
Loss of PGRMC1 Delays the Progression of Hepatocellular Carcinoma via Suppression of Pro-Inflammatory Immune Responses

 ,
,  , , ,
, , ,  , and
, and
Abstract
Simple Summary
Abstract
1. Introduction
2. Results
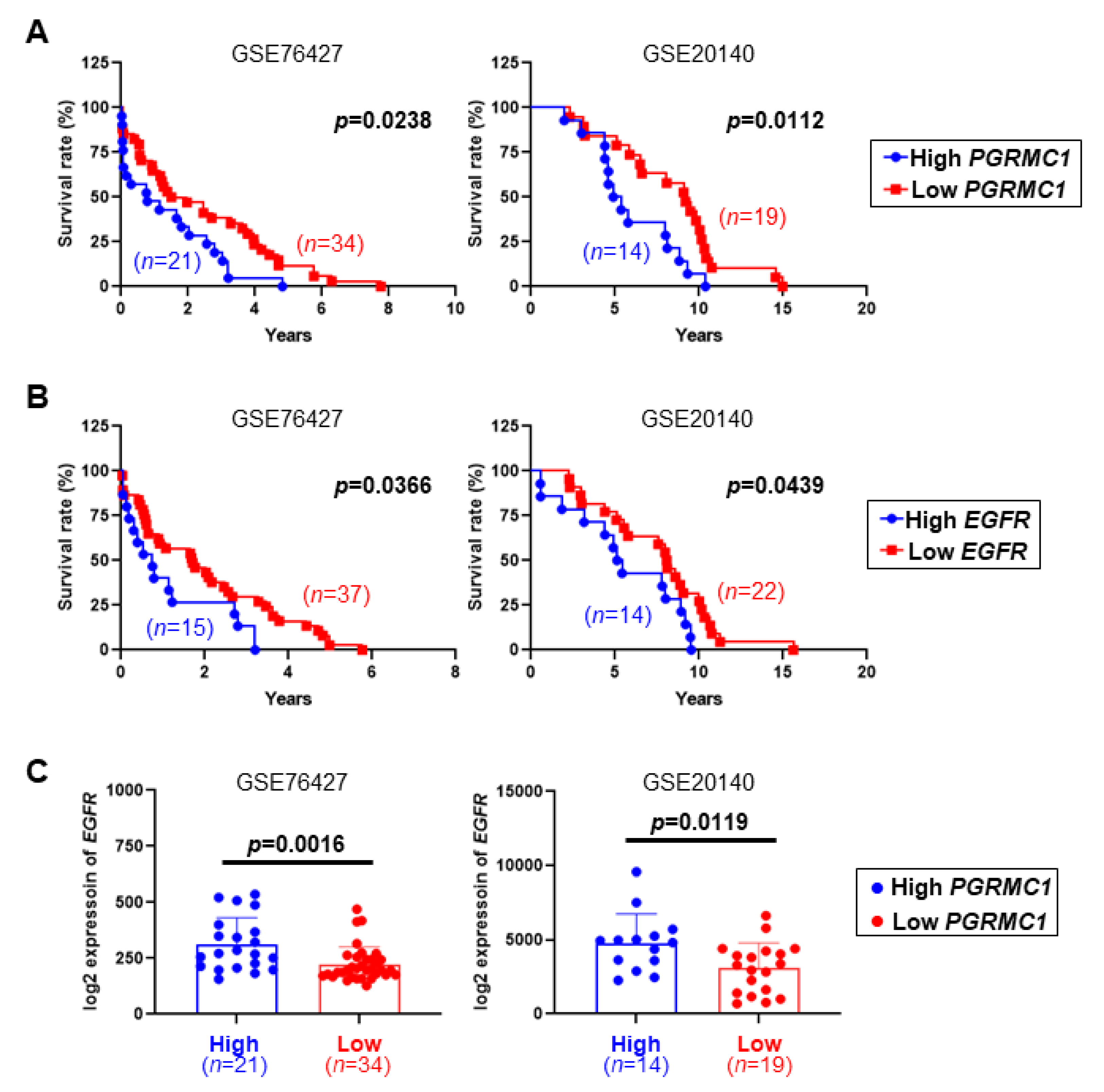
2.1. Low PGRMC1 Transcription Level Is Correlated with Extended Survival and Low EGFR Transcription in HCC
2.2. Loss of Pgrmc1 Suppresses the Progression and Lethality of HCC in a Murine Model
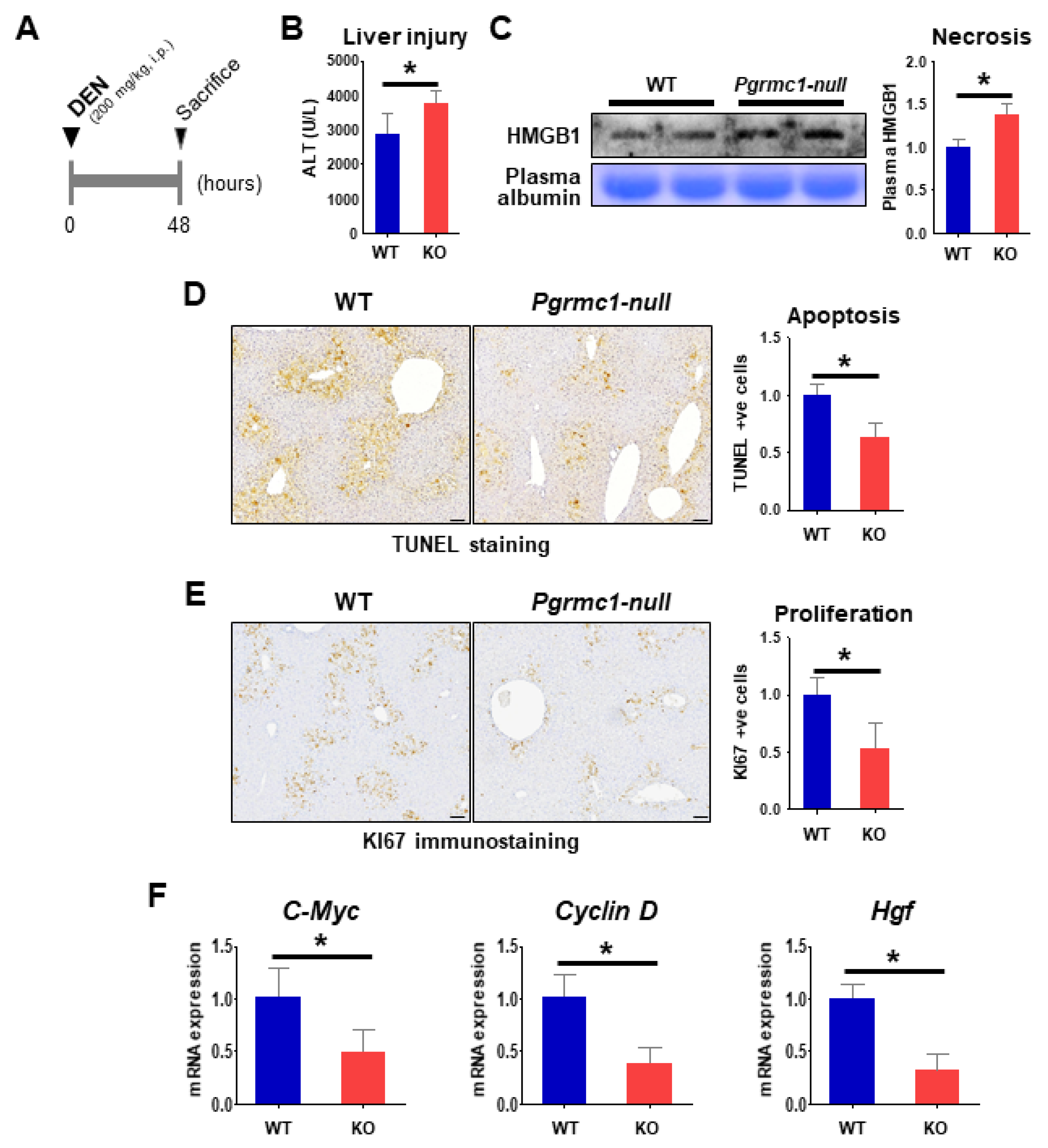
2.3. Loss of Pgrmc1 Suppresses Compensatory Proliferation in DEN-Induced Liver Injury
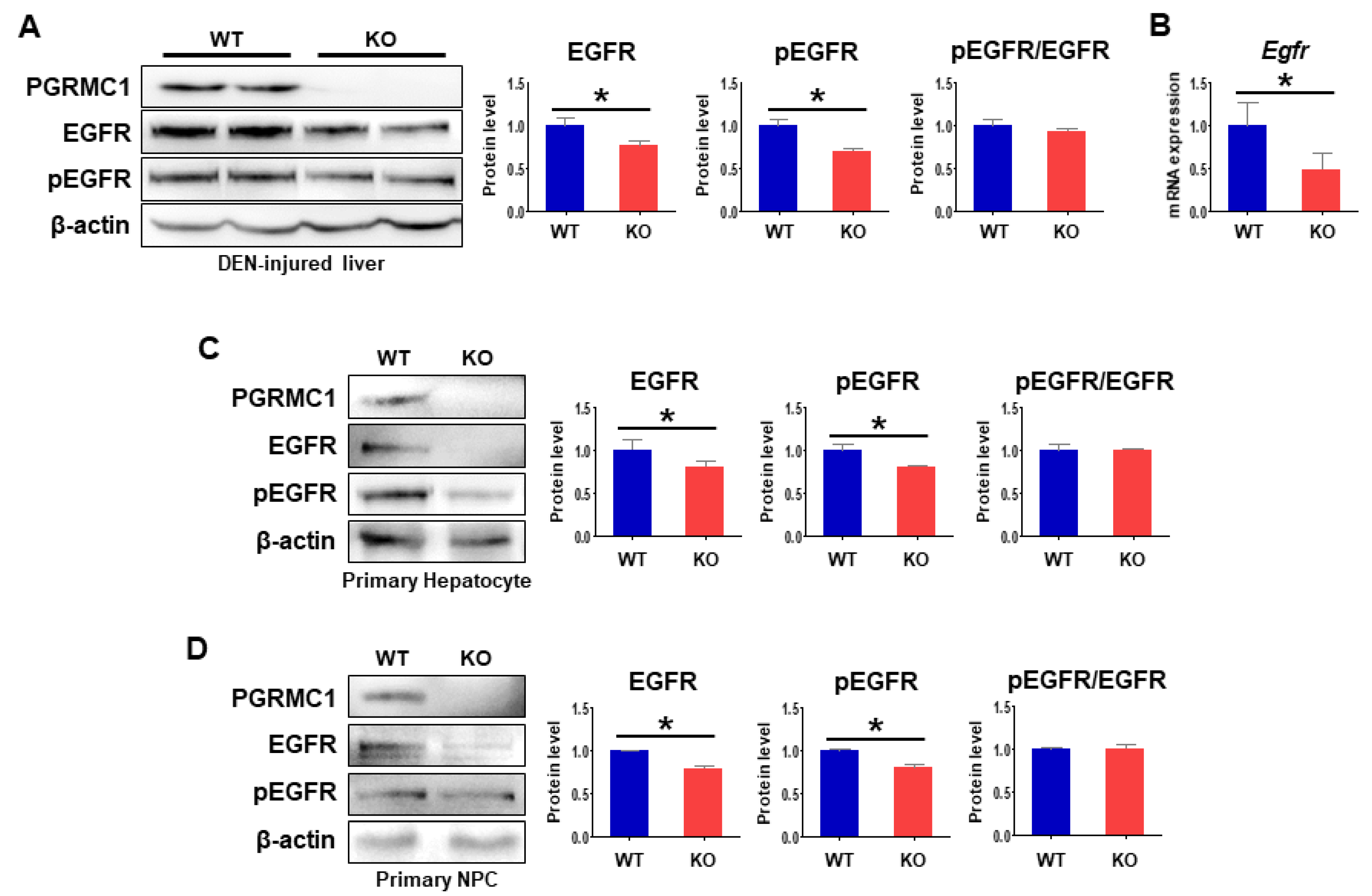
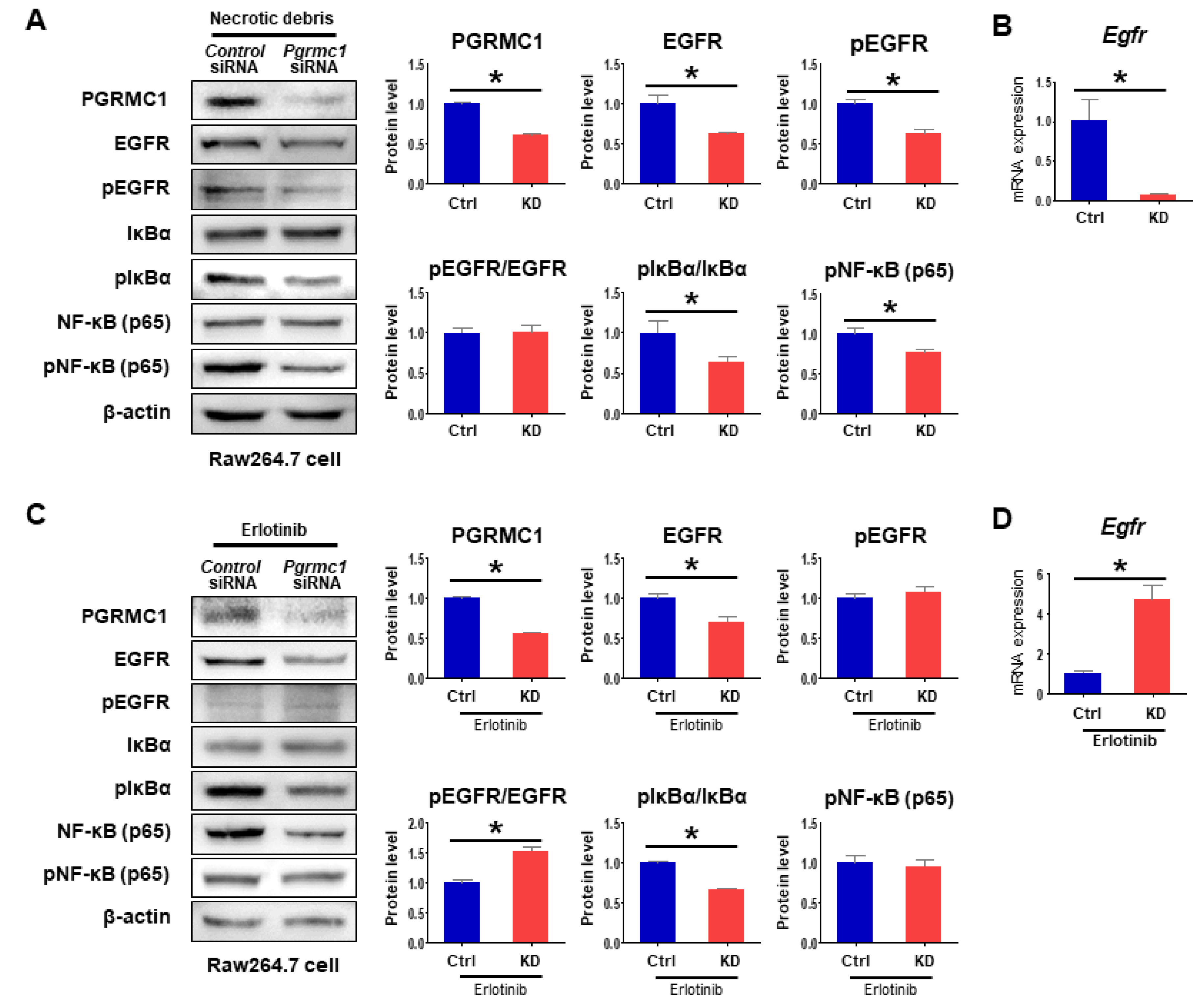
2.4. Loss of Pgrmc1 Suppresses EGFR Activation
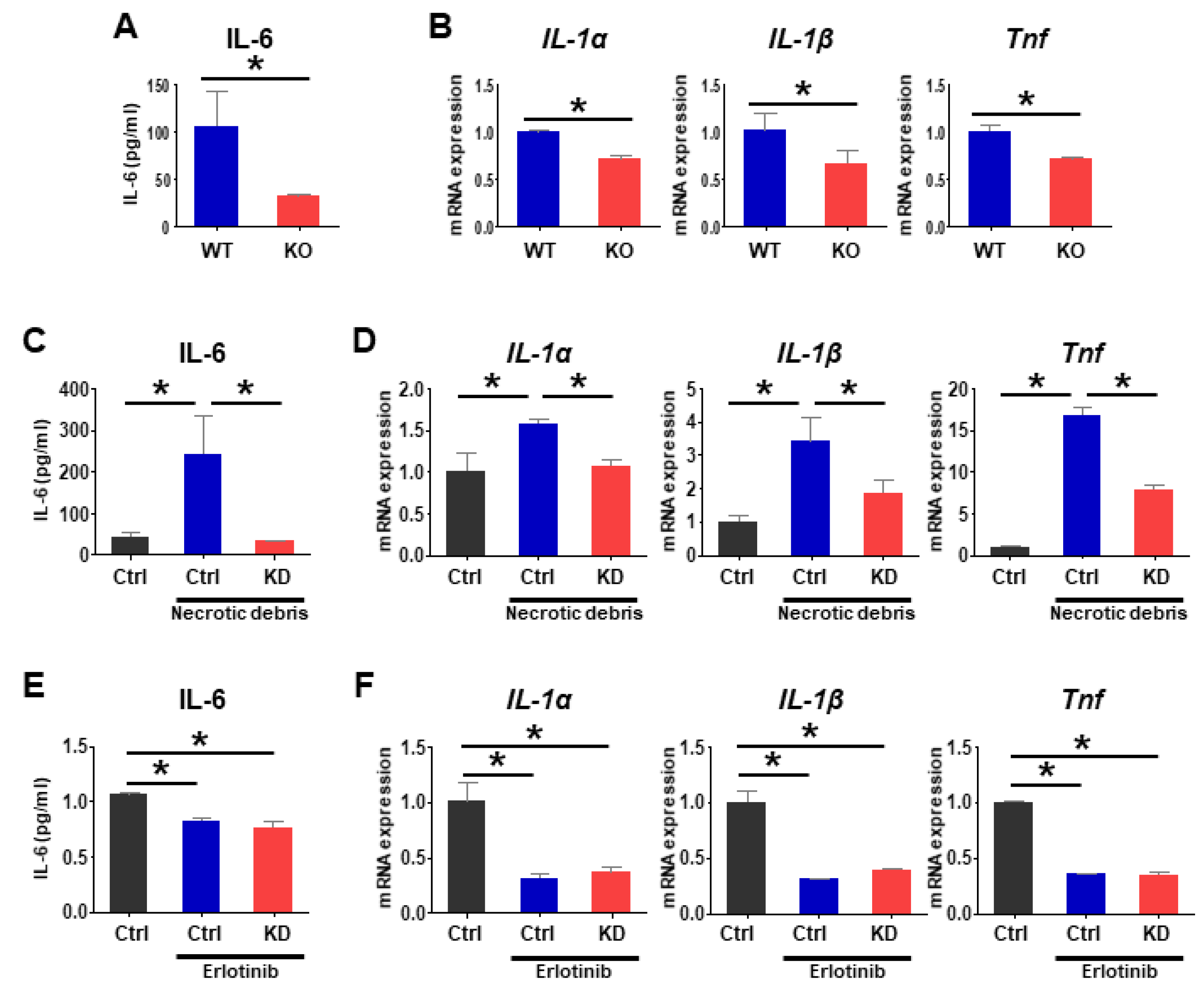
2.5. Loss of Pgrmc1 Suppresses Pro-Inflammatory Response via EGFR Expression
3. Discussion
4. Materials and Methods
4.1. GEO Dataset Analysis
4.2. Animals
4.3. In Vivo Magnetic Resonance Imaging
4.4. RNA Isolation, Reverse Transcription, and qRT-PCR
4.5. Western Blot
4.6. Cell Culture and Gene Knockdown
4.7. Primary Cell Culture
4.8. Alanine Aminotransferase (ALT) Measurement
4.9. Immunostaining
4.10. Enzyme-Linked Immunosorbent Assay (ELISA)
4.11. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Tu, T.; Bühler, S.; Bartenschlager, R. Chronic viral hepatitis and its association with liver cancer. Biol. Chem. 2017, 398, 817–837. [Google Scholar] [CrossRef] [PubMed]
- Capece, D.; Fischietti, M.; Verzella, D.; Gaggiano, A.; Cicciarelli, G.; Tessitore, A.; Zazzeroni, F.; Alesse, E. The inflammatory microenvironment in hepatocellular carcinoma: A pivotal role for tumor-associated macrophages. Biomed. Res. Int. 2013, 2013, 187204. [Google Scholar] [CrossRef]
- Forner, A.; Reig, M.; Bruix, J. Hepatocellular carcinoma. Lancet 2018, 391, 1301–1314. [Google Scholar] [CrossRef]
- Kulik, L.; El-Serag, H.B. Epidemiology and Management of Hepatocellular Carcinoma. Gastroenterology 2019, 156, 477–491. [Google Scholar] [CrossRef] [PubMed]
- Hasskarl, J. Sorafenib: Targeting multiple tyrosine kinases in cancer. Recent Results Cancer Res. 2014, 201, 145–164. [Google Scholar] [PubMed]
- Llovet, J.M.; Ricci, S.; Mazzaferro, V.; Hilgard, P.; Gane, E.; Blanc, J.F.; De Oliveira, A.C.; Santoro, A.; Raoul, J.L.; Forner, A.; et al. Sorafenib in advanced hepatocellular carcinoma. N. Engl. J. Med. 2008, 359, 378–390. [Google Scholar] [CrossRef] [PubMed]
- Kudo, M.; Finn, R.S.; Qin, S.; Han, K.-H.; Ikeda, K.; Piscaglia, F.; Baron, A.; Park, J.-W.; Han, G.; Jassem, J.; et al. Lenvatinib versus sorafenib in first-line treatment of patients with unresectable hepatocellular carcinoma: A randomised phase 3 non-inferiority trial. Lancet 2018, 391, 1163–1173. [Google Scholar] [CrossRef]
- Cheng, A.-L.; Kang, Y.-K.; Chen, Z.; Tsao, C.-J.; Qin, S.; Kim, J.S.; Luo, R.; Feng, J.; Ye, S.; Yang, T.-S.; et al. Efficacy and safety of sorafenib in patients in the Asia-Pacific region with advanced hepatocellular carcinoma: A phase III randomised, double-blind, placebo-controlled trial. Lancet Oncol. 2009, 10, 25–34. [Google Scholar] [CrossRef]
- Ito, Y.; Takeda, T.; Sakon, M.; Tsujimoto, M.; Higashiyama, S.; Noda, K.; Miyoshi, E.; Monden, M.; Matsuura, N. Expression and clinical significance of erb-B receptor family in hepatocellular carcinoma. Br. J. Cancer 2001, 84, 1377–1383. [Google Scholar] [CrossRef]
- Buckley, A.F.; Burgart, L.J.; Sahai, V.; Kakar, S. Epidermal growth factor receptor expression and gene copy number in conventional hepatocellular carcinoma. Am. J. Clin. Pathol. 2008, 129, 245–251. [Google Scholar] [CrossRef]
- Höpfner, M.; Sutter, A.P.; Huether, A.; Schuppan, D.; Zeitz, M.; Scherübl, H. Targeting the epidermal growth factor receptor by gefitinib for treatment of hepatocellular carcinoma. J. Hepatol. 2004, 41, 1008–1016. [Google Scholar] [CrossRef]
- Schiffer, E.; Housset, C.; Cacheux, W.; Wendum, D.; Rey, C.; Poupon, R.; Rosmorduc, O. Gefitinib, an EGFR inhibitor, prevents hepatocellular carcinoma development in the rat liver with cirrhosis. Hepatology 2005, 41, 307–314. [Google Scholar] [CrossRef] [PubMed]
- Whittaker, S.; Marais, R.; Zhu, A.X. The role of signaling pathways in the development and treatment of hepatocellular carcinoma. Oncogene 2010, 29, 4989–5005. [Google Scholar] [CrossRef] [PubMed]
- Philip, P.A.; Mahoney, M.R.; Allmer, C.; Thomas, J.; Pitot, H.C.; Kim, G.; Donehower, R.C.; Fitch, T.; Picus, J.; Erlichman, C. Phase II study of Erlotinib (OSI-774) in patients with advanced hepatocellular cancer. J. Clin. Oncol. 2005, 23, 6657–6663. [Google Scholar] [CrossRef]
- Thomas, M.B.; Chadha, R.; Glover, K.; Wang, X.; Morris, J.; Brown, T.; Rashid, A.; Dancey, J.; Abbruzzese, J.L. Phase 2 study of erlotinib in patients with unresectable hepatocellular carcinoma. Cancer 2007, 110, 1059–1067. [Google Scholar] [CrossRef]
- Zhu, A.X.; Stuart, K.; Blaszkowsky, L.S.; Muzikansky, A.; Reitberg, D.P.; Clark, J.W.; Enzinger, P.C.; Bhargava, P.; Meyerhardt, J.A.; Horgan, K.; et al. Phase 2 study of cetuximab in patients with advanced hepatocellular carcinoma. Cancer 2007, 110, 581–589. [Google Scholar] [CrossRef] [PubMed]
- Ezzoukhry, Z.; Louandre, C.; Trécherel, E.; Godin, C.; Chauffert, B.; Dupont, S.; Diouf, M.; Barbare, J.-C.; Mazière, J.-C.; Galmiche, A. EGFR activation is a potential determinant of primary resistance of hepatocellular carcinoma cells to sorafenib. Int. J. Cancer 2012, 131, 2961–2969. [Google Scholar] [CrossRef]
- Lin, C.-H.; Elkholy, K.H.; Wani, N.A.; Li, D.; Hu, P.; Barajas, J.M.; Yu, L.; Zhang, X.; Jacob, S.T.; Khan, W.N.; et al. Ibrutinib Potentiates Antihepatocarcinogenic Efficacy of Sorafenib by Targeting EGFR in Tumor Cells and BTK in Immune Cells in the Stroma. Mol. Cancer Ther. 2020, 19, 384–396. [Google Scholar] [CrossRef] [PubMed]
- Kabe, Y.; Nakane, T.; Koike, I.; Yamamoto, T.; Sugiura, Y.; Harada, E.; Sugase, K.; Shimamura, T.; Ohmura, M.; Muraoka, K.; et al. Haem-dependent dimerization of PGRMC1/Sigma-2 receptor facilitates cancer proliferation and chemoresistance. Nat. Commun. 2016, 7, 11030. [Google Scholar] [CrossRef] [PubMed]
- McCallum, M.L.; Pru, C.A.; Niikura, Y.; Yee, S.-P.; Lydon, J.P.; Peluso, J.J.; Pru, J.K. Conditional Ablation of Progesterone Receptor Membrane Component 1 Results in Subfertility in the Female and Development of Endometrial Cysts. Endocrinology 2016, 157, 3309–3319. [Google Scholar] [CrossRef]
- Wu, X.-J.; Zhu, Y. Downregulation of nuclear progestin receptor (Pgr) and subfertility in double knockouts of progestin receptor membrane component 1 (pgrmc1) and pgrmc2 in zebrafish. Gen. Comp. Endocrinol. 2020, 285, 113275. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.-J.; Thomas, P.; Zhu, Y. Pgrmc1 Knockout Impairs Oocyte Maturation in Zebrafish. Front. Endocrinol. 2018, 9, 560. [Google Scholar] [CrossRef] [PubMed]
- Clark, N.C.; Pru, C.A.; Yee, S.-P.; Lydon, J.P.; Peluso, J.J.; Pru, J.K. Conditional Ablation of Progesterone Receptor Membrane Component 2 Causes Female Premature Reproductive Senescence. Endocrinology 2017, 158, 640–651. [Google Scholar] [CrossRef]
- Kim, G.; Lee, J.G.; Cheong, S.-A.; Yon, J.-M.; Lee, M.S.; Hong, E.-J.; Baek, I.-J. Progesterone receptor membrane component 1 is required for mammary gland developmentdagger. Biol. Reprod. 2020, 103, 1249–1259. [Google Scholar] [CrossRef]
- Lee, S.R.; Yang, H.; Jo, S.L.; Lee, Y.H.; Lee, H.W.; Park, B.-K.; Hong, E.-J. Suppressed estrogen supply via extra-ovarian progesterone receptor membrane component 1 in menopause. J. Biomed. Res. 2021, 2, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Cahill, M.A.; Medlock, A.E. Thoughts on interactions between PGRMC1 and diverse attested and potential hydrophobic ligands. J. Steroid Biochem. Mol. Biol. 2017, 171, 11–33. [Google Scholar] [CrossRef]
- Thejer, B.M.; Adhikary, P.P.; Teakel, S.L.; Fang, J.; Weston, P.A.; Gurusinghe, S.; Anwer, A.G.; Gosnell, M.; Jazayeri, J.A.; Ludescher, M.; et al. PGRMC1 effects on metabolism, genomic mutation and CpG methylation imply crucial roles in animal biology and disease. BMC Mol. Cell Biol. 2020, 21, 26. [Google Scholar] [CrossRef] [PubMed]
- Thejer, B.M.; Adhikary, P.P.; Kaur, A.; Teakel, S.L.; Van Oosterum, A.; Seth, I.; Pajic, M.; Hannan, K.M.; Pavy, M.; Poh, P.; et al. PGRMC1 phosphorylation affects cell shape, motility, glycolysis, mitochondrial form and function, and tumor growth. BMC Mol. Cell Biol. 2020, 21, 24. [Google Scholar] [CrossRef]
- Lee, S.R.; Heo, J.H.; Jo, S.L.; Kim, G.; Kim, S.J.; Yoo, H.J.; Lee, K.-P.; Kwun, H.-J.; Shin, H.-J.; Baek, I.-J.; et al. Progesterone receptor membrane component 1 reduces cardiac steatosis and lipotoxicity via activation of fatty acid oxidation and mitochondrial respiration. Sci. Rep. 2021, 11, 8781. [Google Scholar] [CrossRef]
- Lee, S.R.; Choi, W.-Y.; Heo, J.H.; Huh, J.; Kim, G.; Lee, K.-P.; Kwun, H.-J.; Shin, H.-J.; Baek, I.-J.; Hong, E.-J. Progesterone increases blood glucose via hepatic progesterone receptor membrane component 1 under limited or impaired action of insulin. Sci. Rep. 2020, 10, 16316. [Google Scholar] [CrossRef] [PubMed]
- Cai, G.; Yang, X.; Ruan, X.; Wang, J.; Fang, Y.; Wei, Y.; Zhang, Y.; Gu, M.; Mueck, A.O. Association of circulating Progesterone Receptor Membrane Component-1 (PGRMC1) with PGRMC1 expression in breast tumour tissue and with clinical breast tumour characteristics. Maturitas 2020, 140, 64–71. [Google Scholar] [CrossRef]
- Ruan, X.; Zhang, Y.; Mueck, A.O.; Willibald, M.; Seeger, H.; Fehm, T.; Brucker, S.; Neubauer, H. Increased expression of progesterone receptor membrane component 1 is associated with aggressive phenotype and poor prognosis in ER-positive and negative breast cancer. Menopause 2017, 24, 203–209. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.R.; Lee, Y.H.; Jo, S.L.; Heo, J.H.; Kim, G.; Lee, G.-S.; An, B.-S.; Baek, I.-J.; Hong, E.-J. Absence of progesterone receptor membrane component 1 reduces migration and metastasis of breast cancer. Cell Commun. Signal. 2021, 19, 42. [Google Scholar] [CrossRef]
- Zhao, Y.; Ruan, X. Identification of PGRMC1 as a Candidate Oncogene for Head and Neck Cancers and Its Involvement in Metabolic Activities. Front. Bioeng. Biotechnol. 2019, 7, 438. [Google Scholar] [CrossRef] [PubMed]
- Hampton, K.K.; Stewart, R.; Napier, D.; Claudio, P.P.; Craven, R.J. PGRMC1 Elevation in Multiple Cancers and Essential Role in Stem Cell Survival. Adv. Lung Cancer 2015, 4, 37–51. [Google Scholar] [CrossRef]
- Ahmed, I.S.; Rohe, H.J.; Twist, K.E.; Craven, R.J. Pgrmc1 (progesterone receptor membrane component 1) associates with epidermal growth factor receptor and regulates erlotinib sensitivity. J. Biol. Chem. 2010, 285, 24775–24782. [Google Scholar] [CrossRef] [PubMed]
- Pedroza, D.A.; Rajamanickam, V.; Subramani, R.; Bencomo, A.; Galvez, A.; Lakshmanaswamy, R. Progesterone receptor membrane component 1 promotes the growth of breast cancers by altering the phosphoproteome and augmenting EGFR/PI3K/AKT signalling. Br. J. Cancer 2020, 123, 1326–1335. [Google Scholar] [CrossRef]
- Zhou, F.; Shang, W.; Yu, X.; Tian, J. Glypican-3: A promising biomarker for hepatocellular carcinoma diagnosis and treatment. Med. Res. Rev. 2018, 38, 741–767. [Google Scholar] [CrossRef]
- Naugler, W.E.; Sakurai, T.; Kim, S.; Maeda, S.; Kim, K.; Elsharkawy, A.M.; Karin, M. Gender disparity in liver cancer due to sex differences in MyD88-dependent IL-6 production. Science 2007, 317, 121–124. [Google Scholar] [CrossRef] [PubMed]
- Shang, N.; Bank, T.; Ding, X.; Breslin, P.; Li, J.; Shi, B.; Qiu, W. Caspase-3 suppresses diethylnitrosamine-induced hepatocyte death, compensatory proliferation and hepatocarcinogenesis through inhibiting p38 activation. Cell Death Dis. 2018, 9, 558. [Google Scholar] [CrossRef]
- Goyal, L.; Muzumdar, M.D.; Zhu, A.X. Targeting the HGF/c-MET pathway in hepatocellular carcinoma. Clin. Cancer Res. 2013, 19, 2310–2318. [Google Scholar] [CrossRef]
- Chen, J.; Li, X.; Cheng, Q.; Ning, D.; Ma, J.; Zhang, Z.; Chen, X.; Jiang, L. Effects of cyclin D1 gene silencing on cell proliferation, cell cycle, and apoptosis of hepatocellular carcinoma cells. J. Cell Biochem. 2018, 119, 2368–2380. [Google Scholar] [CrossRef] [PubMed]
- Qu, A.; Jiang, C.; Cai, Y.; Kim, J.-H.; Tanaka, N.; Ward, J.M.; Shah, Y.M.; Gonzalez, F.J. Role of Myc in hepatocellular proliferation and hepatocarcinogenesis. J. Hepatol. 2014, 60, 331–338. [Google Scholar] [CrossRef] [PubMed]
- He, G.; Karin, M. NF-kappaB and STAT3—Key players in liver inflammation and cancer. Cell Res. 2011, 21, 159–168. [Google Scholar] [CrossRef]
- Sakurai, T.; He, G.; Matsuzawa, A.; Yu, G.-Y.; Maeda, S.; Hardiman, G.; Karin, M. Hepatocyte necrosis induced by oxidative stress and IL-1 alpha release mediate carcinogen-induced compensatory proliferation and liver tumorigenesis. Cancer Cell 2008, 14, 156–165. [Google Scholar] [CrossRef]
- Maeda, S.; Kamata, H.; Luo, J.-L.; Leffert, H.; Karin, M. IKKbeta couples hepatocyte death to cytokine-driven compensatory proliferation that promotes chemical hepatocarcinogenesis. Cell 2005, 121, 977–990. [Google Scholar] [CrossRef]
- Schwabe, R.F.; Brenner, D.A. Mechanisms of Liver Injury. I. TNF-alpha-induced liver injury: Role of IKK, JNK, and ROS pathways. Am. J. Physiol. Liver Physiol. 2006, 290, G583–G589. [Google Scholar]
- Jing, Y.; Sun, K.; Liu, W.; Sheng, D.; Zhao, S.; Gao, L.; Wei, L. Tumor necrosis factor-alpha promotes hepatocellular carcinogenesis through the activation of hepatic progenitor cells. Cancer Lett. 2018, 434, 22–32. [Google Scholar] [CrossRef]
- Luedde, T.; Schwabe, R.F. NF-kappaB in the liver--linking injury, fibrosis and hepatocellular carcinoma. Nat. Rev. Gastroenterol. Hepatol. 2011, 8, 108–118. [Google Scholar] [CrossRef]
- Lian, Q.; Wang, S.; Zhang, G.; Wang, D.; Luo, G.; Tang, J.; Chen, L.; Gu, J. HCCDB: A Database of Hepatocellular Carcinoma Expression Atlas. Genom. Proteom. Bioinform. 2018, 16, 269–275. [Google Scholar] [CrossRef] [PubMed]
- Tessitore, L. Apoptosis and cell proliferation are involved in the initiation of liver carcinogenesis by a subnecrogenic dose of diethylnitrosamine in refed rats. J. Nutr. 2000, 130, 104–110. [Google Scholar] [CrossRef]
- Ying, T.S.; Sarma, D.S.; Farber, E. Role of acute hepatic necrosis in the induction of early steps in liver carcinogenesis by diethylnitrosamine. Cancer Res. 1981, 41, 2096–2102. [Google Scholar] [PubMed]
- Hong, E.-J.; Levasseur, M.-P.; Dufour, C.R.; Perry, M.-C.; Giguère, V. Loss of estrogen-related receptor alpha promotes hepatocarcinogenesis development via metabolic and inflammatory disturbances. Proc. Natl. Acad. Sci. USA 2013, 110, 17975–17980. [Google Scholar] [CrossRef]
- Yu, L.-X.; Ling, Y.; Wang, H.-Y. Role of nonresolving inflammation in hepatocellular carcinoma development and progression. NPJ Precis. Oncol. 2018, 2, 6. [Google Scholar] [CrossRef] [PubMed]
- Mantovani, A.; Allavena, P.; Sica, A.; Balkwill, F.R. Cancer-related inflammation. Nature 2008, 454, 436–444. [Google Scholar] [CrossRef] [PubMed]
- Scaffidi, P.; Misteli, T.; Bianchi, M.E. Release of chromatin protein HMGB1 by necrotic cells triggers inflammation. Nature 2002, 418, 191–195. [Google Scholar] [CrossRef] [PubMed]
- Qin, L.X. Inflammatory immune responses in tumor microenvironment and metastasis of hepatocellular carcinoma. Cancer Microenviron. 2012, 5, 203–209. [Google Scholar] [CrossRef]
- Tian, Z.; Hou, X.; Liu, W.; Han, Z.; Wei, L. Macrophages and hepatocellular carcinoma. Cell Biosci. 2019, 9, 79. [Google Scholar] [CrossRef]
- Cassetta, L.; Pollard, J.W. Targeting macrophages: Therapeutic approaches in cancer. Nat. Rev. Drug Discov. 2018, 17, 887–904. [Google Scholar] [CrossRef]
- Ruffell, B.; Coussens, L.M. Coussens, Macrophages and therapeutic resistance in cancer. Cancer Cell 2015, 27, 462–472. [Google Scholar] [CrossRef] [PubMed]
- Galdiero, M.R.; Bonavita, E.; Barajon, I.; Garlanda, C.; Mantovani, A.; Jaillon, S. Tumor associated macrophages and neutrophils in cancer. Immunobiology 2013, 218, 1402–1410. [Google Scholar] [CrossRef]
- Noy, R.; Pollard, J.W. Tumor-associated macrophages: From mechanisms to therapy. Immunity 2014, 41, 49–61. [Google Scholar] [CrossRef] [PubMed]
- Jiang, J.; Wang, G.-Z.; Wang, Y.; Huang, H.-Z.; Li, W.-T.; Qu, X.-D. Hypoxia-induced HMGB1 expression of HCC promotes tumor invasiveness and metastasis via regulating macrophage-derived IL-6. Exp. Cell Res. 2018, 367, 81–88. [Google Scholar] [CrossRef] [PubMed]
- Karin, M.; Greten, F.R. NF-kappaB: Linking inflammation and immunity to cancer development and progression. Nat. Rev. Immunol. 2005, 5, 749–759. [Google Scholar] [CrossRef]
- Lin, Y.; Higashisaka, K.; Shintani, T.; Maki, A.; Hanamuro, S.; Haga, Y.; Maeda, S.; Tsujino, H.; Nagano, K.; Fujio, Y.; et al. Progesterone receptor membrane component 1 leads to erlotinib resistance, initiating crosstalk of Wnt/beta-catenin and NF-kappaB pathways, in lung adenocarcinoma cells. Sci. Rep. 2020, 10, 4748. [Google Scholar] [CrossRef]
- Cahill, M.A.; Jazayeri, J.A.; Kovacevic, Z.; Richardson, D.R. PGRMC1 regulation by phosphorylation: Potential new insights in controlling biological activity. Oncotarget 2016, 7, 50822–50827. [Google Scholar] [CrossRef]
- Natarajan, A.; Wagner, B.; Sibilia, M. The EGF receptor is required for efficient liver regeneration. Proc. Natl. Acad. Sci. USA 2007, 104, 17081–17086. [Google Scholar] [CrossRef]
- Xia, H.; Dai, X.; Yu, H.; Zhou, S.; Fan, Z.; Wei, G.; Tang, Q.; Gong, Q.; Bi, F. EGFR-PI3K-PDK1 pathway regulates YAP signaling in hepatocellular carcinoma: The mechanism and its implications in targeted therapy. Cell Death Dis. 2018, 9, 269. [Google Scholar] [CrossRef]
- Fuchs, B.C.; Hoshida, Y.; Fujii, T.; Wei, L.; Yamada, S.; Lauwers, G.Y.; McGinn, C.M.; Deperalta, D.K.; Chen, X.; Kuroda, T.; et al. Epidermal growth factor receptor inhibition attenuates liver fibrosis and development of hepatocellular carcinoma. Hepatology 2014, 59, 1577–1590. [Google Scholar] [CrossRef] [PubMed]
- Lanaya, H.; Natarajan, A.; Komposch, K.; Li, L.; Amberg, N.; Chen, L.; Wculek, S.K.; Hammer, M.; Zenz, R.; Peck-Radosavljevic, M.; et al. EGFR has a tumour-promoting role in liver macrophages during hepatocellular carcinoma formation. Nat. Cell Biol. 2014, 16, 972–977. [Google Scholar] [CrossRef]
- Tsai, H.-W.; Ho, C.-L.; Cheng, S.-W.; Lin, Y.-J.; Chen, C.-C.; Cheng, P.-N.; Yen, C.-J.; Chang, T.-T.; Chiang, P.-M.; Chan, S.-H.; et al. Progesterone receptor membrane component 1 as a potential prognostic biomarker for hepatocellular carcinoma. World J. Gastroenterol. 2018, 24, 1152–1166. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.R.; Kwon, S.W.; Kaya, P.; Lee, Y.H.; Lee, J.G.; Kim, G.; Lee, G.-S.; Baek, I.-J.; Hong, E.-J. Loss of progesterone receptor membrane component 1 promotes hepatic steatosis via the induced de novo lipogenesis. Sci. Rep. 2018, 8, 15711. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene Name | Upper Primer (5′–3′) | Lower Primer (5′–3′) | Species |
|---|---|---|---|
| IL-1α | AGT ATC AGC AAC GTC AAG CAA | TCC AGA TCA TGG GTT ATG GAC TG | Mouse |
| IL-1β | GAA ATG CCA CCT TTT GAC AGT G | CTG GAT GCT CTC ATC AGG ACA | Mouse |
| TNF | CCT GTA GCC CAC GTC GTA G | GGG AGT AGA CAA GGT ACA ACC C | Mouse |
| Ccl19 | GGG GTG CTA ATG ATG CGG AA | CCT TAG TGT GGT GAA CAC AAC A | Mouse |
| Ccl21 | GTG ATG GAG GGG GTC AGG A | GGG ATG GGA CAG CCT AAA CT | Mouse |
| Icam1 | GTG ATG CTC AGG TAT CCA TCC A | CAC AGT TCT CAA AGC ACA GCG | Mouse |
| F4/80 | TTG TAC GTG CAA CTC AGG ACT | GAT CCC AGA GTG TTG ATG CAA | Mouse |
| Egfr | GCA TCA TGG GAG AGA ACA ACA | TCA GGA ACC ATT ACT CCA TAG GT | Mouse |
| Rplp0 | GCA GCA GAT CCG CAT GTC GCT CCG | GAG CTG GCA CAG TGA CCT CAC ACG G | Mouse |
| C-Myc | GCT CTC CAT CCT ATG TTG CGG | TCC AAG TAA CTC GGT CAT CAT CT | Mouse |
| Cyclin D | GCG TAC CCT GAC ACC AAT CTC | CTC CTC TTC GCA CTT CTG CTC | Mouse |
| Hgf | TTC ATG TCG CCA TCC CCT ATG | CCC CTG TTC CTG ATA CAC CT | Mouse |
| C-Myc | CCT ACC CTC TCA ACG ACA GC | CTC TGA CCT TTT GCC AGG AG | Human |
| Cyclin D | GCT GCG AAG TGG AAA CCA TC | CCT CCT TCT GCA CAC ATT TGA A | Human |
| Hgf | CTG GTT CCC CTT CAA TAG CA | AAC TCC AGG GCT GAC ATT TG | Human |
| Rplp0 | TCG ACA ATG GCA GCA TCT AC | GCC TTG ACC TTT TCA GCA AG | Human |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lee, S.R.; Lee, J.G.; Heo, J.H.; Jo, S.L.; Ryu, J.; Kim, G.; Yon, J.-M.; Lee, M.S.; Lee, G.-S.; An, B.-S.; et al. Loss of PGRMC1 Delays the Progression of Hepatocellular Carcinoma via Suppression of Pro-Inflammatory Immune Responses. Cancers 2021, 13, 2438. https://doi.org/10.3390/cancers13102438
Lee SR, Lee JG, Heo JH, Jo SL, Ryu J, Kim G, Yon J-M, Lee MS, Lee G-S, An B-S, et al. Loss of PGRMC1 Delays the Progression of Hepatocellular Carcinoma via Suppression of Pro-Inflammatory Immune Responses. Cancers. 2021; 13(10):2438. https://doi.org/10.3390/cancers13102438
Chicago/Turabian StyleLee, Sang R., Jong Geol Lee, Jun H. Heo, Seong Lae Jo, Jihoon Ryu, Globinna Kim, Jung-Min Yon, Myeong Sup Lee, Geun-Shik Lee, Beum-Soo An, and et al. 2021. "Loss of PGRMC1 Delays the Progression of Hepatocellular Carcinoma via Suppression of Pro-Inflammatory Immune Responses" Cancers 13, no. 10: 2438. https://doi.org/10.3390/cancers13102438
APA StyleLee, S. R., Lee, J. G., Heo, J. H., Jo, S. L., Ryu, J., Kim, G., Yon, J.-M., Lee, M. S., Lee, G.-S., An, B.-S., Shin, H.-J., Woo, D.-C., Baek, I.-J., & Hong, E.-J. (2021). Loss of PGRMC1 Delays the Progression of Hepatocellular Carcinoma via Suppression of Pro-Inflammatory Immune Responses. Cancers, 13(10), 2438. https://doi.org/10.3390/cancers13102438