
Chimeric Drug Design with a Noncharged Carrier for Mitochondrial Delivery

 , , ,
, , ,  , ,
, , 

Abstract
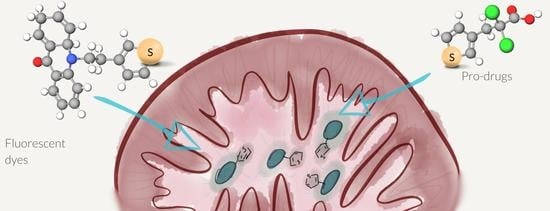
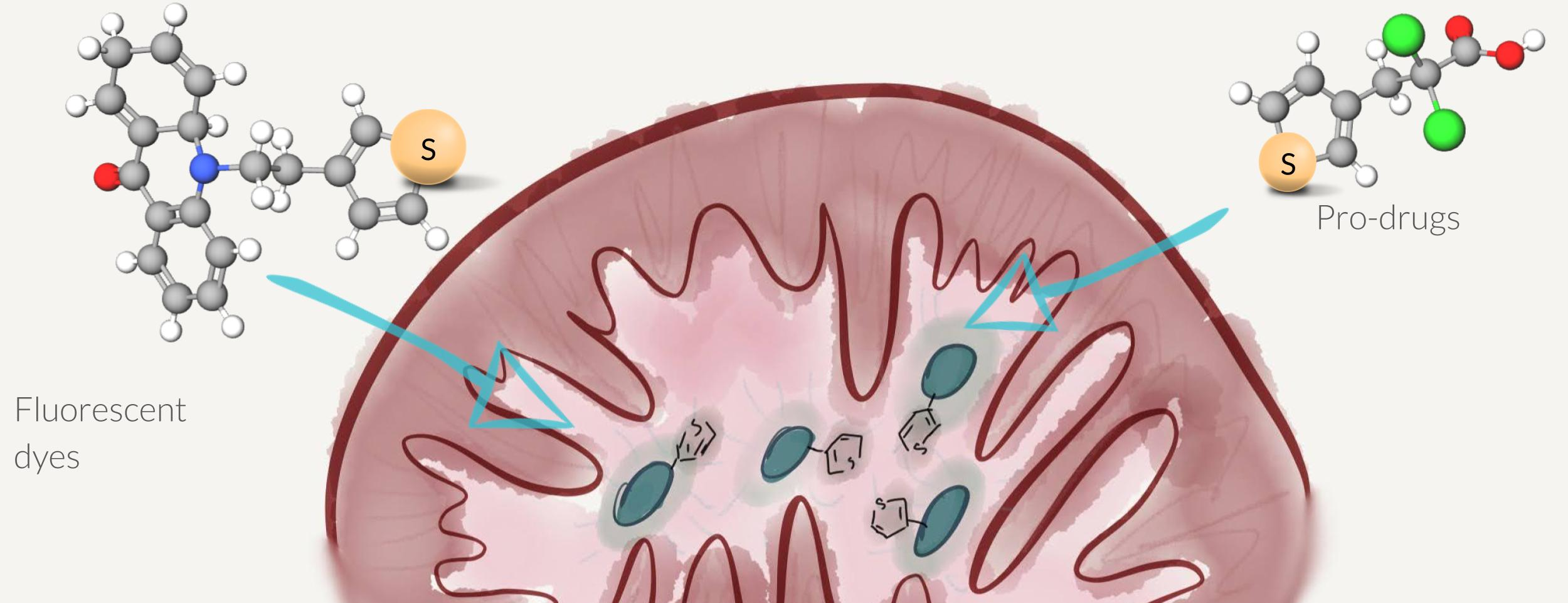{kind=link}
{kind=link}
{kind=link}
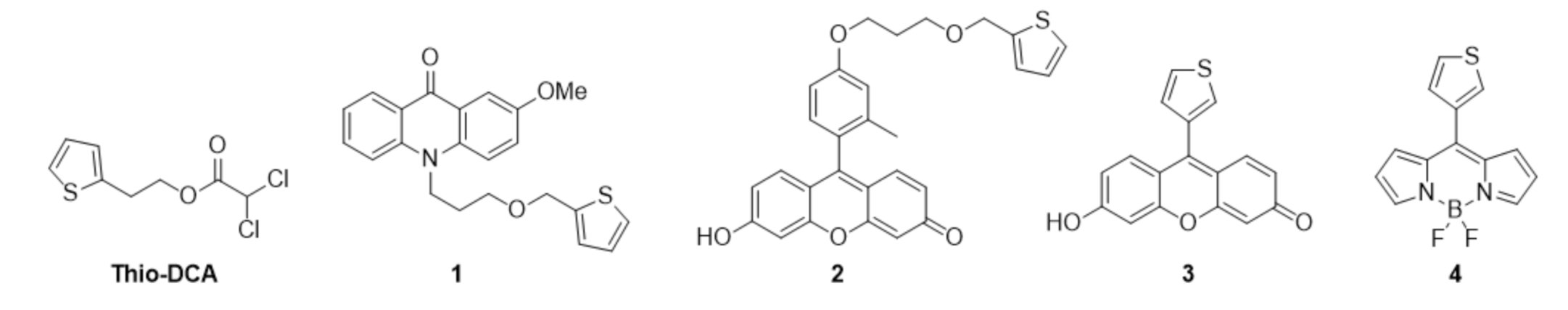{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
2.1. Synthesis of Thiophene-Modified Fluorescent Markers
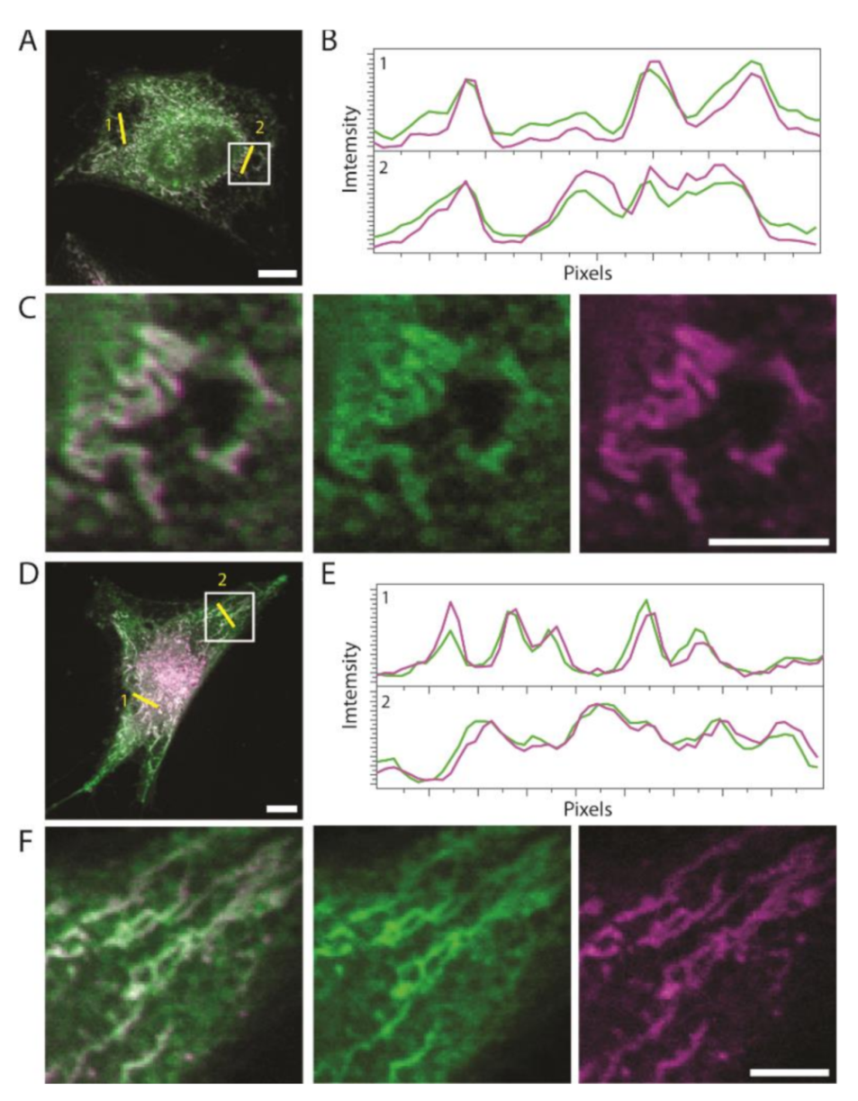
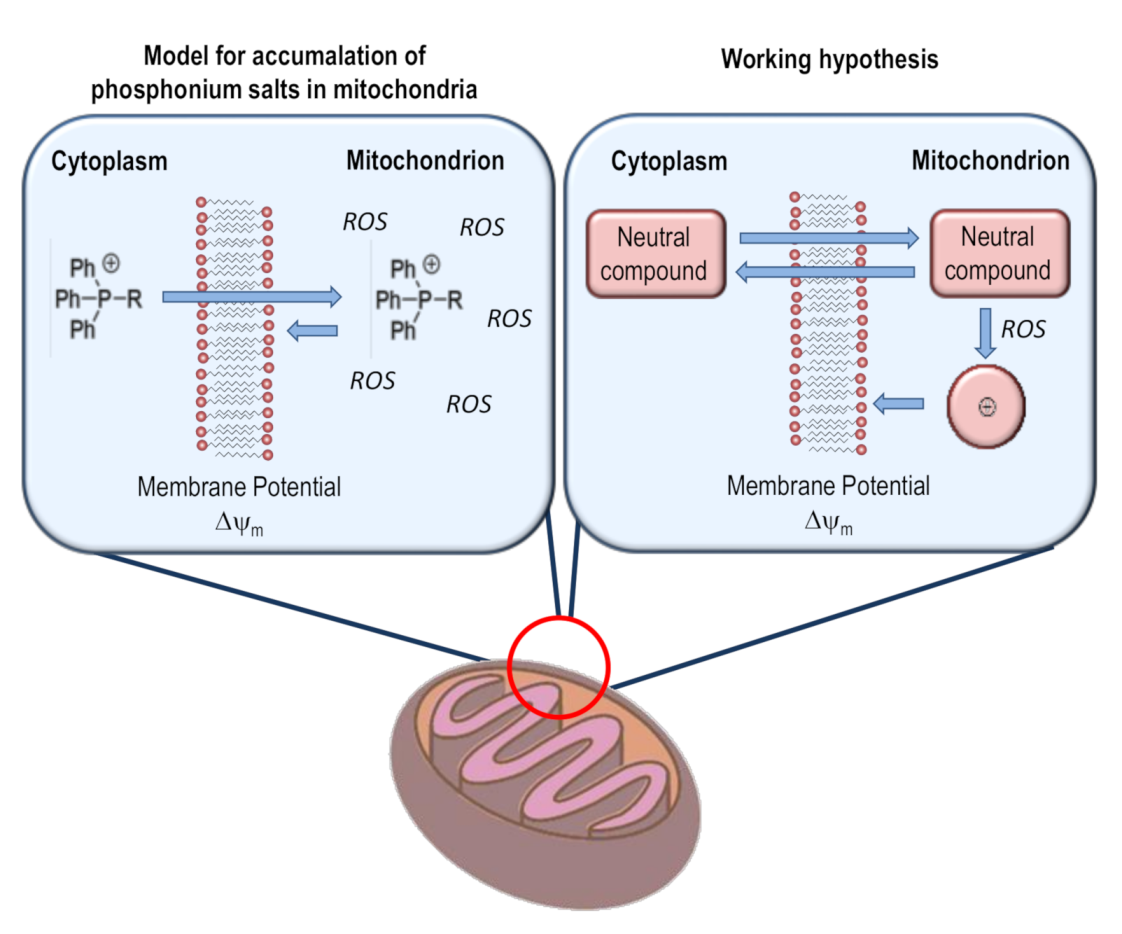
2.2. Thiophene as a General Mitochondrial Delivery Agent
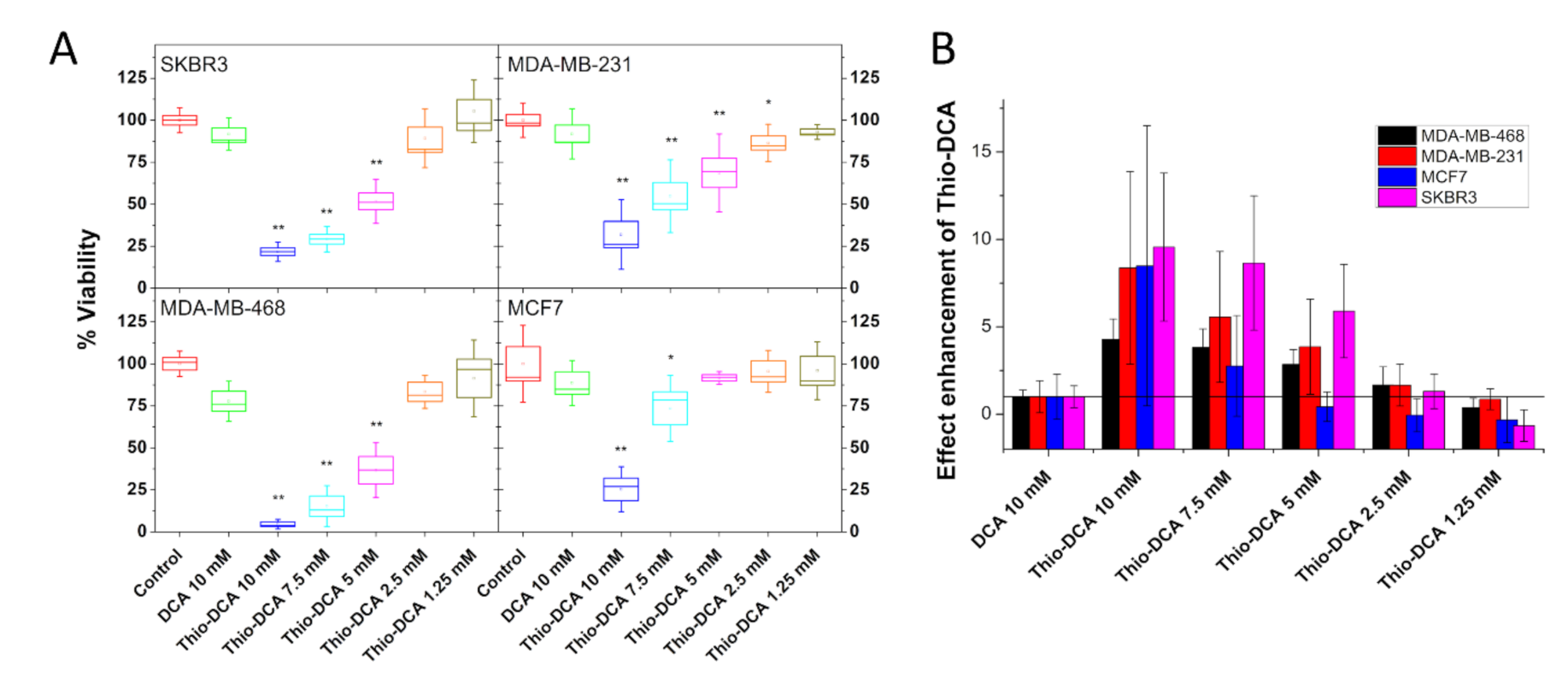
2.3. Thio-DCA, a PDHK Inhibitor with Enhanced Effectiveness
3. Discussion
4. Materials and Methods
4.1. Synthesis Reactions of New Compounds
4.1.1. General Details
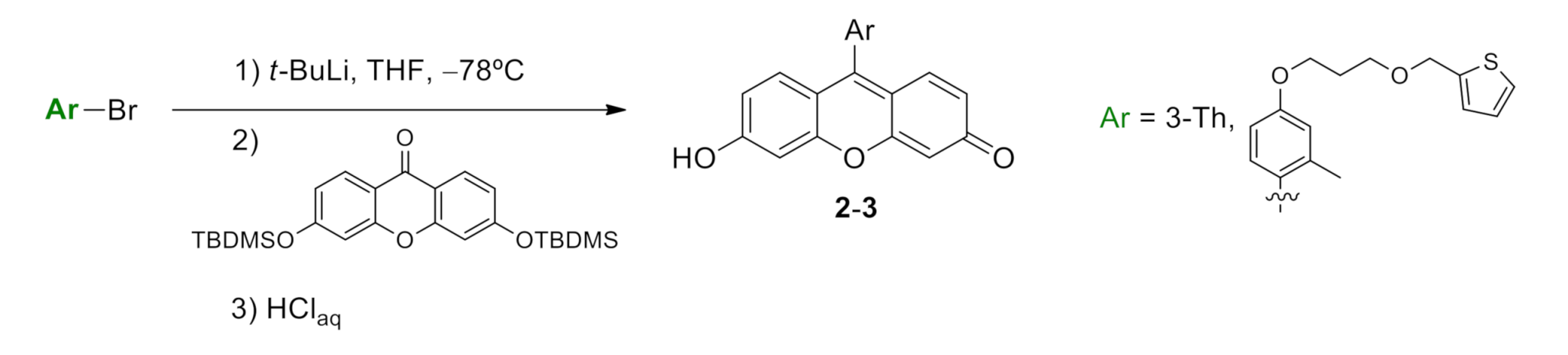
4.1.2. Synthesis of 6-hydroxy-9-(2-methyl-4-(3-(thiophen-2-ylmethoxy)propoxy)phenyl)-3H-xanthen-3-one (compound 2, Scheme S4)
4.1.3. Synthesis of 6-hydroxy-9-(thiophen-3-yl)-3H-xanthen-3-one (compound 3, Scheme S5)
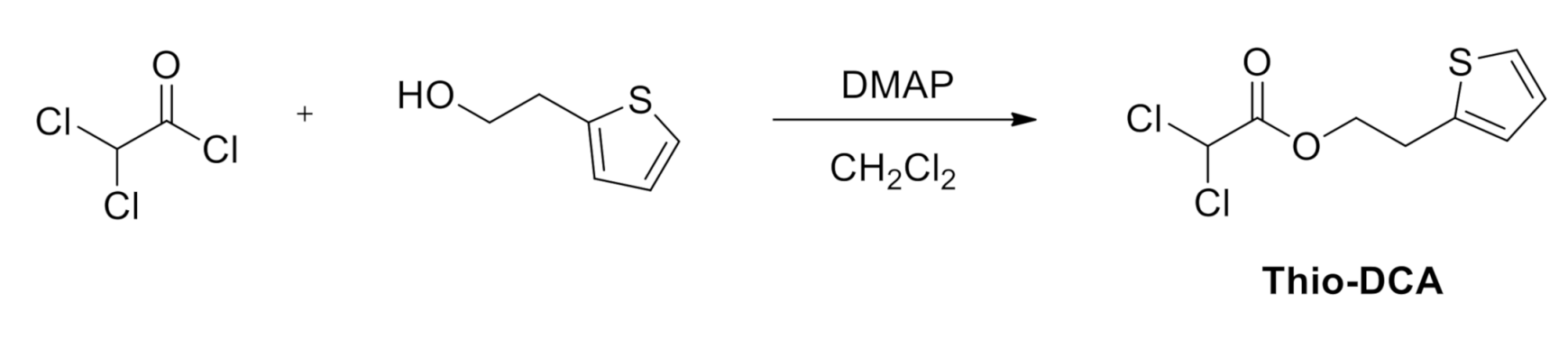
4.1.4. Synthesis of Thio-DCA
4.2. Instrumentation
4.3. Cell Culture
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Scheffler, I.E. Chapter 6: Metabolic Pathways inside Mitochondria. In Mitochondria, 2nd ed.; John Wiley & Sons: Hoboken, NJ, USA, 2007. [Google Scholar]
- Trotta, A.P.; Chipuk, J.E. Mitochondrial dynamics as regulators of cancer biology. Cell. Mol. Life Sci. 2017, 74, 1999–2017. [Google Scholar] [CrossRef]
- Vyas, S.; Zaganjor, E.; Haigis, M.C. Mitochondria and Cancer. Cell 2016, 166, 555–566. [Google Scholar] [CrossRef] [PubMed]
- Lunt, S.Y.; Heiden, M.G.V. Aerobic Glycolysis: Meeting the Metabolic Requirements of Cell Proliferation. Ann. Rev. Cell Dev. Biol. 2011, 27, 441–464. [Google Scholar] [CrossRef] [PubMed]
- Guerra, F.; Arbini, A.A.; Moro, L. Mitochondria and cancer chemoresistance. Biochim. Biophys. Acta 2017, 1858, 686–699. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Chan, D.C. Mitochondrial Dynamics in Regulating the Unique Phenotypes of Cancer and Stem Cells. Cell Metab. 2017, 26, 39–48. [Google Scholar] [CrossRef]
- DeBerardinis, R.J.; Chandel, N.S. Fundamentals of cancer metabolism. Sci. Adv. 2016, 2, e1600200. [Google Scholar] [CrossRef]
- Kalyanaraman, B.; Cheng, G.; Hardy, M.; Ouari, O.; Lopez, M.; Joseph, J.; Zielonka, J.; Dwinell, M.B. A review of the basics of mitochondrial bioenergetics, metabolism, and related signaling pathways in cancer cells: Therapeutic targeting of tumor mitochondria with lipophilic cationic compounds. Redox Biol. 2018, 14, 316–327. [Google Scholar] [CrossRef]
- D’Souza, G.G.M.; Wagle, M.A.; Saxena, V.; Shah, A. Approaches for targeting mitochondria in cancer therapy. Biochim. Biophys. Acta 2011, 1807, 689–696. [Google Scholar] [CrossRef]
- Long, L.; Huang, M.; Wang, N.; Wu, Y.; Wang, K.; Gong, A.; Zhang, Z.; Sessler, J.L. A Mitochondria-Specific Fluorescent Probe for Visualizing Endogenous Hydrogen Cyanide Fluctuations in Neurons. J. Am. Chem. Soc. 2018, 140, 1870–1875. [Google Scholar] [CrossRef]
- Ren, M.; Deng, B.; Zhou, K.; Kong, X.; Wang, J.-Y.; Lin, W. Single Fluorescent Probe for Dual-Imaging Viscosity and H2O2 in Mitochondria with Different Fluorescence Signals in Living Cells. Anal. Chem. 2017, 89, 552–555. [Google Scholar] [CrossRef]
- Zhang, X.; Gao, F. Imaging mitochondrial reactive oxygen species with fluorescent probes: Current applications and challenges. Free Radical Res. 2015, 49, 374–382. [Google Scholar] [CrossRef]
- Kumar, N.; Bhalla, V.; Kumar, M. Development and sensing applications of fluorescent motifs within the mitochondrial environment. Chem. Commun. 2015, 51, 15614–15628. [Google Scholar] [CrossRef] [PubMed]
- Xu, W.; Zeng, Z.; Jiang, J.-H.; Chang, Y.-T.; Yuan, L. Discerning the Chemistry in Individual Organelles with Small-Molecule Fluorescent Probes. Angew. Chem. Int. Ed. 2016, 55, 13658–13699. [Google Scholar] [CrossRef] [PubMed]
- Ripoll, C.; Roldan, M.; Contreras-Montoya, R.; Diaz-Mochon, J.J.; Martin, M.; Ruedas-Rama, M.J.; Orte, A. Mitochondrial pH Nanosensors for Metabolic Profiling of Breast Cancer Cell Lines. Int. J. Mol. Sci. 2020, 21, 3731. [Google Scholar] [CrossRef] [PubMed]
- Sánchez, M.I.; Vida, Y.; Pérez-Inestrosa, E.; Mascareñas, J.L.; Vázquez, M.E.; Sugiura, A.; Martínez-Costas, J. MitoBlue as a tool to analyze the mitochondria-lysosome communication. Sci. Rep. 2020, 10, 3528. [Google Scholar] [CrossRef]
- Reshetnikov, V.; Özkan, H.G.; Daum, S.; Janko, C.; Alexiou, C.; Sauer, C.; Heinrich, M.R.; Mokhir, A. N-Alkylaminoferrocene-Based Prodrugs Targeting Mitochondria of Cancer Cells. Molecules 2020, 25, 2545. [Google Scholar] [CrossRef]
- Ong, H.C.; Hu, Z.; Coimbra, J.T.S.; Ramos, M.J.; Kon, O.L.; Xing, B.; Yeow, E.K.L.; Fernandes, P.A.; García, F. Enabling Mitochondrial Uptake of Lipophilic Dications Using Methylated Triphenylphosphonium Moieties. Inorg. Chem. 2019, 58, 8293–8299. [Google Scholar] [CrossRef]
- Jana, B.; Thomas, A.P.; Kim, S.; Lee, I.S.; Choi, H.; Jin, S.; Park, S.A.; Min, S.K.; Kim, C.; Ryu, J.-H. Self-Assembly of Mitochondria-Targeted Photosensitizer to Increase Photostability and Photodynamic Therapeutic Efficacy in Hypoxia. Chem. A Eur. J. 2020, 26, 10695–10701. [Google Scholar] [CrossRef] [PubMed]
- Murphy, M.P. Selective targeting of bioactive compounds to mitochondria. Trends Biotechnol. 1997, 15, 326–330. [Google Scholar] [CrossRef]
- Lei, E.K.; Kelley, S.O. Delivery and Release of Small-Molecule Probes in Mitochondria Using Traceless Linkers. J. Am. Chem. Soc. 2017, 139, 9455–9458. [Google Scholar] [CrossRef]
- Trapella, C.; Voltan, R.; Melloni, E.; Tisato, V.; Celeghini, C.; Bianco, S.; Fantinati, A.; Salvadori, S.; Guerrini, R.; Secchiero, P.; et al. Design, Synthesis, and Biological Characterization of Novel Mitochondria Targeted Dichloroacetate-Loaded Compounds with Antileukemic Activity. J. Med. Chem. 2016, 59, 147–156. [Google Scholar] [CrossRef]
- Ripcke, J.; Zarse, K.; Ristow, M.; Birringer, M. Small-Molecule Targeting of the Mitochondrial Compartment with an Endogenously Cleaved Reversible Tag. Chembiochem 2009, 10, 1689–1696. [Google Scholar] [CrossRef]
- Pathak, R.K.; Marrache, S.; Harn, D.A.; Dhar, S. Mito-DCA: A Mitochondria Targeted Molecular Scaffold for Efficacious Delivery of Metabolic Modulator Dichloroacetate. ACS Chem. Biol. 2014, 9, 1178–1187. [Google Scholar] [CrossRef] [PubMed]
- Zielonka, J.; Joseph, J.; Sikora, A.; Hardy, M.; Ouari, O.; Vasquez-Vivar, J.; Cheng, G.; Lopez, M.; Kalyanaraman, B. Mitochondria-Targeted Triphenylphosphonium-Based Compounds: Syntheses, Mechanisms of Action, and Therapeutic and Diagnostic Applications. Chem. Rev. 2017, 117, 10043–10120. [Google Scholar] [CrossRef] [PubMed]
- Hu, Z.; Sim, Y.; Kon, O.L.; Ng, W.H.; Ribeiro, A.J.M.; Ramos, M.J.; Fernandes, P.A.; Ganguly, R.; Xing, B.; García, F.; et al. Unique Triphenylphosphonium Derivatives for Enhanced Mitochondrial Uptake and Photodynamic Therapy. Bioconjug. Chem. 2017, 28, 590–599. [Google Scholar] [CrossRef] [PubMed]
- Rhee, W.J.; Bao, G. Slow non-specific accumulation of 2′-deoxy and 2′-O-methyl oligonucleotide probes at mitochondria in live cells. Nucleic Acids Res. 2010, 38, e109. [Google Scholar] [CrossRef]
- Perry, S.W.; Norman, J.P.; Barbieri, J.; Brown, E.B.; Gelbard, H.A. Mitochondrial membrane potential probes and the proton gradient: A practical usage guide. BioTechniques 2011, 50, 98–115. [Google Scholar] [CrossRef]
- Jiang, Z.; Liu, H.; He, H.; Yadava, N.; Chambers, J.J.; Thayumanavan, S. Anionic Polymers Promote Mitochondrial Targeting of Delocalized Lipophilic Cations. Bioconjug. Chem. 2020, 31, 1344–1353. [Google Scholar] [CrossRef]
- Gao, P.; Pan, W.; Li, N.; Tang, B. Fluorescent probes for organelle-targeted bioactive species imaging. Chem. Sci. 2019, 10, 6035–6071. [Google Scholar] [CrossRef]
- Buckman, J.F.; Hernández, H.; Kress, G.J.; Votyakova, T.V.; Pal, S.; Reynolds, I.J. MitoTracker labeling in primary neuronal and astrocytic cultures: Influence of mitochondrial membrane potential and oxidants. J. Neurosci. Meth. 2001, 104, 165–176. [Google Scholar] [CrossRef]
- Xu, Z.; Xu, L. Fluorescent probes for the selective detection of chemical species inside mitochondria. Chem. Commun. 2016, 52, 1094–1119. [Google Scholar] [CrossRef]
- Murphy, M.P. How mitochondria produce reactive oxygen species. Biochem. J. 2009, 417, 1–13. [Google Scholar] [CrossRef]
- Abelha, T.F.; Morris, G.; Lima, S.M.; Andrade, L.H.C.; McLean, A.J.; Alexander, C.; Calvo-Castro, J.; McHugh, C.J. Development of a Neutral Diketopyrrolopyrrole Phosphine Oxide for the Selective Bioimaging of Mitochondria at the Nanomolar Level. Chem. A Eur. J. 2020, 26, 3173–3180. [Google Scholar] [CrossRef]
- Limnios, D.; Kokotos, C.G. 2,2,2-Trifluoroacetophenone as an Organocatalyst for the Oxidation of Tertiary Amines and Azines to N-Oxides. Chem. A Eur. J. 2014, 20, 559–563. [Google Scholar] [CrossRef]
- Copéret, C.; Adolfsson, H.; Khuong, T.-A.V.; Yudin, A.K.; Sharpless, K.B. A Simple and Efficient Method for the Preparation of Pyridine N-Oxides. J. Org. Chem. 1998, 63, 1740–1741. [Google Scholar] [CrossRef]
- Pouzet, P.; Erdelmeier, I.; Ginderow, D.; Mornon, J.-P.; Dansette, P.; Mansuy, D. Thiophene S-oxides: Convenient preparation, first complete structural characterization and unexpected dimerization of one of them, 2,5-diphenylthiophene-1-oxide. J. Chem. Soc. Chem. Commun. 1995, 473–474. [Google Scholar] [CrossRef]
- Gramec, D.; Mašič, L.P.; Dolenc, M.S. Bioactivation Potential of Thiophene-Containing Drugs. Chem. Res. Toxicol. 2014, 27, 1344–1358. [Google Scholar] [CrossRef]
- Dansette, P.M.; Thang, D.C.; Mansuy, H.E.A.D. Evidence for thiophene-s-oxide as a primary reactive metabolite of thiophene in vivo: Formation of a dihydrothiophene sulfoxide mercapturic acid. Biochem. Biophys. Res. Comm. 1992, 186, 1624–1630. [Google Scholar] [CrossRef]
- Herrero-Foncubierta, P.; González-García, M.D.C.; Resa, S.; Paredes, J.M.; Ripoll, C.; Girón, M.D.; Salto, R.; Cuerva, J.M.; Orte, A.; Miguel, D. Simple and non-charged long-lived fluorescent intracellular organelle trackers. Dye. Pigment. 2020, 183, 108649. [Google Scholar] [CrossRef]
- Thimmaiah, K.; Ugarkar, A.G.; Martis, E.F.; Shaikh, M.S.; Coutinho, E.C.; Yergeri, M.C. Drug–DNA Interaction Studies of Acridone-Based Derivatives. Nucleosides Nucleotides Nucleic Acids 2015, 34, 309–331. [Google Scholar] [CrossRef] [PubMed]
- Horobin, R.W. Predicting Mitochondrial Targeting by Small Molecule Xenobiotics Within Living Cells Using QSAR Models. In Mitochondrial Medicine: Volume II, Manipulating Mitochondrial Function; Weissig, V., Edeas, M., Eds.; Springer: New York, NY, USA, 2015; pp. 13–23. [Google Scholar]
- Sullivan, L.B.; Gui, D.Y.; Heiden, M.G.V. Altered metabolite levels in cancer: Implications for tumour biology and cancer therapy. Nat. Rev. Cancer 2016, 16, 680. [Google Scholar] [CrossRef]
- Kankotia, S.; Stacpoole, P.W. Dichloroacetate and cancer: New home for an orphan drug? Biochim. Biophys. Acta Rev. Cancer 2014, 1846, 617–629. [Google Scholar] [CrossRef]
- Holliday, D.L.; Speirs, V. Choosing the right cell line for breast cancer research. Breast Cancer Res. 2011, 13, 215. [Google Scholar] [CrossRef] [PubMed]
- Smith, J.A.; West, R.M.; Allen, M. Acridones and quinacridones: Novel fluorophores for fluorescence lifetime studies. J. Fluoresc. 2004, 14, 151–171. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Garcia, M.C.; Herrero-Foncubierta, P.; Garcia-Fernandez, E.; Orte, A. Building Accurate Intracellular Polarity Maps through Multiparametric Microscopy. Methods Protoc. 2020, 3, 78. [Google Scholar] [CrossRef]
- Gonzalez-Garcia, M.C.; Herrero-Foncubierta, P.; Castro, S.; Resa, S.; Alvarez-Pez, J.M.; Miguel, D.; Cuerva, J.M.; Garcia-Fernandez, E.; Orte, A. Coupled Excited-State Dynamics in N-Substituted 2-Methoxy-9-Acridones. Front. Chem. 2019, 7, 129. [Google Scholar] [CrossRef] [PubMed]
- Martínez-Peragón, A.; Miguel, D.; Jurado, R.; Justicia, J.; Alvarez-Pez, J.M.; Cuerva, J.M.; Crovetto, L. Synthesis and Photophysics of a New Family of Fluorescent 9-alkyl Substituted Xanthenones. Chem. A Eur. J. 2014, 20, 447–455. [Google Scholar] [CrossRef]
- Urano, Y.; Kamiya, M.; Kanda, K.; Ueno, T.; Hirose, K.; Nagano, T. Evolution of Fluorescein as a Platform for Finely Tunable Fluorescence Probes. J. Am. Chem. Soc. 2005, 127, 4888–4894. [Google Scholar] [CrossRef]
- Choi, S.H.; Kim, K.; Lee, J.; Do, Y.; Churchill, D.G. X-ray diffraction, DFT, and spectroscopic study of N,N′-difluoroboryl-5-(2-thienyl)dipyrrin and fluorescence studies of related dipyrromethanes, dipyrrins and BF2-dipyrrins and DFT conformational study of 5-(2-thienyl)dipyrrin. J. Chem. Crystallogr. 2007, 37, 315–331. [Google Scholar] [CrossRef]
- Paredes, J.M.; Crovetto, L.; Rios, R.; Orte, A.; Alvarez-Pez, J.M.; Talavera, E.M. Tuned lifetime, at the ensemble and single molecule level, of a xanthenic fluorescent dye by means of a buffer-mediated excited-state proton exchange reaction. Phys. Chem. Chem. Phys. 2009, 11, 5400–5407. [Google Scholar] [CrossRef]
- Yu, C.; Jiao, L.; Yin, H.; Zhou, J.; Pang, W.; Wu, Y.; Wang, Z.; Yang, G.; Hao, E. α-/β-Formylated Boron–Dipyrrin (BODIPY) Dyes: Regioselective Syntheses and Photophysical Properties. Eur. J. Org. Chem. 2011, 2011, 5460–5468. [Google Scholar] [CrossRef]
- Jiao, L.; Yu, C.; Wang, J.; Briggs, E.A.; Besley, N.A.; Robinson, D.; Ruedas-Rama, M.J.; Orte, A.; Crovetto, L.; Talavera, E.M.; et al. Unusual spectroscopic and photophysical properties of meso-tert-butylBODIPY in comparison to related alkylated BODIPY dyes. RSC Adv. 2015, 5, 89375–89388. [Google Scholar] [CrossRef]
- Kenwood, B.M.; Weaver, J.L.; Bajwa, A.; Poon, I.K.; Byrne, F.L.; Murrow, B.A.; Calderone, J.A.; Huang, L.; Divakaruni, A.S.; Tomsig, J.L.; et al. Identification of a novel mitochondrial uncoupler that does not depolarize the plasma membrane. Mol. Metab. 2014, 3, 114–123. [Google Scholar] [CrossRef] [PubMed]
- Roman, G.; Popek, T.; Lazar, C.; Kiyota, T.; Kluczyk, A.; Konishi, Y. Drug Evolution Concept in Drug Design: 2. Chimera Method. Med. Chem. 2006, 2, 175–189. [Google Scholar] [CrossRef] [PubMed]
- Borsari, C.; Trader, D.J.; Tait, A.; Costi, M.P. Designing Chimeric Molecules for Drug Discovery by Leveraging Chemical Biology. J. Med. Chem. 2020, 63, 1908–1928. [Google Scholar] [CrossRef] [PubMed]
- Yan, C.; Zhang, Y.; Guo, Z. Recent progress on molecularly near-infrared fluorescent probes for chemotherapy and phototherapy. Coord. Chem. Rev. 2021, 427, 213556. [Google Scholar] [CrossRef]
- Kumar, A.; Kant, S.; Singh, S.M. Novel molecular mechanisms of antitumor action of dichloroacetate against T cell lymphoma: Implication of altered glucose metabolism, pH homeostasis and cell survival regulation. Chem. Biol. Interact. 2012, 199, 29–37. [Google Scholar] [CrossRef]
- Ayyanathan, K.; Kesaraju, S.; Dawson-Scully, K.; Weissbach, H. Combination of Sulindac and Dichloroacetate Kills Cancer Cells via Oxidative Damage. PLoS ONE 2012, 7, e39949. [Google Scholar] [CrossRef] [PubMed]
- Ripoll, C.; Roldan, M.; Ruedas-Rama, M.J.; Martin, M.; Ruedas-Rama, M.J.; Orte, A. Mitochondrial pH Nanosensors for Metabolic profiling of breast cancer cell lines. 2020, in preparation. Int. J. Mol. Sci. 2020, 21, 3731. [Google Scholar] [CrossRef]
- Dreiem, A.; Fonnum, F. Thiophene is Toxic to Cerebellar Granule Cells in Culture After Bioactivation by Rat Liver Enzymes. Neurotoxicology 2004, 25, 959–966. [Google Scholar] [CrossRef]
- Kawazoe, Y.; Shimogawa, H.; Sato, A.; Uesugi, M. A Mitochondrial Surface-Specific Fluorescent Probe Activated by Bioconversion. Angew. Chem. Int. Ed. 2011, 50, 5478–5481. [Google Scholar] [CrossRef]
- Jiang, N.; Fan, J.; Liu, T.; Cao, J.; Qiao, B.; Wang, J.; Gao, P.; Peng, X. A near-infrared dye based on BODIPY for tracking morphology changes in mitochondria. Chem. Commun. 2013, 49, 10620–10622. [Google Scholar] [CrossRef]
- Griesbeck, S.; Zhang, Z.; Gutmann, M.; Lühmann, T.; Edkins, R.M.; Clermont, G.; Lazar, A.N.; Haehnel, M.; Edkins, K.; Eichhorn, A.; et al. Water-Soluble Triarylborane Chromophores for One- and Two-Photon Excited Fluorescence Imaging of Mitochondria in Cells. Chem. Eur. J. 2016, 22, 14701–14706. [Google Scholar] [CrossRef]
- Duca, M.; Dozza, B.; Lucarelli, E.; Santi, S.; Di Giorgio, A.; Barbarella, G. Fluorescent labeling of human mesenchymal stem cells by thiophene fluorophores conjugated to a lipophilic carrier. Chem. Commun. 2010, 46, 7948–7950. [Google Scholar] [CrossRef]
- Yang, H.; Li, X.; Cai, Y.; Wang, Q.; Li, W.; Liu, G.; Tang, Y. In silico prediction of chemical subcellular localization via multi-classification methods. MedChemComm 2017, 8, 1225–1234. [Google Scholar] [CrossRef]
- Yang, H.; Lou, C.; Sun, L.; Li, J.; Cai, Y.; Wang, Z.; Li, W.; Liu, G.; Tang, Y. admetSAR 2.0: Web-service for prediction and optimization of chemical ADMET properties. Bioinformatics 2018, 35, 1067–1069. [Google Scholar] [CrossRef] [PubMed]
- Misra, S.K.; Ye, M.; Ostadhossein, F.; Pan, D. Pro-haloacetate Nanoparticles for Efficient Cancer Therapy via Pyruvate Dehydrogenase Kinase Modulation. Sci. Rep. 2016, 6, 28196. [Google Scholar] [CrossRef] [PubMed]
- de Mey, S.; Dufait, I.; Jiang, H.; Corbet, C.; Wang, H.; Van De Gucht, M.; Kerkhove, L.; Law, K.L.; Vandenplas, H.; Gevaert, T.; et al. Dichloroacetate Radiosensitizes Hypoxic Breast Cancer Cells. Int. J. Mol. Sci. 2020, 21, 9367. [Google Scholar] [CrossRef] [PubMed]
- Tataranni, T.; Agriesti, F.; Pacelli, C.; Ruggieri, V.; Laurenzana, I.; Mazzoccoli, C.; Della Sala, G.; Panebianco, C.; Pazienza, V.; Capitanio, N.; et al. Dichloroacetate Affects Mitochondrial Function and Stemness-Associated Properties in Pancreatic Cancer Cell Lines. Cells 2019, 8, 478. [Google Scholar] [CrossRef] [PubMed]
- Rodrigues, A.S.; Correia, M.; Gomes, A.; Pereira, S.L.; Perestrelo, T.; Sousa, M.I.; Ramalho-Santos, J. Dichloroacetate, the Pyruvate Dehydrogenase Complex and the Modulation of mESC Pluripotency. PLoS ONE 2015, 10, e0131663. [Google Scholar] [CrossRef]
- Fernández-Caro, H.; Lostalé-Seijo, I.; Martínez-Calvo, M.; Mosquera, J.; Mascareñas, J.L.; Montenegro, J. Supramolecular caging for cytosolic delivery of anionic probes. Chem. Sci. 2019, 10, 8930–8938. [Google Scholar] [CrossRef] [PubMed]
- Liu, K.K.C.; Zhu, J.; Smith, G.L.; Yin, M.-J.; Bailey, S.; Chen, J.H.; Hu, Q.; Huang, Q.; Li, C.; Li, Q.J.; et al. Highly Selective and Potent Thiophenes as PI3K Inhibitors with Oral Antitumor Activity. ACS Med. Chem. Lett. 2011, 2, 809–813. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Hanson, B.A. Understanding Medicinal Plants. Their Chemistry and Therapeutic Action; The Haworth Press Inc.: Binghamtom, NY, USA, 2005. [Google Scholar]
- Zhang, P.; Liang, D.; Jin, W.; Qu, H.; Cheng, Y.; Li, X.; Ma, Z. Cytotoxic Thiophenes from the Root of Echinops grijisii Hance. Z. Naturforsch. 2009, 64c, 193–196. [Google Scholar] [CrossRef]
- Mohareb, R.M.; Megally Abdo, N.Y. Synthesis and Cytotoxic Evaluation of Pyran, Dihydropyridine and Thiophene Derivatives of 3-Acetylcoumarin. Chem. Pharm. Bull. 2015, 63, 678–687. [Google Scholar] [CrossRef] [PubMed]
- Lisboa, T.; Silva, D.; Duarte, S.; Ferreira, R.; Andrade, C.; Lopes, A.L.; Ribeiro, J.; Farias, D.; Moura, R.; Reis, M.; et al. Toxicity and Antitumor Activity of a Thiophene–Acridine Hybrid. Molecules 2020, 25, 64. [Google Scholar] [CrossRef]
- Nepali, K.; Sharma, S.; Sharma, M.; Bedi, P.M.S.; Dhar, K.L. Rational approaches, design strategies, structure activity relationship and mechanistic insights for anticancer hybrids. Eur. J. Med. Chem. 2014, 77, 422–487. [Google Scholar] [CrossRef] [PubMed]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ripoll, C.; Herrero-Foncubierta, P.; Puente-Muñoz, V.; Gonzalez-Garcia, M.C.; Miguel, D.; Resa, S.; Paredes, J.M.; Ruedas-Rama, M.J.; Garcia-Fernandez, E.; Roldan, M.; et al. Chimeric Drug Design with a Noncharged Carrier for Mitochondrial Delivery. Pharmaceutics 2021, 13, 254. https://doi.org/10.3390/pharmaceutics13020254
Ripoll C, Herrero-Foncubierta P, Puente-Muñoz V, Gonzalez-Garcia MC, Miguel D, Resa S, Paredes JM, Ruedas-Rama MJ, Garcia-Fernandez E, Roldan M, et al. Chimeric Drug Design with a Noncharged Carrier for Mitochondrial Delivery. Pharmaceutics. 2021; 13(2):254. https://doi.org/10.3390/pharmaceutics13020254
Chicago/Turabian StyleRipoll, Consuelo, Pilar Herrero-Foncubierta, Virginia Puente-Muñoz, M. Carmen Gonzalez-Garcia, Delia Miguel, Sandra Resa, Jose M. Paredes, Maria J. Ruedas-Rama, Emilio Garcia-Fernandez, Mar Roldan, and et al. 2021. "Chimeric Drug Design with a Noncharged Carrier for Mitochondrial Delivery" Pharmaceutics 13, no. 2: 254. https://doi.org/10.3390/pharmaceutics13020254
APA StyleRipoll, C., Herrero-Foncubierta, P., Puente-Muñoz, V., Gonzalez-Garcia, M. C., Miguel, D., Resa, S., Paredes, J. M., Ruedas-Rama, M. J., Garcia-Fernandez, E., Roldan, M., Rocha, S., De Keersmaecker, H., Hofkens, J., Martin, M., Cuerva, J. M., & Orte, A. (2021). Chimeric Drug Design with a Noncharged Carrier for Mitochondrial Delivery. Pharmaceutics, 13(2), 254. https://doi.org/10.3390/pharmaceutics13020254