
Description of Si and Al Release from Aluminosilicate in the Acidic Condition Using Density Functional Theory: Protonated Terminal Oxygen

 , ,
, ,
Abstract
:1. Introduction
2. Materials and Methods
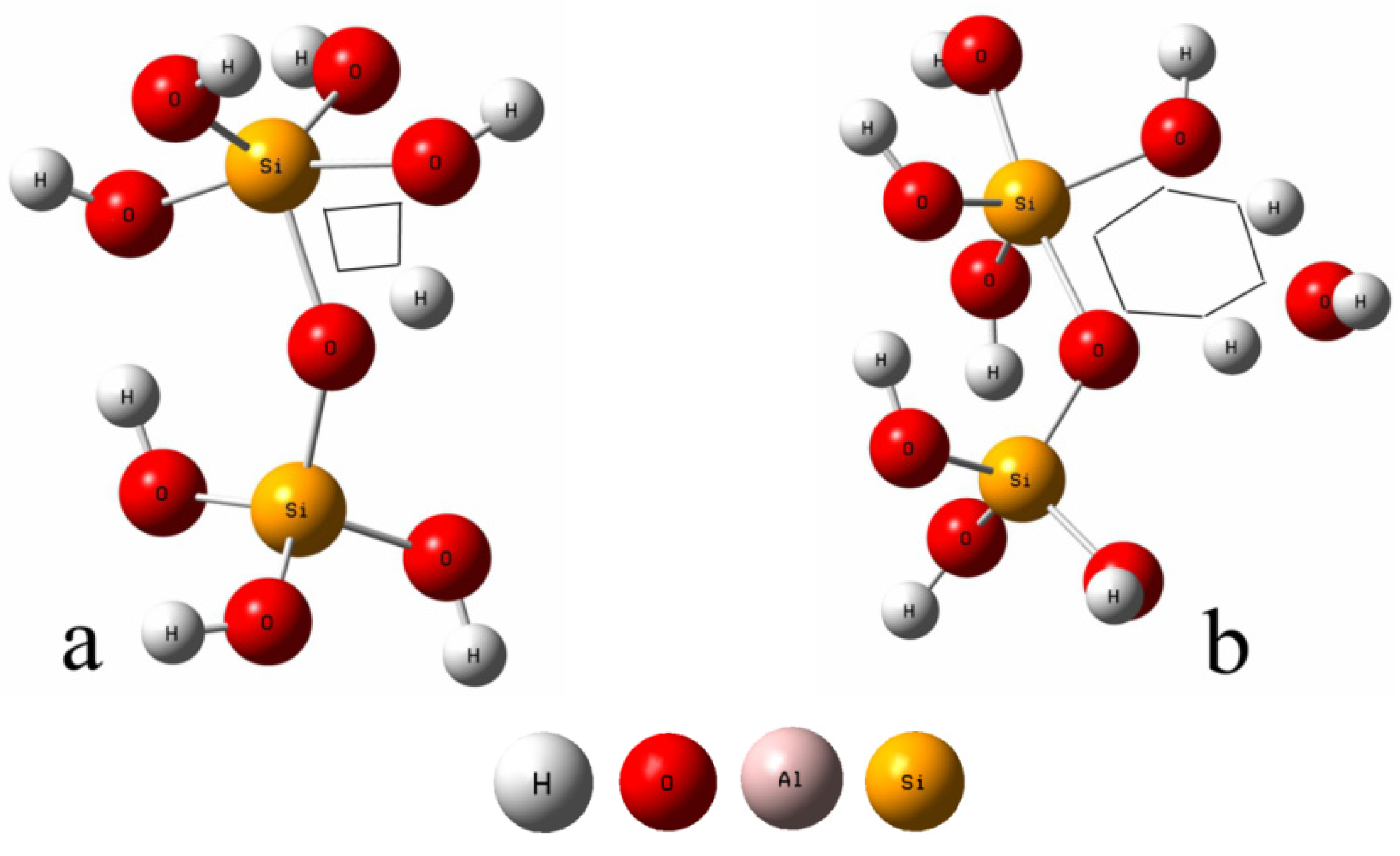
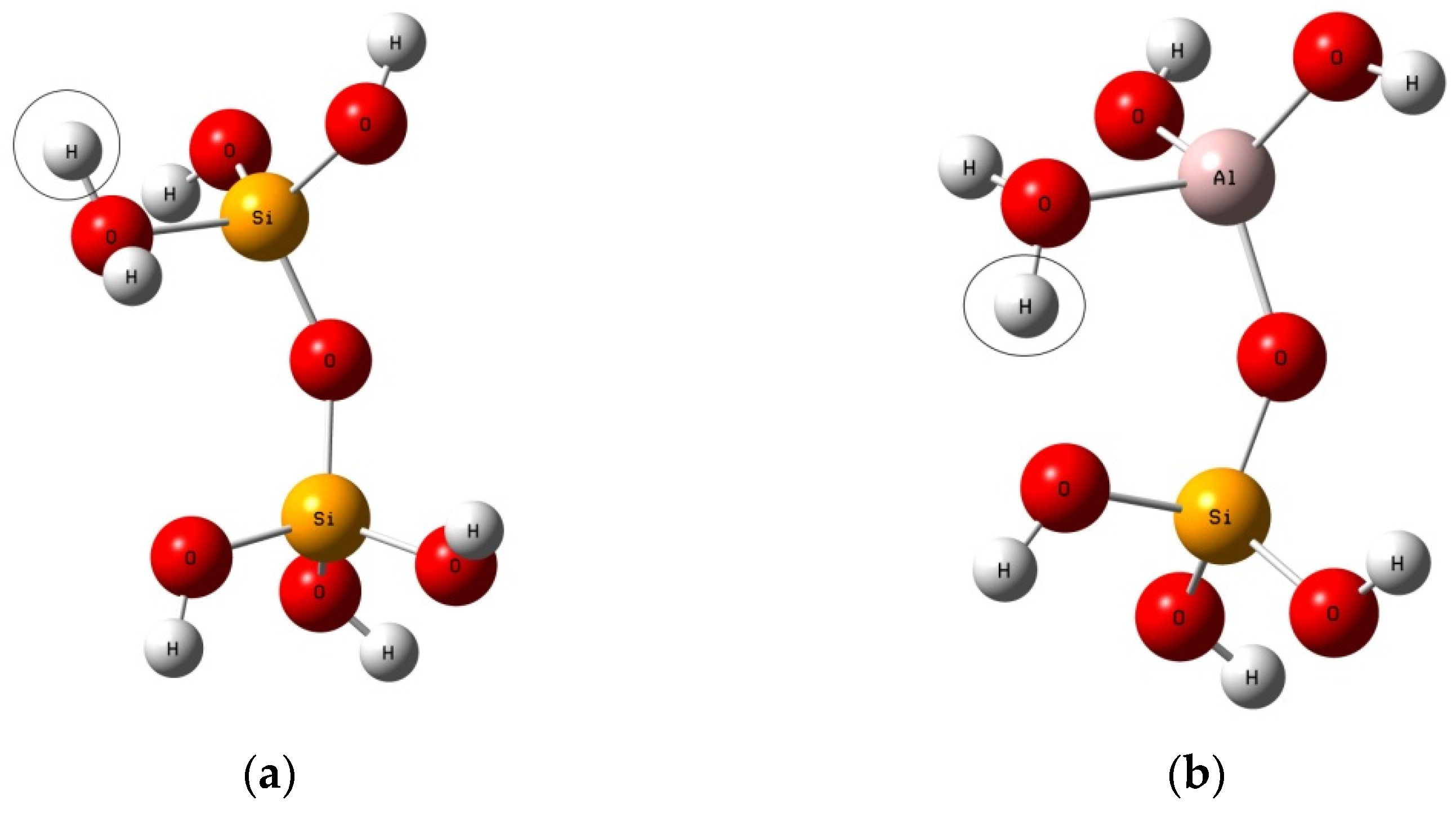
2.1. Reaction Models
2.2. Reaction Rate
3. Computational Methods
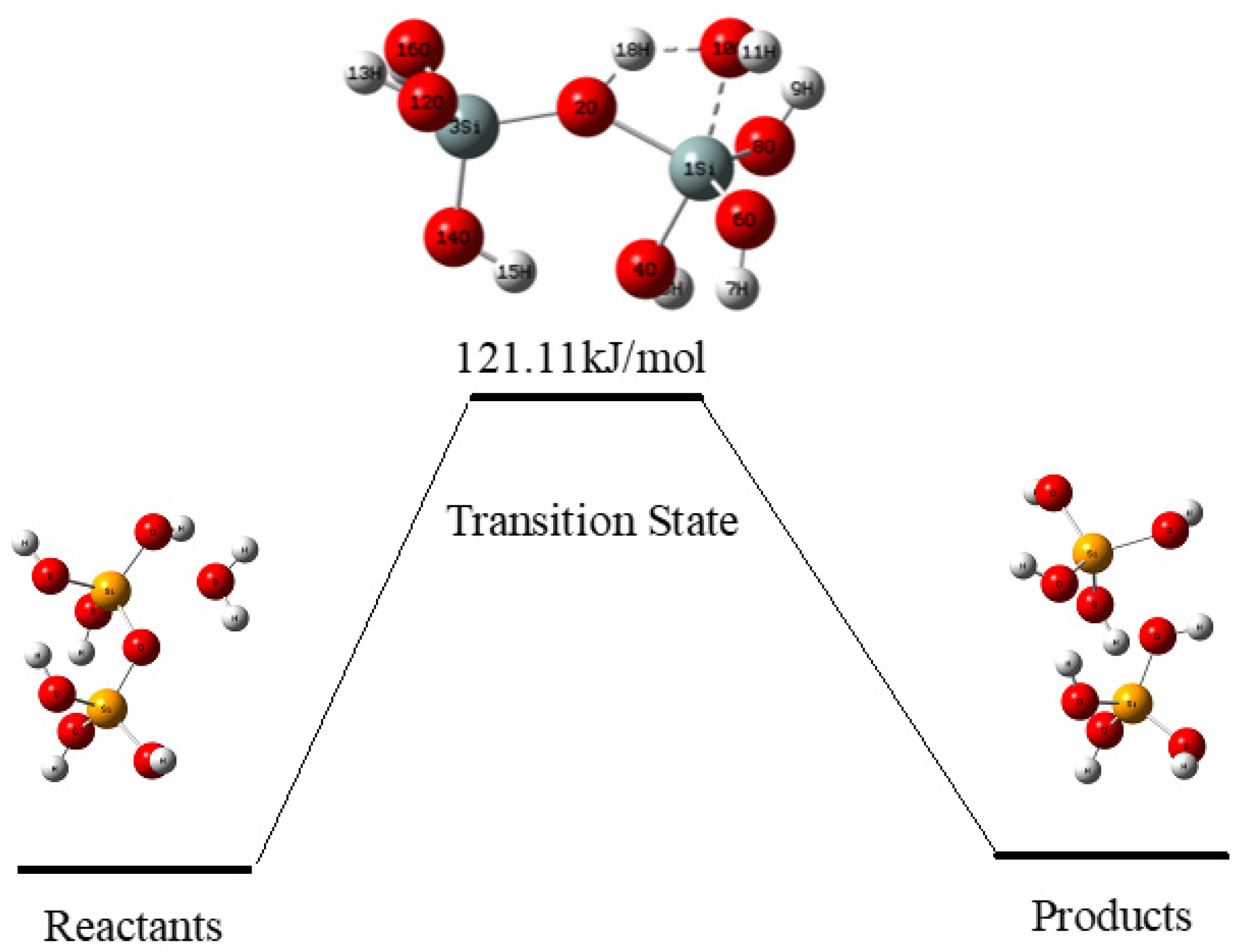
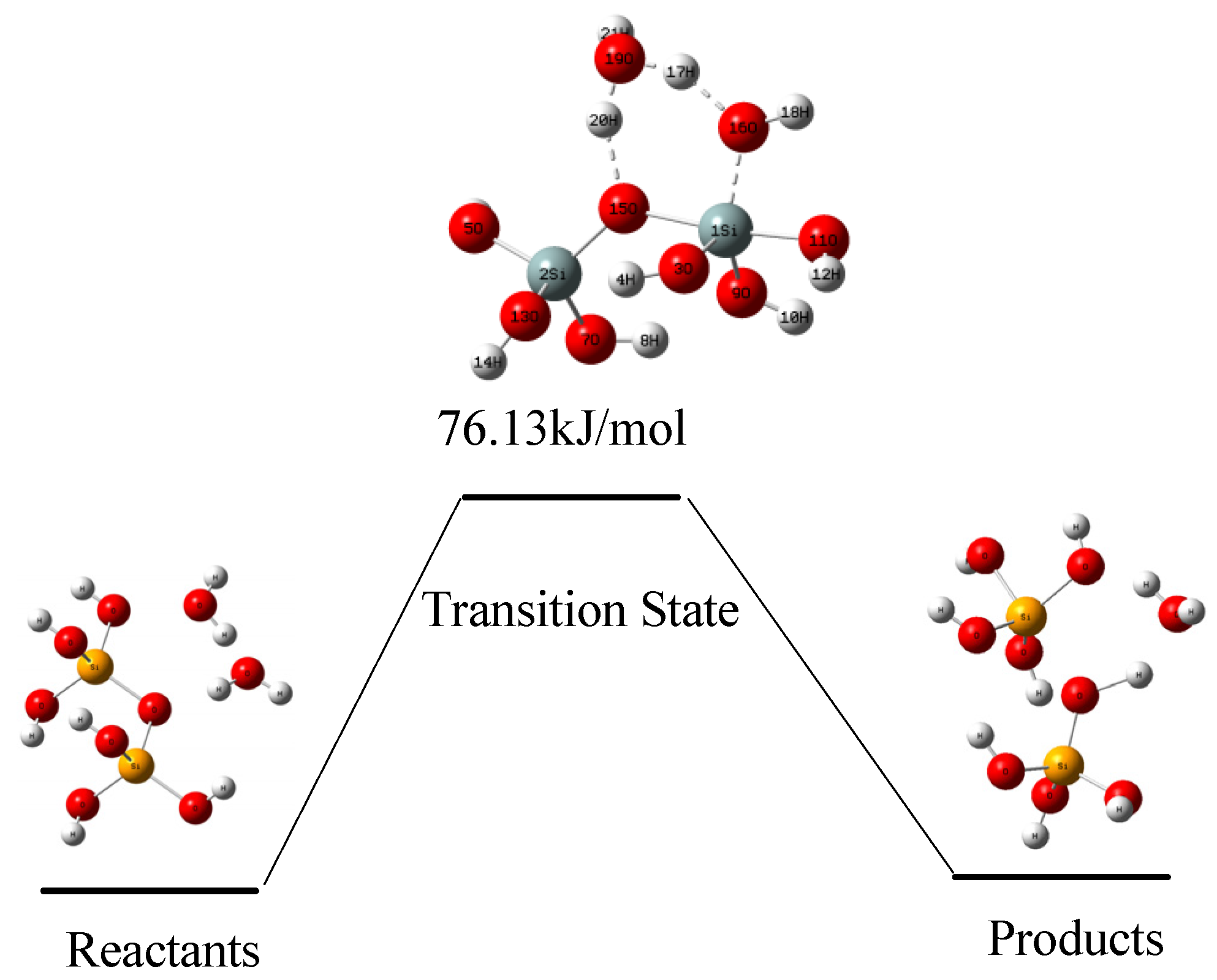
4. Results
4.1. The Configuration of Transition State
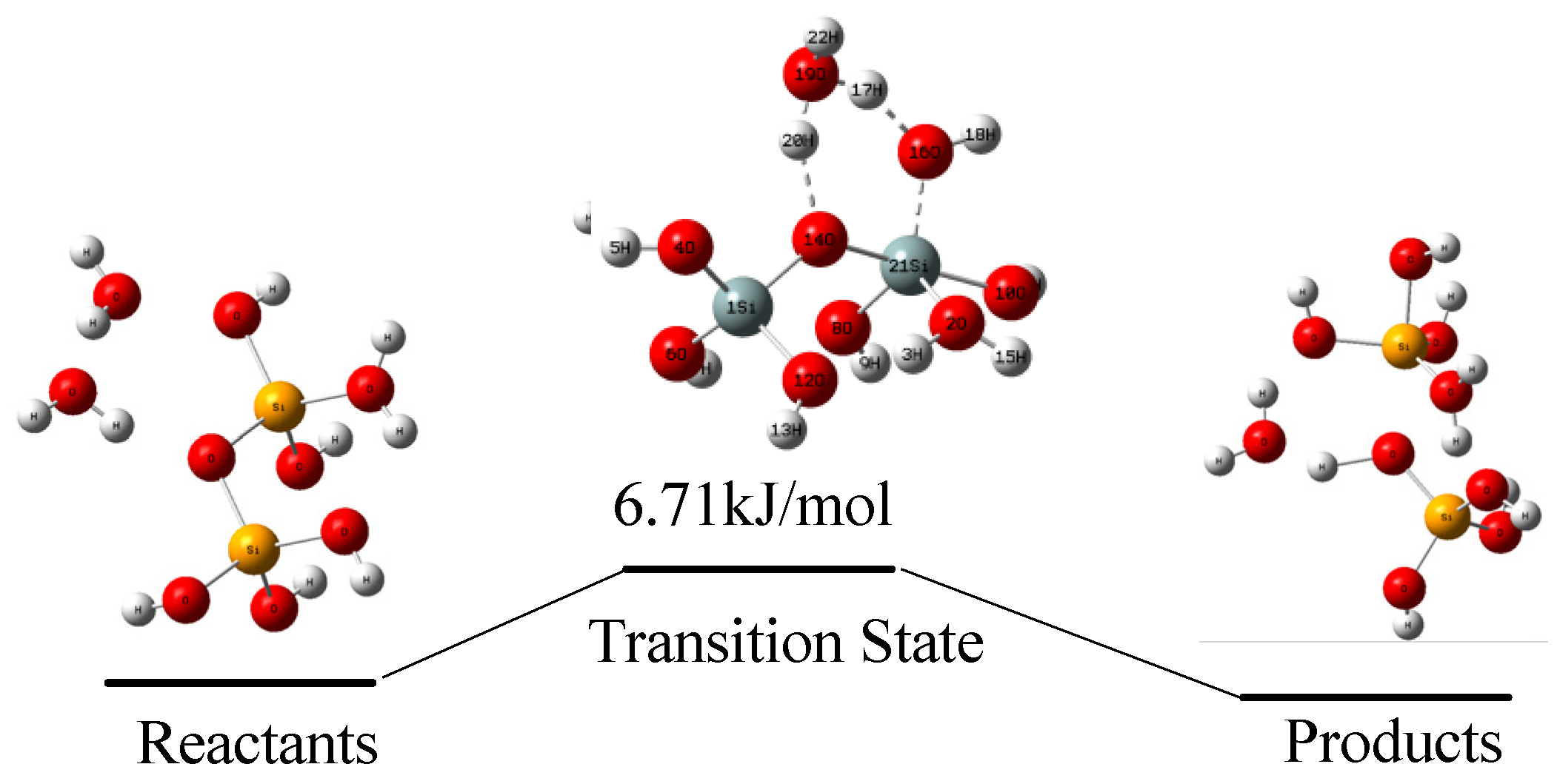
4.2. Hydrolysis Reaction at Siter-O-Si Site
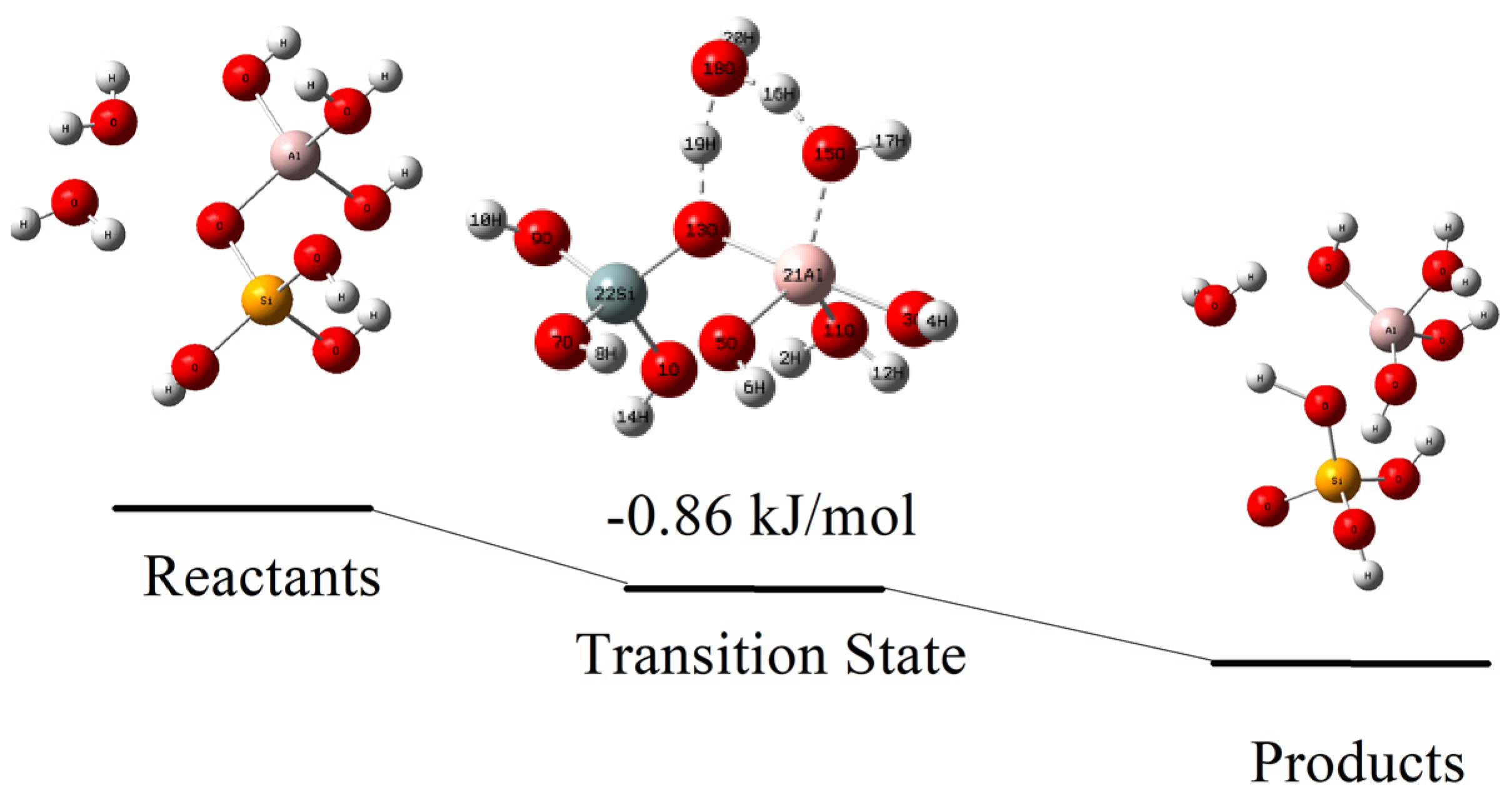
4.3. Hydrolysis Reaction at Alter-O-Si Site
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Huggins, M.L.; Sun, K.-H. Energy Additivity in Oxygen-containing Crystals and Glasses. J. Phys. Chem. 1946, 50, 319–328. [Google Scholar] [CrossRef]
- Zhang, C.; Zhang, Z.; Tan, Y.; Zhong, M. The Effect of Citric Acid on the Kaolin Activation and Mullite Formation. Ceram. Int. 2017, 43, 1466–1471. [Google Scholar] [CrossRef]
- Czech, K.; Banaś, D.; Kubala-Kukuś, A.; Stabrawa, I. The effect of chemical modification on the physico-chemical characteristics of halloysite: FTIR, XRF, and XRD studies. J. Mol. Struct. 2015, 1084, 16–22. [Google Scholar]
- Chaari, I.; Medhioub, M.; Jamoussi, F.; Hamzaoui, A.H. Acid-treated clay materials (Southwestern Tunisia) for removing sodium leuco-vat dye: Characterization, adsorption study and activation mechanism. J. Mol. Struct. 2020, 1223, 128944. [Google Scholar] [CrossRef]
- Lin, S.L.; Yu, Y.L.; Zhang, Z.J.; Zhang, C.Y.; Zhong, M.F.; Wang, L.M.; Lu, S.X.; Xu, W.; Li, N.; Huang, X. The synergistic mechanisms of citric acid and oxalic acid on the rapid dissolution of kaolinite. Appl. Clay Sci. 2020, 196, 105756. [Google Scholar] [CrossRef]
- Steudel, A.; Batenburg, L.F.; Fischer, H.R.; Weidler, P.G.; Emmerich, K. Alteration of Non-Swelling Clay Minerals and Magadiite by Acid Activation. Appl. Clay Sci. 2009, 44, 95–104. [Google Scholar] [CrossRef]
- Franklin, S.P.; Hajash, A.; Dewers, T.A.; Tieh, T.T. The Role of Carboxylic Acids in Albite and Quartz Dissolution: An Experimental Study under Diagenetic Conditions. Geochim. Cosmochim. Acta 1994, 58, 4259–4279. [Google Scholar] [CrossRef]
- Cama, J.; Ganor, J. The Effects of Organic Acids on the Dissolution of Silicate Minerals—A Case Study of Oxalate Catalysis of Kaolinite Dissolution. Geochim. Cosmochim. Acta 2006, 70, 2191–2209. [Google Scholar] [CrossRef]
- Stillings, L.L.; Drever, J.I.; Brantley, S.L.; Sun, Y.T.; Oxburgh, R. Rates of Feldspar Dissolution at pH 3–7 With 0–8 mM Oxalic Acid. Chem. Geol. 1996, 132, 79–89. [Google Scholar] [CrossRef]
- Shikha, N.; Barbara, J.G. Reaction Rates and Dissolution Mechanisms of Quartz as a Function of pH. J. Phys. Chem. A 2008, 112, 2027–2033. [Google Scholar]
- Inna, K.; Andreas, L. Kinetic Monte Carlo Simulations of Silicate Dissolution: Model Complexity and Parametrization. J. Phys. Chem. A 2013, 117, 24894–24906. [Google Scholar]
- Song, K.; Wang, X.; Qian, P.; Zhang, C.; Zhang, Q. Theoretical study of interaction of formamide with kaolinite. Comput. Theor. Chem. 2013, 1020, 72–80. [Google Scholar] [CrossRef]
- Du, M.; Kolchin, A.; Cheng, H. Hydrolysis of a two-membered silica ring on the amorphous silica surface. J. Chem. Phy. 2004, 120, 1044–1054. [Google Scholar] [CrossRef]
- Majid, V.; Alireza, F. DFT investigations for “Fischer” esterification mechanism over silica-propyl-SO3H catalyst: Is the reaction reversible? Comput. Theor. Chem. 2015, 1071, 27–32. [Google Scholar]
- Zhang, C.; Qi, Y.; Qian, P.; Zhong, M.; Wang, L.; Yin, H. Quantum chemical study of the adsorption of water molecules on kaolinite surfaces. Comput. Theor. Chem. 2014, 1046, 10–19. [Google Scholar] [CrossRef]
- Albert, R.; Piero, U.; Mariona, S. Strained Ring Motif at Silica Surfaces: A Quantum Mechanical Study of their Reactivity towards Protic Molecules. Comput. Theor. Chem. 2015, 1074, 168–177. [Google Scholar]
- Christin, P.M.; Shikha, N.; Barbara, J.G. Ab Initio Investigation of Dissolution Mechanisms in Aluminosilicate Minerals. J. Phys. Chem. A 2009, 113, 1343–1352. [Google Scholar]
- Criscenti, L.J.; Kubicki, J.D.; Brantley, S.L. Silicate Glass and Mineral Dissolution: Calculated Reaction Paths and Activation Energies for Hydrolysis of a Q3 Si by H3O+ Using Ab Initio Methods. J. Phys. Chem. A 2006, 110, 198–206. [Google Scholar] [CrossRef]
- Civalleri, B.; Garrone, E.; Ugliengo, P. Ab Initio Study of the Adducts of Small Molecules with the Isolated Hydroxyl of Silica and the Bronsted Site in Zeolites: A Comparison between B3LYP and MP2 Methods. J. Phys. Chem. B 1998, 102, 2373–2382. [Google Scholar] [CrossRef]
- Xiao, Y.T.; Lasaga, A.C. Ab-initio Quantum-mechanical Studies of the Kinetics and Mechanisms of Silicate Dissolution: H+(H3O+) Catalysis. Geochim. Cosmochim. Acta 1994, 58, 5379–5400. [Google Scholar] [CrossRef]
- Felipe, M.A.; Kubicki, J.D.; Rye, D.M. Hydrogen Isotope Exchange Kinetics between H2O and H4SiO4 From Ab Initio Calculations. Geochim. Cosmochim. Acta 2003, 67, 1259–1276. [Google Scholar] [CrossRef]
- Zhao, Y.; Truhlar, D.G. The M06 Suite of Density Functionals for Main Group Thermochemistry, Thermochemical Kinetics, Noncovalent Interactions, Excited States, and Transition Elements: Two New Functionals and Systematic Testing of Four M06-class Functionals and 12 Other Functionals. Theor. Chem. Acc. 2008, 120, 215–241. [Google Scholar]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09; Revision, D.01; Gaussian, Inc.: Wallingford, UK, 2009. [Google Scholar]
- Casey, W.H.; Lasaga, A.C.; Gibbs, G.V. Mechanisms of Silica Dissolution as Inferred from the Kinetic Isotope Effect. Geochim. Cosmochim. Acta 1990, 12, 3369–3378. [Google Scholar] [CrossRef]
- Polster, W.; Barnes, H.L. Kinetics of Quartz Precipitation and Dissolution under Hydrothermal Conditions at 100 to 300 °C. GSA Abstr. Prog. 1992, 24, A206. [Google Scholar]
- Bird, G.; Boon, J.; Stone, T. Silica Transport during Steam Injection into Oil Sands: 1. Dissolution and Precipitation Kinetics of Quartz: New Results and Review of Existing Data. Chem. Geol. 1986, 54, 69–80. [Google Scholar] [CrossRef]
- Welch, S.A.; Ullman, W.J. Feldspar Dissolution in Acidic and Organic Solutions: Compositional and pH Dependence of Dissolution Rate. Geochim. Cosmochim. Acta 1996, 60, 2939–2948. [Google Scholar] [CrossRef]
- Steudel, A.; Batenburg, L.F.; Fischer, H.R.; Weidler, P.G.; Emmerich, K. Alteration of Swelling Clay Minerals and Magadiite by Acid Activation. Appl. Clay Sci. 2009, 44, 105–115. [Google Scholar] [CrossRef]
- Wu, R.; Wang, S.; Wang, L. New Mechanism for the Atmospheric Oxidation of Dimethyl Sulfide. The Importance of Intramolecular Hydrogen Shift in a CH3SCH2OO Radical. Phys. Chem. 2015, 119, 112–117. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Model | Si-Obri | Siter-Obri | Obri-H | 1Si-10O | 1Si-16O | Siter-Obri-Si |
|---|---|---|---|---|---|---|
| TS4 | 1.68016 | 1.80964 | 1.11736 | 1.84944 | -- | 141.62679 |
| TS6 | 1.62984 | 1.84868 | 1.49800 | -- | 1.72330 | 127.61374 |
| Model | Si-Obri | Siter-Obri | Obri-H | 21Si-16O | Siter-Obri-Si |
|---|---|---|---|---|---|
| TS6-1 | 1.65078 | 1.75820 | 1.52743 | 1.71474 | 129.06299 |
| Model | Si-Obri | Alter-Obri | Obri-H | Alter-16O | Alter-15O | Alter-Obri-Si |
|---|---|---|---|---|---|---|
| TS6-2 | 1.64804 | 1.97997 | 1.16485 | 1.89625 | -- | 120.14675 |
| TS6-3 | 1.63210 | 1.94006 | 1.25021 | -- | 1.83037 | 123.71870 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, C.-Y.; Yu, Y.-L.; Yang, H.; Wang, L.-M.; Zhong, M.-F.; Lin, S.-M.; Zhang, Z.-J.; Wu, Y.-Y.; Liu, Y.; Xu, W. Description of Si and Al Release from Aluminosilicate in the Acidic Condition Using Density Functional Theory: Protonated Terminal Oxygen. Sustainability 2022, 14, 14390. https://doi.org/10.3390/su142114390
Zhang C-Y, Yu Y-L, Yang H, Wang L-M, Zhong M-F, Lin S-M, Zhang Z-J, Wu Y-Y, Liu Y, Xu W. Description of Si and Al Release from Aluminosilicate in the Acidic Condition Using Density Functional Theory: Protonated Terminal Oxygen. Sustainability. 2022; 14(21):14390. https://doi.org/10.3390/su142114390
Chicago/Turabian StyleZhang, Chen-Yang, Ya-Ling Yu, Huan Yang, Li-Ming Wang, Ming-Feng Zhong, Shao-Min Lin, Zhi-Jie Zhang, Yun-Ying Wu, Yang Liu, and Wei Xu. 2022. "Description of Si and Al Release from Aluminosilicate in the Acidic Condition Using Density Functional Theory: Protonated Terminal Oxygen" Sustainability 14, no. 21: 14390. https://doi.org/10.3390/su142114390
APA StyleZhang, C.-Y., Yu, Y.-L., Yang, H., Wang, L.-M., Zhong, M.-F., Lin, S.-M., Zhang, Z.-J., Wu, Y.-Y., Liu, Y., & Xu, W. (2022). Description of Si and Al Release from Aluminosilicate in the Acidic Condition Using Density Functional Theory: Protonated Terminal Oxygen. Sustainability, 14(21), 14390. https://doi.org/10.3390/su142114390