
Hedgehog Signaling in Myeloid Malignancies


{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Hedgehog Signaling and Hematopoiesis
3. Hedgehog Signaling in Chronic Myeloid Leukemia
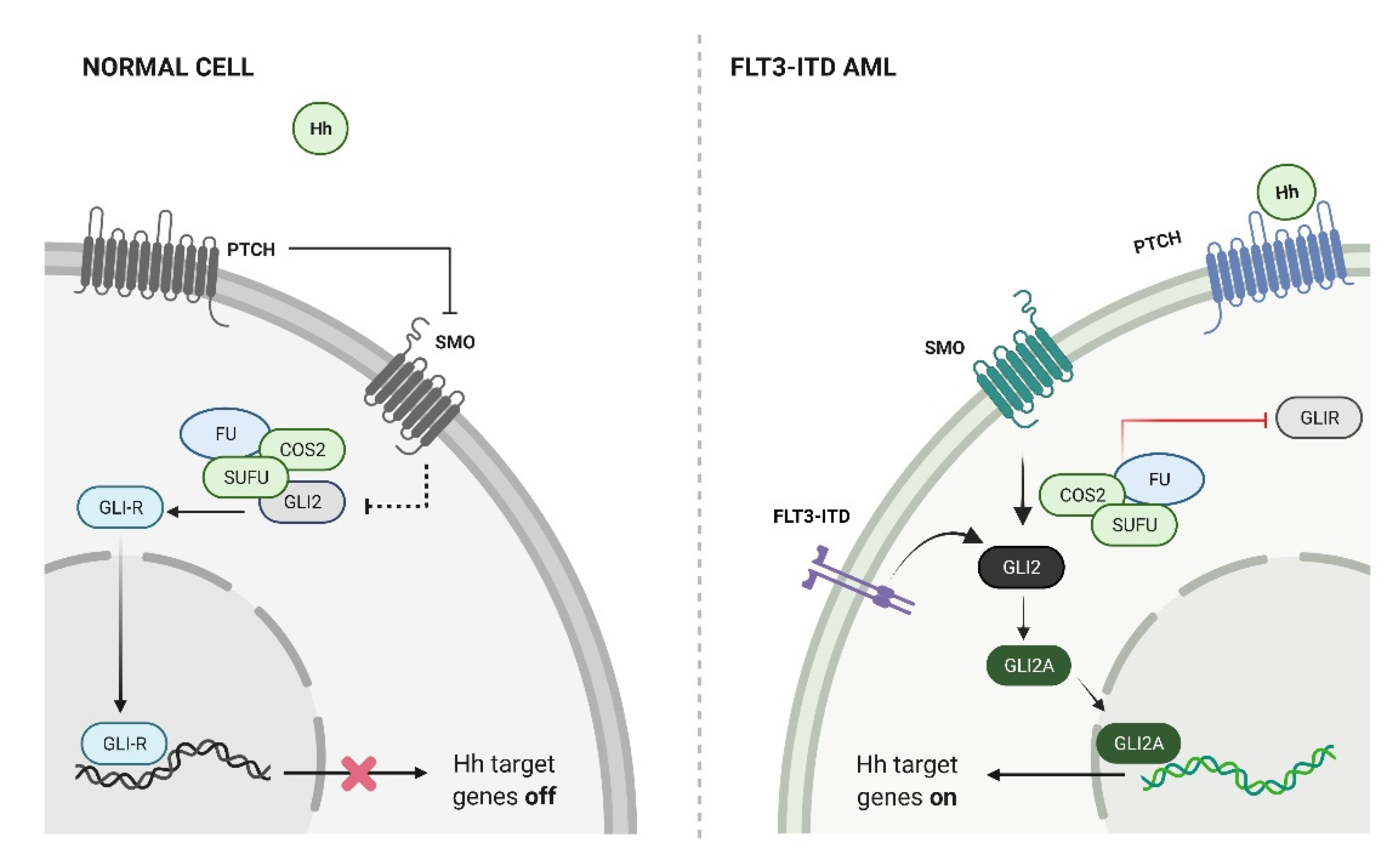
4. Hedgehog Signaling in AML
5. Clinical Studies in AML
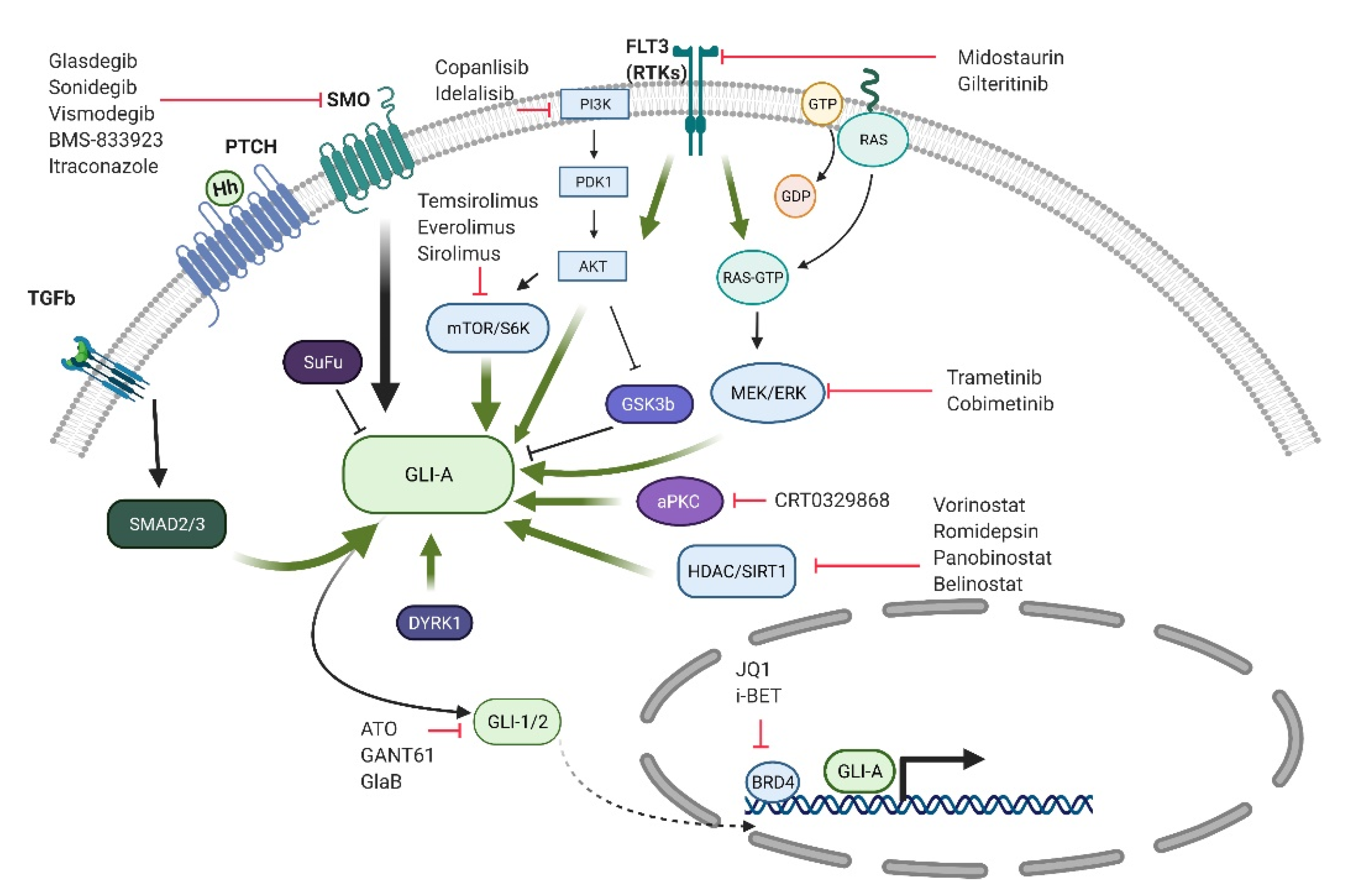
6. GLI Activation and Targeting in Myeloid Malignancies
7. Summary and Future Directions
Author Contributions
Funding
Conflicts of Interest
References
- Nusslein-Volhard, C.; Wieschaus, E. Mutations Affecting Segment Number and Polarity in DROSOPHILA. Nature 1980, 287, 795–801. [Google Scholar] [CrossRef]
- Varjosalo, M.; Taipale, J. Hedgehog: Functions and Mechanisms. Genes Dev. 2008, 22, 2454–2472. [Google Scholar] [CrossRef] [Green Version]
- Ryan, K.E.; Chiang, C. Hedgehog Secretion and Signal Transduction in Vertebrates. J. Biol. Chem. 2012, 287, 17905–17913. [Google Scholar] [CrossRef] [Green Version]
- Aberger, F.; Ruiz i Altaba, A. Context-dependent signal integration by the GLI code: The Oncogenic load, Pathways, Modifiers and Implications for Cancer Therapy. Semin. Cell Dev. Biol. 2014, 33, 93–104. [Google Scholar] [CrossRef]
- Kasper, M.; Regl, G.; Frischauf, A.-M.; Aberger, F. GLI Transcription Factors: Mediators of Oncogenic Hedgehog Signalling. Eur. J. Cancer 2006, 42, 437–445. [Google Scholar] [CrossRef]
- Ruiz i Altaba, A. Gli Proteins Encode Context-Dependent Positive and Negative Functions: Implications for Development and Disease. Development 1999, 126, 3205–3216. [Google Scholar] [CrossRef]
- Lim, Y.; Matsui, W. Hedgehog Signaling in Hematopoiesis. Crit. Rev. Eukaryot. Gene Expr. 2010, 20, 129–139. [Google Scholar] [CrossRef] [Green Version]
- Merchant, A.A.; Matsui, W. Smoothening the Controversial Role of Hedgehog in Hematopoiesis. Cell Stem Cell 2009, 4, 470–471. [Google Scholar] [CrossRef] [Green Version]
- Bhardwaj, G.; Murdoch, B.; Wu, D.; Baker, D.P.; Williams, K.P.; Chadwick, K.; Ling, L.E.; Karanu, F.N.; Bhatia, M. Sonic Hedgehog Induces the Proliferation of Primitive Human Hematopoietic Cells via BMP Regulation. Nat. Immunol. 2001, 2, 172–180. [Google Scholar] [CrossRef]
- Trowbridge, J.J.; Scott, M.P.; Bhatia, M. Hedgehog Modulates Cell Cycle Regulators in Stem Cells to Control Hematopoietic Regeneration. Proc. Natl. Acad. Sci. USA 2006, 103, 14134–14139. [Google Scholar] [CrossRef] [Green Version]
- Dierks, C.; Beigi, R.; Guo, G.-R.; Zirlik, K.; Stegert, M.R.; Manley, P.; Trussell, C.; Schmitt-Graeff, A.; Landwerlin, K.; Veelken, H.; et al. Expansion of Bcr-Abl-Positive Leukemic Stem Cells is Dependent on Hedgehog Pathway Activation. Cancer Cell 2008, 14, 238–249. [Google Scholar] [CrossRef] [Green Version]
- Hofmann, I.; Stover, E.H.; Cullen, D.E.; Mao, J.; Morgan, K.J.; Lee, B.H.; Kharas, M.G.; Miller, P.G.; Cornejo, M.G.; Okabe, R.; et al. Hedgehog Signaling is Dispensable for Adult Murine Hematopoietic Stem Cell Function and Hematopoiesis. Cell Stem Cell 2009, 4, 559–567. [Google Scholar] [CrossRef] [Green Version]
- Gao, J.; Graves, S.; Koch, U.; Liu, S.; Jankovic, V.; Buonamici, S.; El Andaloussi, A.; Nimer, S.D.; Kee, B.L.; Taichman, R.; et al. Hedgehog Signaling is Dispensable for Adult Hematopoietic Stem Cell Function. Cell Stem Cell 2009, 4, 548–558. [Google Scholar] [CrossRef] [Green Version]
- Merchant, A.; Joseph, G.; Wang, Q.; Brennan, S.; Matsui, W. Gli1 Regulates the Proliferation and Differentiation of HSCs and Myeloid Progenitors. Blood 2010, 115, 2391–2396. [Google Scholar] [CrossRef] [Green Version]
- Merchant, A.A.; Matsui, W. Targeting Hedgehog—A cancer stem cell pathway. Clin. Cancer Res. 2010, 16, 3130–3140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sengupta, A.; Banerjee, D.; Chandra, S.; Banerji, S.K.; Ghosh, R.; Roy, R.; Banerjee, S. Deregulation and cross talk among Sonic hedgehog, Wnt, Hox and Notch signaling in chronic myeloid leukemia progression. Leukemia 2007, 21, 949–955. [Google Scholar] [CrossRef] [PubMed]
- Long, B.; Zhu, H.; Zhu, C.; Liu, T.; Meng, W. Activation of the Hedgehog pathway in chronic myelogeneous leukemia patients. J. Exp. Clin. Cancer Res. 2011, 30, 8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Su, W.; Meng, F.; Huang, L.; Zheng, M.; Liu, W.; Sun, H. Sonic hedgehog maintains survival and growth of chronic myeloid leukemia progenitor cells through β-catenin signaling. Exp. Hematol. 2012, 40, 418–427. [Google Scholar] [CrossRef] [PubMed]
- Irvine, D.A.; Zhang, B.; Kinstrie, R.; Tarafdar, A.; Morrison, H.; Campbell, V.L.; Moka, H.A.; Ho, Y.; Nixon, C.; Manley, P.W.; et al. Deregulated hedgehog pathway signaling is inhibited by the smoothened antagonist LDE225 (Sonidegib) in chronic phase chronic myeloid leukaemia. Sci. Rep. 2016, 6, 25476. [Google Scholar] [CrossRef] [Green Version]
- Sadarangani, A.; Pineda, G.; Lennon, K.M.; Chun, H.-J.; Shih, A.; Schairer, A.E.; Court, A.C.; Goff, D.J.; Prashad, S.L.; Geron, I.; et al. GLI2 inhibition abrogates human leukemia stem cell dormancy. J. Transl. Med. 2015, 13, 98. [Google Scholar] [CrossRef] [Green Version]
- Katagiri, S.; Tauchi, T.; Okabe, S.; Minami, Y.; Kimura, S.; Maekawa, T.; Naoe, T.; Ohyashiki, K. Combination of Ponatinib with Hedgehog Antagonist Vismodegib for Therapy-Resistant BCR-ABL1–Positive Leukemia. Clin. Cancer Res. 2013, 19, 1422–1432. [Google Scholar] [CrossRef] [Green Version]
- Zeng, X.; Zhao, H.; Li, Y.; Fan, J.; Sun, Y.; Wang, S.; Wang, Z.; Song, P.; Ju, D. Targeting Hedgehog signaling pathway and autophagy overcomes drug resistance of BCR-ABL-positive chronic myeloid leukemia. Autophagy 2015, 11, 355–372. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Binns, W.; Keeler, R.F.; Balls, L.D. Congenital deformities in lambs, calves, and goats resulting from maternal ingestion of Veratrum californicum: Hare lip, cleft palate, ataxia, and hypoplasia of metacarpal and metatarsal bones. Clin. Toxicol. 1972, 5, 245–261. [Google Scholar] [CrossRef] [PubMed]
- Chiang, C.; Litingtung, Y.; Lee, E.; Young, K.E.; Corden, J.L.; Westphal, H.; Beachy, P.A. Cyclopia and defective axial patterning in mice lacking Sonic hedgehog gene function. Nature 1996, 383, 407–413. [Google Scholar] [CrossRef]
- Cooper, M.K.; Porter, J.A.; Young, K.E.; Beachy, P.A. Teratogen-mediated inhibition of target tissue response to Shh signaling. Science 1998, 280, 1603–1607. [Google Scholar] [CrossRef] [PubMed]
- Okabe, S.; Tauchi, T.; Tanaka, Y.; Katagiri, S.; Ohyashiki, K. Effects of the Hedgehog Inhibitor GDC-0449, Alone or in Combination with Dasatinib, on BCR-ABL-Positive Leukemia Cells. Stem Cells Dev. 2012, 21, 2939–2948. [Google Scholar] [CrossRef] [Green Version]
- Shah, N.P.; Cortes, J.E.; Martinelli, G.; Smith, B.D.; Clarke, E.; Copland, M.; Strauss, L.; Talpaz, M. Dasatinib Plus Smoothened (SMO) Inhibitor BMS-833923 in Chronic Myeloid Leukemia (CML) with Resistance or Suboptimal Response to a Prior Tyrosine Kinase Inhibitor (TKI): Phase I Study CA180323. Blood 2014, 124, 4539. [Google Scholar] [CrossRef]
- LoRusso, P.M.; Rudin, C.M.; Reddy, J.C.; Tibes, R.; Weiss, G.J.; Borad, M.J.; Hann, C.L.; Brahmer, J.R.; Chang, I.; Darbonne, W.C.; et al. Phase I Trial of Hedgehog Pathway Inhibitor Vismodegib (GDC-0449) in Patients with Refractory, Locally Advanced or Metastatic Solid Tumors. Clin. Cancer Res. 2011, 17, 2502–2511. [Google Scholar] [CrossRef] [Green Version]
- Ottmann, O.G.; Stegelmann, F.; Breccia, M.; Steegmann, J.L.; Olavarria, E.; Aimone, P.; Lipton, J.H. Smoothened inhibitor erismodegib combined with nilotinib in patients with chronic myeloid leukemia resistant/intolerant to at least one prior tyrosine kinase inhibitor: A phase 1b study. Leuk. Lymphoma 2021, 62, 739–742. [Google Scholar] [CrossRef] [PubMed]
- Kobune, M.; Takimoto, R.; Murase, K.; Iyama, S.; Sato, T.; Kikuchi, S.; Kawano, Y.; Miyanishi, K.; Sato, Y.; Niitsu, Y.; et al. Drug resistance is dramatically restored by hedgehog inhibitors in CD34+ leukemic cells. Cancer Sci. 2009, 100, 948–955. [Google Scholar] [CrossRef]
- Campbell, V.; Tholouli, E.; Quigley, M.T.; Keilty, J.; Kelly, P.; McGovern, K.; Read, M.; Kutok, J.L.; Byers, R. Evidence That Activated Hedgehog Signaling Predicts for Poor Clinical Outcome in Acute Myeloid Leukemia. ASH Annu. Meet. Abstr. 2012, 120, 1441. [Google Scholar] [CrossRef]
- Kobune, M.; Iyama, S.; Kikuchi, S.; Horiguchi, H.; Sato, T.; Murase, K.; Kawano, Y.; Takada, K.; Ono, K.; Kamihara, Y.; et al. Stromal cells expressing hedgehog-interacting protein regulate the proliferation of myeloid neoplasms. Blood Cancer J. 2012, 2, e87. [Google Scholar] [CrossRef] [PubMed]
- Zahreddine, H.A.; Culjkovic-Kraljacic, B.; Assouline, S.; Gendron, P.; Romeo, A.A.; Morris, S.J.; Cormack, G.; Jaquith, J.B.; Cerchietti, L.; Cocolakis, E.; et al. The sonic hedgehog factor GLI1 imparts drug resistance through inducible glucuronidation. Nature 2014, 511, 90–93. [Google Scholar] [CrossRef] [Green Version]
- Lim, Y.; Gondek, L.; Li, L.; Wang, Q.; Ma, H.; Chang, E.; Huso, D.L.; Foerster, S.; Marchionni, L.; McGovern, K.; et al. Integration of Hedgehog and mutant FLT3 signaling in myeloid leukemia. Sci. Transl. Med. 2015, 7, 291ra296. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wellbrock, J.; Latuske, E.; Kohler, J.; Wagner, K.; Stamm, H.; Vettorazzi, E.; Vohwinkel, G.; Klokow, M.; Uibeleisen, R.; Ehm, P.; et al. Expression of Hedgehog Pathway Mediator GLI Represents a Negative Prognostic Marker in Human Acute Myeloid Leukemia and Its Inhibition Exerts Antileukemic Effects. Clin. Cancer Res. 2015, 21, 2388–2398. [Google Scholar] [CrossRef] [Green Version]
- Xavier-Ferrucio, J.M.; Pericole, F.V.; Lopes, M.R.; Latuf-Filho, P.; Barcellos, K.S.; Dias, A.I.; Campos Pde, M.; Traina, F.; Vassallo, J.; Saad, S.T.; et al. Abnormal Hedgehog pathway in myelodysplastic syndrome and its impact on patients’ outcome. Haematologica 2015, 100, e491–e493. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lau, B.W.; Huh, K.; Madero-Marroquin, R.; De Marchi, F.; Lim, Y.; Wang, Q.; Lobo, F.; Marchionni, L.; Smith, D.B.; DeZern, A.; et al. Hedgehog/GLI1 activation leads to leukemic transformation of myelodysplastic syndrome in vivo and GLI1 inhibition results in antitumor activity. Oncogene 2019, 38, 687–698. [Google Scholar] [CrossRef]
- Fukushima, N.; Minami, Y.; Kakiuchi, S.; Kuwatsuka, Y.; Hayakawa, F.; Jamieson, C.; Kiyoi, H.; Naoe, T. Small-molecule Hedgehog inhibitor attenuates the leukemia-initiation potential of acute myeloid leukemia cells. Cancer Sci. 2016, 107, 1422–1429. [Google Scholar] [CrossRef] [Green Version]
- Tibes, R.; Al-Kali, A.; Oliver, G.R.; Delman, D.H.; Hansen, N.; Bhagavatula, K.; Mohan, J.; Rakhshan, F.; Wood, T.; Foran, J.M.; et al. The Hedgehog pathway as targetable vulnerability with 5-azacytidine in myelodysplastic syndrome and acute myeloid leukemia. J. Hematol. Oncol. 2015, 8, 114. [Google Scholar] [CrossRef] [Green Version]
- Huang, K.; Sun, Z.; Ding, B.; Jiang, X.; Wang, Z.; Zhu, Y.; Meng, F. Suppressing Hedgehog signaling reverses drug resistance of refractory acute myeloid leukemia. Oncol. Targets Ther. 2019, 12, 7477–7488. [Google Scholar] [CrossRef] [Green Version]
- Queiroz, K.C.; Ruela-de-Sousa, R.R.; Fuhler, G.M.; Aberson, H.L.; Ferreira, C.V.; Peppelenbosch, M.P.; Spek, C.A. Hedgehog signaling maintains chemoresistance in myeloid leukemic cells. Oncogene 2010, 29, 6314–6322. [Google Scholar] [CrossRef] [Green Version]
- Freisleben, F.; Behrmann, L.; Thaden, V.; Muschhammer, J.; Bokemeyer, C.; Fiedler, W.; Wellbrock, J. Downregulation of GLI3 Expression Mediates Chemotherapy Resistance in Acute Myeloid Leukemia. Int. J. Mol. Sci. 2020, 21, 5084. [Google Scholar] [CrossRef] [PubMed]
- DiNardo, C.D.; Jonas, B.A.; Pullarkat, V.; Thirman, M.J.; Garcia, J.S.; Wei, A.H.; Konopleva, M.; Döhner, H.; Letai, A.; Fenaux, P.; et al. Azacitidine and Venetoclax in Previously Untreated Acute Myeloid Leukemia. N. Engl. J. Med. 2020, 383, 617–629. [Google Scholar] [CrossRef] [PubMed]
- Zou, J.; Zhou, Z.; Wan, L.; Tong, Y.; Qin, Y.; Wang, C.; Zhou, K. Targeting the Sonic Hedgehog-Gli1 Pathway as a Potential New Therapeutic Strategy for Myelodysplastic Syndromes. PLoS ONE 2015, 10, e0136843. [Google Scholar] [CrossRef] [PubMed]
- Chaudhry, P.; Singh, M.; Triche, T.J.; Guzman, M.; Merchant, A.A. GLI3 repressor determines Hedgehog pathway activation and is required for response to SMO antagonist glasdegib in AML. Blood 2017, 129, 3465–3475. [Google Scholar] [CrossRef]
- Daver, N.; Schlenk, R.F.; Russell, N.H.; Levis, M.J. Targeting FLT3 mutations in AML: Review of current knowledge and evidence. Leukemia 2019, 33, 299–312. [Google Scholar] [CrossRef] [Green Version]
- Latuske, E.-M.; Stamm, H.; Klokow, M.; Vohwinkel, G.; Muschhammer, J.; Bokemeyer, C.; Jücker, M.; Kebenko, M.; Fiedler, W.; Wellbrock, J. Combined inhibition of GLI and FLT3 signaling leads to effective anti-leukemic effects in human acute myeloid leukemia. Oncotarget 2017, 8, 29187. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sekulic, A.; Migden, M.R.; Oro, A.E.; Dirix, L.; Lewis, K.D.; Hainsworth, J.D.; Solomon, J.A.; Yoo, S.; Arron, S.T.; Friedlander, P.A.; et al. Efficacy and safety of vismodegib in advanced basal-cell carcinoma. N. Engl. J. Med. 2012, 366, 2171–2179. [Google Scholar] [CrossRef] [Green Version]
- Robarge, K.D.; Brunton, S.A.; Castanedo, G.M.; Cui, Y.; Dina, M.S.; Goldsmith, R.; Gould, S.E.; Guichert, O.; Gunzner, J.L.; Halladay, J.; et al. GDC-0449-a potent inhibitor of the Hedgehog pathway. Bioorg. Med. Chem. Lett. 2009, 19, 5576–5581. [Google Scholar] [CrossRef] [PubMed]
- Bixby, D.; Noppeney, R.; Lin, T.L.; Cortes, J.; Krauter, J.; Yee, K.; Medeiros, B.C.; Krämer, A.; Assouline, S.; Fiedler, W.; et al. Safety and efficacy of vismodegib in relapsed/refractory acute myeloid leukaemia: Results of a phase Ib trial. Br. J. Haematol. 2019, 185, 595–598. [Google Scholar] [CrossRef]
- Casey, D.; Demko, S.; Shord, S.; Zhao, H.; Chen, H.; He, K.; Putman, A.; Helms, W.; Keegan, P.; Pazdur, R. FDA Approval Summary: Sonidegib for Locally Advanced Basal Cell Carcinoma. Clin. Cancer Res. 2017, 23, 2377–2381. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tibes, R.; Kosiorek, H.E.; Dueck, A.; Palmer, J.; Slack, J.L.; Knight, E.A.; Hashmi, S.K.; Bogenberger, J.M.; Zblewski, D.; Hogan, W.J.; et al. Phase I/IB Study of Azacitidine and Hedgehog Pathway Inhibition with Sonidegib (LDE225) in Myeloid Malignancies. Blood 2017, 130, 2629. [Google Scholar] [CrossRef]
- Vey, N. Addition of the SMO Inhibitor Sonidegib to Azacitidine in Patients with Higher Risk Myelodysplastic Syndrome (MDS) Who Failed to Respond or Lost Response to AZA Alone: Results of a Phase 1-2 Add-on Study By the GFM. Blood 2018, 132, 4368. [Google Scholar] [CrossRef]
- Munchhof, M.J.; Li, Q.; Shavnya, A.; Borzillo, G.V.; Boyden, T.L.; Jones, C.S.; LaGreca, S.D.; Martinez-Alsina, L.; Patel, N.; Pelletier, K.; et al. Discovery of PF-04449913, a Potent and Orally Bioavailable Inhibitor of Smoothened. ACS Med. Chem. Lett. 2012, 3, 106–111. [Google Scholar] [CrossRef] [Green Version]
- Martinelli, G.; Oehler, V.G.; Papayannidis, C.; Courtney, R.; Shaik, M.N.; Zhang, X.; O’Connell, A.; McLachlan, K.R.; Zheng, X.; Radich, J.; et al. Treatment with PF-04449913, an oral smoothened antagonist, in patients with myeloid malignancies: A phase 1 safety and pharmacokinetics study. Lancet Haematol. 2015, 2, e339–e346. [Google Scholar] [CrossRef]
- Sallman, D.A.; Komrokji, R.S.; Sweet, K.L.; Mo, Q.; McGraw, K.L.; Duong, V.H.; Zhang, L.; Nardelli, L.A.; Padron, E.; List, A.F.; et al. A phase 2 trial of the oral smoothened inhibitor glasdegib in refractory myelodysplastic syndromes (MDS). Leuk. Res. 2019, 81, 56–61. [Google Scholar] [CrossRef]
- Minami, Y.; Minami, H.; Miyamoto, T.; Yoshimoto, G.; Kobayashi, Y.; Munakata, W.; Onishi, Y.; Kobayashi, M.; Ikuta, M.; Chan, G.; et al. Phase I study of glasdegib (PF-04449913), an oral smoothened inhibitor, in Japanese patients with select hematologic malignancies. Cancer Sci. 2017, 108, 1628–1633. [Google Scholar] [CrossRef]
- Kent, A.; Vasu, S.; Schatz, D.; Monson, N.; Devine, S.; Smith, C.; Gutman, J.A.; Pollyea, D.A. Glasdegib as maintenance therapy for patients with AML and MDS patients at high risk for postallogeneic stem cell transplant relapse. Blood Adv. 2020, 4, 3102–3108. [Google Scholar] [CrossRef]
- Savona, M.R.; Pollyea, D.A.; Stock, W.; Oehler, V.G.; Schroeder, M.A.; Lancet, J.; McCloskey, J.; Kantarjian, H.M.; Ma, W.W.; Shaik, M.N.; et al. Phase Ib Study of Glasdegib, a Hedgehog Pathway Inhibitor, in Combination with Standard Chemotherapy in Patients with AML or High-Risk MDS. Clin. Cancer Res. 2018, 24, 2294–2303. [Google Scholar] [CrossRef] [Green Version]
- Cortes, J.E.; Douglas Smith, B.; Wang, E.S.; Merchant, A.; Oehler, V.G.; Arellano, M.; DeAngelo, D.J.; Pollyea, D.A.; Sekeres, M.A.; Robak, T.; et al. Glasdegib in combination with cytarabine and daunorubicin in patients with AML or high-risk MDS: Phase 2 study results. Am. J. Hematol. 2018, 93, 1301–1310. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cortes, J.E.; Heidel, F.H.; Hellmann, A.; Fiedler, W.; Smith, B.D.; Robak, T.; Montesinos, P.; Pollyea, D.A.; DesJardins, P.; Ottmann, O.; et al. Randomized comparison of low dose cytarabine with or without glasdegib in patients with newly diagnosed acute myeloid leukemia or high-risk myelodysplastic syndrome. Leukemia 2019, 33, 379–389. [Google Scholar] [CrossRef] [Green Version]
- Cortes, J.E.; Heidel, F.H.; Fiedler, W.; Smith, B.D.; Robak, T.; Montesinos, P.; Candoni, A.; Leber, B.; Sekeres, M.A.; Pollyea, D.A.; et al. Survival outcomes and clinical benefit in patients with acute myeloid leukemia treated with glasdegib and low-dose cytarabine according to response to therapy. J. Hematol. Oncol. 2020, 13, 92. [Google Scholar] [CrossRef] [PubMed]
- Cortes, J.E.; Dombret, H.; Merchant, A.; Tauchi, T.; Di Rienzo, C.G.; Sleight, B.; Zhang, X.; Leip, E.P.; Shaik, N.; Bell, T.; et al. Glasdegib plus intensive/nonintensive chemotherapy in untreated acute myeloid leukemia: BRIGHT AML 1019 Phase III trials. Future Oncol. 2019, 15, 3531–3545. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pietrobono, S.; Gagliardi, S.; Stecca, B. Non-canonical Hedgehog Signaling Pathway in Cancer: Activation of GLI Transcription Factors Beyond Smoothened. Front. Genet. 2019, 10, 556. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Riobo, N.A.; Haines, G.M.; Emerson, C.P., Jr. Protein kinase C-delta and mitogen-activated protein/extracellular signal-regulated kinase-1 control GLI activation in hedgehog signaling. Cancer Res. 2006, 66, 839–845. [Google Scholar] [CrossRef] [Green Version]
- Stecca, B.; Mas, C.; Clement, V.; Zbinden, M.; Correa, R.; Piguet, V.; Beermann, F.; Ruiz, I.A.A. Melanomas require HEDGEHOG-GLI signaling regulated by interactions between GLI1 and the RAS-MEK/AKT pathways. Proc. Natl. Acad. Sci. USA 2007, 104, 5895–5900. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Ding, Q.; Yen, C.J.; Xia, W.; Izzo, J.G.; Lang, J.Y.; Li, C.W.; Hsu, J.L.; Miller, S.A.; Wang, X.; et al. The crosstalk of mTOR/S6K1 and Hedgehog pathways. Cancer Cell 2012, 21, 374–387. [Google Scholar] [CrossRef] [Green Version]
- Dennler, S.; Andre, J.; Verrecchia, F.; Mauviel, A. Cloning of the human GLI2 Promoter: Transcriptional activation by transforming growth factor-beta via SMAD3/beta-catenin cooperation. J. Biol. Chem. 2009, 284, 31523–31531. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Atwood, S.X.; Li, M.; Lee, A.; Tang, J.Y.; Oro, A.E. GLI activation by atypical protein kinase C iota/lambda regulates the growth of basal cell carcinomas. Nature 2013, 494, 484–488. [Google Scholar] [CrossRef] [Green Version]
- Mao, J.; Maye, P.; Kogerman, P.; Tejedor, F.J.; Toftgard, R.; Xie, W.; Wu, G.; Wu, D. Regulation of Gli1 Transcriptional Activity in the Nucleus by Dyrk1. J. Biol. Chem. 2002, 277, 35156–35161. [Google Scholar] [CrossRef] [Green Version]
- Ehe, B.K.; Lamson, D.R.; Tarpley, M.; Onyenwoke, R.U.; Graves, L.M.; Williams, K.P. Identification of a DYRK1A-mediated phosphorylation site within the nuclear localization sequence of the hedgehog transcription factor GLI1. Biochem. Biophys. Res. Commun. 2017, 491, 767–772. [Google Scholar] [CrossRef]
- Lauth, M.; Bergström, Å.; Shimokawa, T.; Tostar, U.; Jin, Q.; Fendrich, V.; Guerra, C.; Barbacid, M.; Toftgård, R. DYRK1B-dependent autocrine-to-paracrine shift of Hedgehog signaling by mutant RAS. Nat. Struct. Mol. Biol. 2010, 17, 718–725. [Google Scholar] [CrossRef] [PubMed]
- Varjosalo, M.; Björklund, M.; Cheng, F.; Syvänen, H.; Kivioja, T.; Kilpinen, S.; Sun, Z.; Kallioniemi, O.; Stunnenberg, H.G.; He, W.-W.; et al. Application of Active and Kinase-Deficient Kinome Collection for Identification of Kinases Regulating Hedgehog Signaling. Cell 2008, 133, 537–548. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, B.; Fei, D.L.; Flaveny, C.A.; Dahmane, N.; Baubet, V.; Wang, Z.; Bai, F.; Pei, X.H.; Rodriguez-Blanco, J.; Hang, B.; et al. Pyrvinium attenuates Hedgehog signaling downstream of smoothened. Cancer Res. 2014, 74, 4811–4821. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Canettieri, G.; Di Marcotullio, L.; Greco, A.; Coni, S.; Antonucci, L.; Infante, P.; Pietrosanti, L.; De Smaele, E.; Ferretti, E.; Miele, E.; et al. Histone deacetylase and Cullin3-REN(KCTD11) ubiquitin ligase interplay regulates Hedgehog signalling through Gli acetylation. Nat. Cell Biol. 2010, 12, 132–142. [Google Scholar] [CrossRef] [PubMed]
- Coni, S.; Antonucci, L.; D’Amico, D.; Di Magno, L.; Infante, P.; De Smaele, E.; Giannini, G.; Di Marcotullio, L.; Screpanti, I.; Gulino, A.; et al. Gli2 acetylation at lysine 757 regulates hedgehog-dependent transcriptional output by preventing its promoter occupancy. PLoS ONE 2013, 8, e65718. [Google Scholar] [CrossRef] [Green Version]
- Zhao, J.; Quan, H.; Xie, C.; Lou, L. NL-103, a novel dual-targeted inhibitor of histone deacetylases and hedgehog pathway, effectively overcomes vismodegib resistance conferred by Smo mutations. Pharmacol. Res. Perspect. 2014, 2, e00043. [Google Scholar] [CrossRef]
- Mirza, A.N.; Fry, M.A.; Urman, N.M.; Atwood, S.X.; Roffey, J.; Ott, G.R.; Chen, B.; Lee, A.; Brown, A.S.; Aasi, S.Z.; et al. Combined inhibition of atypical PKC and histone deacetylase 1 is cooperative in basal cell carcinoma treatment. JCI Insight 2017, 2. [Google Scholar] [CrossRef]
- An, S.S.; Bai, T.R.; Bates, J.H.T.; Black, J.L.; Brown, R.H.; Brusasco, V.; Chitano, P.; Deng, L.; Dowell, M.; Eidelman, D.H.; et al. Airway smooth muscle dynamics: A common pathway of airway obstruction in asthma. Eur. Respir. J. 2007, 29, 834–860. [Google Scholar] [CrossRef]
- Tang, Y.; Gholamin, S.; Schubert, S.; Willardson, M.I.; Lee, A.; Bandopadhayay, P.; Bergthold, G.; Masoud, S.; Nguyen, B.; Vue, N.; et al. Epigenetic targeting of Hedgehog pathway transcriptional output through BET bromodomain inhibition. Nat. Med. 2014, 20, 732–740. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.; Lee, J.J.; Kim, J.; Gardner, D.; Beachy, P.A. Arsenic antagonizes the Hedgehog pathway by preventing ciliary accumulation and reducing stability of the Gli2 transcriptional effector. Proc. Natl. Acad. Sci. USA 2010, 107, 13432–13437. [Google Scholar] [CrossRef] [Green Version]
- Beauchamp, E.M.; Ringer, L.; Bulut, G.; Sajwan, K.P.; Hall, M.D.; Lee, Y.C.; Peaceman, D.; Ozdemirli, M.; Rodriguez, O.; Macdonald, T.J.; et al. Arsenic trioxide inhibits human cancer cell growth and tumor development in mice by blocking Hedgehog/GLI pathway. J. Clin. Investig. 2011, 121, 148–160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Corcoran, R.B.; Scott, M.P. Oxysterols stimulate Sonic hedgehog signal transduction and proliferation of medulloblastoma cells. Proc. Natl. Acad. Sci. USA 2006, 103, 8408–8413. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johnson, J.S.; Meliton, V.; Kim, W.K.; Lee, K.B.; Wang, J.C.; Nguyen, K.; Yoo, D.; Jung, M.E.; Atti, E.; Tetradis, S. Novel oxysterols have pro-osteogenic and anti-adipogenic effects in vitro and induce spinal fusion in vivo. J. Cell. Biochem. 2011, 112, 1673–1684. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nachtergaele, S.; Mydock, L.K.; Krishnan, K.; Rammohan, J.; Schlesinger, P.H.; Covey, D.F.; Rohatgi, R. Oxysterols are allosteric activators of the oncoprotein Smoothened. Nat. Chem. Biol. 2012, 8, 211–220. Available online: http://www.nature.com/nchembio/journal/v8/n2/abs/nchembio.765.html#supplementary-information (accessed on 14 June 2021). [CrossRef] [Green Version]
- Agarwal, J.R.; Wang, Q.; Tanno, T.; Rasheed, Z.; Merchant, A.; Ghosh, N.; Borrello, I.; Huff, C.A.; Parhami, F.; Matsui, W. Activation of Liver X Receptors inhibits of Hedgehog signaling, clonogenic growth, and self-renewal in multiple myeloma. Mol. Cancer Ther. 2014, 13, 1873–1881. [Google Scholar] [CrossRef] [Green Version]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Abraham, A.; Matsui, W. Hedgehog Signaling in Myeloid Malignancies. Cancers 2021, 13, 4888. https://doi.org/10.3390/cancers13194888
Abraham A, Matsui W. Hedgehog Signaling in Myeloid Malignancies. Cancers. 2021; 13(19):4888. https://doi.org/10.3390/cancers13194888
Chicago/Turabian StyleAbraham, Ajay, and William Matsui. 2021. "Hedgehog Signaling in Myeloid Malignancies" Cancers 13, no. 19: 4888. https://doi.org/10.3390/cancers13194888
APA StyleAbraham, A., & Matsui, W. (2021). Hedgehog Signaling in Myeloid Malignancies. Cancers, 13(19), 4888. https://doi.org/10.3390/cancers13194888