CD19-CAR-T Cells Bearing a KIR/PD-1-Based Inhibitory CAR Eradicate CD19+HLA-C1− Malignant B Cells While Sparing CD19+HLA-C1+ Healthy B Cells



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Simple Summary
Abstract
1. Introduction
2. Results
2.1. iKP CAR Doesn’t Affect CAR Expression, Viability, Proliferation and Subsets of CD19-CAR-T Cells
2.2. iKP CAR Functions via PD-1 Signaling Upon Interacting with HLA-C1
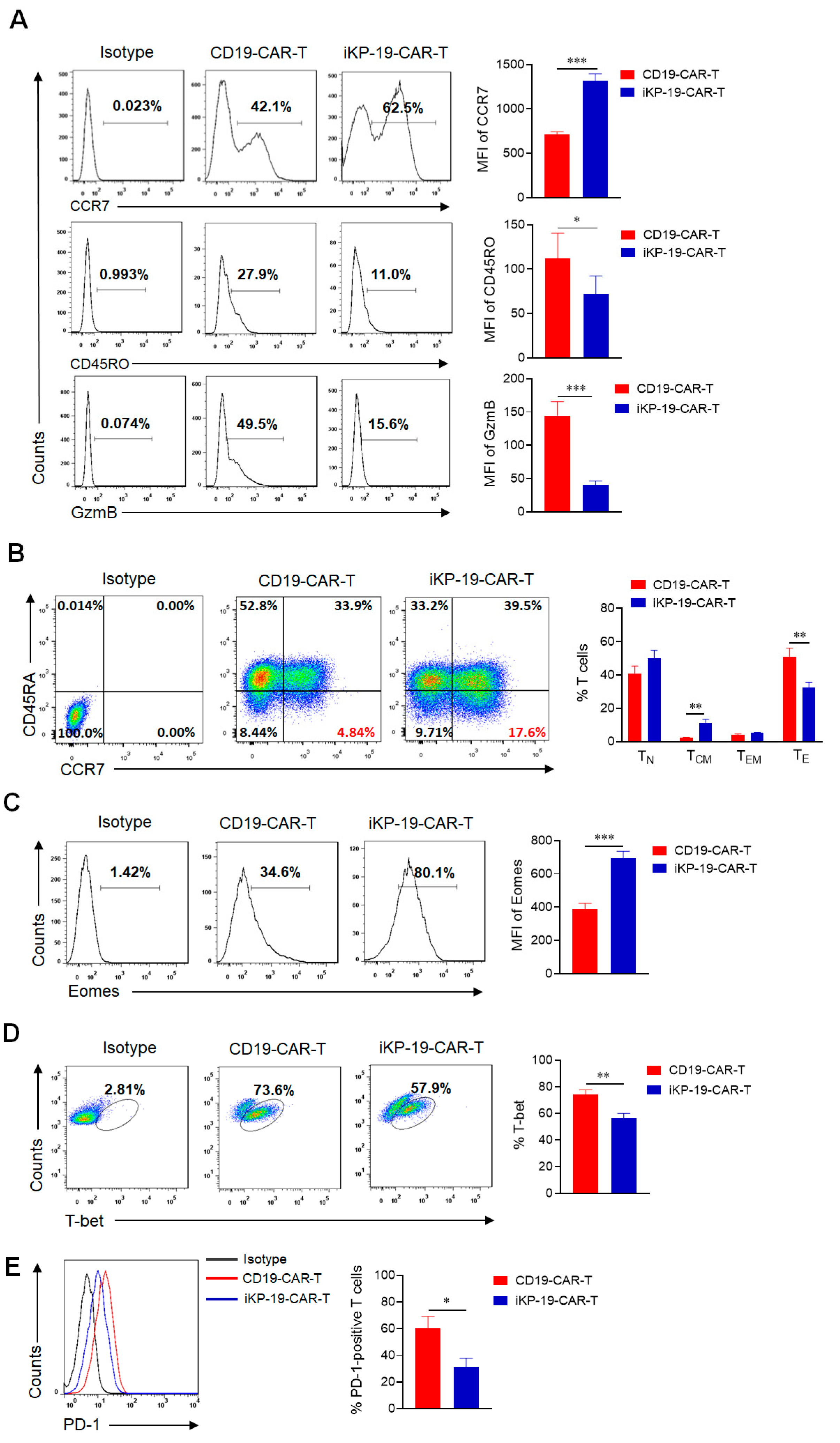
2.3. iKP CAR Renders CD19-CAR-T Cells in Less Differentiated and Less Exhausted State Prior to Antigen Engagement
2.4. CD19-CAR-T Cells Bearing an iKP CAR Eradicate CD19+HLA-C1− Daudi Cells and Present Lower Cytotoxicity on CD19+HLA-C1+ Normal B Cells In Vitro
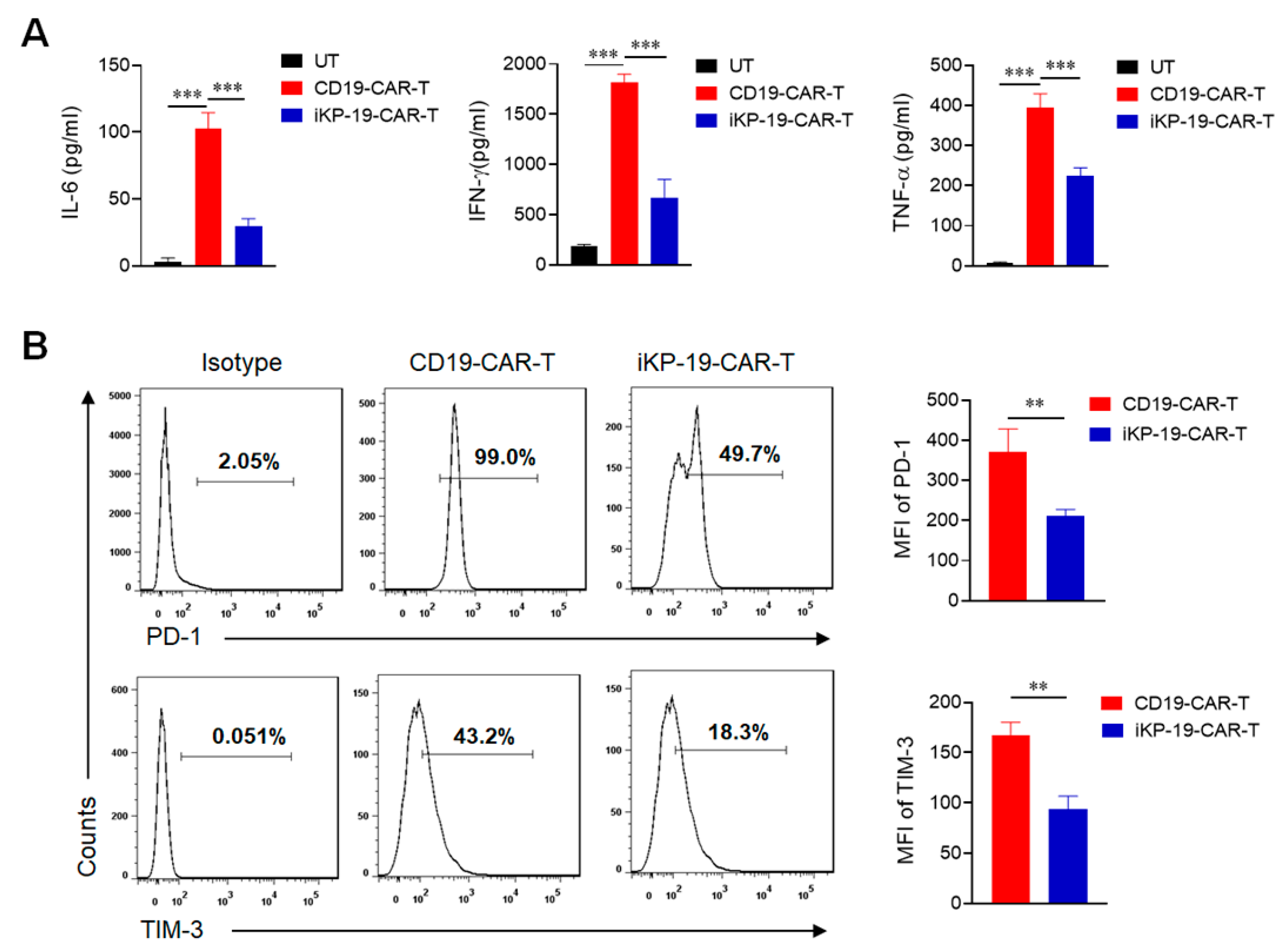
2.5. CD19-CAR-T Cells Bearing an iKP CAR Release Less Cytokines and Express Lower Exhaustion Markers during Lysing Malignant B Cells
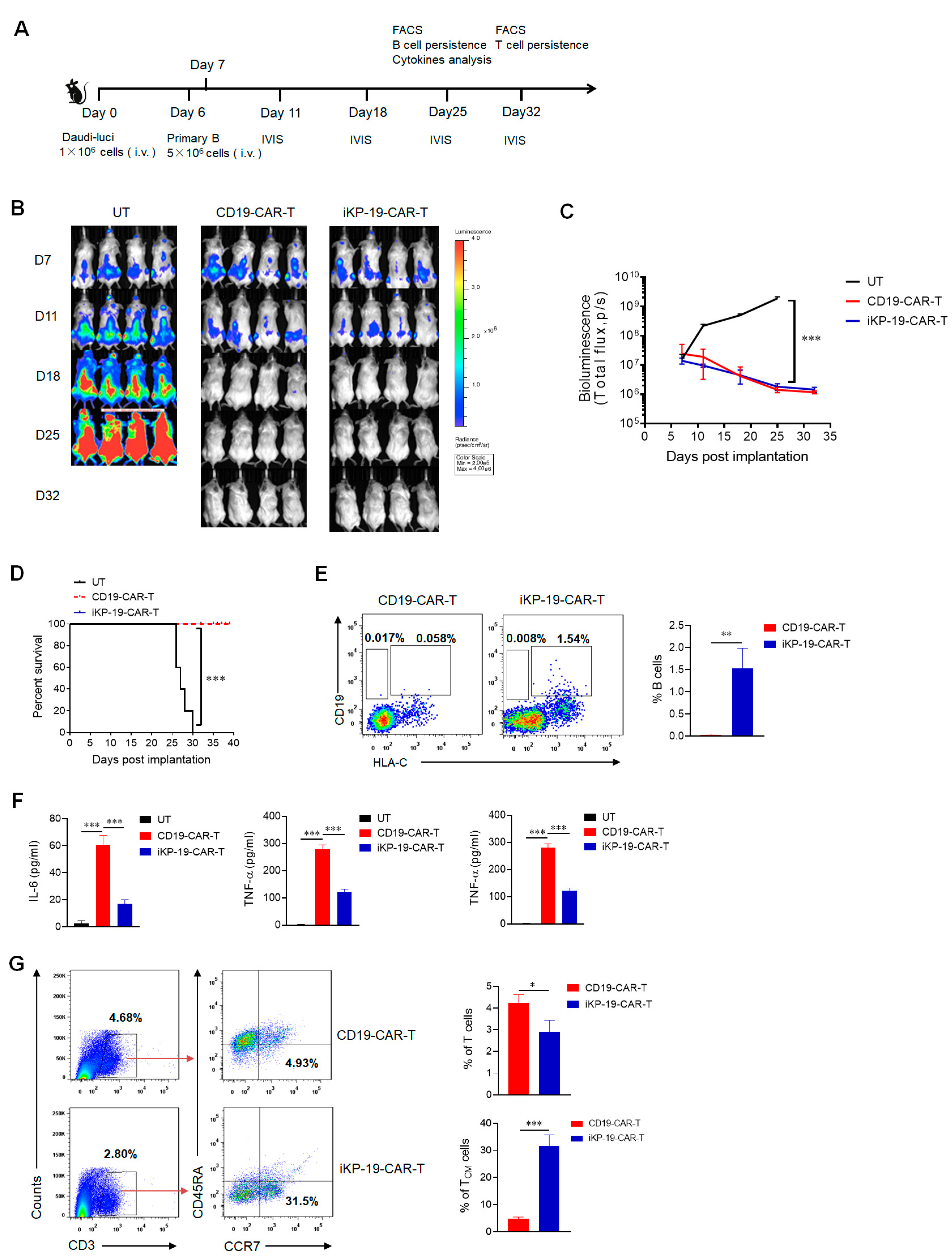
2.6. CD19-CAR-T Cells Bearing an iKP CAR Discern CD19+HLA-C1− Burkitt’s Lymphoma Cell Line and CD19+HLA-C1+ Healthy B Cells In Vivo
3. Discussion
4. Materials and Methods
4.1. Cell Lines
4.2. T-Cell and B-Cell Isolation
4.3. iKP CAR/iKPt CAR Construction
4.4. Lentiviral Vector Production
4.5. iKP-19-CAR-T/iKPt-19-CAR-T Cell Manufacture
4.6. Analysis of iKP CAR Function
4.7. Flow Cytometry
4.8. LDH Release Assay
4.9. Cytokine Assay
4.10. In Vivo Daudi-Derived Xenograft Model
4.11. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Thomas, X.; Paubelle, E. Tisagenlecleucel-T for the treatment of acute lymphocyticleukemia. Expert. Opin. Biol. Ther. 2018, 18, 1095–1106. [Google Scholar] [CrossRef]
- Nair, R.; Neelapu, S.S. The promise of CAR T-cell therapy in aggressive B-cell lymphoma. Best Pract. Res. Clin. Haematol. 2018, 31, 293–298. [Google Scholar] [CrossRef] [PubMed]
- Maude, S.L.; Frey, N.; Shaw, P.A.; Aplenc, R.; Barrett, D.M.; Bunin, N.J.; Chew, A.; Gonzalez, V.E.; Zheng, Z.; Lacey, S.F. Chimeric antigen receptor T cells for sustained remissions in leukemia. N. Engl. J. Med. 2014, 371, 1507–1517. [Google Scholar] [CrossRef] [PubMed]
- Porter, D.L.; Hwang, W.-T.; Frey, N.V.; Lacey, S.F.; Shaw, P.A.; Loren, A.W.; Bagg, A.; Marcucci, K.T.; Shen, A.; Gonzalea, V. Chimeric antigen receptor T cells persist and induce sustained remissions in relapsed refractory chronic lymphocytic leukemia. Sci. Transl. Med. 2015, 7, 303ra139. [Google Scholar] [CrossRef]
- Shah, N.N.; Fry, T.J. Mechanisms of resistance to CAR T cell therapy. Nat. Rev. Clin. Oncol. 2019, 16, 372–385. [Google Scholar] [CrossRef] [PubMed]
- Garfall, A.L.; Maus, M.V.; Hwang, W.-T.; Lacey, S.F.; Mahnke, Y.D.; Melenhorst, J.J.; Zheng, Z.; Vogl, D.T.; Cohen, A.D.; Weiss, B.M. Chimeric antigen receptor T cells against CD19 for multiple myeloma. N. Engl. J. Med. 2015, 373, 1040–1047. [Google Scholar] [CrossRef] [PubMed]
- Grupp, S.A.; Kalos, M.; Barrett, D.; Aplenc, R.; Porter, D.L.; Rheingold, S.R.; Teachey, D.T.; Chew, A.; Hauck, B.; Wright, J.F. Chimeric antigen receptor-modified T cells for acute lymphoid leukemia. N. Engl. J. Med. 2013, 368, 1509–1518. [Google Scholar] [CrossRef]
- Kochenderfer, J.N.; Wilson, W.H.; Janik, J.E.; Dudley, M.E.; Stetler-Stevenson, M.; Feldman, S.A.; Maric, I.; Raffeld, M.; Nathan, D.-A.N.; Lanier, B.J. Eradication of B-lineage cells and regression of lymphoma in a patient treated with autologous T cells genetically engineered to recognize CD19. Blood 2010, 116, 4099–4102. [Google Scholar] [CrossRef]
- Kochenderfer, J.N.; Dudley, M.E.; Feldman, S.A.; Wilson, W.H.; Spaner, D.E.; Maric, I.; Stevenson, M.S.; Phan, G.Q.; Hughes, M.S.; Sherry, R.M. B-cell depletion and remissions of malignancy along with cytokine-associated toxicity in clinical trial of anti-CD19 chimeric-antigen-receptor-transduced T cells. Blood 2012, 119, 2709–2720. [Google Scholar] [CrossRef]
- Oluwole, O.O.; Davila, M.L. At the bedside: Clinical review of chimeric antigen receptor (CAR) T cell therapy for B cell malignancies. J. Leukoc. Biol. 2016, 100, 1265–1272. [Google Scholar] [CrossRef]
- Pennell, C.A.; Barnum, J.L.; McDonald-Hyman, C.S.; Panoskaltsis-Mortari, A.; Riddle, M.J.; Xiong, Z.; Loschi, M.; Thangavelu, G.; Campbell, H.M.; Storlie, M.D. Human CD19-targeted mouse T cells induce B cell aplasia and toxicity in human CD19 transgenic mice. Mol. Ther. 2018, 26, 1423–1434. [Google Scholar] [CrossRef] [PubMed]
- Hill, J.A.; Li, D.; Hay, K.A.; Green, M.L.; Cherian, S.; Chen, X.; Riddell, S.R.; Maloney, D.G.; Boeckh, M.; Turtle, C.J. Infectious complications of CD19-targeted chimeric antigen receptor-modified T-cell immunotherapy. Blood 2018, 131, 121–130. [Google Scholar] [CrossRef] [PubMed]
- Hosen, N.; Matsunaga, Y.; Hasegawa, K.; Matsuno, H.; Nakamura, Y.; Makita, M.; Watanabe, K.; Yoshida, M.; Satoh, K.; Morimoto, S. The activated conformation of integrin β7 is a novel multiple myeloma-specific target for CAR T cell therapy. Nat. Med. 2017, 23, 1436–1443. [Google Scholar] [CrossRef] [PubMed]
- Posey, A.D., Jr.; Schwab, R.D.; Boesteanu, A.C.; Steentoft, C.; Mandel, U.; Engels, B.; Stone, J.D.; Madsen, T.D.; Schreiber, K.; Haines, K.M. Engineered CAR T cells targeting the cancer-associated tn-glycoform of the membrane mucin muc1 control adenocarcinoma. Immunity 2016, 44, 1444–1454. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Li, K.; Jiang, H.; Song, F.; Gao, H.; Pan, X.; Shi, B.; Bi, Y.; Wang, H.; Wang, H. Development of T cells carrying two complementary chimeric antigen receptors against glypican-3 and asialoglycoprotein receptor 1 for the treatment of hepatocellular carcinoma. Cancer Immunol. Immunother. 2017, 66, 475–489. [Google Scholar] [CrossRef]
- Roybal, K.T.; Rupp, L.J.; Morsut, L.; Walker, W.J.; Mcnally, K.A.; Park, J.S.; Lim, W.A. Precision tumor recognition by T cells with combinatorial antigen-sensing circuits. Cell 2016, 164, 770–779. [Google Scholar] [CrossRef]
- Morsut, L.; Roybal, K.T.; Xiong, X.; Gordley, R.M.; Goyle, S.M.; Thomson, M.; Lim, W.A. Engineering customized cell sensing and response behaviors using synthetic notch receptors. Cell 2016, 164, 780–791. [Google Scholar] [CrossRef]
- Roybal, K.T.; Williams, J.Z.; Morsut, L.; Rupp, L.J.; Kolinko, I.; Choe, J.H.; Walker, W.J.; McNally, K.A.; Lim, W.A. Engineering T cells with customized therapeutic response programs using synthetic notch receptors. Cell 2016, 167, 419–432. [Google Scholar] [CrossRef]
- Fedorov, V.D.; Themeli, M.; Sadelain, M. PD-1– and CTLA-4–based inhibitory chimeric antigen receptors (iCARs) divert off-target immunotherapy responses. Sci. Transl. Med. 2013, 5, 215ra172. [Google Scholar] [CrossRef]
- Martinet, L.; Smyth, M.J. Balancing natural killer cell activation through paired receptors. Nat. Rev. Immunol. 2015, 15, 243–254. [Google Scholar] [CrossRef]
- Handgretinger, R.; Lang, P.; Andre, M.C. Exploitation of natural killer cells for the treatment of acute leukemia. Blood 2016, 127, 3341–3349. [Google Scholar] [CrossRef] [PubMed]
- Bryceson, Y.T.; March, M.E.; Ljunggren, H.G.; Long, E.O. Activation, coactivation, and costimulation of resting human natural killer cells. Immunol. Rev. 2006, 214, 73–91. [Google Scholar] [CrossRef] [PubMed]
- Khong, H.T.; Restifo, N.P. Natural selection of tumor variants in the generation of "tumor escape" phenotypes. Nat. Immunol. 2002, 3, 999–1005. [Google Scholar] [CrossRef]
- Tsukahara, T.; Kawaguchi, S.; Torigoe, T.; Asanuma, H.; Nakazawa, E.; Shimozawa, K.; Nabeta, Y.; Kimura, S.; Kaya, M.; Nagoya, S. Prognostic significance of HLA class I expression in osteosarcoma defined by anti-pan HLA class I monoclonal antibody, EMR8-5. Cancer Sci. 2006, 97, 1374–1380. [Google Scholar] [CrossRef] [PubMed]
- Mizukami, Y.; Kono, K.; Maruyama, T.; Watanabe, M.; Kawaguchi, Y.; Kamimura, K.; Fujii, H. Downregulation of HLA class I molecules in the tumor is associated with a poor prognosis in patients with oesophageal squamous cell carcinoma. Br. J. Cancer 2008, 99, 1462–1467. [Google Scholar] [CrossRef]
- Pedoeem, A.; Azoulay-Alfaguter, I.; Strazza, M.; Silverman, G.J.; Mor, A. Programmed death-1 pathway in cancer and autoimmunity. Clin. Immunol. 2014, 153, 145–152. [Google Scholar] [CrossRef]
- Riella, L.V.; Paterson, A.M.; Sharpe, A.H.; Chandraker, A. Role of the PD-1 pathway in the immune response. Am. J. Transplant. 2012, 12, 2575–2587. [Google Scholar] [CrossRef]
- Chikuma, S. Basics of PD-1 in self-tolerance, infection, and cancer immunity. Int. J. Clin. Oncol. 2016, 21, 448–455. [Google Scholar] [CrossRef]
- Gaud, G.; Lesoume, R.; Love, P.E. Regulatory mechanisms in T cell receptor signalling. Nat. Rev. Immunol. 2018, 18, 485–497. [Google Scholar] [CrossRef]
- Wu, X.; Gu, Z.; Chen, Y.; Chen, B.; Chen, W.; Weng, L.; Liu, X. Application of PD-1 blockade in cancer immunotherapy. Comput. Struct. Biotechnol. J. 2019, 17, 661–674. [Google Scholar] [CrossRef]
- Ellery, J.M.; Nicholls, P.J. Alternate signaling pathways from the interleukin-2 receptor. Cytokine Growth Factor Rev. 2002, 13, 27–40. [Google Scholar] [CrossRef]
- Wang, L.H.; Kirken, R.A.; Erwin, R.A.; Yu, C.R.; Farrar, W.L. JAK3, STAT, and MAPK signaling pathways as novel molecular targets for the tyrphostin AG-490 regulation of IL-2-mediated T cell response. J. Immunol. 1999, 162, 3897–3904. [Google Scholar] [PubMed]
- Sheppard, K.-A.; Fitz, L.J.; Lee, J.M.; Benander, C.; George, J.A.; Wooters, J.; Qiu, Y.; Jussif, J.M.; Carter, L.L.; Wood, C.R. PD-1 inhibits T-cell receptor induced phosphorylation of the ZAP70/CD3zeta signalosome and downstream signaling to PKCtheta. FEBS Lett. 2004, 574, 37–41. [Google Scholar] [CrossRef] [PubMed]
- Patsoukis, N.; Brown, J.; Petkova, V.; Liu, F.; Li, L.; Boussiotis, V.A. Selective effects of PD-1 on Akt and Ras pathways regulate molecular compoents of the cell cycle and inhibit T cell proliferation. Sci. Signal. 2012, 5, ra46. [Google Scholar] [CrossRef]
- Parry, R.V.; Chemnitz, J.M.; Frauwirth, K.A.; Lanfranco, A.R.; Braunstein, I.; Kobayashi, S.V.; Linsley, P.S.; Thompson, C.B.; Riley, J.L. CTLA-4 and PD-1 receptors inhibit T-cell activation by distinct mechanisms. Mol. Cell. Biol. 2005, 25, 9543–9553. [Google Scholar] [CrossRef]
- Mahnke, Y.D.; Brodie, T.M.; Sallusto, F.; Roederer, M. The who’s who of T-cell differentiation: Human memory T-cell subsets. Eur. J. Immunol. 2013, 43, 2797–2809. [Google Scholar] [CrossRef]
- Joshi, N.S.; Cui, W.; Chandele, A.; Lee, H.K.; Urso, D.R.; Hagman, J.; Gapin, L.; Kaech, S.M. Inflammation directs memory precursor and short-lived effector CD8(+) T cells fates via the graded expression of T-bet transcription factor. Immunity 2007, 27, 281–295. [Google Scholar] [CrossRef]
- Lee, D.W.; Levine, B.L.; Porter, D.L.; Louis, C.U.; Ahmed, N.; Jensen, M.; Grupp, S.A.; Mackall, C.L. Current concepts in the diagnosis and managment of cytokine release syndrome. Blood 2014, 124, 188–195. [Google Scholar] [CrossRef]
- Kalos, M.; Levine, B.L.; Porter, D.L.; Katz, S.; Grupp, S.A.; Bagg, A.; June, C.H. T cells with chimeric antigen receptors have potent antitumor effects and can establish memory in patients with advanced leukemia. Sci. Transl. Med. 2011, 3, 95ra73. [Google Scholar] [CrossRef]
- Lamers, C.H.; Sleijfer, S.; Vulto, A.G.; Kruit, W.H.; KLiffen, M.; Debets, R.; Gratama, J.W.; Stoter, G.; Oosterwijk, E. Treatment of metastatic renal cell carcinoma with autologous T-lymphocytes genetically retargeted against carbonic anhyfrase IX: First clinical experience. J. Clin. Oncol. 2006, 24, e20–e22. [Google Scholar] [CrossRef] [PubMed]
- Akpek, G.; Lee, S.M.; Anders, V.; Vogelsang, G.B. A high-dose plus steroid regimen for controlling active chronic graft-versus-host disease. Biol. Blood Marrow Transplant. 2001, 7, 495–502. [Google Scholar] [CrossRef] [PubMed]
- Karre, K.; Ljunggren, H.G.; Piontek, G.; Kiessling, R. Selective rejection of H-2-deficient lymphoma variants suggests alternative immune defence strategy. Nature 1986, 319, 675–678. [Google Scholar] [CrossRef] [PubMed]
- Moretta, A.; Bottino, C.; Vitale, M.; Pende, D.; Biassoni, R.; Mingari, M.C.; Moretta, L. Receptors for HLA class I molecules in human natural killer cells. Annu. Rev. Immunol. 1996, 14, 619–648. [Google Scholar] [CrossRef] [PubMed]
- Gomes-Silva, D.; Srinivasan, M.; Sharma, S.; Lee, C.M.; Wagner, D.L.; Davis, T.H.; Rouce, R.H.; Bao, G.; Brenner, M.K.; Mamonkin, M. CD7-edited T cells espressing a CD7-specific CAR for the therapy of T-cell malignancies. Blood 2017, 130, 285–296. [Google Scholar] [CrossRef]
- Mary, E.K.; Manish, J.B.; Gordon, J.F.; Arlene, H.S. PD-1 and Its ligands in tolerance and immunity. Annu. Rev. Immunol. 2008, 26, 677–704. [Google Scholar]
- Ying, Z.; Huang, X.F.; Xiang, X.; Liu, Y.; Kang, X.; Song, Y.; Guo, X.; Liu, H.; Ding, N.; Zhang, T. A safe and potent anti-CD19 CAR T cell therapy. Nat. Med. 2019, 25, 947–953. [Google Scholar] [CrossRef]
- Morgan, R.A.; Yang, J.C.; Kitano, M.; Dudley, M.E.; Laurencot, C.M.; Rosenberg, S.A. Case report of a serious adverse event following the administration of T cells transduced with a chimeric antigen receptor recognizing ERBB2. Mol. Ther. 2010, 18, 843–851. [Google Scholar] [CrossRef]
- Prieto, C.; Stam, R.W.; Agraz-Doblas, A.; Ballerini, P.; Camos, M.; Castano, J.; Marschalek, R.; Bursen, A.; Varela, I.; Bueno, C. Activated Kras cooperates with MLL-AF4 to promote extramedullary engraftment and migration of cord blood CD34+ HSPC but is insufficient to initiate leukemia. Cancer Res. 2016, 76, 2478–2489. [Google Scholar] [CrossRef]
- Mamonkin, M.; Rouce, R.H.; Tashiro, H.; Brenner, M.K. A T-cell-directed chimeric antigen receptor for the selective treatment of T-cell malignancies. Blood 2015, 126, 983–992. [Google Scholar] [CrossRef] [PubMed]
- Song, Y.; Tong, C.; Gao, Y.; Dai, H.; Guo, Y.; Zhao, X. Effective and persistent antitumor activity of HER2-directed CAR-T cells against gastric cancer cells in vitro and xenotransplanted tumors in vivo. Protein Cell 2018, 9, 867–878. [Google Scholar] [CrossRef]







© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tao, L.; Farooq, M.A.; Gao, Y.; Zhang, L.; Niu, C.; Ajmal, I.; Zhou, Y.; He, C.; Zhao, G.; Yao, J.; et al. CD19-CAR-T Cells Bearing a KIR/PD-1-Based Inhibitory CAR Eradicate CD19+HLA-C1− Malignant B Cells While Sparing CD19+HLA-C1+ Healthy B Cells. Cancers 2020, 12, 2612. https://doi.org/10.3390/cancers12092612
Tao L, Farooq MA, Gao Y, Zhang L, Niu C, Ajmal I, Zhou Y, He C, Zhao G, Yao J, et al. CD19-CAR-T Cells Bearing a KIR/PD-1-Based Inhibitory CAR Eradicate CD19+HLA-C1− Malignant B Cells While Sparing CD19+HLA-C1+ Healthy B Cells. Cancers. 2020; 12(9):2612. https://doi.org/10.3390/cancers12092612
Chicago/Turabian StyleTao, Lei, Muhammad Asad Farooq, Yaoxin Gao, Li Zhang, Congyi Niu, Iqra Ajmal, Ying Zhou, Cong He, Guixia Zhao, Jie Yao, and et al. 2020. "CD19-CAR-T Cells Bearing a KIR/PD-1-Based Inhibitory CAR Eradicate CD19+HLA-C1− Malignant B Cells While Sparing CD19+HLA-C1+ Healthy B Cells" Cancers 12, no. 9: 2612. https://doi.org/10.3390/cancers12092612
APA StyleTao, L., Farooq, M. A., Gao, Y., Zhang, L., Niu, C., Ajmal, I., Zhou, Y., He, C., Zhao, G., Yao, J., Liu, M., & Jiang, W. (2020). CD19-CAR-T Cells Bearing a KIR/PD-1-Based Inhibitory CAR Eradicate CD19+HLA-C1− Malignant B Cells While Sparing CD19+HLA-C1+ Healthy B Cells. Cancers, 12(9), 2612. https://doi.org/10.3390/cancers12092612