
Impact of Guidelines Regarding Dihydropyrimidine Dehydrogenase (DPD) Deficiency Screening Using Uracil-Based Phenotyping on the Reduction of Severe Side Effect of 5-Fluorouracil-Based Chemotherapy: A Propension Score Analysis

 , , , and
, , , and
Abstract
1. Introduction
2. Materials and Methods
2.1. Study Design and Patients
2.2. Demographic and Oncologic Data
2.3. Anticancer Treatment
- -
- LV5FU2 alone or included in a protocol with oxaliplatin (FOLFOX), irinotecan (FOLFIRI), or docetaxel (TFOX): leucovorin 400 mg/m2, followed by intravenous 400 mg/m2 5-FU bolus and then continuous 5-FU infusion at the dose of 2400 mg/m2 over 46 h every 14 days; in some simplified FOLFIRINOX protocols, no bolus was administered.
- -
- LV5FU2-dacarbazine (DBZ) protocol: leucovorin 400 mg/m2, followed by intravenous 400 mg/m2 5-FU bolus on days 1 and 2 and then continuous 5-FU infusion at the dose of 1200 mg/m2 over 46 h.
2.4. Chemotherapy-Induced Toxicities
2.5. Time of Assessment
2.6. DPD Phenotyping
2.7. Primary Endpoints
2.8. Secondary Endpoints
2.9. Statistical Analysis
2.10. Ethics
3. Results
3.1. Patients
3.2. Comparison of Severe Toxicities
3.3. DPD Deficiency Prevalence
3.4. DPD Deficiency and Fluoropyrimidines Dose Adjustment
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Longley, D.B.; Harkin, D.P.; Johnston, P.G. 5-Fluorouracil: Mechanisms of Action and Clinical Strategies. Nat. Rev. Cancer 2003, 3, 330–338. [Google Scholar] [CrossRef] [PubMed]
- Gamelin, E.; Boisdron-Celle, M.; Morel, A. Detection of dihydropyrimidine dehydrogenase deficiency before treatment by fluoropyrimidines. Therapie 2007, 62, 99–103. [Google Scholar] [CrossRef] [PubMed]
- Vodenkova, S.; Buchler, T.; Cervena, K.; Veskrnova, V.; Vodicka, P.; Vymetalkova, V. 5-Fluorouracil and Other Fluoropyrimidines in Colorectal Cancer: Past, Present and Future. Pharmacol. Ther. 2020, 206, 107447. [Google Scholar] [CrossRef] [PubMed]
- Walko, C.M.; Lindley, C. Capecitabine: A Review. Clin. Ther. 2005, 27, 23–44. [Google Scholar] [CrossRef] [PubMed]
- Blondy, S.; David, V.; Verdier, M.; Mathonnet, M.; Perraud, A.; Christou, N. 5-Fluorouracil Resistance Mechanisms in Colorectal Cancer: From Classical Pathways to Promising Processes. Cancer Sci. 2020, 111, 3142–3154. [Google Scholar] [CrossRef]
- Adjei, A.A. A Review of the Pharmacology and Clinical Activity of New Chemotherapy Agents for the Treatment of Colorectal Cancer. Br. J. Clin. Pharmacol. 1999, 48, 265–277. [Google Scholar] [CrossRef]
- Milano, G.; Chamorey, A.-L. Clinical Pharmacokinetics of 5-Fluorouracil with Consideration of Chronopharmacokinetics. Chronobiol. Int. 2002, 19, 177–189. [Google Scholar] [CrossRef]
- Mercier, C.; Ciccolini, J. Profiling Dihydropyrimidine Dehydrogenase Deficiency in Patients with Cancer Undergoing 5-Fluorouracil/Capecitabine Therapy. Clin. Colorectal Cancer 2006, 6, 288–296. [Google Scholar] [CrossRef]
- Lunenburg, C.A.T.C.; Henricks, L.M.; Guchelaar, H.-J.; Swen, J.J.; Deenen, M.J.; Schellens, J.H.M.; Gelderblom, H. Prospective DPYD Genotyping to Reduce the Risk of Fluoropyrimidine-Induced Severe Toxicity: Ready for Prime Time. Eur. J. Cancer 2016, 54, 40–48. [Google Scholar] [CrossRef]
- Boisdron-Celle, M.; Capitain, O.; Faroux, R.; Borg, C.; Metges, J.P.; Galais, M.P.; Kaassis, M.; Bennouna, J.; Bouhier-Leporrier, K.; Francois, E.; et al. Prevention of 5-Fluorouracil-Induced Early Severe Toxicity by Pre-Therapeutic Dihydropyrimidine Dehydrogenase Deficiency Screening: Assessment of a Multiparametric Approach. Semin. Oncol. 2017, 44, 13–23. [Google Scholar] [CrossRef]
- Henricks, L.M.; Opdam, F.L.; Beijnen, J.H.; Cats, A.; Schellens, J.H.M. DPYD Genotype-Guided Dose Individualization to Improve Patient Safety of Fluoropyrimidine Therapy: Call for a Drug Label Update. Ann. Oncol. 2017, 28, 2915–2922. [Google Scholar] [CrossRef] [PubMed]
- Diasio, R.B.; Beavers, T.L.; Carpenter, J.T. Familial Deficiency of Dihydropyrimidine Dehydrogenase. Biochemical Basis for Familial Pyrimidinemia and Severe 5-Fluorouracil-Induced Toxicity. J. Clin. Investig. 1988, 81, 47–51. [Google Scholar] [CrossRef] [PubMed]
- Harris, B.E.; Carpenter, J.T.; Diasio, R.B. Severe 5-Fluorouracil Toxicity Secondary to Dihydropyrimidine Dehydrogenase Deficiency. A Potentially More Common Pharmacogenetic Syndrome. Cancer 1991, 68, 499–501. [Google Scholar] [CrossRef]
- Milano, G.; Etienne, M.C. Potential Importance of Dihydropyrimidine Dehydrogenase (DPD) in Cancer Chemotherapy. Pharmacogenetics 1994, 4, 301–306. [Google Scholar] [CrossRef]
- Boisdron-Celle, M.; Remaud, G.; Traore, S.; Poirier, A.L.; Gamelin, L.; Morel, A.; Gamelin, E. 5-Fluorouracil-Related Severe Toxicity: A Comparison of Different Methods for the Pretherapeutic Detection of Dihydropyrimidine Dehydrogenase Deficiency. Cancer Lett. 2007, 249, 271–282. [Google Scholar] [CrossRef]
- Terrazzino, S.; Cargnin, S.; Del Re, M.; Danesi, R.; Canonico, P.L.; Genazzani, A.A. DPYD IVS14+1G>A and 2846A>T Genotyping for the Prediction of Severe Fluoropyrimidine-Related Toxicity: A Meta-Analysis. Pharmacogenomics 2013, 14, 1255–1272. [Google Scholar] [CrossRef]
- Deenen, M.J.; Meulendijks, D.; Cats, A.; Sechterberger, M.K.; Severens, J.L.; Boot, H.; Smits, P.H.; Rosing, H.; Mandigers, C.M.P.W.; Soesan, M.; et al. Upfront Genotyping of DPYD*2A to Individualize Fluoropyrimidine Therapy: A Safety and Cost Analysis. J. Clin. Oncol. 2016, 34, 227–234. [Google Scholar] [CrossRef]
- Launay, M.; Ciccolini, J.; Fournel, C.; Blanquicett, C.; Dupuis, C.; Fakhry, N.; Duffaud, F.; Salas, S.; Lacarelle, B. Upfront dpd deficiency detection to secure 5-fu administration: Part 2- application to head-and-neck cancer patients. Clin. Cancer Drugs 2017, 4, 122–128. [Google Scholar] [CrossRef]
- Mercier, C.; Ciccolini, J. Severe or Lethal Toxicities upon Capecitabine Intake: Is DPYD Genetic Polymorphism the Ideal Culprit? Trends Pharmacol. Sci. 2007, 28, 597–598. [Google Scholar] [CrossRef]
- Meulendijks, D.; Henricks, L.M.; Sonke, G.S.; Deenen, M.J.; Froehlich, T.K.; Amstutz, U.; Largiadèr, C.R.; Jennings, B.A.; Marinaki, A.M.; Sanderson, J.D.; et al. Clinical Relevance of DPYD Variants c.1679T>G, c.1236G>A/HapB3, and c.1601G>A as Predictors of Severe Fluoropyrimidine-Associated Toxicity: A Systematic Review and Meta-Analysis of Individual Patient Data. Lancet Oncol. 2015, 16, 1639–1650. [Google Scholar] [CrossRef]
- Meulendijks, D.; Henricks, L.M.; Jacobs, B.A.W.; Aliev, A.; Deenen, M.J.; de Vries, N.; Rosing, H.; van Werkhoven, E.; de Boer, A.; Beijnen, J.H.; et al. Pretreatment Serum Uracil Concentration as a Predictor of Severe and Fatal Fluoropyrimidine-Associated Toxicity. Br. J. Cancer 2017, 116, 1415–1424. [Google Scholar] [CrossRef] [PubMed]
- Pallet, N.; Hamdane, S.; Garinet, S.; Blons, H.; Zaanan, A.; Paillaud, E.; Taieb, J.; Laprevote, O.; Loriot, M.-A.; Narjoz, C. A Comprehensive Population-Based Study Comparing the Phenotype and Genotype in a Pretherapeutic Screen of Dihydropyrimidine Dehydrogenase Deficiency. Br. J. Cancer 2020, 123, 811–818. [Google Scholar] [CrossRef] [PubMed]
- HAS. Recherche de Déficit en Dihydropyrimidine Déshydrogenase en Vue de Prévenir Certaines Toxicités Sévères Survenant Sous Traitement Comportant des Fluoropyrimidines (5-Fluorouracile), Recommandations et Référentiels; INCa: Boulogne-Billancourt, France; HAS: Saint-Denis, France, 2018. [Google Scholar]
- EMA. EMA Recommendations on DPD Testing Prior to Treatment with Fluorouracil, Capecitabine, Tegafur and Flucytosine. Available online: https://www.ema.europa.eu/en/news/ema-recommendations-dpd-testing-prior-treatment-fluorouracil-capecitabine-tegafur-flucytosine (accessed on 2 August 2022).
- Blazevic, I.; Vaillant, W.; Basso, M.; Salignon, K. Survival and Relative Dose Intensity of 5-Fluorouracil, Oxaliplatin and Irinotecan in Real-Life Treatment of Metastatic Colorectal Cancer. Contemp. Oncol. 2020, 24, 150–156. [Google Scholar] [CrossRef] [PubMed]
- Atkinson, T.M.; Ryan, S.J.; Bennett, A.V.; Stover, A.M.; Saracino, R.M.; Rogak, L.J.; Jewell, S.T.; Matsoukas, K.; Li, Y.; Basch, E. The Association between Clinician-Based Common Terminology Criteria for Adverse Events (CTCAE) and Patient-Reported Outcomes (PRO): A Systematic Review. Support. Care Cancer 2016, 24, 3669–3676. [Google Scholar] [CrossRef] [PubMed]
- Feliu, C.; Millart, H.; Guillemin, H.; Vautier, D.; Binet, L.; Fouley, A.; Djerada, Z. Validation of a Fast UPLC-MS/MS Method for Quantitative Analysis of Opioids, Cocaine, Amphetamines (and Their Derivatives) in Human Whole Blood. Bioanalysis 2015, 7, 2685–2700. [Google Scholar] [CrossRef]
- McCaffrey, D.F.; Ridgeway, G.; Morral, A.R. Propensity Score Estimation with Boosted Regression for Evaluating Causal Effects in Observational Studies. Psychol. Methods 2004, 9, 403–425. [Google Scholar] [CrossRef]
- Nguyen, T.-L.; Collins, G.S.; Spence, J.; Daurès, J.-P.; Devereaux, P.J.; Landais, P.; Le Manach, Y. Double-Adjustment in Propensity Score Matching Analysis: Choosing a Threshold for Considering Residual Imbalance. BMC Med. Res. Methodol. 2017, 17, 78. [Google Scholar] [CrossRef]
- Selim, J.; Jarlier, X.; Clavier, T.; Boujibar, F.; Dusséaux, M.-M.; Thill, J.; Borderelle, C.; Plé, V.; Baste, J.-M.; Besnier, E.; et al. Impact of Opioid-Free Anesthesia After Video-Assisted Thoracic Surgery: A Propensity Score Study. Ann. Thorac. Surg. 2021, 114, 218–224. [Google Scholar] [CrossRef]
- van Kuilenburg, A.B.P. Dihydropyrimidine Dehydrogenase and the Efficacy and Toxicity of 5-Fluorouracil. Eur. J. Cancer 2004, 40, 939–950. [Google Scholar] [CrossRef]
- Launay, M.; Dahan, L.; Manon, D.; Rodallec, A.; Milano, G.; Duluc, M.; Lacarelle, B.; Ciccolini, J.; Seitz, J.-F. Beating the Odds: Efficacy and Toxicity of Dihydropyrimidine Dehydrogenase-Driven Adaptive Dosing of 5-FU in Patients with Digestive Cancer. Br. J. Clin. Pharmacol. 2016, 81, 124–130. [Google Scholar] [CrossRef]
- Tuchman, M.; Stoeckeler, J.S.; Kiang, D.T.; O’Dea, R.F.; Ramnaraine, M.L.; Mirkin, B.L. Familial Pyrimidinemia and Pyrimidinuria Associated with Severe Fluorouracil Toxicity. New Engl. J. Med. 1985, 313, 245–249. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.G.; Ciccolini, J.; Blesius, A.; Dahan, L.; Bagarry-Liegey, D.; Brunet, C.; Varoquaux, A.; Frances, N.; Marouani, H.; Giovanni, A.; et al. DPD-Based Adaptive Dosing of 5-FU in Patients with Head and Neck Cancer: Impact on Treatment Efficacy and Toxicity. Cancer Chemother. Pharmacol. 2011, 67, 49–56. [Google Scholar] [CrossRef]
- Ontario Health (Quality) DPYD Genotyping in Patients Who Have Planned Cancer Treatment With Fluoropyrimidines: A Health Technology Assessment. Ont Health Technol. Assess. Ser. 2021, 21, 1–186.
- Tejedor-Tejada, E.; Rubio Calvo, D.; García Andreo, A. Determination of Plasma Uracil as a Screening for Dihydropyrimidine Dehydrogenase Deficiency: Clinical Application in Oncological Treatments. Eur. J. Hosp. Pharm. 2022. [Google Scholar] [CrossRef] [PubMed]
- Yamashita, T.; Kato, K.; Long, N.K.; Makita, H.; Yonemoto, K.; Iida, K.; Tamaoki, N.; Hatakeyama, D.; Shibata, T. Effects of Smoking and Alcohol Consumption on 5-Fluorouracil-Related Metabolic Enzymes in Oral Squamous Cell Carcinoma. Mol. Clin. Oncol. 2014, 2, 429–434. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Jacobs, B.A.W.; Snoeren, N.; Samim, M.; Rosing, H.; de Vries, N.; Deenen, M.J.; Beijnen, J.H.; Schellens, J.H.M.; Koopman, M.; van Hillegersberg, R. The Impact of Liver Resection on the Dihydrouracil:Uracil Plasma Ratio in Patients with Colorectal Liver Metastases. Eur. J. Clin. Pharmacol. 2018, 74, 737–744. [Google Scholar] [CrossRef]
- de With, M.; Knikman, J.; de Man, F.M.; Lunenburg, C.A.T.C.; Henricks, L.M.; van Kuilenburg, A.B.P.; Maring, J.G.; van Staveren, M.C.; de Vries, N.; Rosing, H.; et al. Dihydropyrimidine Dehydrogenase Phenotyping Using Pretreatment Uracil: A Note of Caution Based on a Large Prospective Clinical Study. Clin. Pharmacol. Ther. 2022, 112, 62–68. [Google Scholar] [CrossRef]
- Dolat, M.; Macaire, P.; Goirand, F.; Vincent, J.; Hennequin, A.; Palmier, R.; Bengrine-Lefevre, L.; Ghiringhelli, F.; Royer, B.; Schmitt, A. Association of 5-FU Therapeutic Drug Monitoring to DPD Phenotype Assessment May Reduce 5-FU Under-Exposure. Pharmaceuticals 2020, 13, E416. [Google Scholar] [CrossRef]
- Gamelin, E.; Boisdron-Celle, M.; Guérin-Meyer, V.; Delva, R.; Lortholary, A.; Genevieve, F.; Larra, F.; Ifrah, N.; Robert, J. Correlation between Uracil and Dihydrouracil Plasma Ratio, Fluorouracil (5-FU) Pharmacokinetic Parameters, and Tolerance in Patients with Advanced Colorectal Cancer: A Potential Interest for Predicting 5-FU Toxicity and Determining Optimal 5-FU Dosage. J. Clin. Oncol. 1999, 17, 1105. [Google Scholar] [CrossRef]
- Konecki, C.; Feliu, C.; Cazaubon, Y.; Giusti, D.; Tonye-Libyh, M.; Brixi, H.; Cadiot, G.; Biron, A.; Djerada, Z. External Evaluation of Population Pharmacokinetic Models and Bayes-Based Dosing of Infliximab. Pharmaceutics 2021, 13, 1191. [Google Scholar] [CrossRef]
- Cazaubon, Y.; Talineau, Y.; Feliu, C.; Konecki, C.; Russello, J.; Mathieu, O.; Djerada, Z. Population Pharmacokinetics Modelling and Simulation of Mitotane in Patients with Adrenocortical Carcinoma: An Individualized Dose Regimen to Target All Patients at Three Months? Pharmaceutics 2019, 11, E566. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.J.; Beumer, J.H.; Chu, E. Therapeutic Drug Monitoring of 5-Fluorouracil. Cancer Chemother. Pharmacol. 2016, 78, 447–464. [Google Scholar] [CrossRef] [PubMed]
- Hashimoto, Y.; Yoshida, Y.; Yamada, T.; Aisu, N.; Yoshimatsu, G.; Yoshimura, F.; Hasegawa, S. Current Status of Therapeutic Drug Monitoring of 5-Fluorouracil Prodrugs. Anticancer Res. 2020, 40, 4655–4661. [Google Scholar] [CrossRef] [PubMed]
- Hodroj, K.; Barthelemy, D.; Lega, J.-C.; Grenet, G.; Gagnieu, M.-C.; Walter, T.; Guitton, J.; Payen-Gay, L. Issues and Limitations of Available Biomarkers for Fluoropyrimidine-Based Chemotherapy Toxicity, a Narrative Review of the Literature. ESMO Open 2021, 6, 100125. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Characteristic | DPD PG (n = 198) | DPD NPG (n = 94) | p Overall |
|---|---|---|---|
| Age (years) mean [min–max] | 66.8 [38.1–88.4] | 62.6 [28.7–88.2] | 0.004 |
| Gender n | 0.213 | ||
| Male | 120 | 49 | |
| Female | 78 | 45 | |
| Stature (cm) [min–max] | 170 [142–196] | 170 [142–196] | 0.986 |
| Weight (kg) [min–max] | 70.0 [39.0–137] | 69.0 [40.0–151] | 0.749 |
| Body surface area (kg/m2) [min–max] | 24.3 [14.3–43.8] | 23.9 [14.8–59.0] | 0.618 |
| GFR CKD-EPI (mL/min) [min–max] | 90.0 [22.0–144] | 97.0 [24.0–141] | 0.071 |
| Primary tumoral location n (%) | 0.080 | ||
| Colon-rectum | 85 (42.9%) | 45 (47.9%) | |
| Pancreas | 55 (27.8%) | 22 (23.4%) | |
| Stomach | 20 (10.1%) | 2 (2.13%) | |
| Oesophagus | 24 (12.1%) | 13 (13.8%) | |
| Neuro-endocrine | 9 (4.55%) | 9 (9.57%) | |
| Small intestine | 5 (2.53%) | 3 (3.19%) | |
| Treatment type n (%) | 0.001 | ||
| Neoadjuvant | 50 (25.3%) | 23 (24.5%) | |
| Adjuvant | 39 (19.7%) | 4 (4.26%) | |
| Palliative | 109 (55.1%) | 67 (71.3%) | |
| Chemotherapy protocol n (%) | 0.014 | ||
| FLOT | 8 (4.04%) | 0 (0.00%) | |
| FOLFIRI | 13 (6.57%) | 16 (17.0%) | |
| FOLFIRINOX | 62 (31.3%) | 21 (22.3%) | |
| FOLFOX | 90 (45.5%) | 41 (43.6%) | |
| LV5FU2 | 17 (8.59%) | 9 (9.57%) | |
| LV5FU2 DBZ | 8 (4.04%) | 7 (7.45%) | |
| Irinotecan: | 0.909 | ||
| No | 123 (62.1%) | 57 (60.6%) | |
| Yes | 75 (37.9%) | 37 (39.4%) | |
| Oxaliplatin: | 0.009 | ||
| No | 38 (19.2%) | 32 (34.0%) | |
| Yes | 160 (80.8%) | 62 (66.0%) | |
| Biotherapy: | 0.962 | ||
| No | 160 (80.8%) | 75 (79.8%) | |
| Yes | 38 (19.2%) | 19 (20.2%) | |
| Radiotherapy: | 0.097 | ||
| No | 168 (84.8%) | 87 (92.6%) | |
| Yes | 30 (15.2%) | 7 (7.45%) |
| Characteristic | DPD PG (n = 198) | DPD NPG (n = 94) | p Overall |
|---|---|---|---|
| Treatment 1: 5-FU dose (bolus + pump) | 2800 [400;4000] | 2800 [2000;2800] | 0.060 |
| Treatment 1: Postponement | 0.313 | ||
| Yes | 24 (12.1%) | 7 (7.45%) | |
| No | 174 (87.9%) | 87 (92.6%) | |
| Treatment 1: 5-FU discontinuation | 0.723 | ||
| Yes | 7 (3.54%) | 2 (2.13%) | |
| No | 191 (96.5%) | 92 (97.9%) | |
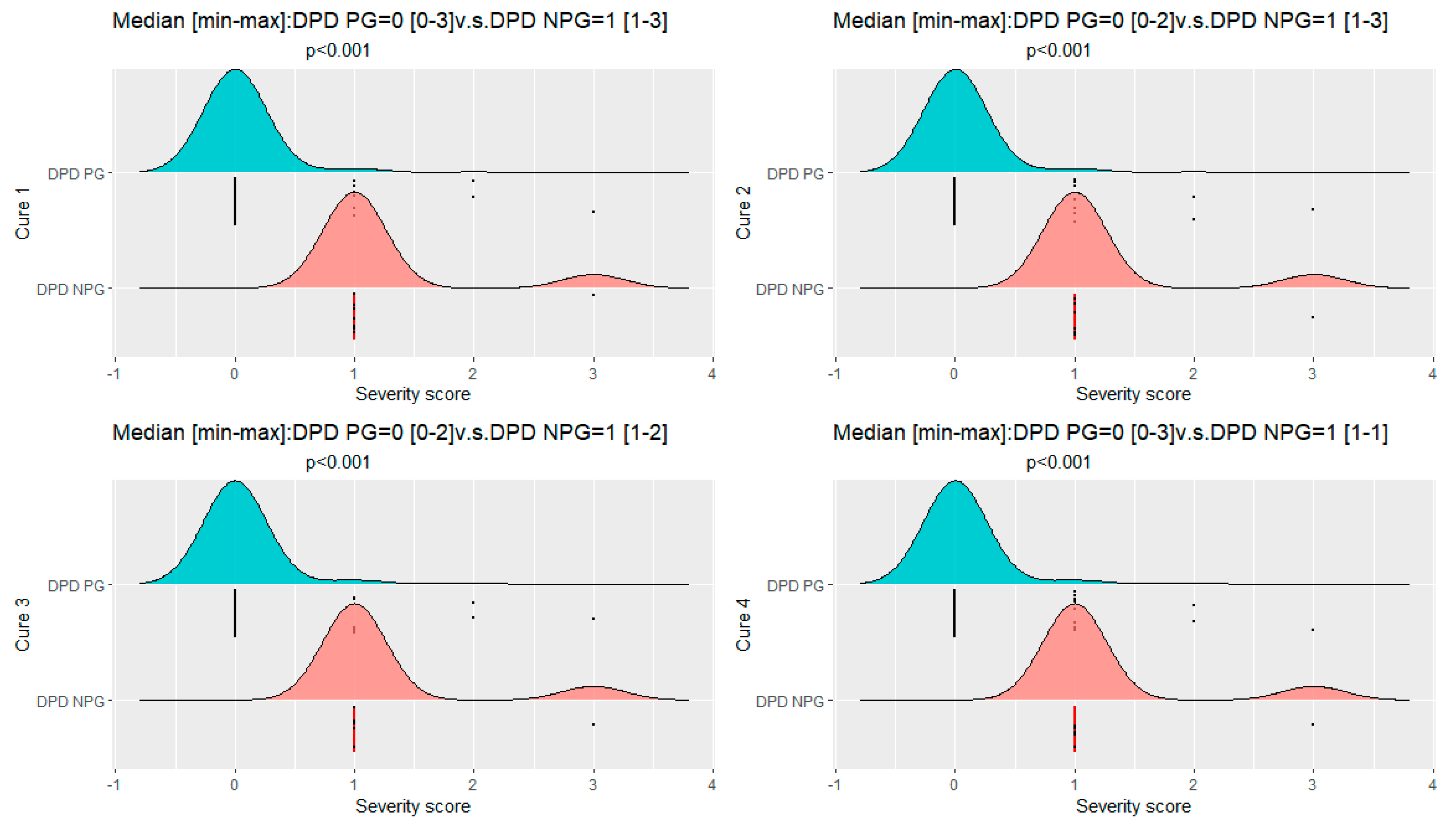
| Treatment 1: severity score median [min–max] | 0.00 [0.00;3.00] | 1.00 [1.00;3.00] | <0.001 |
| Treatment 2: 5-FU dose (bolus + pump) | 2800 [0.00;4000] | 2800 [0.00;2800] | 0.257 |
| Treatment 2: Postponement | 0.606 | ||
| Yes | 22 (11.5%) | 8 (8.70%) | |
| No | 169 (88.5%) | 84 (91.3%) | |
| Treatment 2: 5-FU discontinuation | 0.280 | ||
| Yes | 8 (4.19%) | 1 (1.09%) | |
| No | 183 (95.8%) | 91 (98.9%) | |
| Treatment 2: severity score median [min–max] | 0.00 [0.00;2.00] | 1.00 [1.00;3.00] | <0.001 |
| Treatment 3: 5-FU dose (bolus + pump) | 2800 [0.00;4400] | 2800 [0.00;2800] | 0.352 |
| Treatment 3: Postponement | 0.012 | ||
| Yes | 19 (10.2%) | 1 (1.09%) | |
| No | 167 (89.8%) | 91 (98.9%) | |
| Treatment 3: 5-FU discontinuation | 0.067 | ||
| Yes | 12 (6.45%) | 1 (1.09%) | |
| No | 174 (93.5%) | 91 (98.9%) | |
| Treatment 3: severity score median [min–max] | 0.00 [0.00;2.00] | 1.00 [1.00;2.00] | <0.001 |
| Treatment 4: 5 FU dose (bolus + pump) | 2800 [0.00;4400] | 2800 [0.00;2800] | 0.146 |
| Treatment 4: severity score median [min–max] | 0.00 [0.00;3.00] | 1.00 [1.00;1.00] | <0.001 |
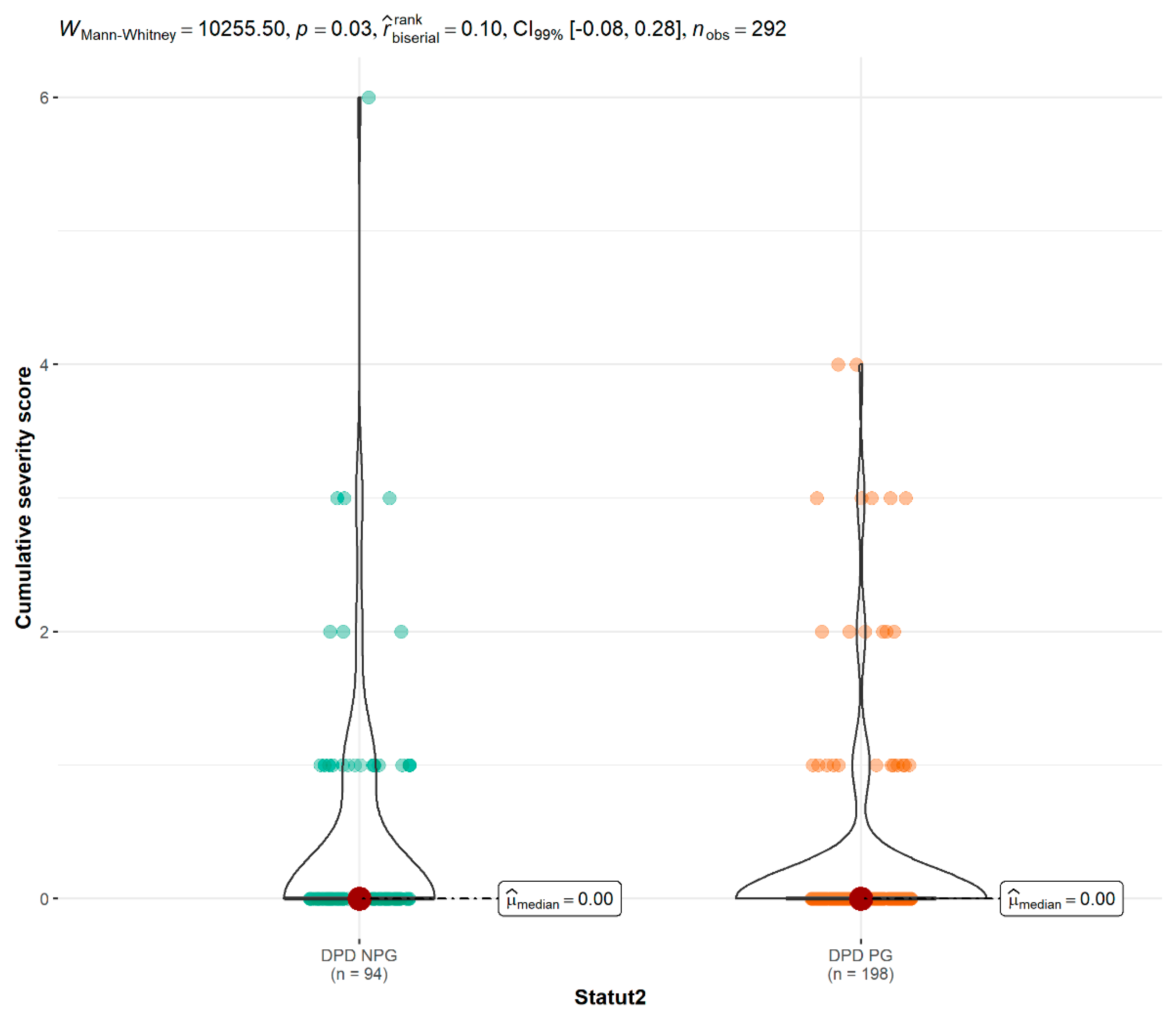
| All Treatment: severity score median [min–max] | 0.00 [0.00;4.00] | 0.00 [0.00;6.00] | 0.028 |
| DPD PG (n = 198) | DPD NPG (n = 94) | |||||||
|---|---|---|---|---|---|---|---|---|
| Cycle 1 | Cycle 2 | Cycle 3 | Cycle 4 | Cycle 1 | Cycle 2 | Cycle 3 | Cycle 4 | |
| Acute toxicity events n | ||||||||
| Neutropenia G3 | 3 | 2 | 4 | 1 | 4 | 1 | 1 | |
| Neutropenia G4 | 1 | 1 | 1 | |||||
| Anemia G3 | 1 | 3 | 1 | 1 | 1 | |||
| Anemia G4 | 1 | |||||||
| Thrombocytopenia G3 | 1 | 2 | ||||||
| Thrombocytopenia G4 | 1 | 1 | ||||||
| Nausea G3 | 1 | 2 | 1 | 1 | 2 | 3 | ||
| Nausea G4 | 1 | 1 | 1 | 1 | ||||
| Diarrhea G3 | 5 | 1 | 1 | 3 | 1 | 4 | 3 | 3 |
| Diarrhea G4 | 1 | |||||||
| Mucite G3 | 1 | 1 | 1 | 2 | ||||
| 5-FU Dose | Cycle 1 n (%) | Cycle 2 n (%) | Cycle 3 n (%) | Cycle 4 n (%) |
|---|---|---|---|---|
| Patients n | 31 | 29 | 27 | 25 |
| Standard dosage | 6 (19%) | 11 (38%) | 14 (52%) | 14 (56%) |
| −25% | 16 (52%) | 10 (34%) | 5 (19%) | 3 (12%) |
| −50% | 3 (10%) | 1 (3%) | 2 (7%) | 3 (12%) |
| −75% | 0 (0%) | 0 (0%) | 0 (0%) | 0 (0%) |
| −100% | 6 (19%) | 7 (24%) | 6 (22%) | 5 (20%) |
| 5-FU Dose | Cycle 1 n (%) | Cycle 2 n (%) | Cycle 3 n (%) | Cycle 4 n (%) |
|---|---|---|---|---|
| Patients n | 31 | 29 | 27 | 25 |
| Standard dosage | 9 (29%) | 17 (59%) | 20 (74%) | 20 (80%) |
| −25% | 21 (68%) | 12 (41%) | 7 (26%) | 4 (16%) |
| −50% | 1 (3%) | 0 (0%) | 0 (0%) | 1 (4%) |
| −75% | 0 (0%) | 0 (0%) | 0 (0%) | 0 (0%) |
| −100% | 0 (0%) | 0 (0%) | 0 (0%) | 0 (0%) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Laures, N.; Konecki, C.; Brugel, M.; Giffard, A.-L.; Abdelli, N.; Botsen, D.; Carlier, C.; Gozalo, C.; Feliu, C.; Slimano, F.; et al. Impact of Guidelines Regarding Dihydropyrimidine Dehydrogenase (DPD) Deficiency Screening Using Uracil-Based Phenotyping on the Reduction of Severe Side Effect of 5-Fluorouracil-Based Chemotherapy: A Propension Score Analysis. Pharmaceutics 2022, 14, 2119. https://doi.org/10.3390/pharmaceutics14102119
Laures N, Konecki C, Brugel M, Giffard A-L, Abdelli N, Botsen D, Carlier C, Gozalo C, Feliu C, Slimano F, et al. Impact of Guidelines Regarding Dihydropyrimidine Dehydrogenase (DPD) Deficiency Screening Using Uracil-Based Phenotyping on the Reduction of Severe Side Effect of 5-Fluorouracil-Based Chemotherapy: A Propension Score Analysis. Pharmaceutics. 2022; 14(10):2119. https://doi.org/10.3390/pharmaceutics14102119
Chicago/Turabian StyleLaures, Nicolas, Céline Konecki, Mathias Brugel, Anne-Lise Giffard, Naceur Abdelli, Damien Botsen, Claire Carlier, Claire Gozalo, Catherine Feliu, Florian Slimano, and et al. 2022. "Impact of Guidelines Regarding Dihydropyrimidine Dehydrogenase (DPD) Deficiency Screening Using Uracil-Based Phenotyping on the Reduction of Severe Side Effect of 5-Fluorouracil-Based Chemotherapy: A Propension Score Analysis" Pharmaceutics 14, no. 10: 2119. https://doi.org/10.3390/pharmaceutics14102119
APA StyleLaures, N., Konecki, C., Brugel, M., Giffard, A.-L., Abdelli, N., Botsen, D., Carlier, C., Gozalo, C., Feliu, C., Slimano, F., Djerada, Z., & Bouché, O. (2022). Impact of Guidelines Regarding Dihydropyrimidine Dehydrogenase (DPD) Deficiency Screening Using Uracil-Based Phenotyping on the Reduction of Severe Side Effect of 5-Fluorouracil-Based Chemotherapy: A Propension Score Analysis. Pharmaceutics, 14(10), 2119. https://doi.org/10.3390/pharmaceutics14102119