and beyond. Immunology 2014, 142, 347–353. [Google Scholar] [CrossRef] [PubMed]
- Chongsathidkiet, P.; Jackson, C.; Koyama, S.; Loebel, F.; Cui, X.; Farber, S.H.; Woroniecka, K.; Elsamadicy, A.A.; Dechant, C.A.; Kemeny, H.R.; et al. Sequestration of T cells in bone marrow in the setting of glioblastoma and other intracranial tumors. Nat. Med. 2018, 24, 1459–1468. [Google Scholar] [CrossRef]
- Wilkinson, D.S.; Chongsathidkiet, P.; Dechant, C.; Fecci, P. Sphingosine-1-phosphate receptor 1 (S1P1) loss mediates T cell sequestration in bone marrow in the setting of intracranial tumors: A novel mode of cancer-induced immunosuppression. J. Immunol. 2019, 202, 138.135. [Google Scholar]
- Wilkinson, D.S.; Champion, C.; Chongsathidkiet, P.; Wang, H.; Laskowitz, D.; Fecci, P. Bone marrow T cell sequestration as a novel mode of CNS immune privilege. J. Immunol. 2020, 204, 78.14. [Google Scholar]
- Rogel, A.; Willoughby, J.E.; Buchan, S.L.; Leonard, H.J.; Thirdborough, S.M.; Al-Shamkhani, A. Akt signaling is critical for memory CD8(+) T-cell development and tumor immune surveillance. Proc. Natl. Acad. Sci. USA 2017, 114, E1178–E1187. [Google Scholar] [CrossRef] [PubMed]
- Joy, A.; Kapoor, M.; Georges, J.; Butler, L.; Chang, Y.; Li, C.; Crouch, A.; Smirnov, I.; Nakada, M.; Hepler, J.; et al. The role of AKT isoforms in glioblastoma: AKT3 delays tumor progression. J. Neuro-Oncol. 2016, 130, 43–52. [Google Scholar] [CrossRef]
- Quambusch, L.; Depta, L.; Landel, I.; Lubeck, M.; Kirschner, T.; Nabert, J.; Uhlenbrock, N.; Weisner, J.; Kostka, M.; Levy, L.M.; et al. Cellular model system to dissect the isoform-selectivity of Akt inhibitors. Nat. Commun. 2021, 12, 5297. [Google Scholar] [CrossRef]
- Abu Eid, R.; Friedman, K.M.; Mkrtichyan, M.; Walens, A.; King, W.; Janik, J.; Khleif, S.N. Akt1 and -2 inhibition diminishes terminal differentiation and enhances central memory CD8(+) T-cell proliferation and survival. Oncoimmunology 2015, 4, e1005448. [Google Scholar] [CrossRef]
- Guedan, S.; Ruella, M.; June, C.H. Emerging Cellular Therapies for Cancer. Annu. Rev. Immunol. 2019, 37, 145–171. [Google Scholar] [CrossRef]
- Kmiecik, J.; Poli, A.; Brons, N.H.; Waha, A.; Eide, G.E.; Enger, P.; Zimmer, J.; Chekenya, M. Elevated CD3+ and CD8+ tumor-infiltrating immune cells correlate with prolonged survival in glioblastoma patients despite integrated immunosuppressive mechanisms in the tumor microenvironment and at the systemic level. J. Neuroimmunol. 2013, 264, 71–83. [Google Scholar] [CrossRef]
- El Andaloussi, A.; Lesniak, M.S. An increase in CD4+CD25+FOXP3+ regulatory T cells in tumor-infiltrating lymphocytes of human glioblastoma multiforme. Neuro-Oncology 2006, 8, 234–243. [Google Scholar] [CrossRef]
- Thornton, A.M.; Shevach, E.M. CD4+CD25+ immunoregulatory T cells suppress polyclonal T cell activation in vitro by inhibiting interleukin 2 production. J. Exp. Med. 1998, 188, 287–296. [Google Scholar] [CrossRef] [PubMed]
- Uyttenhove, C.; Pilotte, L.; Théate, I.; Stroobant, V.; Colau, D.; Parmentier, N.; Boon, T.; Van den Eynde, B.J. Evidence for a tumoral immune resistance mechanism based on tryptophan degradation by indoleamine 2,3-dioxygenase. Nat. Med. 2003, 9, 1269–1274. [Google Scholar] [CrossRef]
- Vitkovic, L.; Maeda, S.; Sternberg, E. Anti-inflammatory cytokines: Expression and action in the brain. Neuroimmunomodulation 2001, 9, 295–312. [Google Scholar] [CrossRef]
- Gong, D.; Shi, W.; Yi, S.J.; Chen, H.; Groffen, J.; Heisterkamp, N. TGFβ signaling plays a critical role in promoting alternative macrophage activation. BMC Immunol. 2012, 13, 31. [Google Scholar] [CrossRef] [PubMed]
- Long, G.V.; Dummer, R.; Hamid, O.; Gajewski, T.F.; Caglevic, C.; Dalle, S.; Arance, A.; Carlino, M.S.; Grob, J.-J.; Kim, T.M.; et al. Epacadostat plus pembrolizumab versus placebo plus pembrolizumab in patients with unresectable or metastatic melanoma (ECHO-301/KEYNOTE-252): A phase 3, randomised, double-blind study. Lancet Oncol. 2019, 20, 1083–1097. [Google Scholar] [CrossRef]
- Singh, K.; Batich, K.A.; Wen, P.Y.; Tan, A.C.; Bagley, S.J.; Lim, M.; Platten, M.; Colman, H.; Ashley, D.M.; Chang, S.M.; et al. Designing Clinical Trials for Combination Immunotherapy: A Framework for Glioblastoma. Clin. Cancer Res. 2021. [Google Scholar] [CrossRef] [PubMed]
- Study of Safety and Tolerability of BCA101 Alone and in Combination with Pembrolizumab in Patients With EGFR-driven Advanced Solid Tumors—Full Text View—ClinicalTrials.gov. Available online: https://clinicaltrials.gov/ct2/show/NCT04429542 (accessed on 30 December 2020).
- Tesselaar, K.; Gravestein, L.A.; van Schijndel, G.M.; Borst, J.; van Lier, R.A. Characterization of murine CD70, the ligand of the TNF receptor family member CD27. J. Immunol. 1997, 159, 4959–4965. [Google Scholar] [PubMed]
- Hintzen, R.Q.; Lens, S.M.; Lammers, K.; Kuiper, H.; Beckmann, M.P.; van Lier, R.A. Engagement of CD27 with its ligand CD70 provides a second signal for T cell activation. J. Immunol. 1995, 154, 2612–2623. [Google Scholar] [PubMed]
- Slaney, C.Y.; Wang, P.; Darcy, P.K.; Kershaw, M.H. CARs versus BiTEs: A Comparison between T Cell-Redirection Strategies for Cancer Treatment. Cancer Discov. 2018, 8, 924–934. [Google Scholar] [CrossRef]
- Hurton, L.V.; Singh, H.; Najjar, A.M.; Switzer, K.C.; Mi, T.; Maiti, S.; Olivares, S.; Rabinovich, B.; Huls, H.; Forget, M.A.; et al. Tethered IL-15 augments antitumor activity and promotes a stem-cell memory subset in tumor-specific T cells. Proc. Natl. Acad. Sci. USA 2016, 113, E7788–E7797. [Google Scholar] [CrossRef]
- Avanzi, M.P.; Yeku, O.; Li, X.; Wijewarnasuriya, D.P.; van Leeuwen, D.G.; Cheung, K.; Park, H.; Purdon, T.J.; Daniyan, A.F.; Spitzer, M.H.; et al. Engineered Tumor-Targeted T Cells Mediate Enhanced Anti-Tumor Efficacy Both Directly and through Activation of the Endogenous Immune System. Cell Rep. 2018, 23, 2130–2141. [Google Scholar] [CrossRef] [PubMed]
- Caruana, I.; Savoldo, B.; Hoyos, V.; Weber, G.; Liu, H.; Kim, E.S.; Ittmann, M.M.; Marchetti, D.; Dotti, G. Heparanase promotes tumor infiltration and antitumor activity of CAR-redirected T lymphocytes. Nat. Med. 2015, 21, 524–529. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.C.; Lo, A.; Scholler, J.; Sun, J.; Majumdar, R.S.; Kapoor, V.; Antzis, M.; Cotner, C.E.; Johnson, L.A.; Durham, A.C.; et al. Targeting fibroblast activation protein in tumor stroma with chimeric antigen receptor T cells can inhibit tumor growth and augment host immunity without severe toxicity. Cancer Immunol. Res. 2014, 2, 154–166. [Google Scholar] [CrossRef] [PubMed]
- Salmon, H.; Idoyaga, J.; Rahman, A.; Leboeuf, M.; Remark, R.; Jordan, S.; Casanova-Acebes, M.; Khudoynazarova, M.; Agudo, J.; Tung, N.; et al. Expansion and Activation of CD103(+) Dendritic Cell Progenitors at the Tumor Site Enhances Tumor Responses to Therapeutic PD-L1 and BRAF Inhibition. Immunity 2016, 44, 924–938. [Google Scholar] [CrossRef]
- Pellegatta, S.; Poliani, P.L.; Stucchi, E.; Corno, D.; Colombo, C.A.; Orzan, F.; Ravanini, M.; Finocchiaro, G. Intra-tumoral dendritic cells increase efficacy of peripheral vaccination by modulation of glioma microenvironment. Neuro-Oncology 2010, 12, 377–388. [Google Scholar] [CrossRef]
- Calzascia, T.; Di Berardino-Besson, W.; Wilmotte, R.; Masson, F.; de Tribolet, N.; Dietrich, P.Y.; Walker, P.R. Cutting edge: Cross-presentation as a mechanism for efficient recruitment of tumor-specific CTL to the brain. J. Immunol. 2003, 171, 2187–2191. [Google Scholar] [CrossRef]
- Barnard, Z.; Wakimoto, H.; Zaupa, C.; Patel, A.P.; Klehm, J.; Martuza, R.L.; Rabkin, S.D.; Curry, W.T., Jr. Expression of FMS-like tyrosine kinase 3 ligand by oncolytic herpes simplex virus type I prolongs survival in mice bearing established syngeneic intracranial malignant glioma. Neurosurgery 2012, 71, 741–748. [Google Scholar] [CrossRef] [PubMed]
- Wick, A.; Felsberg, J.; Steinbach, J.P.; Herrlinger, U.; Platten, M.; Blaschke, B.; Meyermann, R.; Reifenberger, G.; Weller, M.; Wick, W. Efficacy and tolerability of temozolomide in an alternating weekly regimen in patients with recurrent glioma. J. Clin. Oncol. 2007, 25, 3357–3361. [Google Scholar] [CrossRef]
- Heimberger, A.B.; Sun, W.; Hussain, S.F.; Dey, M.; Crutcher, L.; Aldape, K.; Gilbert, M.; Hassenbusch, S.J.; Sawaya, R.; Schmittling, B.; et al. Immunological responses in a patient with glioblastoma multiforme treated with sequential courses of temozolomide and immunotherapy: Case study. Neuro-Oncology 2008, 10, 98–103. [Google Scholar] [CrossRef] [PubMed]
- Duan, L.; Zhang, X.D.; Miao, W.Y.; Sun, Y.J.; Xiong, G.; Wu, Q.; Li, G.; Yang, P.; Yu, H.; Li, H.; et al. PDGFRβ Cells Rapidly Relay Inflammatory Signal from the Circulatory System to Neurons via Chemokine CCL2. Neuron 2018, 100, 183–200.e8. [Google Scholar] [CrossRef] [PubMed]
- Twyman-Saint Victor, C.; Rech, A.J.; Maity, A.; Rengan, R.; Pauken, K.E.; Stelekati, E.; Benci, J.L.; Xu, B.; Dada, H.; Odorizzi, P.M.; et al. Radiation and dual checkpoint blockade activate non-redundant immune mechanisms in cancer. Nature 2015, 520, 373–377. [Google Scholar] [CrossRef]
- Wiranowska, M.; Wilson, T.C.; Bencze, K.S.; Prockop, L.D. A mouse model for the study of blood-brain barrier permeability. J. Neurosci. Methods 1988, 26, 105–109. [Google Scholar] [CrossRef]
- Wiranowska, M. Blood-brain barrier and treatment of central nervous system tumors. J. Fla. Med. Assoc. 1992, 79, 707–710. [Google Scholar]
- Cook, P.R.; Myers, D.; Barker, M.; Jones, J.G. Blood-brain barrier to pertechnetate following drug-induced hypotension. Br. J. Anaesth. 1989, 62, 402–408. [Google Scholar] [CrossRef]
- Wiranowska, M.; Gonzalvo, A.A.; Saporta, S.; Gonzalez, O.R.; Prockop, L.D. Evaluation of blood-brain barrier permeability and the effect of interferon in mouse glioma model. J. Neurooncol. 1992, 14, 225–236. [Google Scholar] [CrossRef]
- Saitou, M.; Furuse, M.; Sasaki, H.; Schulzke, J.D.; Fromm, M.; Takano, H.; Noda, T.; Tsukita, S. Complex phenotype of mice lacking occludin, a component of tight junction strands. Mol. Biol. Cell 2000, 11, 4131–4142. [Google Scholar] [CrossRef]
- Jin, A.Y.; Tuor, U.I.; Rushforth, D.; Kaur, J.; Muller, R.N.; Petterson, J.L.; Boutry, S.; Barber, P.A. Reduced blood brain barrier breakdown in P-selectin deficient mice following transient ischemic stroke: A future therapeutic target for treatment of stroke. BMC Neurosci. 2010, 11, 12. [Google Scholar] [CrossRef] [PubMed]
- Hutchinson, M. Natalizumab: A new treatment for relapsing remitting multiple sclerosis. Clin. Risk Manag. 2007, 3, 259–268. [Google Scholar] [CrossRef] [PubMed]
- Cakir, B.; Xiang, Y.; Tanaka, Y.; Kural, M.H.; Parent, M.; Kang, Y.J.; Chapeton, K.; Patterson, B.; Yuan, Y.; He, C.S.; et al. Engineering of human brain organoids with a functional vascular-like system. Nat. Methods 2019, 16, 1169–1175. [Google Scholar] [CrossRef] [PubMed]
- Pham, M.T.; Pollock, K.M.; Rose, M.D.; Cary, W.A.; Stewart, H.R.; Zhou, P.; Nolta, J.A.; Waldau, B. Generation of human vascularized brain organoids. Neuroreport 2018, 29, 588–593. [Google Scholar] [CrossRef] [PubMed]
- Stamatovic, S.M.; Keep, R.F.; Wang, M.M.; Jankovic, I.; Andjelkovic, A.V. Caveolae-mediated internalization of occludin and claudin-5 during CCL2-induced tight junction remodeling in brain endothelial cells. J. Biol. Chem. 2009, 284, 19053–19066. [Google Scholar] [CrossRef]
- Stamatovic, S.M.; Dimitrijevic, O.B.; Keep, R.F.; Andjelkovic, A.V. Protein kinase Calpha-RhoA cross-talk in CCL2-induced alterations in brain endothelial permeability. J. Biol. Chem. 2006, 281, 8379–8388. [Google Scholar] [CrossRef]
- Ngo, M.T.; Harley, B.A.C. Perivascular signals alter global gene expression profile of glioblastoma and response to temozolomide in a gelatin hydrogel. Biomaterials 2019, 198, 122–134. [Google Scholar] [CrossRef]
- Ozturk, M.S.; Lee, V.K.; Zou, H.; Friedel, R.H.; Intes, X.; Dai, G. High-resolution tomographic analysis of in vitro 3D glioblastoma tumor model under long-term drug treatment. Sci. Adv. 2020, 6, eaay7513. [Google Scholar] [CrossRef] [PubMed]
- Plummer, S.; Wallace, S.; Ball, G.; Lloyd, R.; Schiapparelli, P.; Quinones-Hinojosa, A.; Hartung, T.; Pamies, D. A Human iPSC-derived 3D platform using primary brain cancer cells to study drug development and personalized medicine. Sci. Rep. 2019, 9, 1407. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.W.; Gardner, R.; Porter, D.L.; Louis, C.U.; Ahmed, N.; Jensen, M.; Grupp, S.A.; Mackall, C.L. Current concepts in the diagnosis and management of cytokine release syndrome. Blood 2014, 124, 188–195. [Google Scholar] [CrossRef] [PubMed]
- Leach, D.R.; Krummel, M.F.; Allison, J.P. Enhancement of antitumor immunity by CTLA-4 blockade. Science 1996, 271, 1734–1736. [Google Scholar] [CrossRef]
- Dillard, T.; Yedinak, C.G.; Alumkal, J.; Fleseriu, M. Anti-CTLA-4 antibody therapy associated autoimmune hypophysitis: Serious immune related adverse events across a spectrum of cancer subtypes. Pituitary 2010, 13, 29–38. [Google Scholar] [CrossRef] [PubMed]
- Chodakiewitz, Y.; Brown, S.; Boxerman, J.L.; Brody, J.M.; Rogg, J.M. Ipilimumab treatment associated pituitary hypophysitis: Clinical presentation and imaging diagnosis. Clin. Neurol. Neurosurg. 2014, 125, 125–130. [Google Scholar] [CrossRef]
- Lupi, I.; Brancatella, A.; Cosottini, M.; Viola, N.; Lanzolla, G.; Sgro, D.; Dalmazi, G.D.; Latrofa, F.; Caturegli, P.; Marcocci, C. Clinical heterogeneity of hypophysitis secondary to PD-1/PD-L1 blockade: Insights from four cases. Endocrinol. Diabetes Metab. Case Rep. 2019, 2019. [Google Scholar] [CrossRef] [PubMed]
- Mahzari, M.; Liu, D.; Arnaout, A.; Lochnan, H. Immune checkpoint inhibitor therapy associated hypophysitis. Clin. Med. Insights Endocrinol. Diabetes 2015, 8, 21–28. [Google Scholar] [CrossRef]
- Iwama, S.; De Remigis, A.; Callahan, M.K.; Slovin, S.F.; Wolchok, J.D.; Caturegli, P. Pituitary expression of CTLA-4 mediates hypophysitis secondary to administration of CTLA-4 blocking antibody. Sci. Transl. Med. 2014, 6, 230ra245. [Google Scholar] [CrossRef] [PubMed]
- Bossart, S.; Thurneysen, S.; Rushing, E.; Frontzek, K.; Leske, H.; Mihic-Probst, D.; Nagel, H.W.; Mangana, J.; Goldinger, S.M.; Dummer, R. Case Report: Encephalitis, with Brainstem Involvement, Following Checkpoint Inhibitor Therapy in Metastatic Melanoma. Oncologist 2017, 22, 749–753. [Google Scholar] [CrossRef]
- Robert, L.; Langner-Lemercier, S.; Angibaud, A.; Sale, A.; Thepault, F.; Corre, R.; Lena, H.; Ricordel, C. Immune-related Encephalitis in Two Patients Treated With Immune Checkpoint Inhibitor. Clin. Lung Cancer 2020, 21, e474–e477. [Google Scholar] [CrossRef] [PubMed]
- Cabral, G.; Ladeira, F.; Gil, N. Nivolumab-induced seronegative encephalitis. J. Neuroimmunol. 2020, 347, 577350. [Google Scholar] [CrossRef]
- Astaras, C.; de Micheli, R.; Moura, B.; Hundsberger, T.; Hottinger, A.F. Neurological Adverse Events Associated with Immune Checkpoint Inhibitors: Diagnosis and Management. Curr. Neurol. Neurosci. Rep. 2018, 18, 3. [Google Scholar] [CrossRef]
- Dalakas, M.C. Neurological complications of immune checkpoint inhibitors: What happens when you ‘take the brakes off’ the immune system. Adv. Neurol. Disord. 2018, 11, 1756286418799864. [Google Scholar] [CrossRef]
- Feng, S.; Coward, J.; McCaffrey, E.; Coucher, J.; Kalokerinos, P.; O’Byrne, K. Pembrolizumab-Induced Encephalopathy: A Review of Neurological Toxicities with Immune Checkpoint Inhibitors. J. Thorac. Oncol. 2017, 12, 1626–1635. [Google Scholar] [CrossRef]
- Dalmau, J.; Bataller, L. Limbic encephalitis: The new cell membrane antigens and a proposal of clinical-immunological classification with therapeutic implications. Neurologia 2007, 22, 526–537. [Google Scholar] [PubMed]
- Pillonel, V.; Dunet, V.; Hottinger, A.F.; Berthod, G.; Schiappacasse, L.; Peters, S.; Michielin, O.; Aedo-Lopez, V. Multiple nivolumab-induced CNS demyelination with spontaneous resolution in an asymptomatic metastatic melanoma patient. J. Immunother. Cancer 2019, 7, 336. [Google Scholar] [CrossRef] [PubMed]
- Gettings, E.J.; Hackett, C.T.; Scott, T.F. Severe relapse in a multiple sclerosis patient associated with ipilimumab treatment of melanoma. Mult. Scler. 2015, 21, 670. [Google Scholar] [CrossRef]
- Santomasso, B.; Bachier, C.; Westin, J.; Rezvani, K.; Shpall, E.J. The Other Side of CAR T-Cell Therapy: Cytokine Release Syndrome, Neurologic Toxicity, and Financial Burden. Am. Soc. Clin. Oncol. Educ. Book 2019, 39, 433–444. [Google Scholar] [CrossRef] [PubMed]
- Norelli, M.; Camisa, B.; Barbiera, G.; Falcone, L.; Purevdorj, A.; Genua, M.; Sanvito, F.; Ponzoni, M.; Doglioni, C.; Cristofori, P.; et al. Monocyte-derived IL-1 and IL-6 are differentially required for cytokine-release syndrome and neurotoxicity due to CAR T cells. Nat. Med. 2018, 24, 739–748. [Google Scholar] [CrossRef] [PubMed]
- Brown, C.E.; Alizadeh, D.; Starr, R.; Weng, L.; Wagner, J.R.; Naranjo, A.; Ostberg, J.R.; Blanchard, M.S.; Kilpatrick, J.; Simpson, J.; et al. Regression of Glioblastoma after Chimeric Antigen Receptor T-Cell Therapy. New Engl. J. Med. 2016, 375, 2561–2569. [Google Scholar] [CrossRef]
- Akhavan, D.; Alizadeh, D.; Wang, D.; Weist, M.R.; Shepphird, J.K.; Brown, C.E. CAR T cells for brain tumors: Lessons learned and road ahead. Immunol. Rev. 2019, 290, 60–84. [Google Scholar] [CrossRef]
- Pennisi, M.; Jain, T.; Santomasso, B.D.; Mead, E.; Wudhikarn, K.; Silverberg, M.L.; Batlevi, Y.; Shouval, R.; Devlin, S.M.; Batlevi, C.; et al. Comparing CAR T-cell toxicity grading systems: Application of the ASTCT grading system and implications for management. Blood Adv. 2020, 4, 676–686. [Google Scholar] [CrossRef] [PubMed]
- Gust, J.; Hay, K.A.; Hanafi, L.A.; Li, D.; Myerson, D.; Gonzalez-Cuyar, L.F.; Yeung, C.; Liles, W.C.; Wurfel, M.; Lopez, J.A.; et al. Endothelial Activation and Blood-Brain Barrier Disruption in Neurotoxicity after Adoptive Immunotherapy with CD19 CAR-T Cells. Cancer Discov. 2017, 7, 1404–1419. [Google Scholar] [CrossRef] [PubMed]
- Klinger, M.; Zugmaier, G.; Nagele, V.; Goebeler, M.E.; Brandl, C.; Stelljes, M.; Lassmann, H.; von Stackelberg, A.; Bargou, R.C.; Kufer, P. Adhesion of T Cells to Endothelial Cells Facilitates Blinatumomab-Associated Neurologic Adverse Events. Cancer Res. 2020, 80, 91–101. [Google Scholar] [CrossRef] [PubMed]
- Neelapu, S.S.; Tummala, S.; Kebriaei, P.; Wierda, W.; Gutierrez, C.; Locke, F.L.; Komanduri, K.V.; Lin, Y.; Jain, N.; Daver, N.; et al. Chimeric antigen receptor T-cell therapy—assessment and management of toxicities. Nat. Rev. Clin. Oncol. 2018, 15, 47–62. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Interactor | Behavior | Therapeutic Considerations | References |
|---|---|---|---|
| T cell processes | |||
| LFA-1 | T cell integrin which binds ICAM-1. Promotes T cell capture and rolling in inflammatory and non-inflammatory state. | IL-12 induces LFA-1 expression and can enhance T cell migration in several murine malignancies. | [36,73] |
| VLA-4 (α4β1) | Integrin on T cell which binds VCAM-1 in the inflammatory state and interacts with other transmembrane proteins (JAM-B, JAML, etc.). | IL-12 induces LFA-1 and VLA-4 expression and enhances T cell migration in several murine malignancies. Effect may be malignancy dependent. | [41,42,73] |
| CXCL9: Polarizes T cells to a Th1/Th17 phenotype. | Mediated lymphocyte infiltration and suppresses tumor growth in cutaneous fibrosarcoma. | [134] | |
| CXCR3 (3 ligands) | CXCL10: Only moderately induces CXCR3 internalization and enhances T cell infiltration. | DPP-4 blockade increases TILs but is also tumorigenic (independent of enzymatic function). Combinatorial poly-ICLC enhances CXCL10 expression. | [81,82,83] |
| CXCL11: Binds CXCR3 strongly and induces receptor internalization. | Promotes lineage of regulatory T cells. | [75] | |
| CCR4 | CCL2, CCL22 (and others): Overexpressed on glioma cells, recruits regulatory T cells. | CCR4-CCL22 signaling recruits regulatory T cells. Blockade of CCR4 in vitro can reduce regulatory T cell migration. TMZ can also mitigate production of CCL2. | [135,136] |
| CCR5 | Binds CCL3, CCL4, and CCL5. May help to recruit cytolytic T cells but also regulatory T cells. | CCL4 can help recruit cytolytic CCR5+ T cells in esophageal squamous cell carcinoma but CCL4–CCR5 interaction can enhance the invasion ability of glioblastoma in vitro. CCL5 is also associated with enhanced T cell diapedesis at tricellular junctions. However, CCL5 also binds CD44 on GBM cells to drive proliferation and survival and is produced by perivascular stromal cells such as pericytes. Blockade of CCR5 (maraviroc) may limit cancer-associated fibroblast accumulation. | [54,62,63,91,95] |
| CCR6 | Binds CCL20 expressed at the choroid plexus. CD8+ T cells migrate to CCL20 in murine SAH. | TGF- β promotes CCR6 expression but also is implicated in the promotion of FOXP3+ cells. However, a fraction of the population is CCR6+FOXP3−. CCR6 T cells may also be involved with licensing further recruitment to perivascular spaces. | [115,116] |
| CCR7 | Present on activated CD8 T cells (and central memory T cells). | Interacts with CCL19 and may mediate integrin activation on immune cells or diapedesis. Chemotaxis may be enhanced by a peptide derived from the byproduct of coagulation factor XIIa cleavage. May also promote regulatory T cells. | [117,119,120] |
| Blood–brain barrier processes | |||
| E/P-Selectin | Expressed in inflammatory state only. Binds PSGL-1+ CD8 T cells, slowing them on BBB endothelium. | Expression enhanced in response to inflammatory cytokines (e.g., IL-1 or TNF α). IL-1 has been delivered via CED in rat models of glioma. | [45,137] |
| Claudin-5, PECAM-1 | Commonly expressed proteins involved in sealing tight junctions at BBB. | Modified Clostridium perfringens enterotoxin can reversibly open tight junctions. May drive T cells to transcellular migration. | [93,103,104] |
| ACKR1 | Trafficking of pro-infiltrative chemokines from abluminal to luminal surface of BBB. | IL-1 signaling associated with upregulated expression ACKR1 (along with VCAM-1, ICAM-1). Trialed using CED in rat glioma. | [45] |
| Caveolin-1 | Expressed in endocytic vesicles at BBB and acts as a mediator of transcellular diapedesis. | Regions of BBB rich in CAV-1 are also rich in ICAM-1. Enhancing ICAM-1 on BBB (e.g., via IL-1) may capture more T cells that can undergo para and transcellular diapedesis. | [108] |
| CXCL12 | Acts as a T cell, holding factor cells in perivascular spaces. Expression of CXCR7 on endothelial cells internalizes CXCL12. | IL-17 drives expression of CXCR7 on endothelial cells and CXCR4 on T cells which licenses their entry into the parenchyma. However, CXCL12 may promote CD8+ migration across BCSF barrier—may be a location-specific role. | [131,132,133] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Singh, K.; Hotchkiss, K.M.; Patel, K.K.; Wilkinson, D.S.; Mohan, A.A.; Cook, S.L.; Sampson, J.H. Enhancing T Cell Chemotaxis and Infiltration in Glioblastoma. Cancers 2021, 13, 5367. https://doi.org/10.3390/cancers13215367
Singh K, Hotchkiss KM, Patel KK, Wilkinson DS, Mohan AA, Cook SL, Sampson JH. Enhancing T Cell Chemotaxis and Infiltration in Glioblastoma. Cancers. 2021; 13(21):5367. https://doi.org/10.3390/cancers13215367
Chicago/Turabian StyleSingh, Kirit, Kelly M. Hotchkiss, Kisha K. Patel, Daniel S. Wilkinson, Aditya A. Mohan, Sarah L. Cook, and John H. Sampson. 2021. "Enhancing T Cell Chemotaxis and Infiltration in Glioblastoma" Cancers 13, no. 21: 5367. https://doi.org/10.3390/cancers13215367
APA StyleSingh, K., Hotchkiss, K. M., Patel, K. K., Wilkinson, D. S., Mohan, A. A., Cook, S. L., & Sampson, J. H. (2021). Enhancing T Cell Chemotaxis and Infiltration in Glioblastoma. Cancers, 13(21), 5367. https://doi.org/10.3390/cancers13215367