The Structural Binding Mode of the Four Autotaxin Inhibitor Types that Differentially Affect Catalytic and Non-Catalytic Functions


Abstract
1. Introduction
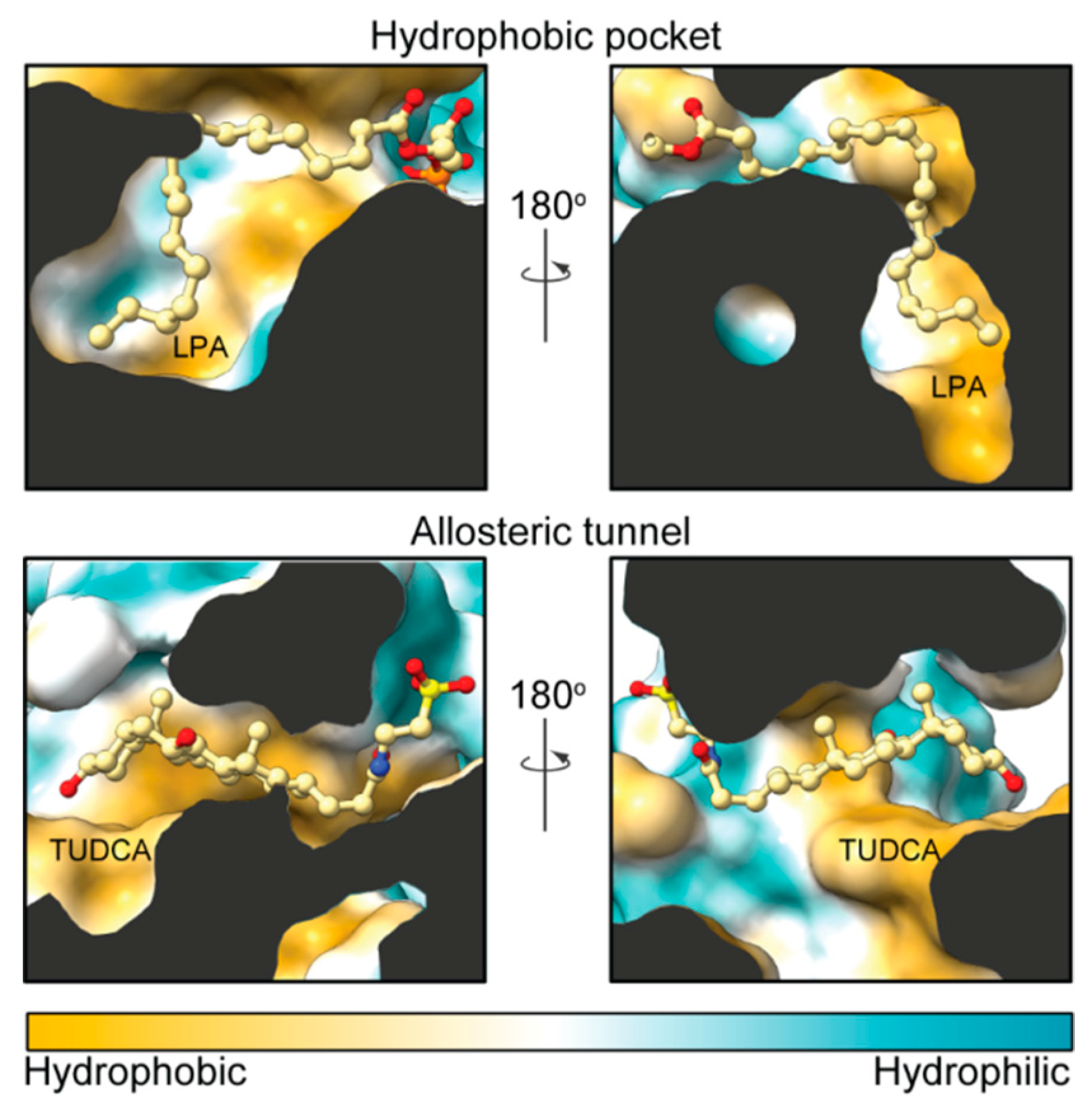
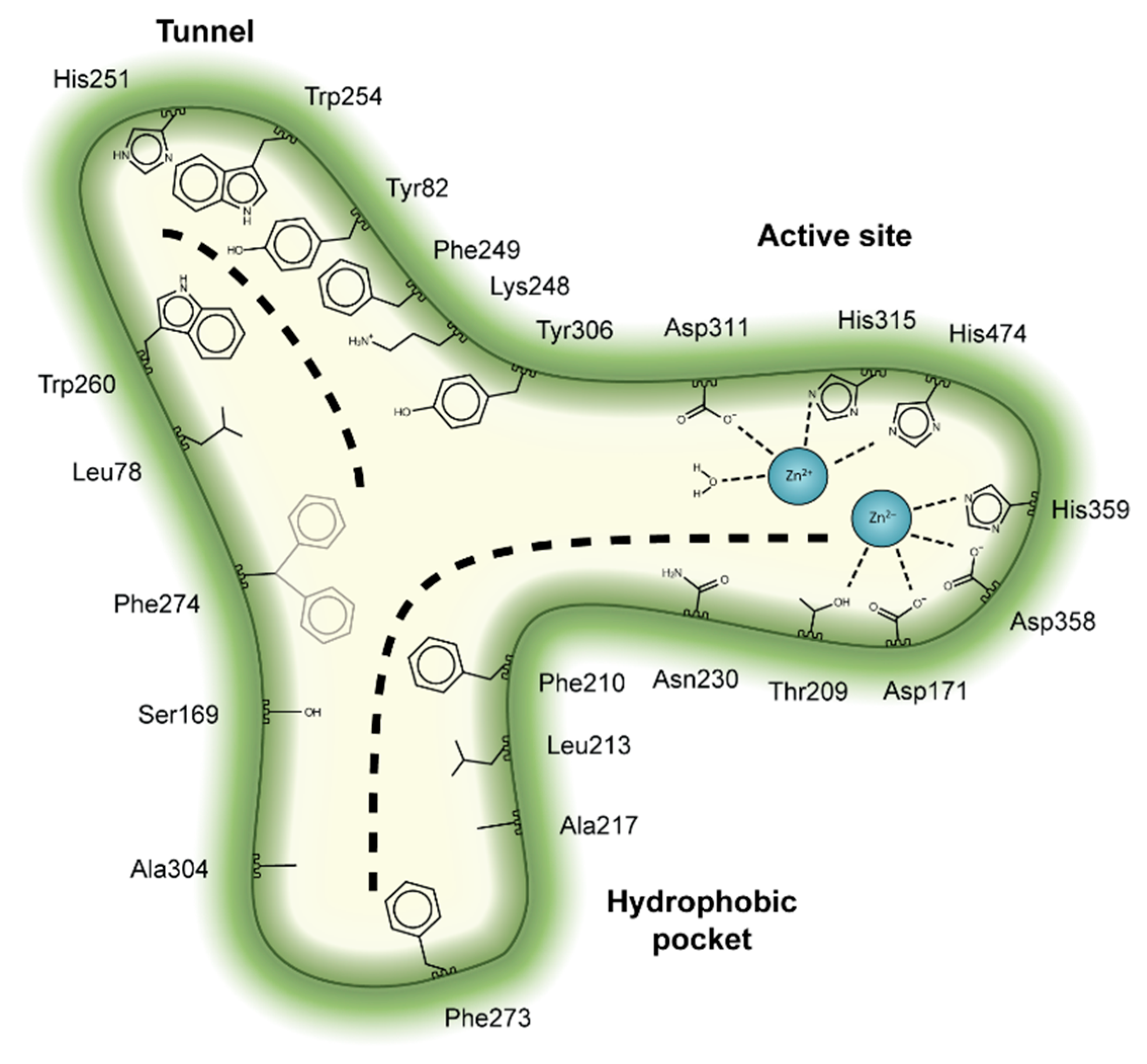
2. Autotaxin Catalytic and Non-Catalytic Functions
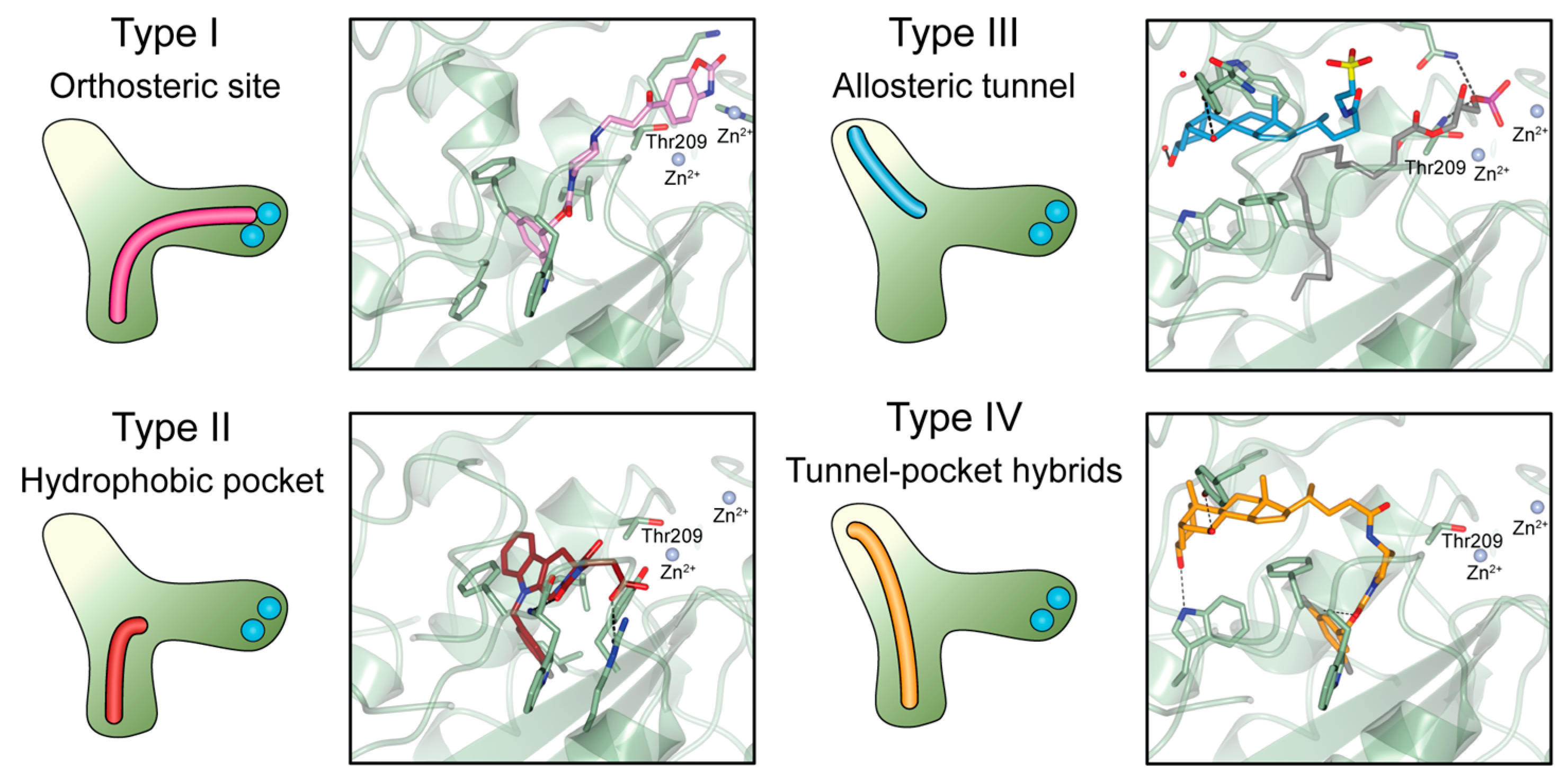
3. The Autotaxin Inhibitor Family
3.1. Type I Inhibitors
3.2. Type II Inhibitors
3.3. Type III Inhibitors
3.4. Type IV Inhibitors
4. Conclusions
Funding
Acknowledgments
Conflicts of Interest
References
- van Corven, E.; Groenink, A.; Jalink, K.; Eichholtz, T.; Moolenaar, W. Lysophosphatidate-induced cell proliferation: Identification and dissection of signaling pathways mediated by G proteins. Cell 1989, 59, 45–54. [Google Scholar] [CrossRef]
- Jalink, K.; Van Corven, E.J.; Moolenaar, W.H. Lysophosphatidic acid, but not phosphatidic acid, is a potent Ca2+-mobilizing stimulus for fibroblasts: Evidence for an extracellular site of action. J. Biol. Chem. 1990, 265, 12232–12239. [Google Scholar] [PubMed]
- Wang, J.; Sun, Y.; Qu, J.; Yan, Y.; Yang, Y.; Cai, H. Roles of LPA receptor signaling in breast cancer. Expert Rev. Mol. Diagn. 2016, 16, 1103–1111. [Google Scholar] [CrossRef] [PubMed]
- Kaffe, E.; Katsifa, A.; Xylourgidis, N.; Ninou, I.; Zannikou, M.; Harokopos, V.; Foka, P.; Dimitriadis, A.; Evangelou, K.; Moulas, A.N.; et al. Hepatocyte autotaxin expression promotes liver fibrosis and cancer. Hepatology 2017, 65, 1369–1383. [Google Scholar] [CrossRef]
- Tager, A.M.; LaCamera, P.; Shea, B.S.; Campanella, G.S.; Selman, M.; Zhao, Z.; Polosukhin, V.; Wain, J.; Karimi-Shah, B.A.; Kim, N.D.; et al. The lysophosphatidic acid receptor LPA1 links pulmonary fibrosis to lung injury by mediating fibroblast recruitment and vascular leak. Nat. Med. 2008, 14, 45–54. [Google Scholar] [CrossRef]
- Kremer, A.E.; Martens, J.J.W.W.; Kulik, W.; Ruëff, F.; Kuiper, E.M.M.; van Buuren, H.R.; van Erpecum, K.J.; Kondrackiene, J.; Prieto, J.; Rust, C.; et al. Lysophosphatidic acid is a potential mediator of cholestatic pruritus. Gastroenterology 2010, 139, 1008–1018. [Google Scholar] [CrossRef]
- Thirunavukkarasu, K.; Tan, B.; Swearingen, C.A.; Rocha, G.; Bui, H.H.; McCann, D.J.; Jones, S.B.; Norman, B.H.; Pfeifer, L.A.; Saha, J.K. Pharmacological characterization of a potent inhibitor of autotaxin in animal models of inflammatory bowel disease and multiple sclerosis. J. Pharmacol. Exp. Ther. 2016, 359, 207–214. [Google Scholar] [CrossRef]
- Barbayiannia, E.; Kaffeb, E.; Aidinisb, V.; Kokotos, G. Autotaxin, a secreted lysophospholipase D, as a promising therapeutic target in chronic inflammation and cancer. Prog. Lipid Res. 2015, 58, 76–96. [Google Scholar] [CrossRef]
- Benesch, M.G.K.; Tang, X.; Venkatraman, G.; Bekele, R.T.; Brindley, D.N. Recent advances in targeting the autotaxin-lysophosphatidate-lipid phosphate phosphatase axis in vivo. J. Biomed. Res. 2016, 30, 272–284. [Google Scholar]
- Chrencik, J.E.; Roth, C.B.; Terakado, M.; Kurata, H.; Omi, R.; Kihara, Y.; Warshaviak, D.; Nakade, S.; Asmar-Rovida, G.; Mileni, M.; et al. Crystal structure of antagonist bound human lysophosphatidic acid receptor 1. Cell 2015, 161, 1633–1643. [Google Scholar] [CrossRef]
- Taniguchi, R.; Inoue, A.; Sayama, M.; Uwamizu, A.; Yamashita, K.; Hirata, K.; Yoshida, M.; Tanaka, Y.; Kato, H.E.; Nakada-Nakura, Y.; et al. Structural insights into ligand recognition by the lysophosphatidic acid receptor LPA6. Nature 2017, 548, 356–360. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Pluhackova, K.; Jiang, Z.; Böckmann, R.A. Binding characteristics of sphingosine-1-phosphate to APOM hints to assisted release mechanism via the APOM calyx-opening. Sci. Rep. 2016, 6, e30655. [Google Scholar] [CrossRef] [PubMed]
- Stanley, N.; Pardo, L.; Fabritiis, G. De The pathway of ligand entry from the membrane bilayer to a lipid G protein-coupled receptor. Sci. Rep. 2016, 6, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Albers, H.M.H.G.; Dong, A.; van Meeteren, L.A.; Egan, D.A.; Sunkara, M.; van Tilburg, E.W.; Schuurman, K.; van Tellingen, O.; Morris, A.J.; Smyth, S.S.; et al. Boronic acid-based inhibitor of autotaxin reveals rapid turnover of LPA in the circulation. Proc. Natl. Acad. Sci. USA 2010, 107, 7257–7262. [Google Scholar] [CrossRef] [PubMed]
- Tomsig, J.L.; Snyder, A.H.; Berdyshev, E.V.; Skobeleva, A.; Mataya, C.; Natarajan, V.; Brindley, D.N.; Lynch, K.R. Lipid phosphate phosphohydrolase type 1 (LPP1) degrades extracellular lysophosphatidic acid in vivo. Biochem. J. 2009, 419, 611–618. [Google Scholar] [CrossRef] [PubMed]
- Tang, X.; Benesch, M.G.K.; Dewald, J.; Zhao, Y.Y.; Patwardhan, N.; Santos, W.L.; Curtis, J.M.; McMullen, T.P.W.; Brindley, D.N. Lipid phosphate phosphatase-1 expression in cancer cells attenuates tumor growth and metastasis in mice. J. Lipid Res. 2014, 55, 2389–2400. [Google Scholar] [CrossRef]
- Jasinska, R.; Zhang, Q.; Pilquil, C.; Singh, I.; Xu, J.; Dewald, J.; Dillon, D.A.; Berthiaume, L.G.; Carman, G.M.; Waggoner, D.W.; et al. Lipid phosphate phosphohydrolase-1 degrades exogenous glycerolipid and sphingolipid phosphate esters. Biochem. J. 1999, 340, 677–686. [Google Scholar] [CrossRef]
- van Meeteren, L.A.; Moolenaar, W.H. Regulation and biological activities of the autotaxin-LPA axis. Prog. Lipid Res. 2007, 46, 145–160. [Google Scholar] [CrossRef]
- Aoki, J.; Taira, A.; Takanezawa, Y.; Kishi, Y.; Hama, K.; Kishimoto, T.; Mizuno, K.; Saku, K.; Taguchi, R.; Arai, H. Serum lysophosphatidic acid is produced through diverse phospholipase pathways. J. Biol. Chem. 2002, 277, 48737–48744. [Google Scholar] [CrossRef]
- Umezu-Goto, M.; Kishi, Y.; Taira, A.; Hama, K.; Dohmae, N.; Takio, K.; Yamori, T.; Mills, G.B.; Inoue, K.; Aoki, J.; et al. Autotaxin has lysophospholipase D activity leading to tumor cell growth and motility by lysophosphatidic acid production. J. Cell Biol. 2002, 158, 227–233. [Google Scholar] [CrossRef]
- Matralis, A.N.; Afantitis, A.; Aidinis, V. Development and therapeutic potential of autotaxin small molecule inhibitors: From bench to advanced clinical trials. Med. Res. Rev. 2019, 39, 976–1013. [Google Scholar] [CrossRef] [PubMed]
- Tokumura, A.; Majima, E.; Kariya, Y.; Tominaga, K.; Kogure, K.; Yasuda, K.; Fukuzawa, K. Identification of human plasma lysophospholipase D, a lysophosphatidic acid-producing enzyme, as autotaxin, a multifunctional phosphodiesterase. J. Biol. Chem. 2002, 277, 39436–39442. [Google Scholar] [CrossRef] [PubMed]
- Jansen, S. Proteolytic maturation and activation of autotaxin (NPP2), a secreted metastasis-enhancing lysophospholipase D. J. Cell Sci. 2005, 118, 3081–3089. [Google Scholar] [CrossRef] [PubMed]
- Hausmann, J.; Keune, W.-J.; Hipgrave Ederveen, A.L.; van Zeijl, L.; Joosten, R.P.; Perrakis, A. Structural snapshots of the catalytic cycle of the phosphodiesterase autotaxin. J. Struct. Biol. 2016, 195, 199–206. [Google Scholar] [CrossRef]
- Hausmann, J.; Kamtekar, S.; Christodoulou, E.; Day, J.E.; Wu, T.; Fulkerson, Z.; Albers, H.M.H.G.H.G.; Van Meeteren, L.A.; Houben, A.J.S.S.; Van Zeijl, L.; et al. Structural basis for substrate discrimination and integrin binding by autotaxin. Nat. Struct. Mol. Biol. 2011, 18, 198–204. [Google Scholar] [CrossRef]
- Keune, W.; Hausmann, J.; Bolier, R.; Tolenaars, D.; Kremer, A.; Heidebrecht, T.; Joosten, R.; Sunkara, M.; Morris, A.; Matas-Rico, E.; et al. Steroid binding to autotaxin links bile salts and lysophosphatidic acid signalling. Nat. Commun. 2016, 7, e11248. [Google Scholar] [CrossRef]
- Nishimasu, H.; Okudaira, S.; Hama, K.; Mihara, E.; Dohmae, N.; Inoue, A.; Ishitani, R.; Takagi, J.; Aoki, J.; Nureki, O. Crystal structure of autotaxin and insight into GPCR activation by lipid mediators. Nat. Struct. Mol. Biol. 2011, 18, 205–212. [Google Scholar] [CrossRef]
- Salgado-Polo, F.; Fish, A.; Matsoukas, M.-T.; Heidebrecht, T.; Keune, W.-J.; Perrakis, A. Lysophosphatidic acid produced by Autotaxin acts as an allosteric modulator of its catalytic efficiency. J. Biol. Chem. 2018, 293, 14312–14327. [Google Scholar] [CrossRef]
- Saunders, L.P.; Cao, W.; Chang, W.C.; Albright, R.A.; Braddock, D.T.; De La Cruz, E.M. Kinetic analysis of autotaxin reveals substrate-specific catalytic pathways and a mechanism for lysophosphatidic acid distribution. J. Biol. Chem. 2011, 286, 30130–30141. [Google Scholar] [CrossRef]
- Jethwa, S.A.; Leah, E.J.; Zhang, Q.; Bright, N.A.; Oxley, D.; Bootman, M.D.; Rudge, S.A.; Wakelam, M.J.O. Exosomes bind to autotaxin and act as a physiological delivery mechanism to stimulate LPA receptor signalling in cells. J. Cell Sci. 2016, 129, 3948–3957. [Google Scholar] [CrossRef]
- Thumser, A.E.A.; Wilton, D.C. The binding of natural and fluorescent lysophospholipids to wild-type and mutant rat liver fatty acid-binding protein and albumin. Biochem. J. 1995, 307, 305–311. [Google Scholar] [CrossRef]
- Leblanc, R.; Lee, S.C.; David, M.; Bordet, J.C.; Norman, D.D.; Patil, R.; Miller, D.; Sahay, D.; Ribeiro, J.; Clezardin, P.; et al. Interaction of platelet-derived autotaxin with tumor integrin alphaVbeta3 controls metastasis of breast cancer cells to bone. Blood 2014, 124, 3141–3150. [Google Scholar] [CrossRef] [PubMed]
- Wu, T.; Vander Kooi, C.; Shah, P.; Charnigo, R.; Huang, C.; Smyth, S.S.; Morris, A.J. Integrin-mediated cell surface recruitment of autotaxin promotes persistent directional cell migration. FASEB J. 2014, 28, 861–870. [Google Scholar] [CrossRef] [PubMed]
- Fulkerson, Z.; Wu, T.; Sunkara, M.; Vander Kooi, C.; Morris, A.J.; Smyth, S.S. Binding of autotaxin to integrins localizes lysophosphatidic acid production to platelets and mammalian cells. J. Biol. Chem. 2011, 286, 34654–34663. [Google Scholar] [CrossRef] [PubMed]
- Pamuklar, Z.; Federico, L.; Liu, S.; Umezu-Goto, M.; Dong, A.; Panchatcharam, M.; Fulerson, Z.; Berdyshev, E.; Natarajan, V.; Fang, X.; et al. Autotaxin/Lysopholipase D and lysophosphatidic acid regulate murine hemostasis and thrombosis. J. Biol. Chem. 2009, 284, 7385–7394. [Google Scholar] [CrossRef] [PubMed]
- Houben, A.J.S.; Van Wijk, X.M.R.; Van Meeteren, L.A.; Van Zeijl, L.; Van De Westerlo, E.M.A.; Hausmann, J.; Fish, A.; Perrakis, A.; Van Kuppevelt, T.H.; Moolenaar, W.H. The polybasic insertion in autotaxin α confers specific binding to heparin and cell surface heparan sulfate proteoglycans. J. Biol. Chem. 2013, 288, 510–519. [Google Scholar] [CrossRef] [PubMed]
- Castagna, D.; Budd, D.C.; Macdonald, S.J.F.F.; Jamieson, C.; Watson, A.J.B.B. Development of Autotaxin Inhibitors: An Overview of the Patent and Primary Literature. J. Med. Chem. 2016, 59, 5604–5621. [Google Scholar] [CrossRef]
- Nikolaou, A.; Kokotou, M.G.; Limnios, D.; Psarra, A.; Kokotos, G. Autotaxin inhibitors: A patent review (2012–2016). Expert Opin. Ther. Pat. 2017, 27, 815–829. [Google Scholar] [CrossRef]
- Lynch, K.R.; Macdonald, T.L. Phosphonate Derivatives as Autotaxin Inhibitors. Patent US 8378100, 19 February 2013. [Google Scholar]
- Kato, K.; Ikeda, H.; Miyakawa, S.; Futakawa, S.; Nonaka, Y.; Fujiwara, M.; Okudaira, S.; Kano, K.; Aoki, J.; Morita, J.; et al. Structural basis for specific inhibition of Autotaxin by a DNA aptamer. Nat. Struct. Mol. Biol. 2016, 23, 395–401. [Google Scholar] [CrossRef]
- Joncour, A.; Desroy, N.; Housseman, C.; Bock, X.; Bienvenu, N.; Cherel, L.; Labeguere, V.; Peixoto, C.; Annoot, D.; Lepissier, L.; et al. Supporting information Discovery, Structure-Activity Relationship and Binding Mode of an Imidazo [1,2-a] pyridine Series of Autotaxin Inhibitors. J. Med. Chem. 2017, 60, 7371–7392. [Google Scholar]
- Ninou, I.; Kaffe, E.; Müller, S.; Budd, D.C.; Stevenson, C.S.; Ullmer, C.; Aidinis, V. Pharmacologic targeting of the ATX/LPA axis attenuates bleomycin-induced pulmonary fibrosis. Pulm. Pharmacol. Ther. 2018, 52, 32–40. [Google Scholar] [CrossRef] [PubMed]
- Ninou, I.; Magkrioti, C.; Aidinis, V. Autotaxin in pathophysiology and pulmonary fibrosis. Front. Med. 2018, 5, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Bhave, S.R.; Dadey, D.Y.A.; Karvas, R.M.; Ferraro, D.J.; Kotipatruni, R.P.; Jaboin, J.J.; Hallahan, A.N.; DeWees, T.A.; Linkous, A.G.; Hallahan, D.E.; et al. Autotaxin inhibition with PF-8380 enhances the radiosensitivity of human and murine glioblastoma cell lines. Front. Oncol. 2013, 3, 1–11. [Google Scholar] [CrossRef]
- Nikolaou, A.; Ninou, I.; Kokotou, M.G.; Kaffe, E.; Afantitis, A.; Aidinis, V.; Kokotos, G. Hydroxamic acids constitute a novel class of autotaxin inhibitors that exhibit in vivo efficacy in a pulmonary fibrosis model. J. Med. Chem. 2018, 61, 3697–3711. [Google Scholar] [CrossRef]
- Lee, S.; Fujiwara, Y.; Liu, J.; Yue, J.; Shimizu, Y.; Norman, D.; Wang, Y.; Tsukahara, R.; Szabo, E.; Patil, R.; et al. Autotaxin, LPA receptors (1 and 5) exert disparate functions in tumor cells versus the host tissue microenvironment in melanoma invasion and metastasis. Mol Cancer Res 2015, 13, 174–185. [Google Scholar] [CrossRef]
- Oikonomou, N.; Mouratis, M.A.; Tzouvelekis, A.; Kaffe, E.; Valavanis, C.; Vilaras, G.; Karameris, A.; Prestwich, G.D.; Bouros, D.; Aidinis, V. Pulmonary autotaxin expression contributes to the pathogenesis of pulmonary fibrosis. Am. J. Respir. Cell Mol. Biol. 2012, 47, 566–574. [Google Scholar] [CrossRef]
- Park, G.Y.; Lee, Y.G.; Berdyshev, E.; Nyenhuis, S.; Du, J.; Fu, P.; Gorshkova, I.A.; Li, Y.; Chung, S.; Karpurapu, M.; et al. Autotaxin production of lysophosphatidic acid mediates allergic asthmatic inflammation. Am. J. Respir. Crit. Care Med. 2013, 188, 928–940. [Google Scholar] [CrossRef]
- Evans, J.F. Methods and Compositions for the Treatment of Metabolic Disorders. Patent WO 2016028686, 25 February 2016. [Google Scholar]
- Stein, A.J.; Bain, G.; Prodanovich, P.; Santini, A.M.; Darlington, J.; Stelzer, N.M.P.; Sidhu, R.S.; Schaub, J.; Goulet, L.; Lonergan, D.; et al. Structural basis for inhibition of human autotaxin by four potent compounds with distinct modes of binding. Mol. Pharmacol. 2015, 88, 982–992. [Google Scholar] [CrossRef]
- Desroy, N.; Housseman, C.; Bock, X.; Joncour, A.; Bienvenu, N.; Cherel, L.; Labeguere, V.; Rondet, E.; Peixoto, C.; Grassot, J.-M.; et al. Discovery of 2-[[2-Ethyl-6-[4-[2-(3-hydroxyazetidin-1-yl)-2-oxoethyl] piperazin-1-yl]-8-methylimidazo [1,2-a] pyridin-3-yl] methylamino]-4-(4-fluorophenyl) thiazole-5-carbonitrile (GLPG1690), a first-in-class autotaxin inhibitor undergoing clinical Ev. J. Med. Chem. 2017, 15, 3580–3590. [Google Scholar] [CrossRef]
- Study to Assess Safety, Tolerability, Pharmacokinetic and Pharmacodynamic Properties of GLPG1690. Available online: https://clinicaltrials.gov/ct2/show/NCT02738801 (accessed on 8 October 2019).
- Benesch, M.G.K.; Tang, X.; Maeda, T.; Ohhata, A.; Zhao, Y.Y.; Kok, B.P.C.; Dewald, J.; Hitt, M.; Curtis, J.M.; McMullen, T.P.W.; et al. Inhibition of autotaxin delays breast tumor growth and lung metastasis in mice. FASEB J. 2014, 28, 2655–2666. [Google Scholar] [CrossRef] [PubMed]
- Benesch, M.G.K.; Zhao, Y.Y.; Curtis, J.M.; McMullen, T.P.W.; Brindley, D.N. Regulation of autotaxin expression and secretion by lysophosphatidate and sphingosine 1-phosphate. J. Lipid Res. 2015, 56, 1134–1144. [Google Scholar] [CrossRef] [PubMed]
- Baker, L.D.; Fujiwara, Y.; Pigg, K.; Tsukahara, R.; Kobatashi, S.; Murofushi, H.; Uchiyama, A.; Murakami-Murofushi, K.; Murph, E.; Mills, G.B.; et al. Carba analogs of cyclic phosphatidic acid are selective inhibitors of autotaxin and cancer cell invasion and metastasis. J. Biol. Chem. 2006, 281, 22786–22793. [Google Scholar] [CrossRef] [PubMed]
- Durgam, G.G.; Virag, T.; Walker, M.D.; Tsukahara, R.; Yasuda, S.; Liliom, K.; van Meeteren, L.A.; Moolenaar, W.H.; Wilke, N.; Siess, W.; et al. Synthesis, structure−activity relationships, and biological evaluation of fatty alcohol phosphates as lysophosphatidic acid receptor ligands, activators of PPARγ, and inhibitors of autotaxin. J. Med. Chem. 2005, 48, 4919–4930. [Google Scholar] [CrossRef]
- Nikitopoulou, I.; Kaffe, E.; Sevastou, I.; Sirioti, I.; Samiotaki, M.; Madan, D.; Prestwich, G.D.; Aidinis, V. A Metabolically-stabilized phosphonate analog of lysophosphatidic acid attenuates collagen-induced arthritis. PLoS ONE 2013, 87, e70941. [Google Scholar] [CrossRef]
- van Meeteren, L.A.; Brinkmann, V.; Saulnier-Blache, J.S.; Lynch, K.R.; Moolenaar, W.H. Anticancer activity of FTY720: Phosphorylated FTY720 inhibits autotaxin, a metastasis-enhancing and angiogenic lysophospholipase D. Cancer Lett. 2008, 266, 203–208. [Google Scholar] [CrossRef]
- Ferry, G.; Moulharat, N.; Pradere, J.-P.; Desos, P.; Try, A.; Genton, A.; Giganti, A.; Beucher-Gaudin, M.; Lonchampt, M.; Bertrand, M.; et al. S32826, A Nanomolar Inhibitor of Autotaxin: Discovery, Synthesis and Applications as a pharmacological tool. J. Pharmacol. Exp. Ther. 2008, 327, 809–819. [Google Scholar] [CrossRef]
- Albers, H.M.H.G.; Hendrickx, L.J.D.; van Tol, R.J.P.; Hausmann, J.; Perrakis, A.; Ovaa, H. Structure-based design of novel boronic acid-based inhibitors of autotaxin. J. Med. Chem. 2011, 54, 4619–4626. [Google Scholar] [CrossRef]
- Jones, S.B.; Pfeifer, L.A.; Bleisch, T.J.; Beauchamp, T.J.; Durbin, J.D.; Klimkowski, V.J.; Hughes, N.E.; Rito, C.J.; Dao, Y.; Gruber, J.M.; et al. Novel autotaxin inhibitors for the treatment of osteoarthritis pain: Lead optimization via structure-based drug design. ACS Med. Chem. Lett. 2016, 7, 857–861. [Google Scholar] [CrossRef]
- Gierse, J.; Thorarensen, A.; Beltey, K.; Bradshaw-pierce, E.; Cortes-burgos, L.; Hall, T.; Johnston, A.; Murphy, M.; Nemirovskiy, O.; Ogawa, S.; et al. A novel autotaxin inhibitor reduced lysophosphatidic acid levels in plasma and the site of inflammation. J. Pharmacol. Exp. Ther. 2010, 2, 310–317. [Google Scholar] [CrossRef]
- Kawaguchi, M.; Okabe, T.; Okudaira, S.; Nishimasu, H.; Ishitani, R.; Kojima, H.; Nureki, O.; Aoki, J.; Nagano, T. Screening and X-ray crystal structure-based optimization of autotaxin (ENPP2) inhibitors, using a newly developed fluorescence probe. ACS Chem. Biol. 2013, 8, 1713–1721. [Google Scholar] [CrossRef]
- Kuttruff, C.A.; Ferrara, M.; Bretschneider, T.; Hoerer, S.; Handschuh, S.; Nosse, B.; Romig, H.; Nicklin, P.; Roth, G.J. Discovery of BI-2545: A novel autotaxin inhibitor that significantly reduces LPA levels in vivo. ACS Med. Chem. Lett. 2017, 8, 1252–1257. [Google Scholar] [CrossRef]
- Keune, W.J.; Potjewyd, F.; Heidebrecht, T.; Salgado-Polo, F.; Macdonald, S.J.F.; Chelvarajan, L.; Abdel Latif, A.; Soman, S.; Morris, A.J.; Watson, A.J.B.; et al. Rational design of autotaxin inhibitors by structural evolution of endogenous modulators. J. Med. Chem. 2017, 60, 2006–2017. [Google Scholar] [CrossRef]
- Katsifa, A.; Kaffe, E.; Nikolaidou-Katsaridou, N.; Economides, A.N.; Newbigging, S.; McKerlie, C.; Aidinis, V. The bulk of autotaxin activity is dispensable for adult mouse life. PLoS ONE 2015, 10, 1–14. [Google Scholar] [CrossRef]
- Weng, J.; Jiang, S.; Ding, L.; Xu, Y.; Zhu, X.; Jin, P. Autotaxin/lysophosphatidic acid signaling mediates obesity-related cardiomyopathy in mice and human subjects. J. Cell. Mol. Med. 2019, 23, 1050–1058. [Google Scholar] [CrossRef]
- Jiang, G.; Madan, D.; Prestwich, D.G. Aromatic phosphonates inhibit the lydophosphodiesterase D activity of autotaxin. Bioorg. Med. Chem. Lett. 2011, 21, 5098–5101. [Google Scholar] [CrossRef]
- Shah, P.; Cheasty, A.; Foxton, C.; Raynham, T.; Farooq, M.; Gutierrez, I.F.; Lejeune, A.; Pritchard, M.; Turnbull, A.; Pang, L.; et al. Discovery of potent inhibitors of the lysophospholipase autotaxin. Bioorg. Med. Chem. Lett. 2016, 26, 5403–5410. [Google Scholar] [CrossRef]
- Bain, G.; Shannon, K.E.; Huang, F.; Darlington, J.; Goulet, L.; Prodanovich, P.; Ma, G.L.; Santini, A.M.; Stein, A.J.; Lonergan, D.; et al. Selective inhibition of autotaxin is efficacious in mouse models of liver fibrosis. J. Pharmacol. Exp. Ther. 2017, 360, 1–13. [Google Scholar] [CrossRef]
- Miller, L.M.; Keune, W.J.; Castagna, D.; Young, L.C.; Duffy, E.L.; Potjewyd, F.; Salgado-Polo, F.; García, P.E.; Semaan, D.; Pritchard, J.M.; et al. Structure-activity relationships of small molecule autotaxin inhibitors with a discrete binding mode. J. Med. Chem. 2017, 60, 722–748. [Google Scholar] [CrossRef]
- Black, K.E.; Berdyshev, E.; Bain, G.; Castelino, F.V.; Shea, B.S.; Probst, C.K.; Fontaine, B.A.; Bronova, I.; Goulet, L.; Lagares, D.; et al. Autotaxin activity increases locally following lung injury, but is not required for pulmonary lysophosphatidic acid production or fibrosis. FASEB J. 2016, 30, 2435–2450. [Google Scholar] [CrossRef]
- Castelino, F.V.; Bain, G.; Pace, V.A.; Black, K.E.; George, L.; Probst, C.K.; Goulet, L.; Lafyatis, R.; Tager, A.M. An Autotaxin-LPA-IL-6 amplification loop drives scleroderma fibrosis. Arthritis Rheumatol. 2016, 68, 2964–2974. [Google Scholar] [CrossRef]
- Roppe, J.R.; Parr, T.A.; Stock, N.S.; Volkots, D.; Hutchinson, J.H. Autotaxin Inhibitors and Uses Thereof. Patent WO 2012166415, 6 December 2012. [Google Scholar]
- Hutchinson, J.H.; Parr, T.A.; Roppe, J.R.; Stock, N.S.; Volkots, D. Heterocyclic autotaxin inhibitors and uses thereof. Patent US 2013029948, 31 January 2013. [Google Scholar]
- Blanque, R.D.N.; Dupont, S.; Cottereaux, C.; Marsais, F.; Lepescheux, L.; Monjardet, A.; Wakselman, E.; Laenen, W.; Russell, V.; van der Aar, E.; et al. Pharmacological Profile and Efficacy of GLPG1690, a Novel ATX Inhibitor for COPD Treatment. 2015. Available online: http://files.glpg.com/docs/website_1/Poster_ERS_2015_final.pdf (accessed on 8 October 2019).
- Maher, T.M.; van der Aar, E.M.; Van de Steen, O.; Allamassey, L.; Desrivot, J.; Dupont, S.; Fagard, L.; Ford, P.; Fieuw, A.; Wuyts, W. Safety, tolerability, pharmacokinetics, and pharmacodynamics of GLPG1690, a novel autotaxin inhibitor, to treat idiopathic pulmonary fibrosis (FLORA): A phase 2a randomised placebo-controlled trial. Lancet Respir. Med. 2018, 6, 627–635. [Google Scholar] [CrossRef]
- Galapagos Announces ISABELA Phase 3 Programin IPF. Available online: http://hugin.info/133350/R/2183965/843660.pdf (accessed on 8 October 2019).
- Zhang, H.; Xu, X.; Gajewiak, J.; Tsukahara, R.; Fujiwara, Y.; Liu, J.; Fells, J.I.; Perygin, D.; Parrill, A.L.; Tigyi, G.; et al. Dual activity lysophosphatidic acid receptor pan-antagonist/autotaxin inhibitor reduces breast cancer cell migration in vitro and causes tumor regression in vivo. Cancer Res. 2009, 69, 5441–5449. [Google Scholar] [CrossRef]
- Gupte, R.; Patil, R.; Liu, J.; Wang, Y.; Lee, S.C.; Fujiwara, Y.; Fells, J.; Bolen, A.L.; Emmons-Thompson, K.; Yates, C.R.; et al. Benzyl and naphthalene methylphosphonic acid inhibitors of autotaxin with anti-invasive and anti-metastatic activity. Chem. Med. Chem. 2011, 6, 922–935. [Google Scholar] [CrossRef]





| Type | Residues Establishing Ligand Contacts (Rat ATX) | |||
|---|---|---|---|---|
| Active Site-Hydrophilic Groove | Hydrophobic Pocket | Allosteric Tunnel | ||
| I | Common | Thr209, Asp311, His474 | - | - |
| Frequent | His315, His359 | Leu213, Phe273, Phe274 *, Trp275 | - | |
| II | Common | - | Leu213, Phe274 *, Trp276 | - |
| Frequent | - | Phe273, Tyr306 | - | |
| III | Common | - | - | Lys248, Phe249, Trp254, Trp260 |
| Frequent | - | - | Phe274 * | |
| IV | Common | - | Leu213, Phe273, Trp275, Tyr306 | Phe249, Trp260 |
| Frequent | - | Phe274 *, Phe210 | His251, Trp254, Phe274 * | |
| Type | Inhibitor | Disease | LPA Inhibition | References |
|---|---|---|---|---|
| I | SBJ-Cpd 1 | Inflammation Multiple sclerosis | >50% | [7] |
| I | PF-8380 | Glioblastoma Liver fibrosis | >90% >90% | [44] [4] |
| I | GK442 | Pulmonary fibrosis | [45] | |
| I | BMP22 | Melanoma metastasis | 50% | [46] |
| II | GWJ-A-23 | Pulmonary fibrosis inflammation | 50% | [47] [48] |
| III | PharmAkea -Cpd A-E | Metabolic disorder | 25–35% | [49] |
| III | PAT-505, PAT-048 | Liver fibrosis Skin fibrosis | 40–80% | [50] |
| IV | GLPG1690 | Pulmonary fibrosis Clinical trials in IPF patients | 84–95% 84–95% | [51] [52] |
| ? | ONO-8430506 | Breast cancer Thyroid cancer | >60% >70% | [53] [54] |
 | Type I Inhibitor | PDB ID | Activity (IC50) | Reference |
|---|---|---|---|---|
 HA-155 | 2xrg | 5.7 nM | [25,60] | |
 PF8380 | 510k | 1.7 nM | [61,62] | |
 FP-Cpd 3 | 5m0e | - | [62] | |
 MK-Cpd 10 | 3wav | 180 nM * | [63] | |
 2BoA | 3waw | 580 nM * | [63] | |
 3BoA | 3wax | 13 nM * | [63] | |
 4BoA | 3way | 22 nM * | [63] | |
 SBJ-Cpd 1 | 5l0b | 520 nM | [61] | |
 SBJ-Cpd 2 | 5l0e | 2.5 nM | [61] | |
 BI-2545 | 5ohi | 2.2 nM | [64] | |
 | Type II Inhibitor | PDB ID | Activity (IC50) | Reference |
|---|---|---|---|---|
 PAT-078 | 4zg6 | 472 nM | [50] | |
 PAT-494 | 4zga | 20 nM | [50] | |
 PAT-352 | 4zg9 | 26 nM | [50] | |
 CRT0273750 | 5lia | 1 nM | [68] | |












{kind=link}
{kind=link}
{kind=link}
{kind=link}
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Salgado-Polo, F.; Perrakis, A. The Structural Binding Mode of the Four Autotaxin Inhibitor Types that Differentially Affect Catalytic and Non-Catalytic Functions. Cancers 2019, 11, 1577. https://doi.org/10.3390/cancers11101577
Salgado-Polo F, Perrakis A. The Structural Binding Mode of the Four Autotaxin Inhibitor Types that Differentially Affect Catalytic and Non-Catalytic Functions. Cancers. 2019; 11(10):1577. https://doi.org/10.3390/cancers11101577
Chicago/Turabian StyleSalgado-Polo, Fernando, and Anastassis Perrakis. 2019. "The Structural Binding Mode of the Four Autotaxin Inhibitor Types that Differentially Affect Catalytic and Non-Catalytic Functions" Cancers 11, no. 10: 1577. https://doi.org/10.3390/cancers11101577
APA StyleSalgado-Polo, F., & Perrakis, A. (2019). The Structural Binding Mode of the Four Autotaxin Inhibitor Types that Differentially Affect Catalytic and Non-Catalytic Functions. Cancers, 11(10), 1577. https://doi.org/10.3390/cancers11101577