
Bempedoic Acid, the First-in-Class Oral ATP Citrate Lyase Inhibitor with Hypocholesterolemic Activity: Clinical Pharmacology and Drug–Drug Interactions



Abstract
1. Introduction
2. Mechanism of Action of Bempedoic Acid
3. Pharmacokinetic Properties
3.1. Absorption
3.2. Distribution
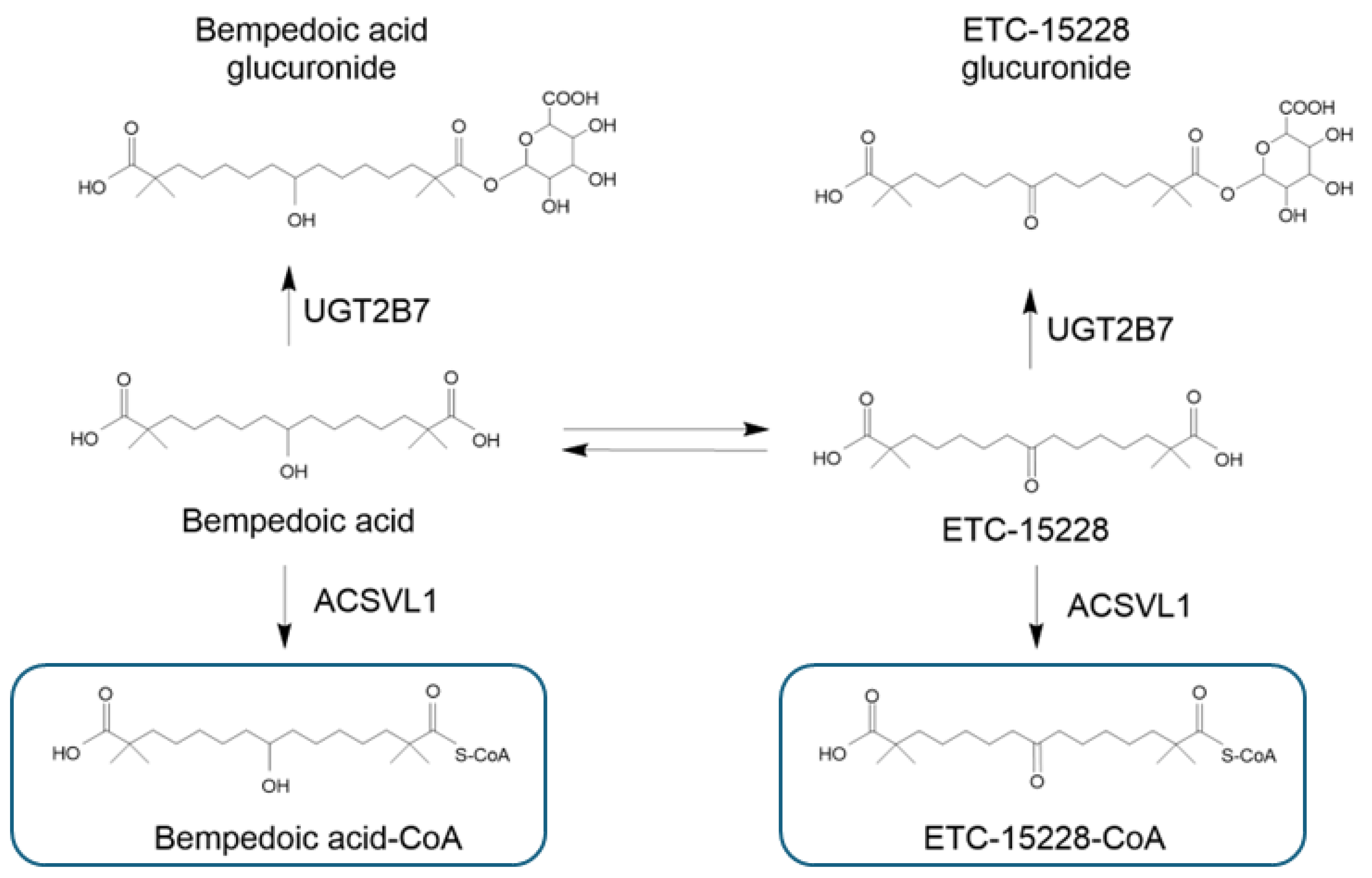
3.3. Metabolism
3.4. Elimination
4. General Considerations on DDI with Bempedoic Acid
5. Pharmacodynamic DDIs with Bempedoic Acid
6. Pharmacokinetics DDIs with Bempedoic Acid
6.1. Potential DDIs with Lipid-Lowering Drugs
6.2. Potential DDIs with Immune-Modulating and Antineoplastic Agents

{kind=link}
{kind=link}
| Drug | Effect on CYP450 and Drug Transporters | Effect on Bempedoic Acid | Effect on Interacting Drug | Expert Opinion |
|---|---|---|---|---|
| Immunosuppressant drugs | ||||
| Cyclosporine | Inhibitor of CYP3A4 and P-gp, OATP1B1/1B3 [36,37,38]. | No significant effect predicted. | No significant effect predicted. | Bempedoic acid inhibits OATP1B1/1B3, but it is not a substrate of these transporters. Thus, no interaction is predicted with OATP1B1/1B3 inhibitors or inducers. |
| Tacrolimus | Metabolized by CYP3A4 and substrate of P-gp [36,37,38]. | No significant effect predicted. | No significant effect predicted. | No drug interaction. |
| Everolimus, sirolimus | Everolimus is a substrate of CYP3A4 and weak inhibitor of P-gp and CYP2D6 [49]. Sirolimus is metabolized by CYP3A4 and is a substrate of P-gp [50]. Both are inhibitors of OATP1B1 [39,40]. | No significant effect predicted. | No significant effect predicted. | Bempedoic acid inhibits OATP1B1/1B3, but it is not a substrate of these transporters. Thus, no interaction is predicted with OATP1B1/1B3 inhibitors or inducers. |
| Leflunomide | Metabolized by CYP1A2, CYP2C19, and CYP3A4. Inhibitor of OAT3 [42]. | Leflunomide could increase the exposure of bempedoic acid. | No significant effect predicted. | It is recommended to have caution when administered with an OAT3 substrate like bempedoic acid–glucuronide. |
| Teriflunomide | Inhibitor of CYP2C8. Inducer of CYP1A2. Inhibitor of OAT3, OATP1B1/B3 [41]. | Teriflunomide could increase the exposure of bempedoic acid. | No significant effect predicted. | The inhibition of OAT3 could increase the AUC of bempedoic acid–glucuronide and potentially the plasma levels of uremic acid and creatinine. |
| Mycophenolate mofetil | Metabolized by UGT1A9. Substrate of OAT1, OAT3, MRP2, and BCRP [44]. | No significant effect predicted. | Bempedoic acid could increase the exposure of mycophenolate mofetil. | Bempedoic acid could reduce the renal excretion of mycophenolate mofetil by competing with OAT1 and OAT3. Both drugs increase uric acid plasma levels. Higher incidence of gout is predicted. |
| Anticancer drugs | ||||
| Tamoxifen | Metabolized by CYP3A4. CYP2D6 metabolism leads to the formation of the active molecule endoxifen [51]. Inhibitor of P-gp and is a substrate of OATP1A2 [52]. | No significant effect predicted. | No significant effect predicted. | No drug interaction. |
| Taxanes (paclitaxel, docetaxel) | Paclitaxel is a substrate of CYP2C8 and CYP3A4. Docetaxel is a substrate of CYP3A4. Both are substrates of OATP1B1/1B3. | No significant effect predicted. | Bempedoic acid could increase the exposure of docetaxel and paclitaxel. | Bempedoic acid inhibits OATP1B1/1B3 and may increase the exposure of docetaxel and paclitaxel. |
| Darolutamide | Metabolized by CYP3A4 and UGT1A9. Substrate of P-gp and BCRP. Inhibitor of OATP1B1/B3 [46]. | No significant effect predicted. | No significant effect predicted. | Co-administration of rosuvastatin with darolutamide should be avoided unless there is no alternative therapy. Consider drugs which are not substrates for OATP1B1 and OATP1B3. Bempedoic acid inhibits OATP1B1/1B3, but it is not a substrate of these transporters. Thus, no interaction is predicted with OATP1B1/1B3 inhibitors or inducers. |
| Imatinib, crizotinib and tucatinib | Inhibitor of CYP3A4 and P-gp. Imatinib inhibits CYP2D6, UGT2B17, and, partially, UGT1A1 [53]. Tucatinib could induce CYP2B6, CYP3A4, CYP2C9, CYP2C19, or UGT1A1, and inhibits MRP2, OCT1, and OAT3 [48]. | Tucatinib could increase the exposure of bempedoic acid. | No significant effect predicted. | The inhibition of OAT3 could increase the AUC of bempedoic acid–glucuronide and potentially the plasma levels of uremic acid and creatinine. |
| Enzalutamide | Metabolized by CYP2C8 and CYP3A4. Inducer of CYP3A4, CYP2B6, CYP2C9, CYP2C19, UGT1A1, MRP2, OATP1B1, and P-gp [47]. Possible inhibition of MRP2, OCT1, and OAT3. | Enzalutamide could increase the exposure of bempedoic acid. | No significant effect predicted. | Induction of OATP1B1 is not expected to potentiate the hepatic uptake and activity of bempedoic acid. Enzalutamide is an OAT3 inhibitor. This could increase the AUC of bempedoic acid–glucuronide and potentially the plasma levels of uremic acid and creatinine. |
6.3. Potential DDIs with Antiviral, Antibiotic, and Antifungal Agents
| Drug | Effect on CYP450 and Drug Transporters | Effect on Bempedoic Acid | Effect on Interacting Drug | Expert Opinion |
|---|---|---|---|---|
| Antivirals, antibiotics, antifungals | ||||
| Dolutegravir/lamivudine/abacavir | No inhibition of CYP450. Dolutegravir inhibits OAT1 and OAT3. | Dolutegravir may increase plasma concentrations of drugs excreted through OAT3, such as bempedoic acid. | No significant effect predicted. | The inhibition of OAT3 could increase the AUC of bempedoic acid–glucuronide and potentially the plasma levels of uremic acid and creatinine. |
| Atazanavir and ritonavir | Potent inhibitors of CYP3A4. Inhibitors of glucuronidation [61,62]. | Possible reduction in glucuronide metabolite of bempedoic acid. | No significant effect predicted. | Inhibition of glucuronidation could prolong the half-life of bempedoic acid. |
| Lopinavir and ritonavir | Potent inhibitors of CYP3A4. Inhibitors of OATP1B1 and OATP1B3 [54,55]. | No significant effect predicted. | No significant effect predicted. | Bempedoic acid inhibits OATP1B1/1B3, but it is not a substrate of these transporters. Thus, no interaction is predicted with OATP1B1/1B3 inhibitors or inducers. |
| Asunaprevir, glecaprevir, grazoprevir and voxilaprevir | Substrate of OATP1B1 and OATP1B3 | No significant effect predicted. | Possible increase in their exposure. | Bempedoic acid, by inhibiting OATP1B1/1B3, may increase the exposure of anti-HCV drugs. |
| Antibiotic | ||||
| Rifampicin | Single dose inhibits OATP1B1 and OATP1B3. Strong inducer of CYP3A4, CYP2C19, P-gp and UGT [57]. | No significant effect predicted. | No significant effect predicted. | Bempedoic acid inhibits OATP1B1/1B3, but it is not a substrate of these transporters. Thus, no interaction is predicted with OATP1B1/1B3 inhibitors or inducers. The induction of UGT could increase the metabolism of bempedoic acid. |
| Erythromycin and clarithromycin | Inhibitors of CYP3A4 and P-gp. Inhibitors of OATP1B1 and OATP1B3 [58,59,60,63]. | No significant effect predicted. | No significant effect predicted. | Bempedoic acid inhibits OATP1B1/1B3, but it is not a substrate of these transporters. Thus, no interaction is predicted with OATP1B1/1B3 inhibitors or inducers. |
| Antifungal | ||||
| Ketoconazole, itraconazole and voriconazole | Potent inhibitors of CYP3A4, P-gp, and BCRP. Ketoconazole is a potent inhibitor of OATP1B1, OATP1B3, OAT3, OCT1, and OCT2, and a weak inhibitor of OAT [64]. | No significant effect predicted. | No significant effect predicted. | An increase in systemic exposure of bempedoic acid–glucuronide may be expected by OAT3 inhibition of ketoconazole. Itraconazole and voriconazole do not appear to inhibit this transporter [65,66]. |
6.4. Potential DDIs with Antidiabetic Agents
| Drug | Effect on CYP450 and Drug Transporters | Effect on Bempedoic Acid | Effect on Interacting Drug | Expert Opinion |
|---|---|---|---|---|
| Antidiabetic drugs | ||||
| Metformin | Metabolized by UGT1A1 and partially by UGT1A3 and CYP3A4. Substrate of MATE-1, MATE-2K, and OCT2 [69]. | No significant effect predicted. | No significant effect predicted. | No pharmacokinetic and pharmacodynamic interaction observed between metformin and bempedoic acid [15,19]. |
| Glyburide | Substrate of CYP2C9. Substrate and inhibitor of OATP1B1/1B3. | No significant effect predicted. | Possible increase in glyburide exposure. | Bempedoic acid inhibits OATP1B1/1B3, but it is not a substrate of these transporters. Thus, no significant interaction is predicted. |
| Repaglinide | Substrate of CYP2C8 and OATP1B1/1B3. | No significant effect predicted. | Possible increase in repaglinide exposure. | Bempedoic acid inhibits OATP1B1/1B3, but it is not a substrate of these transporters. Thus, no significant interaction is predicted. |
| SGLT2 (ertugliflozin, dapagliflozin, empagliflozin, canagliflozin) | Metabolized by UGT. Empagliflozin is a substrate of OAT3 and OATP1B1/1B3 [67,71]. The dapagliflozin metabolite is a substrate of OAT3 [72]. | No significant effect predicted. | No significant effect predicted. | Although empagliflozin is a substrate for OAT3, OATP1B1, and OATP1B3, no significant interaction is predicted with bempedoic acid. Indeed, the strong OAT3 inhibitor probenecid showed a minimal effect on the exposure of empagliflozin [67]. |
| DPP4 inhibitors (sitagliptin, vildagliptin, saxagliptin, linagliptin, alogliptin) | Saxagliptin is metabolized by CYP3A4/5. Linagliptin is a weak inhibitor of CYP3A4 and substrate of P-gp [73]. Sitagliptin is a substrate of P-gp and OAT3 [68]. | No significant effect predicted. | Possible increase in sitagliptin exposure. | Bempedoic acid, by inhibiting the OAT3, could reduce the clearance of sitagliptin. No interactions are predicted with other DDP4 inhibitors. |
| GLP1 agonists (liraglutide, exenatide, semaglutide) | Proteolytic metabolism. | No significant effect predicted. | No significant effect predicted. | No drug interaction. |
6.5. Potential DDIs of Bempedoic Acid with Cardiovascular Drugs
6.6. Potential DDIs of Bempedoic Acid with Antidepressant, Antipsychotic, and Antiepileptic Drugs
6.7. Bempedoic Acid and Uric Acid: Potential DDI
7. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Cholesterol Treatment Trialists’ (CTT) Collaborators; Baigent, C.; Blackwell, L.; Emberson, J.; Holland, L.E.; Reith, C.; Bhala, N.; Peto, R.; Barnes, E.H.; Keech, A.; et al. Efficacy and safety of more intensive lowering of LDL cholesterol: A meta-analysis of data from 170,000 participants in 26 randomised trials. Lancet 2010, 376, 1670–1681. [Google Scholar]
- Cholesterol Treatment Trialists’ (CTT) Collaborators; Mihaylova, B.; Emberson, J.; Blackwell, L.; Keech, A.; Simes, J.; Barnes, E.H.; Voysey, M.; Gray, A.; Collins, R.; et al. The effects of lowering LDL cholesterol with statin therapy in people at low risk of vascular disease: Meta-analysis of individual data from 27 randomised trials. Lancet 2012, 380, 581–590. [Google Scholar] [CrossRef]
- Kastelein, J.J.; Akdim, F.; Stroes, E.S.; Zwinderman, A.H.; Bots, M.L.; Stalenhoef, A.F.; Visseren, F.L.; Sijbrands, E.J.; Trip, M.D.; Stein, E.A.; et al. Simvastatin with or without ezetimibe in familial hypercholesterolemia. N. Engl. J. Med. 2008, 358, 1431–1443. [Google Scholar] [CrossRef]
- Sabatine, M.S.; Giugliano, R.P.; Keech, A.C.; Honarpour, N.; Wiviott, S.D.; Murphy, S.A.; Kuder, J.F.; Wang, H.; Liu, T.; Wasserman, S.M.; et al. Evolocumab and Clinical Outcomes in Patients with Cardiovascular Disease. N. Engl. J. Med. 2017, 376, 1713–1722. [Google Scholar] [CrossRef]
- Schwartz, G.G.; Steg, P.G.; Szarek, M.; Bhatt, D.L.; Bittner, V.A.; Diaz, R.; Edelberg, J.M.; Goodman, S.G.; Hanotin, C.; Harrington, R.A.; et al. Alirocumab and Cardiovascular Outcomes after Acute Coronary Syndrome. N. Engl. J. Med. 2018, 379, 2097–2107. [Google Scholar] [CrossRef]
- Silverman, M.G.; Ference, B.A.; Im, K.; Wiviott, S.D.; Giugliano, R.P.; Grundy, S.M.; Braunwald, E.; Sabatine, M.S. Association Between Lowering LDL-C and Cardiovascular Risk Reduction Among Different Therapeutic Interventions: A Systematic Review and Meta-analysis. JAMA 2016, 316, 1289–1297. [Google Scholar] [CrossRef]
- Ference, B.A.; Braunwald, E.; Catapano, A.L. The LDL cumulative exposure hypothesis: Evidence and practical applications. Nat. Rev. Cardiol. 2024, 21, 701–716. [Google Scholar] [CrossRef]
- Gaba, P.; O’Donoghue, M.L.; Park, J.G.; Wiviott, S.D.; Atar, D.; Kuder, J.F.; Im, K.; Murphy, S.A.; De Ferrari, G.M.; Gaciong, Z.A.; et al. Association Between Achieved Low-Density Lipoprotein Cholesterol Levels and Long-Term Cardiovascular and Safety Outcomes: An Analysis of FOURIER-OLE. Circulation 2023, 147, 1192–1203. [Google Scholar] [CrossRef]
- Pinkosky, S.L.; Newton, R.S.; Day, E.A.; Ford, R.J.; Lhotak, S.; Austin, R.C.; Birch, C.M.; Smith, B.K.; Filippov, S.; Groot, P.H.E.; et al. Liver-specific ATP-citrate lyase inhibition by bempedoic acid decreases LDL-C and attenuates atherosclerosis. Nat. Commun. 2016, 7, 13457. [Google Scholar] [CrossRef]
- Banach, M.; Duell, P.B.; Gotto, A.M., Jr.; Laufs, U.; Leiter, L.A.; Mancini, G.B.J.; Ray, K.K.; Flaim, J.; Ye, Z.; Catapano, A.L. Association of Bempedoic Acid Administration With Atherogenic Lipid Levels in Phase 3 Randomized Clinical Trials of Patients With Hypercholesterolemia. JAMA Cardiol. 2020, 5, 1124–1135. [Google Scholar] [CrossRef]
- Ballantyne, C.M.; Laufs, U.; Ray, K.K.; Leiter, L.A.; Bays, H.E.; Goldberg, A.C.; Stroes, E.S.; MacDougall, D.; Zhao, X.; Catapano, A.L. Bempedoic acid plus ezetimibe fixed-dose combination in patients with hypercholesterolemia and high CVD risk treated with maximally tolerated statin therapy. Eur. J. Prev. Cardiol. 2020, 27, 593–603. [Google Scholar] [CrossRef] [PubMed]
- Ray, K.K.; Nicholls, S.J.; Li, N.; Louie, M.J.; Brennan, D.; Lincoff, A.M.; Nissen, S.E.; CLEAR OUTCOMES Committees and Investigators. Efficacy and safety of bempedoic acid among patients with and without diabetes: Prespecified analysis of the CLEAR Outcomes randomised trial. Lancet Diabetes Endocrinol. 2024, 12, 19–28. [Google Scholar] [CrossRef] [PubMed]
- Nissen, S.E.; Lincoff, A.M.; Brennan, D.; Ray, K.K.; Mason, D.; Kastelein, J.J.P.; Thompson, P.D.; Libby, P.; Cho, L.; Plutzky, J.; et al. Bempedoic Acid and Cardiovascular Outcomes in Statin-Intolerant Patients. N. Engl. J. Med. 2023, 388, 1353–1364. [Google Scholar] [CrossRef] [PubMed]
- Nissen, S.E.; Menon, V.; Nicholls, S.J.; Brennan, D.; Laffin, L.; Ridker, P.; Ray, K.K.; Mason, D.; Kastelein, J.J.P.; Cho, L.; et al. Bempedoic Acid for Primary Prevention of Cardiovascular Events in Statin-Intolerant Patients. JAMA 2023, 330, 131–140. [Google Scholar] [CrossRef] [PubMed]
- Nilemdo: Summary of Product Characteristics. 2024. Available online: https://www.ema.europa.eu/en/documents/product-information/nilemdo-epar-product-information_en.pdf (accessed on 15 September 2024).
- Nilemdo. 2024. Available online: https://www.ema.europa.eu/en/documents/assessment-report/nilemdo-epar-public-assessment-report_en.pdf (accessed on 15 September 2024).
- Hanselman, J.C.; MacDougall, D.; Emery, M.G.; Sasiela, W.; Amore, B.M. A phase 1 drugdrug interaction study assessing the effects of steady state probenicid on single-dose bempedoic acid pharmacokinetics in healthy subjects. Clin. Pharmacol. Ther. 2020, 107, S43. [Google Scholar]
- Amore, B.M.; Sasiela, W.J.; Ries, D.K.; Tresh, P.; Emery, M.G. Pharmacokinetics of bempedoic acid in patients with renal impairment. Clin. Transl. Sci. 2022, 15, 789–798. [Google Scholar] [CrossRef]
- Emery, M.G.; Hanselman, J.H.; MacDougall, D.; Amore, B.; Sasiela, W.; McGonigal, J. Effect of bempedoic acid on the pharmacokinetics and bempedoic acid (BA) effect on metformin (MET) pharmacokinetics (PK) and pharmacodynamics (PD) in patients with type 2 diabetes (T2D): In vitro–in vivo correlation. J. Clin. Pharmacol. Ther. 2020, 107, S43. [Google Scholar]
- Amore, B.M.; Cramer, C.; MacDougall, D.; Emery, M.G. The Disposition and Metabolism of Bempedoic Acid, a Potent Inhibitor of ATP Citrate Lyase, in Healthy Human Subjects. Drug Metab. Dispos. 2023, 51, 599–609. [Google Scholar] [CrossRef]
- Oniciu, D.C.; Dasseux, J.L.; Yang, J.; Mueller, R.; Pop, E.; Denysenko, A.; Duan, C.; Huang, T.B.; Zhang, L.; Krause, B.R.; et al. Influence of various central moieties on the hypolipidemic properties of long hydrocarbon chain diols and diacids. J. Med. Chem. 2006, 49, 334–348. [Google Scholar] [CrossRef]
- Nexletol. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2020/211616s000lbl.pdf (accessed on 15 September 2024).
- Ferri, N.; Corsini, A. Mechanism of bempedoic acid induced cholelithiasis: A role for statins to limit this adverse effect? Pharmacol. Res. 2023, 196, 106900. [Google Scholar] [CrossRef]
- Caroli-Bosc, F.X.; Le Gall, P.; Pugliese, P.; Delabre, B.; Caroli-Bosc, C.; Demarquay, J.F.; Delmont, J.P.; Rampal, P.; Montet, J.C. Role of fibrates and HMG-CoA reductase inhibitors in gallstone formation: Epidemiological study in an unselected population. Dig. Dis. Sci. 2001, 46, 540–544. [Google Scholar] [CrossRef] [PubMed]
- Semwal, R.B.; Semwal, D.K.; Vermaak, I.; Viljoen, A. A comprehensive scientific overview of Garcinia cambogia. Fitoterapia 2015, 102, 134–148. [Google Scholar] [CrossRef] [PubMed]
- Bhatt, D.L.; Steg, P.G.; Miller, M.; Brinton, E.A.; Jacobson, T.A.; Ketchum, S.B.; Doyle, R.T., Jr.; Juliano, R.A.; Jiao, L.; Granowitz, C.; et al. Cardiovascular Risk Reduction with Icosapent Ethyl for Hypertriglyceridemia. N. Engl. J. Med. 2019, 380, 11–22. [Google Scholar] [CrossRef] [PubMed]
- Mach, F.; Baigent, C.; Catapano, A.L.; Koskinas, K.C.; Casula, M.; Badimon, L.; Chapman, M.J.; De Backer, G.G.; Delgado, V.; Ference, B.A.; et al. 2019 ESC/EAS Guidelines for the management of dyslipidaemias: Lipid modification to reduce cardiovascular risk. Eur. Heart J. 2020, 41, 111–188. [Google Scholar] [CrossRef] [PubMed]
- Group, S.C.; Link, E.; Parish, S.; Armitage, J.; Bowman, L.; Heath, S.; Matsuda, F.; Gut, I.; Lathrop, M.; Collins, R. SLCO1B1 variants and statin-induced myopathy—A genomewide study. N. Engl. J. Med. 2008, 359, 789–799. [Google Scholar] [CrossRef]
- Niemi, M.; Pasanen, M.K.; Neuvonen, P.J. Organic anion transporting polypeptide 1B1: A genetically polymorphic transporter of major importance for hepatic drug uptake. Pharmacol. Rev. 2011, 63, 157–181. [Google Scholar] [CrossRef]
- Corsini, A.; Bellosta, S. Drug-drug interaction with statins. Expert. Rev. Clin. Pharmacol. 2008, 1, 105–113. [Google Scholar] [CrossRef]
- Prueksaritanont, T.; Subramanian, R.; Fang, X.; Ma, B.; Qiu, Y.; Lin, J.H.; Pearson, P.G.; Baillie, T.A. Glucuronidation of statins in animals and humans: A novel mechanism of statin lactonization. Drug Metab. Dispos. 2002, 30, 505–512. [Google Scholar] [CrossRef]
- Prueksaritanont, T.; Tang, C.; Qiu, Y.; Mu, L.; Subramanian, R.; Lin, J.H. Effects of fibrates on metabolism of statins in human hepatocytes. Drug Metab. Dispos. 2002, 30, 1280–1287. [Google Scholar] [CrossRef]
- Bellosta, S.; Corsini, A. Statin drug interactions and related adverse reactions. Expert Opin. Drug Saf. 2012, 11, 933–946. [Google Scholar] [CrossRef]
- Ferri, N.; Bellosta, S.; Baldessin, L.; Boccia, D.; Racagni, G.; Corsini, A. Pharmacokinetics interactions of monoclonal antibodies. Pharmacol. Res. 2016, 111, 592–599. [Google Scholar] [CrossRef] [PubMed]
- Leqvio: Summary of Product Characteristics. 2024. Available online: https://www.ema.europa.eu/en/documents/product-information/leqvio-epar-product-information_en.pdf (accessed on 15 September 2024).
- Hebert, M.F. Contributions of hepatic and intestinal metabolism and P-glycoprotein to cyclosporine and tacrolimus oral drug delivery. Adv. Drug Deliv. Rev. 1997, 27, 201–214. [Google Scholar] [CrossRef] [PubMed]
- Yigitaslan, S.; Erol, K.; Cengelli, C. The Effect of P-Glycoprotein Inhibition and Activation on the Absorption and Serum Levels of Cyclosporine and Tacrolimus in Rats. Adv. Clin. Exp. Med. 2016, 25, 237–242. [Google Scholar] [CrossRef] [PubMed]
- Cakaloglu, Y.; Tredger, J.M.; Devlin, J.; Williams, R. Importance of cytochrome P-450IIIA activity in determining dosage and blood levels of FK 506 and cyclosporine in liver transplant recipients. Hepatology 1994, 20, 309–316. [Google Scholar] [CrossRef] [PubMed]
- Farasyn, T.; Crowe, A.; Hatley, O.; Neuhoff, S.; Alam, K.; Kanyo, J.; Lam, T.T.; Ding, K.; Yue, W. Preincubation With Everolimus and Sirolimus Reduces Organic Anion-Transporting Polypeptide (OATP)1B1- and 1B3-Mediated Transport Independently of mTOR Kinase Inhibition: Implication in Assessing OATP1B1- and OATP1B3-Mediated Drug-Drug Interactions. J. Pharm. Sci. 2019, 108, 3443–3456. [Google Scholar] [CrossRef]
- Picard, N.; Levoir, L.; Lamoureux, F.; Yee, S.W.; Giacomini, K.M.; Marquet, P. Interaction of sirolimus and everolimus with hepatic and intestinal organic anion-transporting polypeptide transporters. Xenobiotica 2011, 41, 752–757. [Google Scholar] [CrossRef]
- Teriflunomide: Summary of Product Characteristics. Available online: https://www.ema.europa.eu/en/documents/product-information/teriflunomide-mylan-epar-product-information_en.pdf (accessed on 15 September 2024).
- Arava: Summary of Product Characteristics. Available online: https://www.ema.europa.eu/en/documents/product-information/arava-epar-product-information_en.pdf (accessed on 15 September 2024).
- Choe, J.Y.; Kim, S.K. Association between serum uric acid and inflammation in rheumatoid arthritis: Perspective on lowering serum uric acid of leflunomide. Clin. Chim. Acta 2015, 438, 29–34. [Google Scholar] [CrossRef]
- Mycophenolate Mofetil: Summary of Product Characteristics. Available online: https://www.ema.europa.eu/en/documents/product-information/cellcept-epar-product-information_en.pdf (accessed on 15 September 2024).
- Ding, X.; Zhu, X.; Zhang, Y.; Zhang, L.; Cheng, M.; Yu, Y.; Xu, L.; Li, G. Influence of serum uric acid levels in response to the conversion from mycophenolate mofetil to mizoribine in kidney transplant recipients. Transplant. Proc. 2013, 45, 190–193. [Google Scholar] [CrossRef]
- Nubeqa: Summary of Product Characteristics. Available online: https://www.ema.europa.eu/en/documents/product-information/nubeqa-epar-product-information_en.pdf (accessed on 15 September 2024).
- Xtandi: Summary of Product Characteristics. Available online: https://www.ema.europa.eu/en/documents/product-information/xtandi-epar-product-information_en.pdf (accessed on 15 September 2024).
- Tukysa: Summary of Product Characteristics. Available online: https://www.ema.europa.eu/en/documents/product-information/tukysa-epar-product-information_en.pdf (accessed on 15 September 2024).
- Afinitor: Summary of Product Characteristics. 2024. Available online: https://www.ema.europa.eu/en/documents/product-information/afinitor-epar-product-information_en.pdf (accessed on 15 September 2024).
- Rapamune: Summary of Product Characteristics. 2024. Available online: https://www.ema.europa.eu/en/documents/product-information/rapamune-epar-product-information_en.pdf (accessed on 15 September 2024).
- Tamoxifene, R. Available online: https://farmaci.agenziafarmaco.gov.it/aifa/servlet/PdfDownloadServlet?pdfFileName=footer_001561_033688_RCP.pdf&retry=0&sys=m0b1l3 (accessed on 15 September 2024).
- Keller, D.N.; Medwid, S.J.; Ross, C.D.; Wigle, T.J.; Kim, R.B. Impact of organic anion transporting polypeptide, P-glycoprotein, and breast cancer resistance protein transporters on observed tamoxifen and endoxifen concentration and adverse effects. Pharmacogenet Genom. 2023, 33, 10–18. [Google Scholar] [CrossRef]
- Glivec: Summary of Product Characteristics. Available online: https://www.ema.europa.eu/en/documents/product-information/glivec-epar-product-information_en.pdf (accessed on 15 September 2024).
- Kaletra: Summary of Product Characteristics. Available online: https://www.ema.europa.eu/en/documents/product-information/kaletra-epar-product-information_en.pdf (accessed on 15 September 2024).
- Annaert, P.; Ye, Z.W.; Stieger, B.; Augustijns, P. Interaction of HIV protease inhibitors with OATP1B1, 1B3, and 2B1. Xenobiotica 2010, 40, 163–176. [Google Scholar] [CrossRef]
- Tivicay: Summary of Product Characteristics. Available online: https://www.ema.europa.eu/en/documents/product-information/tivicay-epar-product-information_en.pdf (accessed on 15 September 2024).
- Howard, P.; Twycross, R.; Grove, G.; Charlesworth, S.; Mihalyo, M.; Wilcock, A. Rifampin (INN Rifampicin). J. Pain. Symptom Manag. 2015, 50, 891–895. [Google Scholar] [CrossRef] [PubMed]
- Ketek: Summary of Product Characteristics. Available online: https://www.ema.europa.eu/en/documents/product-information/ketek-epar-product-information_en.pdf (accessed on 15 September 2024).
- Higgins, J.W.; Ke, A.B.; Zamek-Gliszczynski, M.J. Clinical CYP3A inhibitor alternatives to ketoconazole, clarithromycin and itraconazole, are not transported into the liver by hepatic organic anion transporting polypeptides and organic cation transporter 1. Drug Metab. Dispos. 2014, 42, 1780–1784. [Google Scholar] [CrossRef] [PubMed]
- Hirano, M.; Maeda, K.; Shitara, Y.; Sugiyama, Y. Drug-drug interaction between pitavastatin and various drugs via OATP1B1. Drug Metab. Dispos. 2006, 34, 1229–1236. [Google Scholar] [CrossRef] [PubMed]
- Atazanavir: Summary of Product Characteristics. Available online: https://www.ema.europa.eu/en/documents/product-information/atazanavir-krka-epar-product-information_en.pdf (accessed on 15 September 2024).
- Norvir: Summary of Product Characteristics. Available online: https://www.ema.europa.eu/en/documents/product-information/norvir-epar-product-information_en.pdf (accessed on 15 September 2024).
- Claritromicina, R. Available online: https://farmaci.agenziafarmaco.gov.it/aifa/servlet/PdfDownloadServlet?pdfFileName=footer_004852_044779_RCP.pdf&retry=0&sys=m0b1l3 (accessed on 15 September 2024).
- Ketoconazole: Summary of Product Characteristics. Available online: https://www.ema.europa.eu/en/documents/product-information/ketoconazole-hra-epar-product-information_en.pdf (accessed on 15 September 2024).
- Vfend: Summary of Product Characteristics. Available online: https://www.ema.europa.eu/en/documents/product-information/vfend-epar-product-information_en.pdf (accessed on 15 September 2024).
- Fungitraxx: Summary of Product Characteristics. Available online: https://www.ema.europa.eu/en/documents/product-information/fungitraxx-epar-product-information_en.pdf (accessed on 15 September 2024).
- Macha, S.; Koenen, R.; Sennewald, R.; Schone, K.; Hummel, N.; Riedmaier, S.; Woerle, H.J.; Salsali, A.; Broedl, U.C. Effect of gemfibrozil, rifampicin, or probenecid on the pharmacokinetics of the SGLT2 inhibitor empagliflozin in healthy volunteers. Clin. Ther. 2014, 36, 280–290.e281. [Google Scholar] [CrossRef] [PubMed]
- Chu, X.Y.; Bleasby, K.; Yabut, J.; Cai, X.; Chan, G.H.; Hafey, M.J.; Xu, S.; Bergman, A.J.; Braun, M.P.; Dean, D.C.; et al. Transport of the dipeptidyl peptidase-4 inhibitor sitagliptin by human organic anion transporter 3, organic anion transporting polypeptide 4C1, and multidrug resistance P-glycoprotein. J. Pharmacol. Exp. Ther. 2007, 321, 673–683. [Google Scholar] [CrossRef]
- Shin, E.; Shin, N.; Oh, J.H.; Lee, Y.J. High-Dose Metformin May Increase the Concentration of Atorvastatin in the Liver by Inhibition of Multidrug Resistance-Associated Protein 2. J. Pharm. Sci. 2017, 106, 961–967. [Google Scholar] [CrossRef]
- Zhou, G.; Myers, R.; Li, Y.; Chen, Y.; Shen, X.; Fenyk-Melody, J.; Wu, M.; Ventre, J.; Doebber, T.; Fujii, N.; et al. Role of AMP-activated protein kinase in mechanism of metformin action. J. Clin. Investig. 2001, 108, 1167–1174. [Google Scholar] [CrossRef]
- Jardiance: Summary of Product Characteristics. Available online: https://www.ema.europa.eu/en/documents/product-information/jardiance-epar-product-information_en.pdf (accessed on 15 September 2024).
- Obermeier, M.; Yao, M.; Khanna, A.; Koplowitz, B.; Zhu, M.; Li, W.; Komoroski, B.; Kasichayanula, S.; Discenza, L.; Washburn, W.; et al. In vitro characterization and pharmacokinetics of dapagliflozin (BMS-512148), a potent sodium-glucose cotransporter type II inhibitor, in animals and humans. Drug Metab. Dispos. 2010, 38, 405–414. [Google Scholar] [CrossRef]
- Trajenta: Summary of Product Characteristics. Available online: https://www.ema.europa.eu/en/documents/product-information/trajenta-epar-product-information_en.pdf (accessed on 15 September 2024).
- Ferri, N.; Colombo, E.; Tenconi, M.; Baldessin, L.; Corsini, A. Drug-Drug Interactions of Direct Oral Anticoagulants (DOACs): From Pharmacological to Clinical Practice. Pharmaceutics 2022, 14, 1120. [Google Scholar] [CrossRef]
- Ferri, N.; Corsini, A. Nuovi anticoagulanti orali: Considerazioni di farmacologia clinica. G. Ital. Di Di Cardiol. 2015, 16, 3S–16S. [Google Scholar]
- Bellosta, S.; Corsini, A. Statin drug interactions and related adverse reactions: An update. Expert Opin. Drug Saf. 2018, 17, 25–37. [Google Scholar] [CrossRef] [PubMed]
- Wang, E.J.; Barecki-Roach, M.; Johnson, W.W. Quantitative characterization of direct P-glycoprotein inhibition by St John’s wort constituents hypericin and hyperforin. J. Pharm. Pharmacol. 2004, 56, 123–128. [Google Scholar] [CrossRef] [PubMed]
- Smith, N.F.; Acharya, M.R.; Desai, N.; Figg, W.D.; Sparreboom, A. Identification of OATP1B3 as a high-affinity hepatocellular transporter of paclitaxel. Cancer Biol. Ther. 2005, 4, 815–818. [Google Scholar] [CrossRef] [PubMed]
- Leung, N.; Yip, K.; Pillinger, M.H.; Toprover, M. Lowering and Raising Serum Urate Levels: Off-Label Effects of Commonly Used Medications. Mayo Clin. Proc. 2022, 97, 1345–1362. [Google Scholar] [CrossRef] [PubMed]
- Caspi, D.; Lubart, E.; Graff, E.; Habot, B.; Yaron, M.; Segal, R. The effect of mini-dose aspirin on renal function and uric acid handling in elderly patients. Arthritis Rheum. 2000, 43, 103–108. [Google Scholar] [CrossRef]
- Ohtsu, N.; Anzai, N.; Fukutomi, T.; Kimura, T.; Sakurai, H.; Endou, H. Human renal urate transpoter URAT1 mediates the transport of salicylate. Nihon Jinzo Gakkai Shi 2010, 52, 499–504. [Google Scholar]
- El-Sheikh, A.A.; van den Heuvel, J.J.; Koenderink, J.B.; Russel, F.G. Effect of hypouricaemic and hyperuricaemic drugs on the renal urate efflux transporter, multidrug resistance protein 4. Br. J. Pharmacol. 2008, 155, 1066–1075. [Google Scholar] [CrossRef]
- Ben Salem, C.; Slim, R.; Fathallah, N.; Hmouda, H. Drug-induced hyperuricaemia and gout. Rheumatology 2017, 56, 679–688. [Google Scholar] [CrossRef]
- Zhang, N.; Zhang, Z.; Yang, Y.; Xu, Y.; Li, G.; Liu, T. Ticagrelor-related gout: An underestimated side effect. Int. J. Cardiol. 2015, 192, 11–13. [Google Scholar] [CrossRef]
- Jutabha, P.; Anzai, N.; Wempe, M.F.; Wakui, S.; Endou, H.; Sakurai, H. Apical voltage-driven urate efflux transporter NPT4 in renal proximal tubule. Nucleosides Nucleotides Nucleic Acids 2011, 30, 1302–1311. [Google Scholar] [CrossRef]
- Hagos, Y.; Stein, D.; Ugele, B.; Burckhardt, G.; Bahn, A. Human renal organic anion transporter 4 operates as an asymmetric urate transporter. J. Am. Soc. Nephrol. 2007, 18, 430–439. [Google Scholar] [CrossRef] [PubMed]
- Stamp, L.; Searle, M.; O’Donnell, J.; Chapman, P. Gout in solid organ transplantation: A challenging clinical problem. Drugs 2005, 65, 2593–2611. [Google Scholar] [CrossRef]
- Bahn, A.; Hagos, Y.; Reuter, S.; Balen, D.; Brzica, H.; Krick, W.; Burckhardt, B.C.; Sabolic, I.; Burckhardt, G. Identification of a new urate and high affinity nicotinate transporter, hOAT10 (SLC22A13). J. Biol. Chem. 2008, 283, 16332–16341. [Google Scholar] [CrossRef] [PubMed]
- Hansen, J.M.; Fogh-Andersen, N.; Leyssac, P.P.; Strandgaard, S. Glomerular and tubular function in renal transplant patients treated with and without ciclosporin A. Nephron 1998, 80, 450–457. [Google Scholar] [CrossRef]
- Kanbay, M.; Akcay, A.; Huddam, B.; Usluogullari, C.A.; Arat, Z.; Ozdemir, F.N.; Haberal, M. Influence of cyclosporine and tacrolimus on serum uric acid levels in stable kidney transplant recipients. Transplant. Proc. 2005, 37, 3119–3120. [Google Scholar] [CrossRef] [PubMed]
- Hosoyamada, M.; Takiue, Y.; Shibasaki, T.; Saito, H. The effect of testosterone upon the urate reabsorptive transport system in mouse kidney. Nucleosides Nucleotides Nucleic Acids 2010, 29, 574–579. [Google Scholar] [CrossRef]
- Gouni-Berthold, I.; Wouter Jukema, J.; Koskinas, K.; Ray, K.; Averna, M.; Stulnig, T.; Vanassche, T.; Climente, M.; Lamparter, M.; Soronen, J.; et al. Real-world effectiveness and safety of bempedoic acid in Europe: Final 2-year results from the MILOS German cohort. In Proceedings of the DGK Hertztage, Hamburg, Germany, 26–28 September 2024. [Google Scholar]
- Ray, K.K.; Bays, H.E.; Catapano, A.L.; Lalwani, N.D.; Bloedon, L.T.; Sterling, L.R.; Robinson, P.L.; Ballantyne, C.M.; Trial, C.H. Safety and Efficacy of Bempedoic Acid to Reduce LDL Cholesterol. N. Engl. J. Med. 2019, 380, 1022–1032. [Google Scholar] [CrossRef]

| Target | ATP-citrate lyase (ACLY) |
| Prodrug | Yes, active metabolite is bempedoic acid–CoA produced in the liver by the very-long-chain acyl–CoA synthetase 1 (ACSVL1) |
| IC50 | 10 µmol/L |
| Bioavailability | 95% |
| Effect of food | Not influenced |
| Vd | 18 L |
| Proteins bound | 99% |
| Cmax | 24.8 ± 6.9 μg/mL |
| AUC | 348 ± 120 μg·h/mL |
| Tmax | 3.5 h |
| Half-life time | 19–21 h |
| Metabolism | UGT2B7 no CYP450 |
| Substrate/inhibitor P-gp | No |
| Substrate of other transporters | OAT3 |
| Inhibitor of other transporters | OAT2, OAT3, OATP1B1, OATP1B3 |
| Renal excretion | 70% |
| Posology | 180 mg OD |
| Drug | Effect on CYP450 and Drug Transporters | Effect on Bempedoic Acid | Effect on Interacting Drug | Expert Opinion |
|---|---|---|---|---|
| Hypocholesterolemic drugs | ||||
| Statins (pravastatin, simvastatin, atorvastatin, rosuvastatin) | All substrate of OATP1B1 [30]. | No significant effect predicted. | A 2-fold increase in AUC simvastatin 40 mg. A 1.4-fold increase in AUC atorvastatina 80 mg. A 1.4-fold increase in AUC pravastatin 80 mg. A 1.5-fold increase of AUC rosuvastatina 40 mg [15]. | Do not exceed 40 mg of simvastatin with bempedoic acid. Use rosuvastatin, atorvastatin, or other statins [15]. |
| Fibrates (fenofibrate, gemfibrozil) | Gemfibrozil inhibits UGT2B7 [31,32,33]. | Possible reduction in glucuronide metabolite of bempedoic acid. | No significant effect predicted. | Avoid gemfibrozil. Fenofibrate causes cholelithiasis. Possible increased risk of cholelithiasis with bempedoic acid [13]. |
| mAbs anti-PCSK9 (evolocumab, alirocumab) | No effect on CYP450 and drug transporters [34]. | No significant effect predicted. | Bempedoic acid increases PCSK9 expression and potentially the clearance of evolocumab and alirocumab [34]. | This interaction is not considered clinically relevant. |
| siRNA anti-PCSK9 (inclisiran) | No effect on CYP450 and drug transporters [35]. | No significant effect predicted. | No significant effect predicted. | No drug interaction. |
| Icosapent ethyl (omega 3) | Icosapent ethyl or EPA could compete with bempedoic acid for ASCVL1 [9]. | Icosapent ethyl could interfere with the activation of bempedoic acid by ASCVL1. | No significant effect predicted. | Possible reduction in the hypocholesterolemic effect of bempedoic acid in the presence of Icosapent ethyl. |
| Drug | Effect on CYP450 and Drug Transporters | Effect on Bempedoic Acid | Effect on Interacting Drug | Expert Opinion |
|---|---|---|---|---|
| Anticoagulant drugs | ||||
| Warfarin | Metabolized by CYP3A4, 2C9, 2C19, and 1A2. Not a substrate for P-gp. | No significant effect predicted. | No significant effect predicted. | Monitor INR in patients taking warfarin and bempedoic acid. |
| DOAC (dabigatran, apixaban, rivaroxaban, edoxaban) | All are substrates of P-gp. Apixaban and rivaroxaban are metabolized by CYP3A4 and are substrates of BCRP and ABCG2 [75]. | No significant effect predicted. | No significant effect predicted. | No drug interaction. |
| Antihypertensive drugs | ||||
| Bosentan | Substrate of OATP1B1 and OATP1B3 [15]. | No significant effect predicted. | Possible increase in bosentan exposure. | Bempedoic acid inhibits OATP1B1 and OATP1B3, increasing the exposure of bosentan. |
| Antiarrhythmic drugs | ||||
| Amiodarone | Weak inhibitor of P-gp and moderate inhibitor of CYP3A4. | No significant effect predicted. | No significant effect predicted. | No drug interaction. |
| Dronedarone | Inhibitor of P-gp e CYP3A4. | No significant effect predicted. | No significant effect predicted. | No drug interaction. |
| Digoxin | Substrate of P-gp. | No significant effect predicted. | No significant effect predicted. | No drug interaction. |
| Diltiazem, verapamil, | Inhibitor of P-gp and moderate inhibitor of CYP3A4. | No significant effect predicted. | No significant effect predicted. | No drug interaction. |
| Drug | Effect on CYP450 and Drug Transporters | Effect on Bempedoic Acid | Effect on Interacting Drug | Expert Opinion |
|---|---|---|---|---|
| Antidepressive drugs | ||||
| SSRI (fluoxetine, paroxetine, sertraline, venlafaxine, citalopram, escitalopram) | Fluoxetine, paroxetine, and sertraline inhibit CYP2D6. | No significant effect predicted. | No significant effect predicted. | No drug interaction. |
| TCA (amitriptyline, imipramine, clomipramine, amoxapine, desipramine, nortriptyline) | Metabolized by CYP3A4, CYP2C9, and CYP2D6. | No significant effect predicted. | No significant effect predicted. | No drug interaction. |
| Antipsychotic drugs | ||||
| Haloperidol, clozapine, perphenazine, risperidone, quetiapine, chlorpromazine | Metabolized by CYP3A4 and 2D6. Clozapine also by CYP2C19. | No significant effect predicted. | No significant effect predicted. | No drug interaction. Clozapine and olanzapine cause dyslipidemias: possible treatment with bempedoic acid. |
| Antiepileptic drugs | ||||
| Carbamazepine, phenobarbital, phenytoin, levetiracetam | Potent inducer of CYP3A4 and P-gp. Levetiracetam has a minor effect. | No significant effect predicted. | No significant effect predicted. | No drug interaction. |
| Phytotherapies | ||||
| St John’s Wort | Potent inducer of CYP3A4 and P-gp. Iperforin inhibits P-gp, OATP1B1, and OATP1B3 [77,78]. | Uncertain effect. | No significant effect predicted. | Co-administration is possible. |
| Drug | Mechanism |
|---|---|
| Aspirin (low dose) | At low doses (60–300 mg), aspirin reduces uric acid excretion and can induce hyperuricemia, while higher doses are uricosuric [80]. At low doses, aspirin acts as an exchange substrate and facilitates urate reabsorption, whereas at high doses it acts as an inhibitor of urate reabsorption [81]. Aspirin interacts with MRP4 [82], OAT1, and OAT3 [83]. |
| Ticagrelor | Ticagrelor and its metabolite are weak inhibitors of the urate transporter URAT1 and the OAT3 transporter. The metabolite also inhibits OAT1 [84]. |
| Cancer chemotherapy | Massive cytotoxic effect of tumor cells, with consequent release of urate [83]. |
| Diuretics | Loop and thiazide diuretics inhibit OAT1- and OAT3-mediated uric acid secretion [85]. Furosemide and hydrochlorothiazide are substrates of the uric acid transporter MRP4 [82]. Reduction in plasma volume [83]. Hydrochlorothiazide increases uric acid reuptake via the OAT4 transporter [86]. |
| Cyclosporine | Increased reabsorption of uric acid in the proximal tubule via the OAT1 transporter [87,88]. Decreased glomerular filtration rate secondary to vasoconstriction of afferent arterioles [89]. |
| Tacrolimus | Reduce acid uric excretion [90]. |
| Mycophenolate mofetil | Increased uric acid due to the inhibition of IMPDH (Inosine-5′-monophosphate dehydrogenase) [45]. |
| Testosterone | Increases uric acid reabsorption through induction of Smct1 (SLC5A8) [91]. |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ferri, N.; Colombo, E.; Corsini, A. Bempedoic Acid, the First-in-Class Oral ATP Citrate Lyase Inhibitor with Hypocholesterolemic Activity: Clinical Pharmacology and Drug–Drug Interactions. Pharmaceutics 2024, 16, 1371. https://doi.org/10.3390/pharmaceutics16111371
Ferri N, Colombo E, Corsini A. Bempedoic Acid, the First-in-Class Oral ATP Citrate Lyase Inhibitor with Hypocholesterolemic Activity: Clinical Pharmacology and Drug–Drug Interactions. Pharmaceutics. 2024; 16(11):1371. https://doi.org/10.3390/pharmaceutics16111371
Chicago/Turabian StyleFerri, Nicola, Elisa Colombo, and Alberto Corsini. 2024. "Bempedoic Acid, the First-in-Class Oral ATP Citrate Lyase Inhibitor with Hypocholesterolemic Activity: Clinical Pharmacology and Drug–Drug Interactions" Pharmaceutics 16, no. 11: 1371. https://doi.org/10.3390/pharmaceutics16111371
APA StyleFerri, N., Colombo, E., & Corsini, A. (2024). Bempedoic Acid, the First-in-Class Oral ATP Citrate Lyase Inhibitor with Hypocholesterolemic Activity: Clinical Pharmacology and Drug–Drug Interactions. Pharmaceutics, 16(11), 1371. https://doi.org/10.3390/pharmaceutics16111371