Simple Summary
Based on the four molecular subtypes of pancreatic cancer described by Bailey et al. 2016, in the present article we match the molecular, histology and microenvironment features of pancreatic cancer. This approach may help to understand the molecular basis of this kind of tumor, and how their microenvironment may affect treatment response. Moreover, we compile information about crucial factors that could serve as potential targets for drug design to achieve higher anti-tumor activity, and how histological evaluation of tumor microenvironment could provide first signs about treatment response.
Abstract
In the last decade, several studies based on whole transcriptomic and genomic analyses of pancreatic tumors and their stroma have come to light to supplement histopathological stratification of pancreatic cancers with a molecular point-of-view. Three main molecular studies: Collisson et al. 2011, Moffitt et al. 2015 and Bailey et al. 2016 have found specific gene signatures, which identify different molecular subtypes of pancreatic cancer and provide a comprehensive stratification for both a personalized treatment or to identify potential druggable targets. However, the routine clinical management of pancreatic cancer does not consider a broad molecular analysis of each patient, due probably to the lack of target therapies for this tumor. Therefore, the current treatment decision is taken based on patients´ clinicopathological features and performance status. Histopathological evaluation of tumor samples could reveal many other attributes not only from tumor cells but also from their microenvironment specially about the presence of pancreatic stellate cells, regulatory T cells, tumor-associated macrophages, myeloid derived suppressor cells and extracellular matrix structure. In the present article, we revise the four molecular subtypes proposed by Bailey et al. and associate each subtype with other reported molecular subtypes. Moreover, we provide for each subtype a potential description of the tumor microenvironment that may influence treatment response according to the gene expression profile, the mutational landscape and their associated histology.
1. Introduction
Pancreatic cancer (PC) incidence has increased in developed countries and its trend for 2030 is to be higher reaching the second cause of cancer-related deaths [1]. When tumors are <2 cm in size, the 5-year survival rate is around 50% while for tumors <1 cm could average as much as 100% [2]. Regrettably, pancreatic cancer symptoms are often misdiagnosed and commonly treated ambulatory that leads to a late diagnosis with metastatic disease in distant organs; then, their 5-year survival rate decreased to 3% [3,4]. PC could be detected by elevated levels of CA19-9 serum marker [5]. However, CA19-9 is not PC specific and its levels are also high after biliary obstruction [6]. Recently, CA19-9 serum level in combination with IGF-1 and albumin have increased the sensitivity up to 93.6% and the specificity up to 95% to identify PC patients [7].
Risk factors of PC are cigarette smoking associated to 20–25% cases [8,9], chronic pancreatitis (4%) [10], diabetes (30%) [11], and some infectious agents like Helicobacterpylori (65%) presents an increased risk [12].
Surgical resection is considered the best treatment approach against PC. Histopathological characteristics of resected tumor like margins of resection (R), differentiation of tumor cells (G) or lymph-node status (N) could predict patient prognosis [13]. After resection, adjuvant treatment will depend on patient performance status but is usually based on gemcitabine [14], 5-fluorouracil and their combination with other cytotoxics [15]. The combination of gemcitabine with capecitabine exhibited better survival rates [16]. Other regimens based on FOLFIRINOX (folinic acid, 5-fluorouracil, irinotecan and oxaliplatin) or gemcitabine in combination with nano-albumin-bound paclitaxel (nab-paclitaxel) is administered to borderline resectable tumors as neoadjuvant treatment [17]. Another option for R1 patients, borderline resectable or locally advanced unresectable tumors is chemoradiotherapy [18,19]. However, PC exhibits chemoresistance due to a complex link between tumor cells and their microenvironment [20]. The tumor microenvironment TME is referred to the local environment where tumors developed [21]. The TME of PC is composed by tumor cells, extracellular matrix and stromal cells (pancreatic stellate cells (PSCs), regulatory T cells (Tregs), tumor-associated macrophages (TAMs), and myeloid derived suppressor cells (MDSCs). All these TME components foster high levels of hypoxia, which confer several metabolic advantages to tumor cells. Some studies have shown how PC microenvironment regulates proliferation, invasion and metastasis, chemoresistance and immune evasion [22,23].
Microenvironment of PC has immunosuppressive characteristics that lead to escape from the immune system, which enhances tumor progression. Cell populations from TME could induce the deposition of extracellular molecules such as matrix metalloproteinases, extracellular matrix molecules, growth factors and transforming growth factor β (TGF-β) to maintain the enabling microenvironment [24]. PSCs are able to produce a collagenous stroma and its role is crucial in both normal pancreas and tumor development. PSCs cooperate with cancer cells to build a perfect niche of tumor development with invasive abilities [25]. While in a normal physiological state, PSCs are quiescent and express nestin, vimentin, GFAP and desmin; activated PSCs become myofibroblast-like cells with overexpression of collagen type I and III, fibronectin and laminin, and present high proliferation and migration abilities [26]. Furthermore, activated PSCs allow pancreatic cancer-related fibrosis [27]. Interestingly, PSCs with an activated phenotype present expression of α-SMA [26], while CD10 positive PSCs have the ability to promote an invasive phenotype in PC [28]. There are subgroups of PSCs that interact with pancreatic β-cells called islet stellate cells. These islets are able to induce β-cell apoptosis, inhibit their proliferation, and diminish β-cell function [29,30]. Indeed, activated PSCs could impair pancreatic islet endocrine function. Since PSCs are glucose intolerant, this cell population is involved in development of type 2 diabetes, which directly promotes invasive phenotype because high levels of glucose are related to epithelial-to-mesenchymal transition [29,31]. PSCs promote also epithelial-to-mesenchymal transition of PC tumor cells [26].
Another TME cell population is Tregs that are a CD4+T cells subpopulation generated from naïve T cells with expression of FOXP3 and CD25.They are present in TME and they are able to suppress autoimmunity in physiological conditions, but also inhibit anti-tumor immune response. This fact could be explained because Tregs can impact tumor-associated CD11c+ dendritic cells to inhibit the activation of CD8+ T cells [32]. Recently, it has been described how Tregs induce differentiation of cancer-associated fibroblasts that promote tumor development [33]. Shevchenko et al. reported that gemcitabine at low doses reduce Tregs population in PC and provide a modest increase in survival [34]. Another study described how neutrophil-lymphocyte ratio and a high proportion of Treg cells in pancreatic tumors are potential pathological biomarkers for poor outcome [35]. On the other hand, TAMs imply a subtype of immune cells that are commonly expressed in several solid tumors, and closely related to cancer-derived inflammation. Specially M2-polarized TAMs (CD163+ cells) are those cells with direct implication in cancer-derived inflammation; in addition, M2-polarized TAMs enable epithelial-to-mesenchymal transition through activation of TLR4/IL-10 signaling and inhibition of E-cadherin [36]. Additionally, TAMs promote angiogenesis, dedifferentiation and stem-cell phenotype in PC [37]. TAMs show several tumor-prone characteristics based on the release of some immunosuppressive and angiogenic cytokines [38]. In PC, TAMs present a high expression of BMP-4, BMP-7, TGF-β1 and TGF-β2 [39]. The infiltration of M2-polarized TAMs in PC stroma seems to be preferentially located in the body and tail of the pancreas, and similarly with other TME cell populations, M2-polarized TAM confers shorter overall survival to patients [40]. It has been reported how metformin could reduce desmoplasia of PC, reversion of epithelial-to-mesenchymal transition, and tumor-related inflammation via modulation of the AMPK/STAT3 that decreases levels of IL-1β and hampers infiltration of M2-polarized TAMs [41]. Indeed, metformin in combination with simvastatin and digoxin is being evaluated in a phase IB trial to target crucial factors for PC development like PDX1 or BIRC5 (NCT03889795) [42]. In PC, M2-polarized TAM population rises significantly after gemcitabine administration, and it has been reported how aspirin could improve the effects of gemcitabine and decrease not only M2-polarized TAMs but also MDSCs populations [43]. MDSCs are a very heterogeneous immature myeloid cell population derived from the myeloid lineage that act as a multipotent progenitor cells. They are involved in development of obesity and several pathologies like autoimmune disease, chronic inflammation and tumorigenesis [44], and they are characterized by the expression of CD11b and Gr-1 surface markers [45]. Sangaletti et al. revealed how downregulation of SPARC decreased immunosuppression and reverted invasive phenotype triggered by the presence of MDSCs [46]. Immunosuppression by MDSCs is also driven by downregulation of JAK3, MHC class II and STAT5 that inhibit activation of T-cells or induce their apoptosis [44]. Another study revealed that the pro-inflammatory microenvironment that promotes MDSCs proliferation and recruitment is produced by a cytokine cascade, which includes the release of IL-1β, IL-6, IL-13, IL-17, TNF-α, TGF-β and VEGF [47]. Similarly, other factors such as ARG-1, COX-2, Cybb, Cytochrome b-245, iNOS2 and PAUF are related to MDSCs activation [48]. MDSCs also impair chemo- and radiotherapy response. In fact, high levels of proinflammatory cytokines associated with MDSCs have been found in previously treated versus untreated patients or healthy samples [49]. Since radiotherapy induces the release of lactate by tumor cells that lead to activation of MDSCs, a proposed treatment strategy would imply target lactate to increase radiotherapy response [50]. Another mechanism described recently against MDSCs is by omega 3 administration due to its anti-inflammatory properties. Indeed, the combination of omega 3 with gemcitabine drastically stabilizes Tregs population and decreases MDSCs in advanced PC [51]. Other treatment approaches, like anti-CXCR2, have been described to counteract MDSCs [52], or an anti-ENO1 that limits the invasion of MDSCs and causes the subsequent disruption of TME interactions with tumor cells, which may improve survival or PC patients [53].
Other studies have proposed the administration of triterpenoid to hamper the activation of MDSCs and their immunosuppressive action in PC, which lead to an increased immune surveillance and immune response [54,55]. Therefore, diminish MDSCs population is a potential target for the treatment of PC and increase its chemoradiosensitivity.
Molecularly, PC is characterized by inactivating mutations in TP53, RB, CDKN1A, CDKN2A, or those associated to heritable PC such as BRCA1 and BRCA2 [56,57]. The effect of tumor suppressor genes could be blinded by overexpression of other oncogenes like MYC, CCNE1 or RAF [58]. Broad genomic analysis has revealed different subtypes of PC. Bailey et al., identified four molecular subtypes of PC associated with specific histopathological characteristics: Squamous, progenitor, immunogenic and aberrantly differentiated endocrine/exocrine (ADEX) [59]. The squamous molecular subtype is associated to squamous differentiation, TP63N activation, inflammation, hypoxia, metabolic reprogramming, dense extracellular matrix, TGF-β and WNT signaling pathway, increased proliferation, activation of MYC, autophagy and enhanced RNA processing. Progenitor and immunogenic subtypes share several characteristics like activation of FOXA2 and FOXA3 networks, xenobiotic metabolism, fatty acid oxidation and mucins metabolism. However, immunogenic subtype also involves the presence of several immune pathways like B cell, CD4 and CD8 T cell signaling, TLR signaling, and other factors associated with antigen presentation. ADEX is characterized by upregulation of NR5A2, RBPJL transcriptional regulation pathway, exocrine differentiation, development of pancreatic β-cell, epithelial cells differentiation and presents mutations in KRAS. Moffitt et al., proposed other molecular subtypes based on analyses of patient prognostic and gene expression profile of tumors (classical or basal-like tumors) and stroma (normal or activated stroma) compared with healthy samples [60]. Collisson et al., also proposed three molecular subtypes by a transcriptional profile analysis: Classical, which overlaps with Moffitt´s classical subtype and exhibit high expression of molecules necessary for cell adhesion and epithelial morphology; quasi-mesenchymal subtype that shows high expression levels of genes associated to mesenchymal morphology; and exocrine-like subtype with overexpression of genes required for digestive enzymes release [61]. These diverse molecular subtypes exhibit several similarities with each other and highlight several opportunities for novel therapeutic strategies and targeted drug design.
TME of PC should be different according to different molecular subtypes and their response to chemotherapy should be different too. In the present article, we review the four molecular subtypes of pancreatic cancer proposed by Bailey et al., and we used them as backbone to supplement their features with other PC molecular subtypes proposed by Moffitt et al., and Collisson et al. Furthermore, we match each molecular subtype to a specific tumor histology and microenvironment, and how each complex network may influence treatment response.
2. Squamous Subtype
Squamous and adenosquamous tumors represent 1–4% of all PCs, are often generated in the head of the pancreas [62], and present the worst survival ratios even detected at early stages [63]. These tumors frequently carry TP53 mutations and present 3p loss compared to adenocarcinomas (Figure 1 and Table 1). Losses in 3p21.1–11.1 imply downregulation in several anti-oncogenic genes like ROBO1, ROBO2, WNT5A, FHIT. The squamous histology also presents upregulation of the WNT/β-Catenin signaling pathway and downregulation of some chromatin modification factors [64]. These tumors are characterized by several cell layers with irregular borders, protruding intercellular junctions, high eosinophilic cytoplasm and keratin deposition, with denotes a rich and dense extracellular matrix (Figure 1 and Table 1) [65].
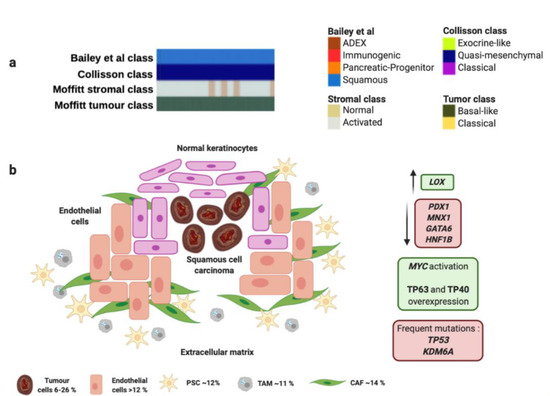
Figure 1.
Squamous molecular profile and microenvironment. (a) Squamous subtype is associated with specific histological characteristics such as squamous and adenosquamous carcinomas. (b) Squamous subtype proposed by Bailey et al., matches perfectly with "quasi-mesenchymal" tumor subtype proposed by Collisson et al., and “basal-like” tumors and “activated” stroma subtypes proposed by Moffitt et al. Green boxes contain upregulated significant factors and activated pathways associated to squamous molecular subtype. Red boxes contain downregulated significant factors (up) and most common mutations found in squamous molecular subtype (down). Percentage of each subcellular population has been obtained from Peng et al., [86]. Abbreviations: TAM: Tumor-associated macrophage; CAF: Cancer-associated fibroblast; PSC: Pancreatic stellate cell.

Table 1.
Molecular features of each pancreatic cancer subtype according to their main subcellular population.
The squamous molecular subtype proposed by Bailey et al., presents MYC activation and overexpression of proteins TP63 and CD7 (TP40) as well as its target factors [59]. This molecular subtype was associated with presence of TP53 and KDM6A mutations [59,66].
Interestingly, overexpression of TP63 in combination with TP53 mutation is able to induce epithelial-to-mesenchymal transition to promote aggressive phenotype [67]. Squamous tumors also express integrins α6β1, α6β4 and EGF. Concerning epigenetic modifications, this subtype exhibits an hypermethylator phenotype that downregulates protein expression of PDX1, MNX1, GATA6 or HNF1B (Figure 1 and Table 1). However, it presents upregulation of LOX, which is associated with metastasis development [68]. Squamous molecular subtype exhibit an hypoxic, inflamed and autophagic microenvironment that correlates with squamous histology [59]. Moreover, Moffitt et al. found a squamous subtype characterized by overexpression associated with several components of the extracellular matrix like laminins and keratins [60]. This matrix composition allows an oncogenic microenvironment that fosters proliferation of PC tumor cells. This dense extracellular matrix generates a physical barrier for drug delivery that leads to a chemoresistant phenotype [69]. This rich extracellular matrix contributes to generation of hypoxic conditions and expression of hypoxia-inducible factor 1 (HIF1) [70], and overexpression of MUC1, VEGF and PDGF that enhance the endothelial tube formation, proliferation and migration ability [71]. The squamous molecular subtype also presents activation of TGF-β signaling pathway [59], and overexpression of EGFR and S100A2 [72]. These findings support the presence and important role of PSCs in the development of squamous tumor microenvironment since cytokines TGF-β, PDGF, Angiotensin II could bind to the PSCs receptors and activate downstream signaling pathways such as ERK, c-Jun, P38, MAPK or JAK-STAT to promote the activation and proliferation of PSCs. Moreover, activated PSCs can produce and secrete to stroma a variety of growth factors through paracrine mechanism, to activate EGFR, PI3K-AKT, and mTOR signaling pathways, and then promote proliferation and inhibition of apoptosis of PC cells [73,74]. One study has reported that squamous PC cells are able to recruit neutrophils that convert PSCs into tumor-associated fibroblasts (TAFs) that overexpress inflammatory cytokines like IL1A and CXCL1 [75]. TAFs also contribute to generate a rich tumor prone extracellular matrix by secretion of fibroblast activating protein (FAP) that participate in angiogenesis and affect the activity of IFN-γ and TNF-α [76].
On the other hand, the squamous molecular subtype proposed by Bailey et al. linked with “basal-like” and “activated” stroma signatures proposed by Moffitt et al. (Figure 1 and Table 1). The “basal-like” and “activated” stroma signatures also exhibited the worst survivals compared to “classical” and “normal” stroma subgroups, which goes in accordance with both squamous molecular subtype of Bailey et al., and the squamous histology of PC [60]. “Basal-like” subtype presents KRASG12D mutation, while “activated” stroma subtype expresses SPARC, WNT family members WNT2 and WNT5A, gelatinase B (MMP9), stromelysin 3 (MMP11), and FAP that contribute to tumor development [60]. Furthermore, “activated” stroma subgroup is characterized by other factors like the chemokine ligands CCL13 and CCL18, and integrin ITGAM, which are associated with the presence of macrophages [60]. This fact suggests the important role of macrophages in the tumor microenvironment to promote angiogenesis and chemoresistance [77]. In this sense, it has been described how TAMs enable chemoresistance to gemcitabine and nab-paclitaxel by expression of insulin growth factor (IGF) that activates IGF-1 receptors [78]. It has been reported that PSCs also promote gemcitabine resistance mediated by secretion of deoxycytidine that modulate nucleoside transporters [79]. Moreover, PSCs reduce the sensitivity of PC cells to radiotherapy by regulation of epithelial-to-mesenchymal transition and increasing cancer stem cell markers [80]. Furthermore, these authors provide a rationale for the use of an anti-TGF-β neutralizing antibody to inhibit epithelial-to-mesenchymal transition and cancer stem cells to sensitize cells to radiation and reduce tumors [80]. Other cell population like tumor associated fibroblasts can secrete proinflammatory factors like IL-6, which activates STAT3, and secrete CXCL12 that inhibit T-cells infiltration in tumors [81]. In addition, CXCL12 in combination with anti-PD-L1 immunotherapy has shown a synergic anti-tumor effect and is considered a potential target for those PCs with high FAP expression [82].
Unfortunately, Bailey et al., support that squamous molecular subtype exhibits the highest expression of PD-L1, PD-L2 and IDO1 [59], thus, they sustain that these tumors are not good candidates for immunotherapy administration [83,84].
However, the combination of IDO1 inhibitor and anti-PD-1/PD-L1 treatment could achieve promising results and become a potential strategy to induce intratumoral T-cell infiltration [85]. In the light of the foregoing, the microenvironment of the squamous molecular subtype presents a complex interaction between different cell populations and extracellular components to confer chemoresistance of PC.
3. Progenitor and Immunogenic Subtypes
The progenitor molecular subtype proposed by Bailey et al. presents activation on transcriptional networks that exhibit high expression of the proteins HES1, HNF1A, HNF1B, HNF4A, HNF4G, FOXA2, FOXA3, LAMA1, MNX1 and EP300 (Figure 2 and Table 1) [59]. These transcription factors are considered crucial for determination and development of the pancreatic endoderm. While the squamous subtype presented downregulation of PDX1, which is also critical for pancreatic development, the progenitor subtype overexpress PDX1 (Figure 2 and Table 1) [88]. Moreover, progenitor molecular subtype expresses multiple genes necessary for the metabolism of mucins, fatty acids, steroid hormones, and also involves drug metabolism. Indeed, progenitor molecular subtype overexpresses MUC5AC and MUC1 (Figure 2 and Table 1) [59]. Interestingly, both progenitor and immunogenic molecular subtypes share the same histology derived from premalignant lesions like mucinous non-cystic adenocarcinomas and intraductal papillary mucinous neoplasms (IPMN), characterized by a high mucin production (Figure 2 and Table 1) [106], different epithelial-to-mesenchymal transition markers [107], and immunohistochemical staining of CDX2 [89,90].
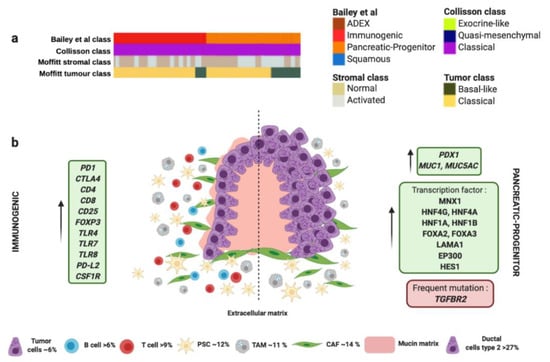
Figure 2.
Immunogenic and progenitor molecular profiles and their microenvironment. (a) Both subtypes proposed by Bailey et al., match perfectly with “classical” tumor subtype proposed by Collisson et al., and “classical” tumor subtype proposed by Moffitt et al. (b) Both subtypes share the same histology derived from premalignant lesions like mucinous non-cystic adenocarcinomas and intraductal papillary mucinous neoplasms (IPMN) characterized by high mucin production. Dotted line separates schematic representation of two subtypes. Green boxes contain upregulated significant factors associated to each subtype. Red box contains the most characteristic gene mutation in progenitor molecular subtype. Pink color represents mucin production. Percentage of each subcellular population has been obtained from Peng et al., [86]. Abbreviations: TAM: tumor-associated macrophage; CAF: cancer-associated fibroblast; PSC: pancreatic stellate cell.4. Microenvironment of ADEX Subtype.
The immunogenic molecular subtype also presents many other similarities in the gene expression profile with the progenitor subtype; however, immunogenic subtype exhibit a significantly higher immune infiltrate [59]. Gene expression analysis identified upregulation in those genes associated with nine different populations of immune cells [108], being the most significant those involved with B and T cells infiltration (CD4, CD8, CD25, FOXP3) (Figure 2 and Table 1). Furthermore, immunogenic subtype presents activation of CTLA4 and PD1, which suggests that this molecular subtype could be feasible treated with immunecheckpoints inhibitors [59]. In contrast, the pancreatic progenitor subtype presents the lowest levels of CTLA4, PD-L1, PD-L2, and IDO1 [59]. The immunogenic subtype also has enrichment in genes for Toll-like receptors that play a key role in the innate immune system. These genes include TLR4, TLR7, TLR8, PD-L2 and CSF1R (Figure 2 and Table 1) [59]. These receptors usually are expressed on sentinel cells such as macrophages and dendritic cells, which reveal the strong link between the immunogenic molecular subtype with the presence of immune cell population in the TME (Figure 2 and Table 1). In this sense, TAMs present in PC, especially those M2-type macrophages can promote neovasculature formation to feed tumor cells and induce chemoresistance, proliferation, invasion and metastasis of PC tumor cells [77]. TAMs could be recruited to neoplasic foci by cytokines and vascular endothelial growth factors (VEGF). Once there, TAMs promote proliferation of PC cells through the release of growth factors like Il10 or TGF-β [97]. In addition, the presence of TAMs in the TME is associated with chemoresistance [78].
On the other hand, the immunogenic molecular subtype of PC present inflammation, caused by abundant Tregs infiltration and high expression of cytotoxic T lymphocyte-associated protein 4 (CTLA-4). These factors promote tumor immunosuppression through the inhibition of the killing ability of effector cells [95]. The presence of CD8+ T cells in immunogenic subtype can decrease the capability of anti-tumor immune cells due to the ability of CD8+T cells to express FAS-ligand and secrete perforin and granzymes with cytolytic effect [96]. Despite the immunosuppressive skills of immunogenic subtype, patients with this kind of tumor present the best outcome out of the four molecular subtypes [59].
Interestingly, both progenitor and immunogenic subtypes correlate with “classical” tumor subtype proposed by Moffitt et al., which also share the expression of extracellular mucin in more than 10% (Figure 2 and Table 1) [60]. Similarly to immunogenic molecular subtype, “classical” tumors present better outcome compared to those “basal-like” tumors [60]. Remarkably, progenitor and immunogenic molecular subtypes by Bailey et al. and “classical” tumor subtype by Moffitt et al., correlate with the same gene expression signature of “classical” tumor subtype proposed by Collisson et al. (Figure 2 and Table 1) [109]. This fact supports the presence of the G12V variant of KRAS mutation that is associated with the “classical” subtype [60]. “Classical” molecular subtype also presents enrichment in genes associated with GATA6 overexpression responsible for promoting epithelial cell differentiation [60]. However, other authors propose the loss of GATA6 expression in tumors confers a shorter survival and they have a poor response to adjuvant treatment based on 5-fluorouracil (5-FU)/leucovorin [91]. In this sense, “classical” molecular subtype tumors also exhibit poor response to platinum based therapy [60]. Therefore, the adjuvant treatment used for the management of immunogenic/progenitor/classical molecular subtype tumors would be more aggressive to overcome the chemoresistance. For this, immunotherapy has arisen as one of the most effective anti-tumor treatments. PC overexpress important factors for targeted therapies, such as MUC-1, CEA, PSC, VEGF, MSLN and mutation KRAS [110]. However, the most important targets for immunotherapy currently are PD-1, PD-L1 and CTLA-4. In animal models, anti-PD-1 or anti-PD-L1 treatment boosted infiltration of CD8+ T cells in the tumor and enhanced efficacy of immunocheckpoint inhibitors.
Regretfully, immunotherapy against PC has presented no benefit in clinical trials so far [111,112]. Currently, the failure of anti-PD-1/anti-PD-L1 based therapy is mainly attributed to the lack of T-cell infiltration in the TME of PC. Therefore, the immunogenic molecular subtype that presents elevated genes for B- and T-cell infiltration is a potential responder for immunotherapy. Other type of immunotherapy based on CAR-T cells has obtained promising results. Some clinical trials have focused on modified CAR-T cells able to target MUC1 or Mesothelin (MSLN) [113]. Although MUC1 is overexpressed in approximately 85% of PC, those tumors present overexpression of IDO1, COX1/2, and Gal-9 that conferred resistance to these anti-MUC1 CAR-T cells [114]. However, the use of inhibitors of IDO1, COX1/2, and Gal-9 overcame this resistance in combination with anti-MUC1 CAR-T cells [114]. In this concern, a phase I/II trial has been designed with resectable PC patients´ T cells modified to allow identification and kill of MUC1 positive tumor cells (NCT02587689). CAR-T cells could be engineered to target other antigens such as Mesothelin, and they have achieved encouraging results in solid malignancies [115], which has established the bases for a subsequent clinical trial with anti-MSLN CAR-T cells (NCT04203459). Although CAR-T cell therapy has achieved favorable results, it is crucial to evaluate the potential side effects on T-cell exhaustion.
The ADEX molecular subtype by Bailey et al., is defined by the orchestrated expression of factors necessary for the exocrine and endocrine pancreatic cell-fate determination. ADEX presents upregulation in transcription factors involved in healing and regeneration after pancreatitis events such as RBPJL, NR5A2, MIST1 and their associated downstream cascade factors (Figure 3 and Table 1) [100,101]. This molecular subtype exhibits an expression profile associated with extremely rare acinar histopathology (Figure 3 and Table 1) [59,63]. Macroscopically, this histology shows large tumors, frequently presented in encapsulated form and well circumscribed. It also presents cystic evolution, necrosis, and upper digestive hemorrhage [116]. In clinical practice, diagnosis of acinar carcinomas is performed by immunohistochemical evaluation of pancreatic enzymes like trypsin and chymotrypsin that are produced in the rough endoplasmic reticulum of acinar cells. It also presents high specificity for BCL-10 immunostaining [65]. Furthermore, somatic mutational index of acinar subtype is higher than adenocarcinoma being the most frequent mutations in the following genes: FAT1, FAT3, and FAT4 (57%), BRCA2 (42%), SMAD4 (26%), JAK1 (17%), RB1 (13%), TP53 (13%), CTNNB1 (11%), APC (9%), ARID1A (9%), GNAS (9%), MLL3 (9%), PTEN (9%), RNF43 (4%) and MEN1 (4%); however, KRAS mutations are not often observed in acinar subtype (Figure 3 and Table 1) [98,99]. In this sense, Bailey et al. found in ADEX molecular subtype high expression levels of INS, NEUROD1, NKX2-2 and MAFA, which are associated with endocrine differentiation and sudden occurrence of diabetes type MODY (Figure 3 and Table 1).
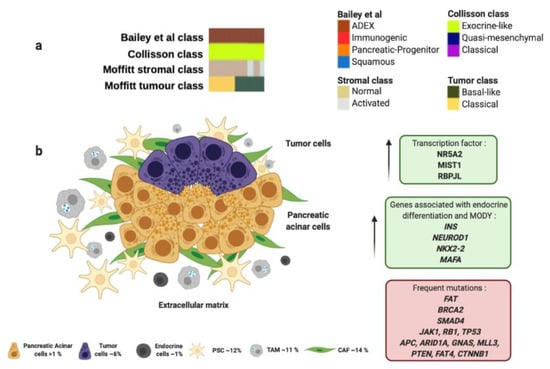
Figure 3.
ADEX molecular profile and microenvironment. (a) ADEX subtype proposed by Bailey et al., matches perfectly with “exocrine-like” subtype proposed by Collisson et al., and most of “normal stroma” subtype cases analyzed by Moffitt et al. (b) ADEX molecular subtype is associated with the rare acinar histology. Green boxes contain upregulated transcription factors and other genes involved in ADEX molecular subtype. Red box contains genes commonly mutated in ADEX. Percentage of each subcellular population has been obtained from Peng et al., [86]. Abbreviations: TAM: tumor-associated macrophage; CAF: cancer-associated fibroblast; PSC: pancreatic stellate cell; MODY: maturity onset diabetes of the young.
Importantly, most PC-derived cell lines present enrichment in genes found in ADEX molecular subtype like AMY2B, CEL, INS, PRSS1 and PRSS3 [59].
ADEX molecular subtype proposed by Bailey et al., matches perfectly with “exocrine-like” subtype proposed by Collisson et al., which shows relatively high expression of tumor cell-derived digestive enzymes like ELA3A and CFTR, which controls Cl− and Na+ ions, and water flux (Figure 3 and Table 1) [61]. In addition, ADEX molecular subtype correlates with “normal stroma” subtype proposed by Moffitt et al., which exhibit longer survival rates (Figure 3 and Table 1) [60]. “Normal stroma” is also distinguished by high expression levels of PSCs markers: Smooth muscle actin (ACTA2), Desmin (DES) and Vimentin (VIM) [60,102]. This fact highlights the importance of the PSCs population in the ADEX microenvironment. PSCs role go in accordance with ADEX origin, since they present expression markers of stem and progenitor cells needed to maintain differentiation and regeneration of pancreas [117]. Indeed, PSCs are necessary to trigger desmoplastic reaction by the synthesize of large amounts of extracellular matrix proteins, such as collagens, and amplify endostatin production of cancer cells that contributes to an hypoxic milieu [103,118]. Moverover, PSCs produce VEGF to increase endothelial cell growth in the peritumoral stroma [103]. All the above supports the role of PSCs in induction of fibrosis [119], that impairs drug delivery, stimulates epithelial-to-mesenchymal transition, and increases genetic instability leading to a more chemoresistant tumor [104]. Furthermore, PSCs play a crucial role in islet cell dysfunction since PSCs decrease insulin expression and an increased apoptosis of pancreatic β-cells that connects to initiation of diabetes [105]. One study supported that PC cells cultured with extracellular matrix proteins produced by PSCs were able to develop resistance to 5-FU, cisplatin and doxorubicin [120]. As above mentioned, the presence of PSCs in ADEX molecular subtype could imply resistance to gemcitabine [79] and a reduced sensitivity to radiotherapy [80].
4. Conclusions and Future Perspectives
The match between molecular features at genomic, epigenomic and expression levels combined with the biological behavior of PC is bringing new therapeutic strategies for the clinical management of these patients. In addition, the study of the TME could explain chemoresistance of PC previously attributed only to tumor cells. The TME and especially the role of its immune cell population in tumor initiation, development and immune evasion is changing the paradigm of PC diagnosis and treatment approach with the identification of new molecular subtypes (Table 2).
Table 2.
On-going clinical trials that include different subtypes of pancreatic cancer.
Although treatment strategies based on immunocheckpoint inhibitors have shown disappointing results, new studies are ongoing with novel drug design to improve patient response, and to avoid tumor immune evasion. In this concern, a randomized clinical trial (NCT02030860) aimed to evaluate paricalcitol that target vitamin D metabolism in the TME in combination with gemcitabine plus nab-paclitaxel in the neoadjuvant scenario for resectable PC patients. Another phase I/II clinical trial is designed to evaluate the safety and tolerability of the microenvironment modifier drug, L-DOS47, in combination with doxorubicin. The L-DOS47 is reported to neutralize the acid extracellular matrix that protects the tumor (NCT04203641) [121]. Recently, other treatments like GVAX or CRS-207 have appeared with promising results against PC [122]. GVAX is a vaccine based on granulocyte-macrophage colony-stimulating factor that inhibits Tregs and induces T cells; while CRS-207 is a vaccine based on live-attenuated Listeria monocytogenes–expressing mesothelin that has been described to induce innate and adaptive immune response against tumor cells and has achieved longer survivals of PC patients (NCT03161379; NCT02451982) [122]. Other drugs are based on nucleic acid, this is the case of olaptesed pegol a L-RNA Spiegelmer that exhibits a high affinity against CXCL12, a factor previously described responsible for TAMs recruitment (NCT03168139) [123]. These new treatment approaches highlight the importance of targeting not only tumor cells but also TME. Although translational research and drug design are working synergistically to improve outcome of PC patients, clinical research must not ignore that patients might be stratified not only based on performance status of patients and/or the molecular characteristics of tumors, but also according to their TME features to obtain better treatment responses and longer survivals.
Author Contributions
Conceptualization, J.M.-U. and J.G.-F. writing—original draft preparation, J.M.-U. figures and tables, M.M.-G.; writing—review and editing, M.M.-G. and J.M.-U. visualization, M.M.-G. supervision, J.G.-F. funding acquisition, J.G.-F. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Acknowledgments
We especially thank oncologist Aberto Orta-Ruiz (MD, PhD) from the Medical Oncology Department (Fundacion Jimenez Diaz University Hospital) for his appreciated revision, suggestions and criticism for the present review article. All figures have been designed with BioRender.com.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Rahib, L.; Smith, B.D.; Aizenberg, R.; Rosenzweig, A.B.; Fleshman, J.M.; Matrisian, L.M. Projecting Cancer Incidence and Deaths to 2030: The Unexpected Burden of Thyroid, Liver, and Pancreas Cancers in the United States. Cancer Res. 2014, 74, 2913–2921. [Google Scholar] [CrossRef] [PubMed]
- Tamm, E.P.; Bhosale, P.R.; Vikram, R.; de Almeida Marcal, L.P.; Balachandran, A. Imaging of Pancreatic Ductal Adenocarcinoma: State of the Art. World J. Radiol. 2013, 5, 98–105. [Google Scholar] [CrossRef] [PubMed]
- Kelsen, D.P.; Portenoy, R.; Thaler, H.; Tao, Y.; Brennan, M. Pain as a Predictor of Outcome in Patients with Operable Pancreatic Carcinoma. Surgery 1997, 122, 53–59. [Google Scholar] [CrossRef]
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer Statistics, 2019. CA Cancer J. Clin. 2019, 69, 7–34. [Google Scholar] [CrossRef] [PubMed]
- Duffy, M.J.; Sturgeon, C.; Lamerz, R.; Haglund, C.; Holubec, V.L.; Klapdor, R.; Nicolini, A.; Topolcan, O.; Heinemann, V. Tumor Markers in Pancreatic Cancer: A European Group on Tumor Markers (EGTM) Status Report. Ann. Oncol. 2010, 21, 441–447. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.-E.; Lee, K.T.; Lee, J.K.; Paik, S.W.; Rhee, J.C.; Choi, K.W. Clinical Usefulness of Carbohydrate Antigen 19-9 as a Screening Test for Pancreatic Cancer in an Asymptomatic Population. J. Gastroenterol. Hepatol. 2004, 19, 182–186. [Google Scholar] [CrossRef]
- Ferri, M.J.; Saez, M.; Figueras, J.; Fort, E.; Sabat, M.; López-Ben, S.; de Llorens, R.; Aleixandre, R.N.; Peracaula, R. Improved Pancreatic Adenocarcinoma Diagnosis in Jaundiced and Non-Jaundiced Pancreatic Adenocarcinoma Patients through the Combination of Routine Clinical Markers Associated to Pancreatic Adenocarcinoma Pathophysiology. PLoS ONE 2016, 11, e0147214. [Google Scholar] [CrossRef]
- Blackford, A.; Parmigiani, G.; Kensler, T.W.; Wolfgang, C.; Jones, S.; Zhang, X.; Parsons, D.W.; Lin, J.C.-H.; Leary, R.J.; Eshleman, J.R.; et al. Genetic Mutations Associated with Cigarette Smoking in Pancreatic Cancer. Cancer Res. 2009, 69, 3681–3688. [Google Scholar] [CrossRef]
- Bosetti, C.; Lucenteforte, E.; Silverman, D.T.; Petersen, G.; Bracci, P.M.; Ji, B.T.; Negri, E.; Li, D.; Risch, H.A.; Olson, S.H.; et al. Cigarette Smoking and Pancreatic Cancer: An Analysis from the International Pancreatic Cancer Case-Control Consortium (Panc4). Ann. Oncol. 2012, 23, 1880–1888. [Google Scholar] [CrossRef]
- Lowenfels, A.B.; Maisonneuve, P.; DiMagno, E.P.; Elitsur, Y.; Gates, L.K.; Perrault, J.; Whitcomb, D.C. Hereditary Pancreatitis and the Risk of Pancreatic Cancer. International Hereditary Pancreatitis Study Group. J. Natl. Cancer Inst. 1997, 89, 442–446. [Google Scholar] [CrossRef]
- Chari, S.T.; Leibson, C.L.; Rabe, K.G.; Timmons, L.J.; Ransom, J.; de Andrade, M.; Petersen, G.M. Pancreatic Cancer-Associated Diabetes Mellitus: Prevalence and Temporal Association with Diagnosis of Cancer. Gastroenterology 2008, 134, 95–101. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.-Z.; Wang, R.; Chen, H.-N.; Hu, J.-K. Cytotoxin-Associated Gene A-Negative Strains of Helicobacter Pylori as a Potential Risk Factor of Pancreatic Cancer: A Meta-Analysis Based on Nested Case-Control Studies. Pancreas 2015, 44, 1340–1344. [Google Scholar] [CrossRef] [PubMed]
- Neoptolemos, J.P.; Urrutia, R.; Abbruzzese, J.L.; Büchler, M.W. Pancreatic Cancer; Springer: Berlin/Heidelberg, Germany, 2018; ISBN 9781493971930. [Google Scholar]
- Oettle, H.; Post, S.; Neuhaus, P.; Gellert, K.; Langrehr, J.; Ridwelski, K.; Schramm, H.; Fahlke, J.; Zuelke, C.; Burkart, C.; et al. Adjuvant Chemotherapy with Gemcitabine vs. Observation in Patients Undergoing Curative-Intent Resection of Pancreatic Cancer: A Randomized Controlled Trial. J. Am. Med. Assoc. 2007, 297, 267–277. [Google Scholar] [CrossRef] [PubMed]
- Neoptolemos, J.P.; Stocken, D.D.; Bassi, C.; Ghaneh, P.; Cunningham, D.; Goldstein, D.; Padbury, R.; Moore, M.J.; Gallinger, S.; Mariette, C.; et al. Adjuvant Chemotherapy with Fluorouracil plus Folinic Acid vs. Gemcitabine Following Pancreatic Cancer Resection: A Randomized Controlled Trial. JAMA J. Am. Med. Assoc. 2010, 304, 1073–1081. [Google Scholar] [CrossRef] [PubMed]
- Neoptolemos, J.P.; Palmer, D.; Ghaneh, P.; Valle, J.W.; Cunningham, D.; Wadsley, J.; Meyer, T.; Anthoney, A.; Glimelius, B.; Falk, S.; et al. ESPAC-4: A Multicenter, International, Open-Label Randomized Controlled Phase III Trial of Adjuvant Combination Chemotherapy of Gemcitabine (GEM) and Capecitabine (CAP) versus Monotherapy Gemcitabine in Patients with Resected Pancreatic Ductal Adenocarcin. J. Clin. Oncol. 2016, 34. [Google Scholar] [CrossRef]
- Vera, R.; Dotor, E.; Feliu, J.; González, E.; Laquente, B.; Macarulla, T.; Martínez, E.; Maurel, J.; Salgado, M.; Manzano, J.L. SEOM Clinical Guideline for the Treatment of Pancreatic Cancer. Clin. Transl. Oncol. 2016, 18, 1172–1178. [Google Scholar] [CrossRef]
- Mukherjee, S.; Hurt, C.N.; Bridgewater, J.; Falk, S.; Cummins, S.; Wasan, H.; Crosby, T.; Jephcott, C.; Roy, R.; Radhakrishna, G.; et al. Gemcitabine-Based or Capecitabine-Based Chemoradiotherapy for Locally Advanced Pancreatic Cancer (SCALOP): A Multicentre, Randomised, Phase 2 Trial. Lancet Oncol. 2013, 14, 317–326. [Google Scholar] [CrossRef]
- Hammel, P.; Huguet, F.; Van Laethem, J.-L.; Goldstein, D.; Glimelius, B.; Artru, P.; Borbath, I.; Bouche, O.; Shannon, J.; André, T.; et al. Comparison of Chemoradiotherapy (CRT) and Chemotherapy (CT) in Patients with a Locally Advanced Pancreatic Cancer (LAPC) Controlled after 4 Months of Gemcitabine with or without Erlotinib: Final Results of the International Phase III LAP 07 Study. J. Clin. Oncol. 2013, 31. [Google Scholar] [CrossRef]
- Zeng, S.; Pöttler, M.; Lan, B.; Grützmann, R.; Pilarsky, C.; Yang, H. Chemoresistance in Pancreatic Cancer. Int. J. Mol. Sci. 2019, 20, 4504. [Google Scholar] [CrossRef]
- Ioannides, C.G.; Whiteside, T.L. T Cell Recognition of Human Tumors: Implications for Molecular Immunotherapy of Cancer. Clin. Immunol. Immunopathol. 1993, 66, 91–106. [Google Scholar] [CrossRef]
- Apte, M.V.; Xu, Z.; Pothula, S.; Goldstein, D.; Pirola, R.C.; Wilson, J.S. Pancreatic Cancer: The Microenvironment Needs Attention Too! Pancreatology 2015, 15, S32–S38. [Google Scholar] [CrossRef] [PubMed]
- Chang, J.H.; Jiang, Y.; Pillarisetty, V.G. Role of Immune Cells in Pancreatic Cancer from Bench to Clinical Application: An Updated Review. Medicine 2016, 95, e5541. [Google Scholar] [CrossRef] [PubMed]
- Roberts, N.J.; Norris, A.L.; Petersen, G.M.; Bondy, M.L.; Brand, R.; Gallinger, S.; Kurtz, R.C.; Olson, S.H.; Rustgi, A.K.; Schwartz, A.G.; et al. Whole Genome Sequencing Defines the Genetic Heterogeneity of Familial Pancreatic Cancer. Cancer Discov. 2016, 6, 166–175. [Google Scholar] [CrossRef] [PubMed]
- Pothula, S.P.; Pirola, R.C.; Wilson, J.S.; Apte, M.V. Pancreatic Stellate Cells: Aiding and Abetting Pancreatic Cancer Progression. Pancreatology 2020, 20, 409–418. [Google Scholar] [CrossRef] [PubMed]
- Xue, R.; Jia, K.; Wang, J.; Yang, L.; Wang, Y.; Gao, L.; Hao, J. A Rising Star in Pancreatic Diseases: Pancreatic Stellate Cells. Front. Physiol. 2018, 9, 754. [Google Scholar] [CrossRef]
- Che, M.; Kweon, S.-M.; Teo, J.-L.; Yuan, Y.-C.; Melstrom, L.G.; Waldron, R.T.; Lugea, A.; Urrutia, R.A.; Pandol, S.J.; Lai, K.K.Y. Targeting the CBP/β-Catenin Interaction to Suppress Activation of Cancer-Promoting Pancreatic Stellate Cells. Cancers 2020, 12, 1476. [Google Scholar] [CrossRef]
- Ikenaga, N.; Ohuchida, K.; Mizumoto, K.; Cui, L.; Kayashima, T.; Morimatsu, K.; Moriyama, T.; Nakata, K.; Fujita, H.; Tanaka, M. CD10+ Pancreatic Stellate Cells Enhance the Progression of Pancreatic Cancer. Gastroenterology 2010, 139, 1041–1051. [Google Scholar] [CrossRef]
- Zha, M.; Xu, W.; Zhai, Q.; Li, F.; Chen, B.; Sun, Z. High Glucose Aggravates the Detrimental Effects of Pancreatic Stellate Cells on Beta-Cell Function. Int. J. Endocrinol. 2014, 2014, 165612. [Google Scholar] [CrossRef]
- Li, F.-F.; Chen, B.-J.; Li, W.; Li, L.; Zha, M.; Zhou, S.; Bachem, M.G.; Sun, Z.-L. Islet Stellate Cells Isolated from Fibrotic Islet of Goto-Kakizaki Rats Affect Biological Behavior of Beta-Cell. J. Diabetes Res. 2016, 2016, 6924593. [Google Scholar] [CrossRef]
- Lee, E.; Ryu, G.R.; Ko, S.-H.; Ahn, Y.-B.; Song, K.-H. A Role of Pancreatic Stellate Cells in Islet Fibrosis and β-Cell Dysfunction in Type 2 Diabetes Mellitus. Biochem. Biophys. Res. Commun. 2017, 485, 328–334. [Google Scholar] [CrossRef]
- Jang, J.-E.; Hajdu, C.H.; Liot, C.; Miller, G.; Dustin, M.L.; Bar-Sagi, D. Crosstalk between Regulatory T Cells and Tumor-Associated Dendritic Cells Negates Anti-Tumor Immunity in Pancreatic Cancer. Cell Rep. 2017, 20, 558–571. [Google Scholar] [CrossRef] [PubMed]
- Aykut, B.; Chen, R.; Miller, G. Regulatory T Cells Keep Pancreatic Cancer at Bay. Cancer Discov. 2020, 10, 345–347. [Google Scholar] [CrossRef] [PubMed]
- Shevchenko, I.; Karakhanova, S.; Soltek, S.; Link, J.; Bayry, J.; Werner, J.; Umansky, V.; Bazhin, A.V. Low-Dose Gemcitabine Depletes Regulatory T Cells and Improves Survival in the Orthotopic Panc02 Model of Pancreatic Cancer. Int. J. Cancer 2013, 133, 98–107. [Google Scholar] [CrossRef] [PubMed]
- Cheng, H.; Luo, G.; Lu, Y.; Jin, K.; Guo, M.; Xu, J.; Long, J.; Liu, L.; Yu, X.; Liu, C. The Combination of Systemic Inflammation-Based Marker NLR and Circulating Regulatory T Cells Predicts the Prognosis of Resectable Pancreatic Cancer Patients. Pancreatology 2016, 16, 1080–1084. [Google Scholar] [CrossRef]
- Liu, C.-Y.; Xu, J.-Y.; Shi, X.-Y.; Huang, W.; Ruan, T.-Y.; Xie, P.; Ding, J.-L. M2-Polarized Tumor-Associated Macrophages Promoted Epithelial-Mesenchymal Transition in Pancreatic Cancer Cells, Partially through TLR4/IL-10 Signaling Pathway. Lab. Invest. 2013, 93, 844–854. [Google Scholar] [CrossRef]
- Meng, F.; Li, C.; Li, W.; Gao, Z.; Guo, K.; Song, S. Interaction between Pancreatic Cancer Cells and Tumor-Associated Macrophages Promotes the Invasion of Pancreatic Cancer Cells and the Differentiation and Migration of Macrophages. IUBMB Life 2014, 66, 835–846. [Google Scholar] [CrossRef]
- Malmberg, K.-J. Effective Immunotherapy against Cancer: A Question of Overcoming Immune Suppression and Immune Escape? Cancer Immunol. Immunother. 2004, 53, 879–892. [Google Scholar] [CrossRef]
- Shen, Z.; Seppänen, H.; Kauttu, T.; Vainionpää, S.; Ye, Y.; Wang, S.; Mustonen, H.; Puolakkainen, P. Vasohibin-1 Expression Is Regulated by Transforming Growth Factor-β/Bone Morphogenic Protein Signaling Pathway Between Tumor-Associated Macrophages and Pancreatic Cancer Cells. J. Interferon. Cytokine Res. 2013, 33, 428–433. [Google Scholar] [CrossRef]
- Hu, H.; Hang, J.-J.; Han, T.; Zhuo, M.; Jiao, F.; Wang, L.-W. The M2 Phenotype of Tumor-Associated Macrophages in the Stroma Confers a Poor Prognosis in Pancreatic Cancer. Tumour Biol. 2016, 37, 8657–8664. [Google Scholar] [CrossRef]
- Incio, J.; Suboj, P.; Chin, S.M.; Vardam-Kaur, T.; Liu, H.; Hato, T.; Babykutty, S.; Chen, I.; Deshpande, V.; Jain, R.K.; et al. Metformin Reduces Desmoplasia in Pancreatic Cancer by Reprogramming Stellate Cells and Tumor-Associated Macrophages. PLoS ONE 2015, 10, e0141392. [Google Scholar] [CrossRef]
- Liu, S.-H.; Yu, J.; Creeden, J.F.; Sutton, J.M.; Markowiak, S.; Sanchez, R.; Nemunaitis, J.; Kalinoski, A.; Zhang, J.-T.; Damoiseaux, R.; et al. Repurposing Metformin, Simvastatin and Digoxin as a Combination for Targeted Therapy for Pancreatic Ductal Adenocarcinoma. Cancer Lett. 2020, 491, 97–107. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Li, Y.; Niu, Z.; Zong, Y.; Wang, M.; Yao, L.; Lu, Z.; Liao, Q.; Zhao, Y. Atorvastatin (Lipitor) Attenuates the Effects of Aspirin on Pancreatic Cancerogenesis and the Chemotherapeutic Efficacy of Gemcitabine on Pancreatic Cancer by Promoting M2 Polarized Tumor Associated Macrophages. J. Exp. Clin. Cancer Res. 2016, 35, 33. [Google Scholar] [CrossRef] [PubMed]
- Gabrilovich, D.I.; Nagaraj, S. Myeloid-Derived Suppressor Cells as Regulators of the Immune System. Nat. Rev. Immunol. 2009, 9, 162–174. [Google Scholar] [CrossRef] [PubMed]
- Talmadge, J.E.; Gabrilovich, D.I. History of Myeloid-Derived Suppressor Cells. Nat. Rev. Cancer 2013, 13, 739–752. [Google Scholar] [CrossRef] [PubMed]
- Sangaletti, S.; Talarico, G.; Chiodoni, C.; Cappetti, B.; Botti, L.; Portararo, P.; Gulino, A.; Consonni, F.M.; Sica, A.; Randon, G.; et al. SPARC Is a New Myeloid-Derived Suppressor Cell Marker Licensing Suppressive Activities. Front. Immunol. 2019, 10. [Google Scholar] [CrossRef] [PubMed]
- Karakhanova, S.; Link, J.; Heinrich, M.; Shevchenko, I.; Yang, Y.; Hassenpflug, M.; Bunge, H.; von Ahn, K.; Brecht, R.; Mathes, A.; et al. Characterization of Myeloid Leukocytes and Soluble Mediators in Pancreatic Cancer: Importance of Myeloid-Derived Suppressor Cells. Oncoimmunology 2015, 4, e998519. [Google Scholar] [CrossRef] [PubMed]
- Song, J.; Lee, J.; Kim, J.; Jo, S.; Kim, Y.J.; Baek, J.E.; Kwon, E.-S.; Lee, K.-P.; Yang, S.; Kwon, K.-S.; et al. Pancreatic Adenocarcinoma Up-Regulated Factor (PAUF) Enhances the Accumulation and Functional Activity of Myeloid-Derived Suppressor Cells (MDSCs) in Pancreatic Cancer. Oncotarget 2016, 7, 51840–51853. [Google Scholar] [CrossRef]
- Markowitz, J.; Brooks, T.R.; Duggan, M.C.; Paul, B.K.; Pan, X.; Wei, L.; Abrams, Z.; Luedke, E.; Lesinski, G.B.; Mundy-Bosse, B.; et al. Patients with Pancreatic Adenocarcinoma Exhibit Elevated Levels of Myeloid-Derived Suppressor Cells upon Progression of Disease. Cancer Immunol. Immunother. 2015, 64, 149–159. [Google Scholar] [CrossRef]
- Yang, X.; Lu, Y.; Hang, J.; Zhang, J.; Zhang, T.; Huo, Y.; Liu, J.; Lai, S.; Luo, D.; Wang, L.; et al. Lactate-Modulated Immunosuppression of Myeloid-Derived Suppressor Cells Contributes to the Radioresistance of Pancreatic Cancer. Cancer Immunol. Res. 2020, 8, 1440–1451. [Google Scholar] [CrossRef]
- Isherwood, J.; Arshad, A.; Chung, W.Y.; Runau, F.; Cooke, J.; Pollard, C.; Howells, L.; Fishwick, J.; Thompson, J.; Metcalfe, M.; et al. Myeloid Derived Suppressor Cells Are Reduced and T Regulatory Cells Stabilised in Patients with Advanced Pancreatic Cancer Treated with Gemcitabine and Intravenous Omega 3. Ann. Transl. Med. 2020, 8, 172. [Google Scholar] [CrossRef]
- Ijichi, H.; Chytil, A.; Gorska, A.E.; Aakre, M.E.; Bierie, B.; Tada, M.; Mohri, D.; Miyabayashi, K.; Asaoka, Y.; Maeda, S.; et al. Inhibiting Cxcr2 Disrupts Tumor-Stromal Interactions and Improves Survival in a Mouse Model of Pancreatic Ductal Adenocarcinoma. J. Clin. Invest. 2011, 121, 4106–4117. [Google Scholar] [CrossRef] [PubMed]
- Cappello, P.; Tonoli, E.; Curto, R.; Giordano, D.; Giovarelli, M.; Novelli, F. Anti-α-Enolase Antibody Limits the Invasion of Myeloid-Derived Suppressor Cells and Attenuates Their Restraining Effector T Cell Response. Oncoimmunology 2016, 5, e1112940. [Google Scholar] [CrossRef] [PubMed]
- Nagaraj, S.; Youn, J.-I.; Weber, H.; Iclozan, C.; Lu, L.; Cotter, M.J.; Meyer, C.; Becerra, C.R.; Fishman, M.; Antonia, S.; et al. Anti-Inflammatory Triterpenoid Blocks Immune Suppressive Function of MDSCs and Improves Immune Response in Cancer. Clin. Cancer Res. 2010, 16, 1812–1823. [Google Scholar] [CrossRef] [PubMed]
- Ling, X.; Konopleva, M.; Zeng, Z.; Ruvolo, V.; Stephens, L.C.; Schober, W.; McQueen, T.; Dietrich, M.; Madden, T.L.; Andreeff, M. The Novel Triterpenoid C-28 Methyl Ester of 2-Cyano-3, 12-Dioxoolen-1, 9-Dien-28-Oic Acid Inhibits Metastatic Murine Breast Tumor Growth through Inactivation of STAT3 Signaling. Cancer Res. 2007, 67, 4210–4218. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Useros, J.; Garcia-Foncillas, J. Can Molecular Biomarkers Change the Paradigm of Pancreatic Cancer Prognosis? Biomed. Res. Int. 2016, 2016, 4873089. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Useros, J.; Garcia-Foncillas, J. The Role of BRCA2 Mutation Status as Diagnostic, Predictive, and Prognosis Biomarker for Pancreatic Cancer. Biomed. Res. Int. 2016, 2016, 1869304. [Google Scholar] [CrossRef]
- Sabharwal, S.S.; Schumacker, P.T. Mitochondrial ROS in Cancer: Initiators, Amplifiers or an Achilles’ Heel? Nat. Rev. Cancer 2014, 14, 709–721. [Google Scholar] [CrossRef] [PubMed]
- Bailey, P.; Chang, D.K.; Nones, K.; Johns, A.L.; Patch, A.-M.; Gingras, M.-C.; Miller, D.K.; Christ, A.N.; Bruxner, T.J.C.; Quinn, M.C.; et al. Genomic Analyses Identify Molecular Subtypes of Pancreatic Cancer. Nature 2016, 531, 47–52. [Google Scholar] [CrossRef]
- Moffitt, R.A.; Marayati, R.; Flate, E.L.; Volmar, K.E.; Loeza, S.G.H.; Hoadley, K.A.; Rashid, N.U.; Williams, L.A.; Eaton, S.C.; Chung, A.H.; et al. Virtual Microdissection Identifies Distinct Tumor- and Stroma-Specific Subtypes of Pancreatic Ductal Adenocarcinoma. Nat. Genet. 2015, 47, 1168–1178. [Google Scholar] [CrossRef]
- Collisson, E.A.; Sadanandam, A.; Olson, P.; Gibb, W.J.; Truitt, M.; Gu, S.; Cooc, J.; Weinkle, J.; Kim, G.E.; Jakkula, L.; et al. Subtypes of Pancreatic Ductal Adenocarcinoma and Their Differing Responses to Therapy. Nat. Med. 2011, 17, 500–503. [Google Scholar] [CrossRef]
- Feng, Y.-F.; Chen, J.-Y.; Chen, H.-Y.; Wang, T.-G.; Shi, D.; Lu, Y.-F.; Pan, Y.; Shao, C.-W.; Yu, R.-S. 110 Patients with Adenosquamous Carcinomas of the Pancreas (PASC): Imaging Differentiation of Small (≤3 Cm) versus Large (>3 Cm) Tumors. Abdom. Radiol. (N. Y.) 2019, 44, 2466–2473. [Google Scholar] [CrossRef] [PubMed]
- Niger, M.; Prisciandaro, M.; Antista, M.; Monica, M.A.T.; Cattaneo, L.; Prinzi, N.; Manglaviti, S.; Nichetti, F.; Brambilla, M.; Torchio, M.; et al. One Size Does Not Fit All for Pancreatic Cancers: A Review on Rare Histologies and Therapeutic Approaches. World J. Gastrointest. Oncol. 2020, 12, 833–849. [Google Scholar] [CrossRef] [PubMed]
- Fang, Y.; Su, Z.; Xie, J.; Xue, R.; Ma, Q.; Li, Y.; Zhao, Y.; Song, Z.; Lu, X.; Li, H.; et al. Genomic Signatures of Pancreatic Adenosquamous Carcinoma (PASC). J. Pathol. 2017, 243, 155–159. [Google Scholar] [CrossRef] [PubMed]
- Bosman, F.T.; Carneiro, F.; Hruban, R.H.; Theise, N.D. WHO Classification of Tumours of the Digestive System—NLM Catalog—NCBI; IARC Press: Lyon, France, 2010. [Google Scholar]
- Cancer Genome Atlas Research Network. By The Cancer Genome Atlas Research Network. Integrated Genomic Characterization of Pancreatic Ductal Adenocarcinoma. Cancer Cell 2017, 32, 185–203. [Google Scholar] [CrossRef] [PubMed]
- Engelmann, D.; Pützer, B.M. Emerging from the Shade of P53 Mutants: N-Terminally Truncated Variants of the P53 Family in EMT Signaling and Cancer Progression. Sci. Signal 2014, 7, re9. [Google Scholar] [CrossRef] [PubMed]
- Miller, B.W.; Morton, J.P.; Pinese, M.; Saturno, G.; Jamieson, N.B.; McGhee, E.; Timpson, P.; Leach, J.; McGarry, L.; Shanks, E.; et al. Targeting the LOX/Hypoxia Axis Reverses Many of the Features That Make Pancreatic Cancer Deadly: Inhibition of LOX Abrogates Metastasis and Enhances Drug Efficacy. EMBO Mol. Med. 2015, 7, 1063–1076. [Google Scholar] [CrossRef]
- Hosein, A.N.; Brekken, R.A.; Maitra, A. Pancreatic Cancer Stroma: An Update on Therapeutic Targeting Strategies. Nat. Rev. Gastroenterol. Hepatol. 2020, 17, 487–505. [Google Scholar] [CrossRef]
- Bristow, R.G.; Hill, R.P. Hypoxia and Metabolism. Hypoxia, DNA Repair and Genetic Instability. Nat. Rev. Cancer 2008, 8, 180–192. [Google Scholar] [CrossRef]
- Kitamoto, S.; Yokoyama, S.; Higashi, M.; Yamada, N.; Takao, S.; Yonezawa, S. MUC1 Enhances Hypoxia-Driven Angiogenesis through the Regulation of Multiple Proangiogenic Factors. Oncogene 2013, 32, 4614–4621. [Google Scholar] [CrossRef]
- Biankin, A.V.; Kench, J.G.; Colvin, E.K.; Segara, D.; Scarlett, C.J.; Nguyen, N.Q.; Chang, D.K.; Morey, A.L.; Lee, C.-S.; Pinese, M.; et al. Expression of S100A2 Calcium-Binding Protein Predicts Response to Pancreatectomy for Pancreatic Cancer. Gastroenterology 2009, 137, 558–568. [Google Scholar] [CrossRef]
- Mews, P.; Phillips, P.; Fahmy, R.; Korsten, M.; Pirola, R.; Wilson, J.; Apte, M. Pancreatic Stellate Cells Respond to Inflammatory Cytokines: Potential Role in Chronic Pancreatitis. Gut 2002, 50, 535–541. [Google Scholar] [CrossRef] [PubMed]
- Masamune, A.; Satoh, M.; Kikuta, K.; Suzuki, N.; Shimosegawa, T. Activation of JAK-STAT Pathway Is Required for Platelet-Derived Growth Factor-Induced Proliferation of Pancreatic Stellate Cells. World J. Gastroenterol. 2005, 11, 3385–3391. [Google Scholar] [CrossRef] [PubMed]
- Somerville, T.D.; Biffi, G.; Daßler-Plenker, J.; Hur, S.K.; He, X.-Y.; Vance, K.E.; Miyabayashi, K.; Xu, Y.; Maia-Silva, D.; Klingbeil, O.; et al. Squamous Trans-Differentiation of Pancreatic Cancer Cells Promotes Stromal Inflammation. Elife 2020, 9, e53381. [Google Scholar] [CrossRef] [PubMed]
- Öhlund, D.; Handly-Santana, A.; Biffi, G.; Elyada, E.; Almeida, A.S.; Ponz-Sarvise, M.; Corbo, V.; Oni, T.E.; Hearn, S.A.; Lee, E.J.; et al. Distinct Populations of Inflammatory Fibroblasts and Myofibroblasts in Pancreatic Cancer. J. Exp. Med. 2017, 214, 579–596. [Google Scholar] [CrossRef]
- Lankadasari, M.B.; Mukhopadhyay, P.; Mohammed, S.; Harikumar, K.B. TAMing Pancreatic Cancer: Combat with a Double Edged Sword. Mol. Cancer 2019, 18, 48. [Google Scholar] [CrossRef]
- Ireland, L.; Santos, A.; Ahmed, M.S.; Rainer, C.; Nielsen, S.R.; Quaranta, V.; Weyer-Czernilofsky, U.; Engle, D.D.; Perez-Mancera, P.A.; Coupland, S.E.; et al. Chemoresistance in Pancreatic Cancer Is Driven by Stroma-Derived Insulin-Like Growth Factors. Cancer Res. 2016, 76, 6851–6863. [Google Scholar] [CrossRef]
- Dalin, S.; Sullivan, M.R.; Lau, A.N.; Grauman-Boss, B.; Mueller, H.S.; Kreidl, E.; Fenoglio, S.; Luengo, A.; Lees, J.A.; Vander Heiden, M.G.; et al. Deoxycytidine Release from Pancreatic Stellate Cells Promotes Gemcitabine Resistance. Cancer Res. 2019, 79, 5723–5733. [Google Scholar] [CrossRef]
- Al-Assar, O.; Demiciorglu, F.; Lunardi, S.; Gaspar-Carvalho, M.M.; McKenna, W.G.; Muschel, R.M.; Brunner, T.B. Contextual Regulation of Pancreatic Cancer Stem Cell Phenotype and Radioresistance by Pancreatic Stellate Cells. Radiother. Oncol. 2014, 111, 243–251. [Google Scholar] [CrossRef]
- Panni, R.Z.; Sanford, D.E.; Belt, B.A.; Mitchem, J.B.; Worley, L.A.; Goetz, B.D.; Mukherjee, P.; Wang-Gillam, A.; Link, D.C.; Denardo, D.G.; et al. Tumor-Induced STAT3 Activation in Monocytic Myeloid-Derived Suppressor Cells Enhances Stemness and Mesenchymal Properties in Human Pancreatic Cancer. Cancer Immunol. Immunother. 2014, 63, 513–528. [Google Scholar] [CrossRef]
- Feig, C.; Jones, J.O.; Kraman, M.; Wells, R.J.B.; Deonarine, A.; Chan, D.S.; Connell, C.M.; Roberts, E.W.; Zhao, Q.; Caballero, O.L.; et al. Targeting CXCL12 from FAP-Expressing Carcinoma-Associated Fibroblasts Synergizes with Anti-PD-L1 Immunotherapy in Pancreatic Cancer. Proc. Natl. Acad. Sci. USA 2013, 110, 20212–20217. [Google Scholar] [CrossRef]
- Jenkins, R.W.; Barbie, D.A.; Flaherty, K.T. Mechanisms of Resistance to Immune Checkpoint Inhibitors. Br. J. Cancer 2018, 118, 9–16. [Google Scholar] [CrossRef]
- Holmgaard, R.B.; Zamarin, D.; Munn, D.H.; Wolchok, J.D.; Allison, J.P. Indoleamine 2,3-Dioxygenase Is a Critical Resistance Mechanism in Antitumor T Cell Immunotherapy Targeting CTLA-4. J. Exp. Med. 2013, 210, 1389–1402. [Google Scholar] [CrossRef] [PubMed]
- Blair, A.B.; Kleponis, J.; Thomas, D.L.; Muth, S.T.; Murphy, A.G.; Kim, V.; Zheng, L. IDO1 Inhibition Potentiates Vaccine-Induced Immunity against Pancreatic Adenocarcinoma. J. Clin. Investig. 2019, 129, 1742–1755. [Google Scholar] [CrossRef] [PubMed]
- Peng, J.; Sun, B.-F.; Chen, C.-Y.; Zhou, J.-Y.; Chen, Y.-S.; Chen, H.; Liu, L.; Huang, D.; Jiang, J.; Cui, G.-S.; et al. Single-Cell RNA-Seq Highlights Intra-Tumoral Heterogeneity and Malignant Progression in Pancreatic Ductal Adenocarcinoma. Cell Res. 2019, 29, 725–738. [Google Scholar] [CrossRef] [PubMed]
- Shi, S.; Xu, J.; Zhang, B.; Ji, S.; Xu, W.; Liu, J.; Jin, K.; Liang, D.; Liang, C.; Liu, L.; et al. VEGF Promotes Glycolysis in Pancreatic Cancer via HIF1α Up-Regulation. Curr. Mol. Med. 2016, 16, 394–403. [Google Scholar] [CrossRef]
- Hale, M.A.; Kagami, H.; Shi, L.; Holland, A.M.; Elsässer, H.-P.; Hammer, R.E.; MacDonald, R.J. The Homeodomain Protein PDX1 Is Required at Mid-Pancreatic Development for the Formation of the Exocrine Pancreas. Dev. Biol. 2005, 286, 225–237. [Google Scholar] [CrossRef]
- Patra, K.C.; Bardeesy, N.; Mizukami, Y. Diversity of Precursor Lesions For Pancreatic Cancer: The Genetics and Biology of Intraductal Papillary Mucinous Neoplasm. Clin. Transl. Gastroenterol. 2017, 8, e86. [Google Scholar] [CrossRef]
- Baker, M.L.; Seeley, E.S.; Pai, R.; Suriawinata, A.A.; Mino-Kenudson, M.; Zamboni, G.; Klöppel, G.; Longnecker, D.S. Invasive Mucinous Cystic Neoplasms of the Pancreas. Exp. Mol. Pathol. 2012, 93, 345–349. [Google Scholar] [CrossRef]
- Martinelli, P.; Carrillo-de Santa Pau, E.; Cox, T.; Sainz, B.; Dusetti, N.; Greenhalf, W.; Rinaldi, L.; Costello, E.; Ghaneh, P.; Malats, N.; et al. GATA6 Regulates EMT and Tumour Dissemination, and Is a Marker of Response to Adjuvant Chemotherapy in Pancreatic Cancer. Gut 2017, 66, 1665–1676. [Google Scholar] [CrossRef]
- Suh, H.; Pillai, K.; Morris, D.L. Mucins in Pancreatic Cancer: Biological Role, Implications in Carcinogenesis and Applications in Diagnosis and Therapy. Am. J. Cancer Res. 2017, 7, 1372–1383. [Google Scholar]
- Pompella, L.; Tirino, G.; Pappalardo, A.; Caterino, M.; Ventriglia, A.; Nacca, V.; Orditura, M.; Ciardiello, F.; De Vita, F. Pancreatic Cancer Molecular Classifications: From Bulk Genomics to Single Cell Analysis. Int. J. Mol. Sci. 2020, 21, 2814. [Google Scholar] [CrossRef] [PubMed]
- Cheng, H.; Luo, G.; Jin, K.; Fan, Z.; Huang, Q.; Gong, Y.; Xu, J.; Yu, X.; Liu, C. Kras Mutation Correlating with Circulating Regulatory T Cells Predicts the Prognosis of Advanced Pancreatic Cancer Patients. Cancer Med. 2020, 9, 2153–2159. [Google Scholar] [CrossRef] [PubMed]
- Bauer, C.A.; Kim, E.Y.; Marangoni, F.; Carrizosa, E.; Claudio, N.M.; Mempel, T.R. Dynamic Treg Interactions with Intratumoral APCs Promote Local CTL Dysfunction. J. Clin. Invest. 2014, 124, 2425–2440. [Google Scholar] [CrossRef] [PubMed]
- Trivedi, P.M.; Fynch, S.; Kennedy, L.M.; Chee, J.; Krishnamurthy, B.; O’Reilly, L.A.; Strasser, A.; Kay, T.W.H.; Thomas, H.E. Soluble FAS Ligand Is Not Required for Pancreatic Islet Inflammation or Beta-Cell Destruction in Non-Obese Diabetic Mice. Cell Death Discov. 2019, 5, 136. [Google Scholar] [CrossRef] [PubMed]
- Sideras, K.; Braat, H.; Kwekkeboom, J.; van Eijck, C.H.; Peppelenbosch, M.P.; Sleijfer, S.; Bruno, M. Role of the Immune System in Pancreatic Cancer Progression and Immune Modulating Treatment Strategies. Cancer Treat. Rev. 2014, 40, 513–522. [Google Scholar] [CrossRef] [PubMed]
- Jiao, Y.; Yonescu, R.; Offerhaus, G.J.A.; Klimstra, D.S.; Maitra, A.; Eshleman, J.R.; Herman, J.G.; Poh, W.; Pelosof, L.; Wolfgang, C.L.; et al. Whole-Exome Sequencing of Pancreatic Neoplasms with Acinar Differentiation. J. Pathol. 2014, 232, 428–435. [Google Scholar] [CrossRef]
- Furukawa, T.; Sakamoto, H.; Takeuchi, S.; Ameri, M.; Kuboki, Y.; Yamamoto, T.; Hatori, T.; Yamamoto, M.; Sugiyama, M.; Ohike, N.; et al. Whole Exome Sequencing Reveals Recurrent Mutations in BRCA2 and FAT Genes in Acinar Cell Carcinomas of the Pancreas. Sci. Rep. 2015, 5, 8829. [Google Scholar] [CrossRef]
- Von Figura, G.; Morris, J.P.; Wright, C.V.E.; Hebrok, M. Nr5a2 Maintains Acinar Cell Differentiation and Constrains Oncogenic Kras-Mediated Pancreatic Neoplastic Initiation. Gut 2014, 63, 656–664. [Google Scholar] [CrossRef]
- Hale, M.A.; Swift, G.H.; Hoang, C.Q.; Deering, T.G.; Masui, T.; Lee, Y.-K.; Xue, J.; MacDonald, R.J. The Nuclear Hormone Receptor Family Member NR5A2 Controls Aspects of Multipotent Progenitor Cell Formation and Acinar Differentiation during Pancreatic Organogenesis. Development 2014, 141, 3123–3133. [Google Scholar] [CrossRef]
- Mathison, A.; Liebl, A.; Bharucha, J.; Mukhopadhyay, D.; Lomberk, G.; Shah, V.; Urrutia, R. Pancreatic Stellate Cell Models for Transcriptional Studies of Desmoplasia-Associated Genes. Pancreatology 2010, 10, 505–516. [Google Scholar] [CrossRef]
- Erkan, M.; Reiser-Erkan, C.; Michalski, C.W.; Deucker, S.; Sauliunaite, D.; Streit, S.; Esposito, I.; Friess, H.; Kleeff, J. Cancer-Stellate Cell Interactions Perpetuate the Hypoxia-Fibrosis Cycle in Pancreatic Ductal Adenocarcinoma. Neoplasia 2009, 11, 497–508. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Li, Y.; Ahmad, A.; Banerjee, S.; Azmi, A.S.; Kong, D.; Sarkar, F.H. Pancreatic Cancer: Understanding and Overcoming Chemoresistance. Nat. Rev. Gastroenterol. Hepatol. 2011, 8, 27–33. [Google Scholar] [CrossRef] [PubMed]
- Mekapogu, A.R.; Pothula, S.P.; Pirola, R.C.; Wilson, J.S.; Apte, M.V. Multifunctional Role of Pancreatic Stellate Cells in Pancreatic Cancer. Ann. Pancreat. Cancer 2019, 2. [Google Scholar] [CrossRef]
- Franses, J.W.; Basar, O.; Kadayifci, A.; Yuksel, O.; Choz, M.; Kulkarni, A.S.; Tai, E.; Vo, K.D.; Arora, K.S.; Desai, N.; et al. Improved Detection of Circulating Epithelial Cells in Patients with Intraductal Papillary Mucinous Neoplasms. Oncologist 2018, 23, 1260. [Google Scholar] [CrossRef]
- Lahat, G.; Lubezky, N.; Loewenstein, S.; Nizri, E.; Gan, S.; Pasmanik-Chor, M.; Hayman, L.; Barazowsky, E.; Ben-Haim, M.; Klausner, J.M. Epithelial-to-Mesenchymal Transition (EMT) in Intraductal Papillary Mucinous Neoplasm (IPMN) Is Associated with High Tumor Grade and Adverse Outcomes. Ann. Surg. Oncol. 2014, 21, S750–S757. [Google Scholar] [CrossRef]
- Rooney, M.S.; Shukla, S.A.; Wu, C.J.; Getz, G.; Hacohen, N. Molecular and Genetic Properties of Tumors Associated with Local Immune Cytolytic Activity. Cell 2015, 160, 48–61. [Google Scholar] [CrossRef]
- Yachida, S.; Jones, S.; Bozic, I.; Antal, T.; Leary, R.; Fu, B.; Kamiyama, M.; Hruban, R.H.; Eshleman, J.R.; Nowak, M.A.; et al. Distant Metastasis Occurs Late during the Genetic Evolution of Pancreatic Cancer. Nature 2010, 467, 1114–1117. [Google Scholar] [CrossRef]
- A Phase I/II Study of a MUC1 Peptide Pulsed Autologous Dendritic Cell Vaccine as Adjuvant Therapy in Patients with Resected Pancreatic and Biliary Tumors—PubMed. Available online: https://pubmed.ncbi.nlm.nih.gov/19129927/ (accessed on 11 November 2020).
- Hu, G.; He, N.; Cai, C.; Cai, F.; Fan, P.; Zheng, Z.; Jin, X. HDAC3 Modulates Cancer Immunity via Increasing PD-L1 Expression in Pancreatic Cancer. Pancreatology 2019, 19, 383–389. [Google Scholar] [CrossRef]
- Park, H.; Bang, J.-H.; Nam, A.-R.; Eun Park, J.; Hua Jin, M.; Bang, Y.-J.; Oh, D.-Y. Prognostic Implications of Soluble Programmed Death-Ligand 1 and Its Dynamics during Chemotherapy in Unresectable Pancreatic Cancer. Sci. Rep. 2019, 9, 11131. [Google Scholar] [CrossRef]
- Saka, D.; Gökalp, M.; Piyade, B.; Cevik, N.C.; Arik Sever, E.; Unutmaz, D.; Ceyhan, G.O.; Demir, I.E.; Asimgil, H. Mechanisms of T-Cell Exhaustion in Pancreatic Cancer. Cancers 2020, 12, 2274. [Google Scholar] [CrossRef]
- Yazdanifar, M.; Zhou, R.; Grover, P.; Williams, C.; Bose, M.; Moore, L.J.; Wu, S.-T.; Maher, J.; Dreau, D.; Mukherjee, A.P. Overcoming Immunological Resistance Enhances the Efficacy of A Novel Anti-TMUC1-CAR T Cell Treatment against Pancreatic Ductal Adenocarcinoma. Cells 2019, 8, 1070. [Google Scholar] [CrossRef] [PubMed]
- Beatty, G.L.; Haas, A.R.; Maus, M.V.; Torigian, D.A.; Soulen, M.C.; Plesa, G.; Chew, A.; Zhao, Y.; Levine, B.L.; Albelda, S.M.; et al. Mesothelin-Specific Chimeric Antigen Receptor MRNA-Engineered T Cells Induce Anti-Tumor Activity in Solid Malignancies. Cancer Immunol. Res. 2014, 2, 112–120. [Google Scholar] [CrossRef] [PubMed]
- Diaz Sanchez, A.; Ponferrada Diaz, Á.; Senosiain Labiano, M.; Huerta Madrigal, A. Hemorragia digestiva alta como presentación de un carcinoma acinar pancreático. Gastroenterol. Hepatol. 2006, 29, 380. [Google Scholar] [CrossRef]
- Kordes, C.; Sawitza, I.; Götze, S.; Häussinger, D. Stellate Cells from Rat Pancreas Are Stem Cells and Can Contribute to Liver Regeneration. PLoS ONE 2012, 7, e51878. [Google Scholar] [CrossRef] [PubMed]
- Tang, D.; Wang, D.; Yuan, Z.; Xue, X.; Zhang, Y.; An, Y.; Chen, J.; Tu, M.; Lu, Z.; Wei, J.; et al. Persistent Activation of Pancreatic Stellate Cells Creates a Microenvironment Favorable for the Malignant Behavior of Pancreatic Ductal Adenocarcinoma. Int. J. Cancer 2013, 132, 993–1003. [Google Scholar] [CrossRef]
- Masamune, A.; Kikuta, K.; Watanabe, T.; Satoh, K.; Hirota, M.; Shimosegawa, T. Hypoxia Stimulates Pancreatic Stellate Cells to Induce Fibrosis and Angiogenesis in Pancreatic Cancer. Am. J. Physiol. Gastrointest. Liver Physiol. 2008, 295, G709–G717. [Google Scholar] [CrossRef]
- Miyamoto, H.; Murakami, T.; Tsuchida, K.; Sugino, H.; Miyake, H.; Tashiro, S. Tumor-Stroma Interaction of Human Pancreatic Cancer: Acquired Resistance to Anticancer Drugs and Proliferation Regulation Is Dependent on Extracellular Matrix Proteins. Pancreas 2004, 28, 38–44. [Google Scholar] [CrossRef]
- Tian, B.; Wong, W.Y.; Uger, M.D.; Wisniewski, P.; Chao, H. Development and Characterization of a Camelid Single Domain Antibody-Urease Conjugate That Targets Vascular Endothelial Growth Factor Receptor 2. Front. Immunol. 2017, 8, 956. [Google Scholar] [CrossRef]
- Le, D.T.; Wang-Gillam, A.; Picozzi, V.; Greten, T.F.; Crocenzi, T.; Springett, G.; Morse, M.; Zeh, H.; Cohen, D.; Fine, R.L.; et al. Safety and Survival With GVAX Pancreas Prime and Listeria Monocytogenes–Expressing Mesothelin (CRS-207) Boost Vaccines for Metastatic Pancreatic Cancer. J. Clin. Oncol. 2015, 33, 1325–1333. [Google Scholar] [CrossRef]
- Roccaro, A.M.; Sacco, A.; Purschke, W.G.; Moschetta, M.; Buchner, K.; Maasch, C.; Zboralski, D.; Zöllner, S.; Vonhoff, S.; Mishima, Y.; et al. SDF-1 Inhibition Targets the Bone Marrow Niche for Cancer Therapy. Cell Rep. 2014, 9, 118–128. [Google Scholar] [CrossRef]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).