Hepatitis C Virus and Cellular Stress Response: Implications to Molecular Pathogenesis of Liver Diseases

Abstract
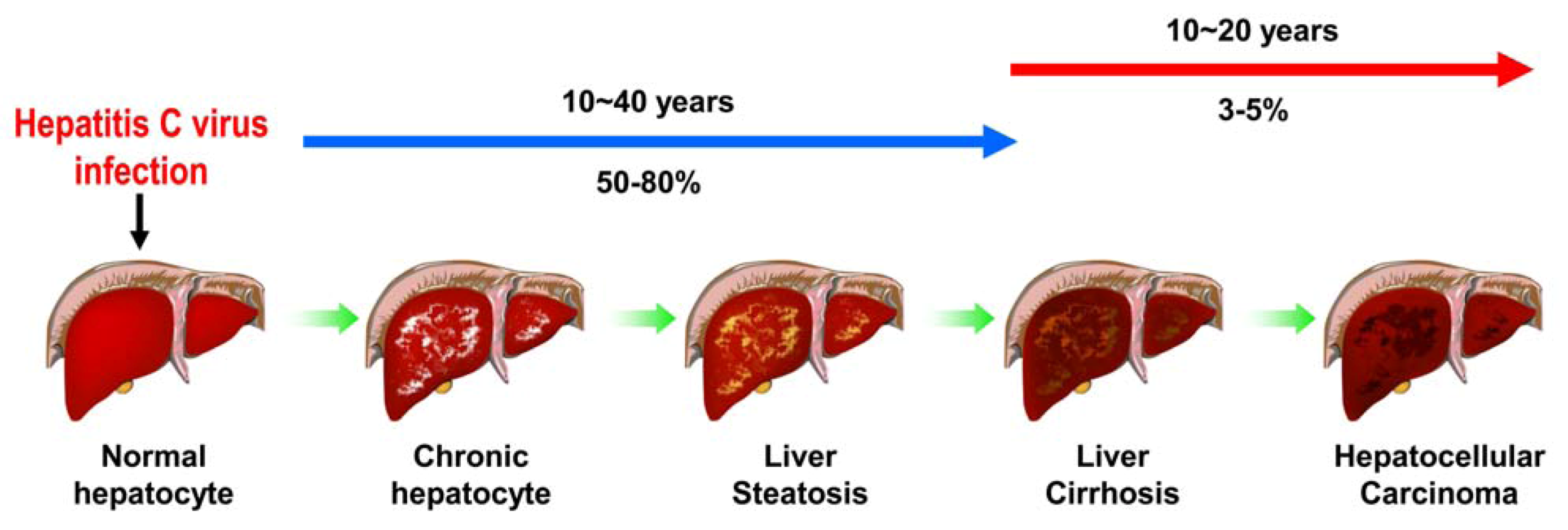
:1. Introduction
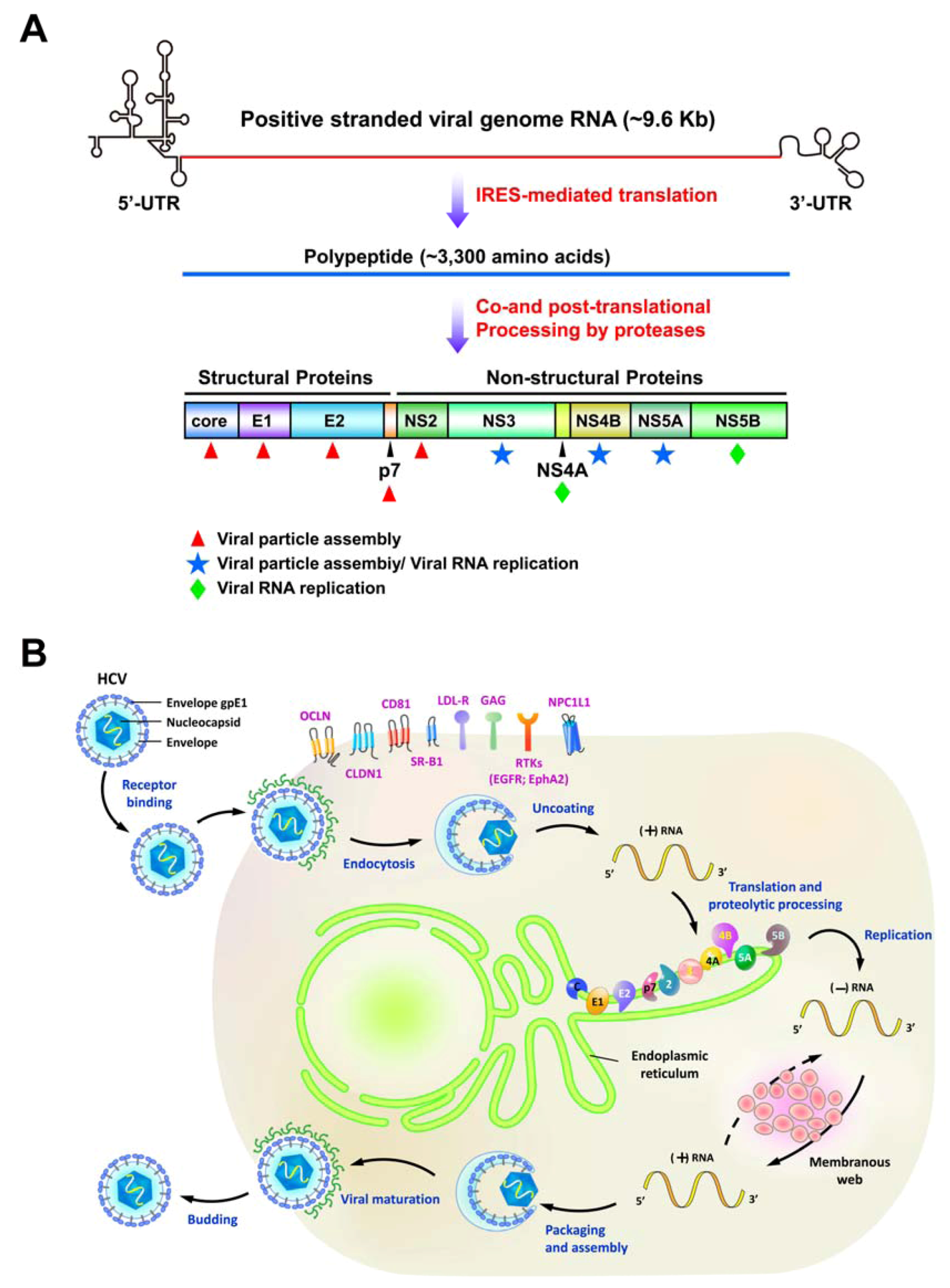
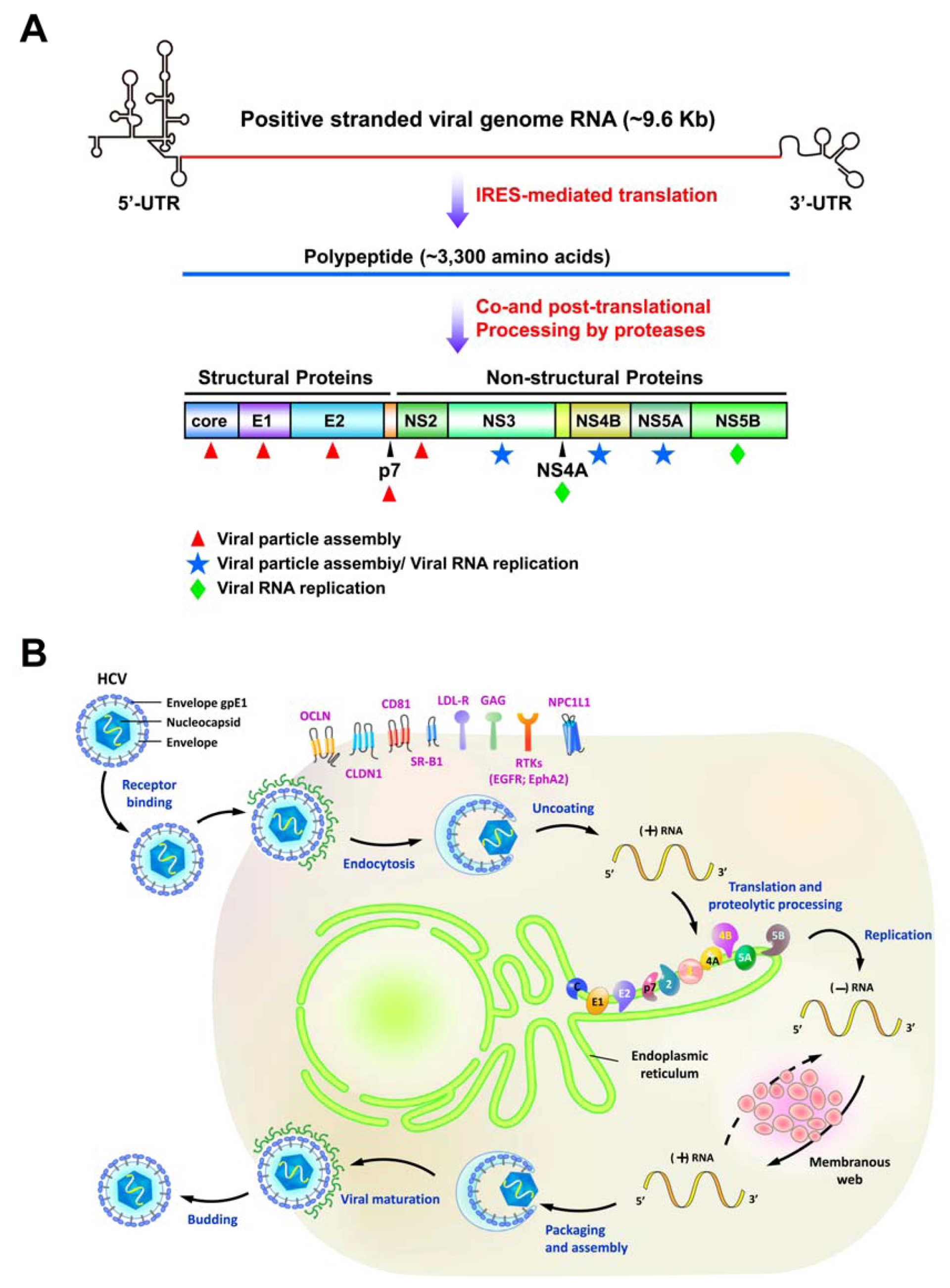
2. Various Cellular Responses Activated by HCV
2.1. ER stress and UPR
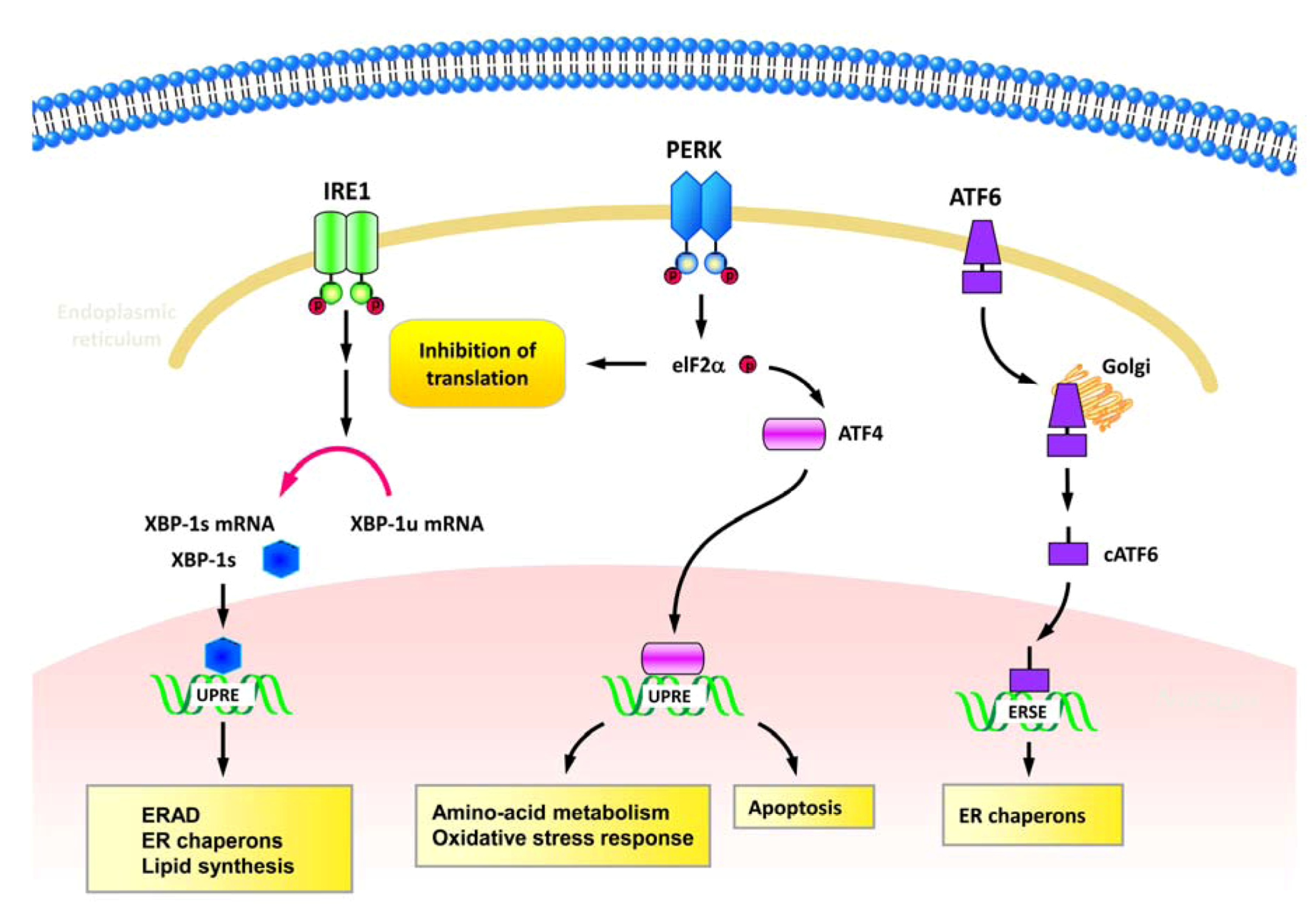

{kind=link}
{kind=link}
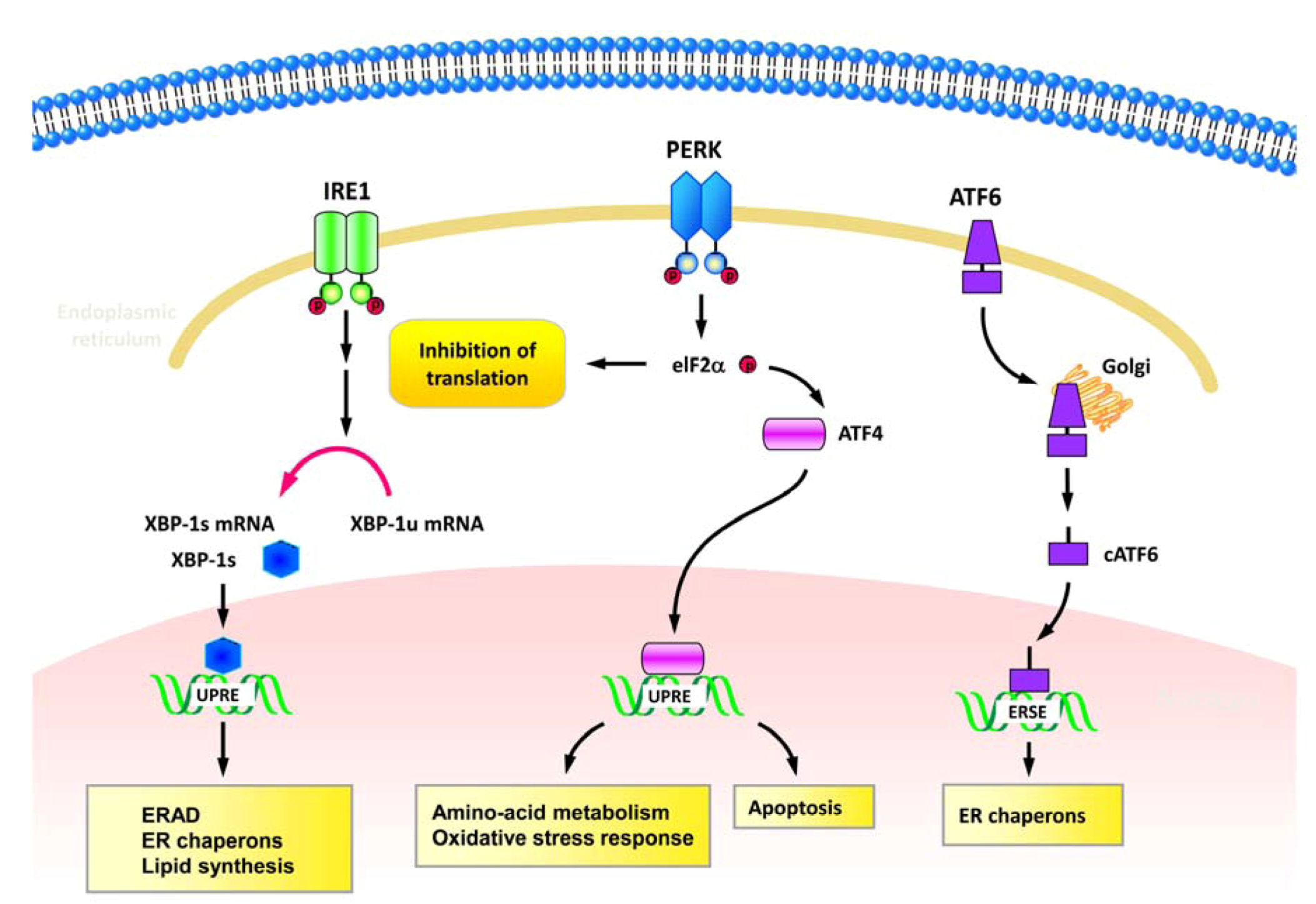{kind=link}
{kind=link}
| Approach/Model | Characteristics | Functional impacts | Reference |
|---|---|---|---|
| Overexpression of HCV NS4B/ Yeast-two hybrid; Coimmuno-precipitation in human cervical cancer cell, HeLa |
| Modulation of ATF6-mediated UPR | Tong et al. [41] |
| HCV-Con1 (1b) replicon transfection/ Human hepatoma cell Huh7 |
| Activation of cap-independent and cap-dependent translation | Tardif et al. [42] |
| Overexpression of HCV E2/ Human embryonic kidney (HEK) 293 and HeLa cell lines |
| Establishment of persistent infection by E2-mediated counteraction against ER stress | Pavio et al. [48] |
| Overexpression of HCV NS4B/Huh7 and HeLa |
| Benefit to viral RNA replication | Zheng et al. [44] |
| Overexpression of HCV E1 and E2/ HeLa and Mouse embryonic fibroblast (MEF) |
| Activation of UPR and ERAD by HCV | Chan and Egan [47] |
| HCV-JFH1 (2a) viral RNA transfection/ Human hepatoma cell Huh7.5-1 |
| Promotion of viral RNA replication; Activation of autophagy | Sir et al. [50] |
| Overexpression of HCV NS4B/ Human hepatic cell lines Hep3B, HepG2, and Huh7 |
| Modulation of intracellular NF-κB signaling | Li et al. [43] |
| HCV-H77c (1a) infection/ Chimeric SCID/Alb-uPA mice transplanted with human hepatocytes |
| Sensitization of the infected cells to apoptosis | Joyce et al. [53] |
| Overexpression of HCV NS2; full-length and subgenomic HCV (1b) replicons transfection/ Huh7 and Huh7.5 |
| Modulation of IRES-mediated translation | Von derm Bussche et al. [46] |
| Liver biopsy specimens from patients with chronic HCV infection |
| Modulations of inflammation and apoptosis | Asselah et al. [55] |
| HCV-JFH1 (2a) infection/Huh7 |
| Promotion of viral RNA replication; Activation of autophagy; Suppression of antiviral innate immunity | Ke and Chen [51] |
| HCV-JFH1 (2a) infection/Huh7; HCV-transgenic mice |
| Counteracting cellular ER stress and adaptation of UPR | Merquiol et al. [54] |
| HCV-JFH1 (2a) infection/ Huh7 and Huh7.5-1 |
| Increment of HCV envelope glycoproteins degrdation | Saeed et al. [52] |
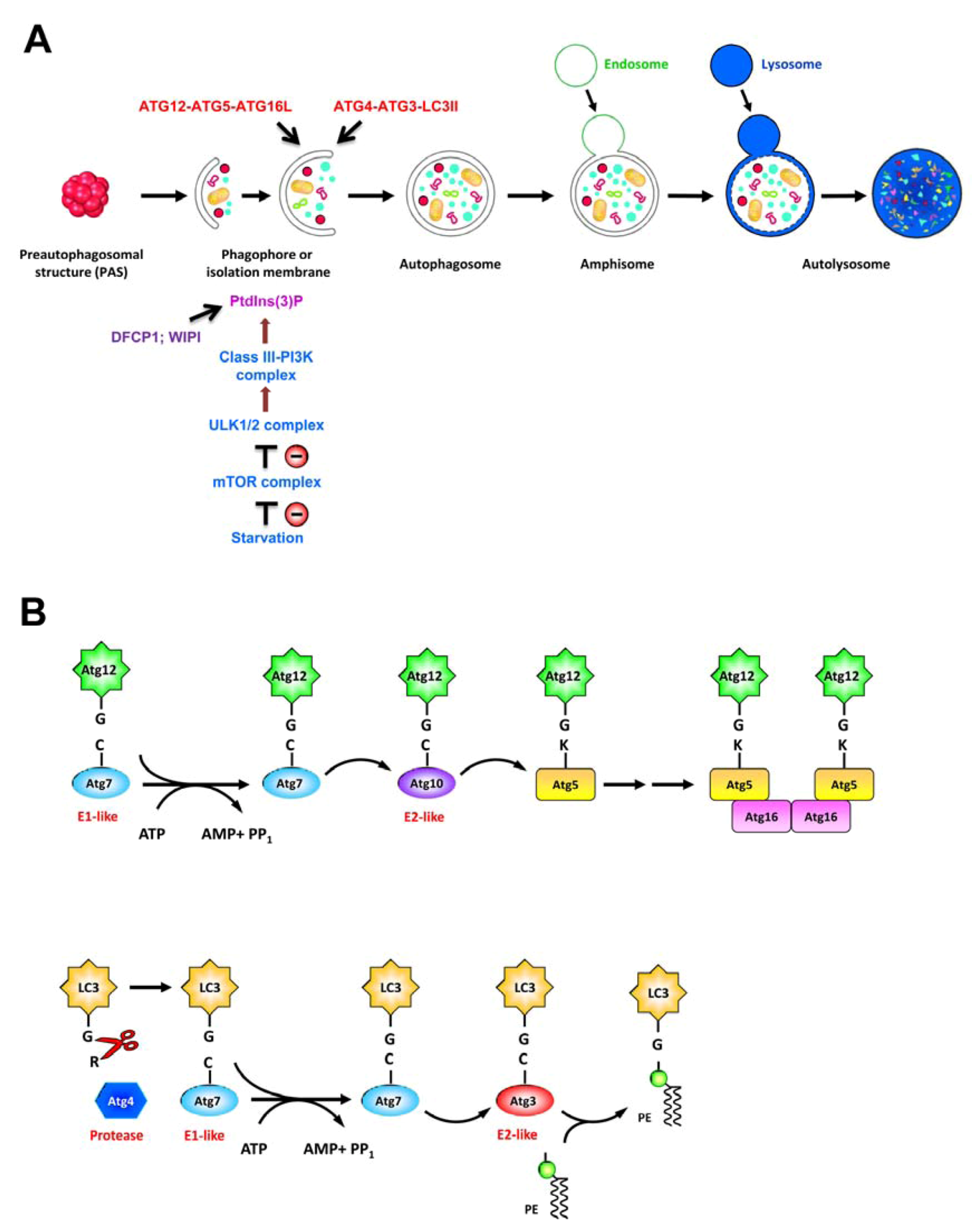
2.2. Autophagy
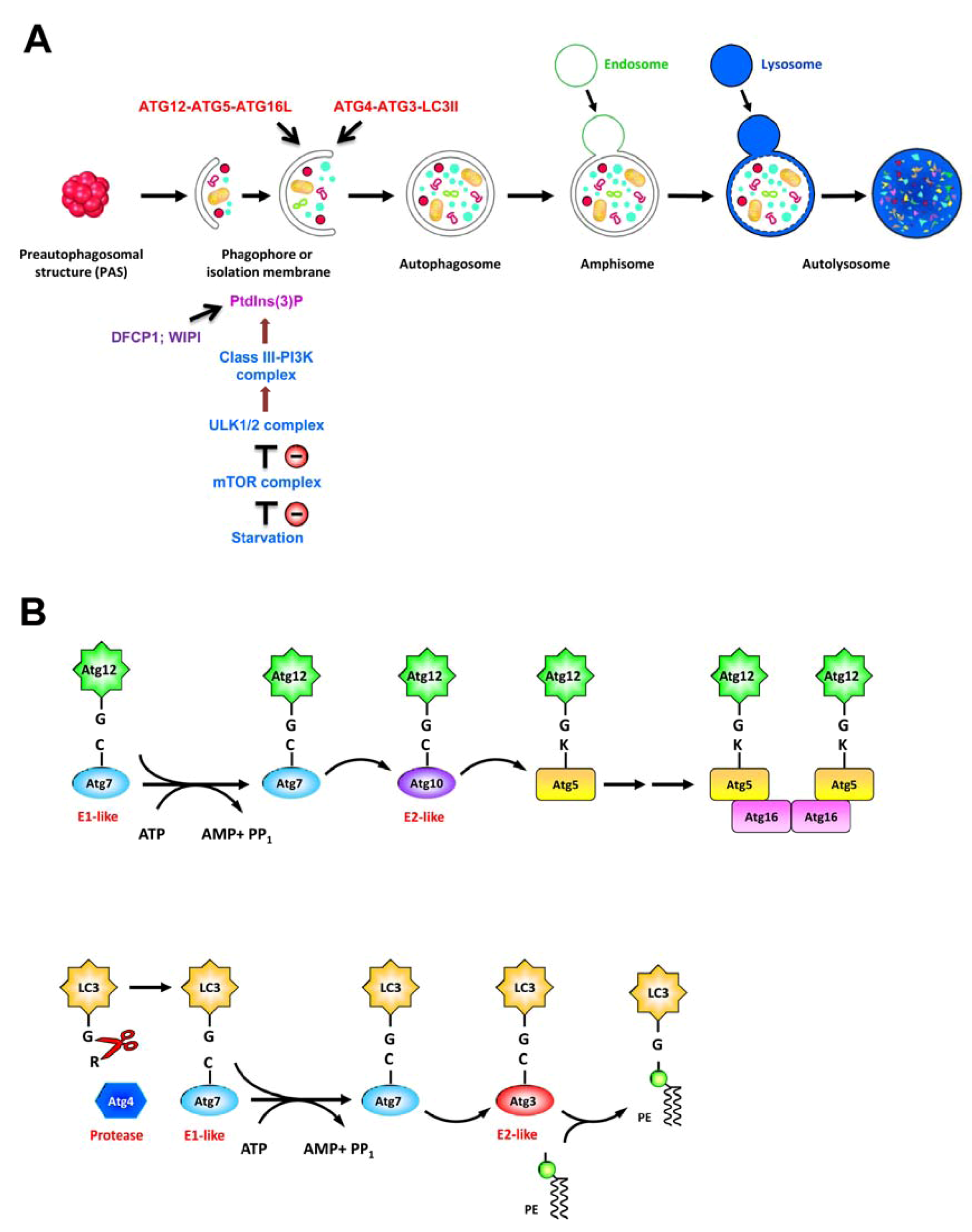
| Approach/Model | Characteristics | Functional impacts | Reference |
|---|---|---|---|
| HCV-H77 (1a) viral RNA transfection/ Immortalized human hepatocytes (IHH) |
| Viral RNA replication | Ait-Goughoulte et al. [73] |
| HCV-JFH1 (2a) viral RNA transfection/ Huh7.5 |
| Viral RNA replication | Sir et al. [50] |
| HCV-JFH1 (2a) infection/ Huh7 |
| Translation of incoming viral RNA | Dreux et al. [74] |
| HCV-JFH1 (2a) infection/ Huh7.5-1 |
| Viral particle assembly | Tanida et al. [82] |
| HCV-JFH1 (2a) infection/ Huh7 |
| Organization of replication site | Guevin et al. [90] |
| HCV-JFH1 (2a) infection/ Huh7 |
| Viral RNA replication; Suppression of antiviral innate immunity | Ke and Chen [51] |
| HCV-H77 (1a); HCV-JFH1 (2a) infection/ IHH |
| Viral RNA replication; Suppression of antiviral innate immunity | Shrivastava et al. [80] |
| HCV-JC1 (2a) infection; overexpression of HCV NS4B/Huh7.5 |
| Organization of viral replication site | Su et al. [87] |
| HCV-Con1 (1b) and JFH1 (2a) Replicon RNA transfection / Huh7 |
| Protection of host cells from viral infection-induced death | Taguwa et al. [81] |
| HCV-JFH1 (2a) replicon RNA transfection/ Huh7.5 |
| Replication site of viral RNA | Sir et al. [79] |
| HCV-JFH1 (2a) full-length and subgenomic HCV replicon RNA transfection/ Huh7; HCV-transgenic mice |
| Regulation of oxidative response.Mitochondria-mediated cytopathic effects | Chu et al. [91] |
| HCV-JFH1 (2a) infection/ IHH |
| Viral RNA replication | Shrivastava et al. [88] |
| HCV-Con1 (1b) and JFH1 (2a) replicon RNA transfection / Huh7 and Huh7.5-1 |
| Metabolism of LDs; Regulation of lipid storage | Vescovo et al. [92] |
| HCV-JFH1 (2a) infection/ Huh7.5 |
| Promoting viral particle production; Regulation of antiviral response | Gregoire et al. [89] |
| HCV-JFH1 (2a) infection/ Huh7; Huh7.5 |
| No apparent role of HCV-induced autophagosomal membrane in HCV replication | Mohl et al. [86] |
| HCV-JFH1 (2a) viral RNA transfection/ Huh7 |
| Dysregulation of glucose homeostasis; Induction of insulin resistance | Das et al. [93] |
2.3. Apoptosis
| Approach/Model | Characteristics | Functional impacts | Reference |
|---|---|---|---|
| Overexpression of HCV core/ Human breast cancer cell line MCF7 |
| Inhibition of TNF-α-mediated apoptosis | Ray et al. [111] |
| Overexpression of HCV core/ HeLa and HepG2 cell lines |
| Activation of TNF-α-induced apoptotic signaling | Zhu et al. [110] |
| Overexpression of HCV NS5A/ Monkey kidney cell line COS7 and Hep3B |
| Inhibition of p53 downstream apoptotic signaling | Lan et al. [114] |
| Transfection of HCV viral replicon RN/ Huh7 |
| Inhibition of TNF-α-mediated extrinsic apoptosis | Lee et al. [112] |
| Overexpression of HCV core/ Huh7 and HepG2; HCV core-transgenic mice |
| Activation of apoptotic cell death | Benali-Furet et al. [104] |
| Overexpression of HCV NS3/ Huh7, HepG2, and HEK293 |
| Promotion of caspase 8-mediated apoptosis | Prikhod'ko et al. [109] |
| Baculovirus-mediated expression of HCV E1/ Insect Sf9 cell |
| Activation of apoptosis | Ciccaglione et al. [106] |
| Overexpression of HCV E2/ Huh7 |
| Sensitization of the cells to apoptosis | Chiou et al. [105] |
| Overexpression of HCV NS4A; subgenomic HCV (1b) replicons transfection/ Huh7 |
| Promotion of mitochondria-mediated intrinsic apoptotic cell death | Nomura-Takigaw et al. [108] |
| Overexpression of HCV core/ HEK293T |
| Promotion of mitochondria-mediated intrinsic apoptotic cell death | Lee et al. [107] |
| Overexpression of HCV NS3/ Huh7 and HeLa cells |
| Inhibition of apoptosis by HCV NS3 required serine protease activity | Tanaka et al. [113] |
| HCV- chimeric J6/JFH1 (2a) infection/ Huh7.5-1 |
| Activation of mitochondria-mediated intrinsic apoptosis by HCV infection | Deng et al. [115] |
| HCV-JFH1 (2a) infection/ Huh7 and LH86 cell lines |
| Activation of death receptor-mediated extrinsic apoptosis by HCV infection | Zhu et al. [117] |
| HCV-JFH1 (2a) infection; replicon viral RNA transfection/ Huh7 |
| Sensitization of the virus-infected cells to TRAIL-mediated extrinsic apoptotic pathway | Deng et al. [116] |
2.4. Cell Cycle Arrest and DNA Damage Mitogenic Signaling, and PI3K Pathway
| Approach/Model | Characteristics | Functional impacts | Reference |
|---|---|---|---|
| Retrovirus infection-mediated expression of HCV core/ HepG2 and HeLa cell lines |
| Modulation of RB/E2F-1-mediated cell cycle progression | Hassan et al. [122] |
| Transfection of HCV viral replicon RNA / Huh7 |
| Promotion of G1/S transition | Munakata et al. [123] |
| HCV H77S (1a) and JFH1 (2a) infection/ Huh7 and Huh7.5-1 |
| Interference with RB-mediated cell cycle progression | Munakata et al. [124] |
| HCV H77S (1a) and JFH1 (2a) infection/ Huh7 and Huh7.5-1 |
| Modulation of host gene expression by regulating RB abundance | McGivern et al. [125] |
| HCV+ PBMC; HCV- PBMC/ B cell-derived HCV infection/ Raji cells HCV JFH1 (2a) infection/ Huh7; HCV core transgenic mice |
| Perturbing mitotic checkpoint; Increasing chromosome instability | Machida et al. [126] |
| HCV+ PBMC; HCV- PBMC/ B cell-derived HCV infection/ Raji cells |
| Inhibiting DNA repair process; Potentiating chromosomal instability | Machida et al. [130] |
| HCV- chimeric H77S/JFH1 (1a/2a) infection/ Huh7.5-1 |
| Interfering with G2/M progression | Kannan et al. [127] |
| Overexpression of HCV NS3/4A/ Huh7 |
| Interference with DNA repair process; Sensitization of the cells to DNA damage | Lai et al. [128] |
| Transfection of HCV subgenomic HCV (1b) replicons; HCV JFH1 (2a) infection / Huh7 |
| Promotion of HCV viral RNA replication | Ariumi et al. [129] |
| Vaccinia virus-mediated expression of HCV NS5A/ HeLa |
| Interference with ERK signaling; Implication to HCV pathogenesis | Tan et al. [132] |
| Overexpression of HCV NS5A/ HeLa; NIH3T3 |
| Interference with ERK signaling | Georgopoulou et al. [133] |
| Overexpression of HCV NS5A; Transfection of subgenomic HCV replicon / Cos7; 293T; Huh7 |
| Interruption ERK pathway; Inhibition of MAPK-mediated transcription | Macdonald et al. [134] |
| Vaccinia virus-mediated expression of HCV NS5A; Transfection of subgenomic HCV replicon / HeLa S3; Huh7 |
| Interruption p38MAPK pathway; Inhibition of cap-dependent protein translation | He et al. [135,136] |
| Overexpression of HCV NS5A/ 293T |
| Modulation of TNF signaling; Implication to HCV pathogenesis | Park et al. [138] |
| Tet-Off-mediated expression of HCV NS5A/ HeLa; |
| Activation of PI3K and AKT signaling; Implication of HCV pathogenesis | He et al. [139] |
| Overexpression of HCV NS5A; Transfection of subgenomic HCV replicon / Cos7; 293T; Huh7 |
| Activation of PI3K and AKT signaling; Implication of HCV pathogenesis | Street et al. [140] |
3. Implication of HCV-Induced Cellular Responses on the Pathogenesis of HCV-associated Liver Diseases
3.1. ER stress and UPR
3.2. Autophagy
3.3. Apoptosis
3.4. Cell Cycle Arrest and DNA Damage, Mitogenic signaling, and PI3K pathway
4. Conclusion and Future Directions
Conflict of Interest
Acknowledgements
References and Notes
- Chisari, F.V. Unscrambling hepatitis C virus-host interactions. Nature 2005, 436, 930–932. [Google Scholar] [CrossRef]
- Moradpour, D.; Penin, F.; Rice, C.M. Replication of hepatitis C virus. Nat. Rev. Microbiol. 2007, 5, 453–463. [Google Scholar] [CrossRef]
- Bartosch, B.; Vitelli, A.; Granier, C.; Goujon, C.; Dubuisson, J.; Pascale, S.; Scarselli, E.; Cortese, R.; Nicosia, A.; Cosset, F.L. Cell entry of hepatitis C virus requires a set of co-receptors that include the CD81 tetraspanin and the SR-B1 scavenger receptor. J. Biol. Chem. 2003, 278, 41624–41630. [Google Scholar]
- Evans, M.J.; von Hahn, T.; Tscherne, D.M.; Syder, A.J.; Panis, M.; Wolk, B.; Hatziioannou, T.; McKeating, J.A.; Bieniasz, P.D.; Rice, C.M. Claudin-1 is a hepatitis C virus co-receptor required for a late step in entry. Nature 2007, 446, 801–805. [Google Scholar] [CrossRef]
- Ploss, A.; Evans, M.J.; Gaysinskaya, V.A.; Panis, M.; You, H.; de Jong, Y.P.; Rice, C.M. Human occludin is a hepatitis C virus entry factor required for infection of mouse cells. Nature 2009, 457, 882–886. [Google Scholar] [CrossRef]
- Zhang, J.; Randall, G.; Higginbottom, A.; Monk, P.; Rice, C.M.; McKeating, J.A. CD81 is required for hepatitis C virus glycoprotein-mediated viral infection. J. Virol. 2004, 78, 1448–1455. [Google Scholar]
- Helle, F.; Dubuisson, J. Hepatitis C virus entry into host cells. Cell. Mol. Life. Sci. 2008, 65, 100–112. [Google Scholar] [CrossRef]
- Lupberger, J.; Zeisel, M.B.; Xiao, F.; Thumann, C.; Fofana, I.; Zona, L.; Davis, C.; Mee, C.J.; Turek, M.; Gorke, S.; Royer, C.; Fischer, B.; Zahid, M.N.; Lavillette, D.; Fresquet, J.; Cosset, F.-L.; Rothenberg, S.M.; Pietschmann, T.; Patel, A.H.; Pessaux, P.; Doffoel, M.; Raffelsberger, W.; Poch, O.; McKeating, J.A.; Brino, L.; Baumert, T.F. EGFR and EphA2 are host factors for hepatitis C virus entry and possible targets for antiviral therapy. Nat. Med. 2011, 17, 589–595. [Google Scholar] [CrossRef]
- Sainz, B.; Barretto, N.; Martin, D.N.; Hiraga, N.; Imamura, M.; Hussain, S.; Marsh, K.A.; Yu, X.; Chayama, K.; Alrefai, W.A.; Uprichard, S.L. Identification of the Niemann-Pick C1-like 1 cholesterol absorption receptor as a new hepatitis C virus entry factor. Nat. Med. 2012, 18, 281–285. [Google Scholar] [CrossRef]
- Appel, N.; Zayas, M.; Miller, S.; Krijnse-Locker, J.; Schaller, T.; Friebe, P.; Kallis, S.; Engel, U.; Bartenschlager, R. Essential role of domain III of nonstructural protein 5A for hepatitis C virus infectious particle assembly. PLoS Pathog. 2008, 4, e1000035. [Google Scholar] [CrossRef]
- Masaki, T.; Suzuki, R.; Murakami, K.; Aizaki, H.; Ishii, K.; Murayama, A.; Date, T.; Matsuura, Y.; Miyamura, T.; Wakita, T.; Suzuki, T. Interaction of hepatitis C virus nonstructural protein 5A with core protein is critical for the production of infectious virus particles. J. Virol. 2008, 82, 7964–7976. [Google Scholar]
- Miyanari, Y.; Atsuzawa, K.; Usuda, N.; Watashi, K.; Hishiki, T.; Zayas, M.; Bartenschlager, R.; Wakita, T.; Hijikata, M.; Shimotohno, K. The lipid droplet is an important organelle for hepatitis C virus production. Nat. Cell. Biol. 2007, 9, 1089–1097. [Google Scholar] [CrossRef]
- Tellinghuisen, T.L.; Foss, K.L.; Treadaway, J.C.; Rice, C.M. Identification of residues required for RNA replication in domains II and III of the hepatitis C virus NS5A protein. J. Virol. 2008, 82, 1073–1083. [Google Scholar] [CrossRef]
- Zoulim, F.; Chevallier, M.; Maynard, M.; Trepo, C. Clinical consequences of hepatitis C virus infection. Rev. Med. Virol. 2003, 13, 57–68. [Google Scholar] [CrossRef]
- Gao, B.; Hong, F.; Radaeva, S. Host factors and failure of interferon-alpha treatment in hepatitis C virus. Hepatology 2004, 39, 880–890. [Google Scholar] [CrossRef]
- Egger, D.; Wolk, B.; Gosert, R.; Bianchi, L.; Blum, H.E.; Moradpour, D.; Bienz, K. Expression of hepatitis C virus proteins induces distinct membrane alterations including a candidate viral replication complex. J. Virol. 2002, 76, 5974–5984. [Google Scholar] [CrossRef]
- Gosert, R.; Egger, D.; Lohmann, V.; Bartenschlager, R.; Blum, H.E.; Bienz, K.; Moradpour, D. Identification of the hepatitis C virus RNA replication complex in Huh-7 cells harboring subgenomic replicons. J. Virol. 2003, 77, 5487–5492. [Google Scholar] [CrossRef]
- Gosert, R.; Kanjanahaluethai, A.; Egger, D.; Bienz, K.; Baker, S.C. RNA replication of mouse hepatitis virus takes place at double-membrane vesicles. J Virol 2002, 76, 3697–3708. [Google Scholar]
- Jones, D.M.; McLauchlan, J. Hepatitis C virus: assembly and release of virus particles. J. Biol. Chem. 2010, 285, 22733–22739. [Google Scholar] [CrossRef]
- Op De Beeck, A.; Cocquerel, L.; Dubuisson, J. Biogenesis of hepatitis C virus envelope glycoproteins. J. Gen. Virol. 2001, 82, 2589–2595. [Google Scholar]
- Ron, D.; Walter, P. Signal integration in the endoplasmic reticulum unfolded protein response. Nat. Rev. Mol. Cell. Biol. 2007, 8, 519–529. [Google Scholar] [CrossRef]
- Cox, J.S.; Shamu, C.E.; Walter, P. Transcriptional induction of genes encoding endoplasmic reticulum resident proteins requires a transmembrane protein kinase. Cell 1993, 73, 1197–1206. [Google Scholar] [CrossRef]
- Credle, J.J.; Finer-Moore, J.S.; Papa, F.R.; Stroud, R.M.; Walter, P. On the mechanism of sensing unfolded protein in the endoplasmic reticulum. Proc. Natl. Acad. Sci. USA 2005, 102, 18773–18784. [Google Scholar]
- Zhou, J.; Liu, C.Y.; Back, S.H.; Clark, R.L.; Peisach, D.; Xu, Z.; Kaufman, R.J. The crystal structure of human IRE1 luminal domain reveals a conserved dimerization interface required for activation of the unfolded protein response. Proc. Natl. Acad. Sci. USA 2006, 103, 14343–14348. [Google Scholar]
- Calfon, M.; Zeng, H.; Urano, F.; Till, J.H.; Hubbard, S.R.; Harding, H.P.; Clark, S.G.; Ron, D. IRE1 couples endoplasmic reticulum load to secretory capacity by processing the XBP-1 mRNA. Nature 2002, 415, 92–96. [Google Scholar] [CrossRef]
- Yoshida, H.; Oku, M.; Suzuki, M.; Mori, K. pXBP1(U) encoded in XBP1 pre-mRNA negatively regulates unfolded protein response activator pXBP1(S) in mammalian ER stress response. J. Cell. Biol. 2006, 172, 565–575. [Google Scholar] [CrossRef]
- Niwa, M.; Patil, C.K.; DeRisi, J.; Walter, P. Genome-scale approaches for discovering novel nonconventional splicing substrates of the Ire1 nuclease. Genome Biol 2005, 6, R3. [Google Scholar]
- Yoshida, H.; Matsui, T.; Yamamoto, A.; Okada, T.; Mori, K. XBP1 mRNA is induced by ATF6 and spliced by IRE1 in response to ER stress to produce a highly active transcription factor. Cell 2001, 107, 881–891. [Google Scholar] [CrossRef]
- Chen, X.; Shen, J.; Prywes, R. The luminal domain of ATF6 senses endoplasmic reticulum (ER) stress and causes translocation of ATF6 from the ER to the Golgi. J. Biol. Chem. 2002, 277, 13045–13052. [Google Scholar] [CrossRef]
- Haze, K.; Yoshida, H.; Yanagi, H.; Yura, T.; Mori, K. Mammalian transcription factor ATF6 is synthesized as a transmembrane protein and activated by proteolysis in response to endoplasmic reticulum stress. Mol. Biol. Cell. 1999, 10, 3787–3799. [Google Scholar]
- Ye, J.; Rawson, R.B.; Komuro, R.; Chen, X.; Dave, U.P.; Prywes, R.; Brown, M.S.; Goldstein, J.L. ER stress induces cleavage of membrane-bound ATF6 by the same proteases that process SREBPs. Mol. Cell. 2000, 6, 1355–1364. [Google Scholar] [CrossRef]
- Zhang, K.; Shen, X.; Wu, J.; Sakaki, K.; Saunders, T.; Rutkowski, D.T.; Back, S.H.; Kaufman, R.J. Endoplasmic reticulum stress activates cleavage of CREBH to induce a systemic inflammatory response. Cell 2006, 124, 587–599. [Google Scholar] [CrossRef]
- Shen, J.; Chen, X.; Hendershot, L.; Prywes, R. ER stress regulation of ATF6 localization by dissociation of BiP/GRP78 binding and unmasking of Golgi localization signals. Dev. Cell. 2002, 3, 99–111. [Google Scholar] [CrossRef]
- Harding, H.P.; Zhang, Y.; Zeng, H.; Novoa, I.; Lu, P.D.; Calfon, M.; Sadri, N.; Yun, C.; Popko, B.; Paules, R.; Stojdl, D.F.; Bell, J.C.; Hettmann, T.; Leiden, J.M.; Ron, D. An integrated stress response regulates amino acid metabolism and resistance to oxidative stress. Mol. Cell. 2003, 11, 619–633. [Google Scholar] [CrossRef]
- Harding, H.P.; Zhang, Y.; Ron, D. Protein translation and folding are coupled by an endoplasmic-reticulum-resident kinase. Nature 1999, 397, 271–274. [Google Scholar] [CrossRef]
- Jiang, H.Y.; Wek, S.A.; McGrath, B.C.; Scheuner, D.; Kaufman, R.J.; Cavener, D.R.; Wek, R.C. Phosphorylation of the alpha subunit of eukaryotic initiation factor 2 is required for activation of NF-kappaB in response to diverse cellular stresses. Mol. Cell. Biol. 2003, 23, 5651–5663. [Google Scholar] [CrossRef]
- Lu, P.D.; Harding, H.P.; Ron, D. Translation reinitiation at alternative open reading frames regulates gene expression in an integrated stress response. J. Cell. Biol. 2004, 167, 27–33. [Google Scholar] [CrossRef]
- Novoa, I.; Zeng, H.; Harding, H.P.; Ron, D. Feedback inhibition of the unfolded protein response by GADD34-mediated dephosphorylation of eIF2alpha. J. Cell. Biol. 2001, 153, 1011–1022. [Google Scholar] [CrossRef]
- Novoa, I.; Zhang, Y.; Zeng, H.; Jungreis, R.; Harding, H.P.; Ron, D. Stress-induced gene expression requires programmed recovery from translational repression. EMBO J. 2003, 22, 1180–1187. [Google Scholar] [CrossRef]
- Smith, M.H.; Ploegh, H.L.; Weissman, J.S. Road to ruin: targeting proteins for degradation in the endoplasmic reticulum. Science 2011, 334, 1086–1090. [Google Scholar] [CrossRef]
- Tong, W.Y.; Nagano-Fujii, M.; Hidajat, R.; Deng, L.; Takigawa, Y.; Hotta, H. Physical interaction between hepatitis C virus NS4B protein and CREB-RP/ATF6beta. Biochem. Biophys. Res. Commun. 2002, 299, 366–372. [Google Scholar] [CrossRef]
- Tardif, K.D.; Mori, K.; Siddiqui, A. Hepatitis C virus subgenomic replicons induce endoplasmic reticulum stress activating an intracellular signaling pathway. J. Virol. 2002, 76, 7453–7459. [Google Scholar] [CrossRef]
- Li, S.; Ye, L.; Yu, X.; Xu, B.; Li, K.; Zhu, X.; Liu, H.; Wu, X.; Kong, L. Hepatitis C virus NS4B induces unfolded protein response and endoplasmic reticulum overload response-dependent NF-kappaB activation. Virology 2009, 391, 257–264. [Google Scholar] [CrossRef]
- Zheng, Y.; Gao, B.; Ye, L.; Kong, L.; Jing, W.; Yang, X.; Wu, Z. Hepatitis C virus non-structural protein NS4B can modulate an unfolded protein response. J. Microbiol. 2005, 43, 529–536. [Google Scholar]
- Zhao, P.; Han, T.; Guo, J.J.; Zhu, S.L.; Wang, J.; Ao, F.; Jing, M.Z.; She, Y.L.; Wu, Z.H.; Ye, L.B. HCV NS4B induces apoptosis through the mitochondrial death pathway. Virus. Res. 2012, 169, 1–7. [Google Scholar] [CrossRef]
- von dem Bussche, A.; Machida, R.; Li, K.; Loevinsohn, G.; Khander, A.; Wang, J.; Wakita, T.; Wands, J.R.; Li, J. Hepatitis C virus NS2 protein triggers endoplasmic reticulum stress and suppresses its own viral replication. J. Hepatol. 2010, 53, 797–804. [Google Scholar] [CrossRef]
- Chan, S.W.; Egan, P.A. Hepatitis C virus envelope proteins regulate CHOP via induction of the unfolded protein response. FASEB J. 2005, 19, 1510–1512. [Google Scholar]
- Pavio, N.; Romano, P.R.; Graczyk, T.M.; Feinstone, S.M.; Taylor, D.R. Protein synthesis and endoplasmic reticulum stress can be modulated by the hepatitis C virus envelope protein E2 through the eukaryotic initiation factor 2alpha kinase PERK. J. Virol. 2003, 77, 3578–3585. [Google Scholar]
- Wakita, T.; Pietschmann, T.; Kato, T.; Date, T.; Miyamoto, M.; Zhao, Z.; Murthy, K.; Habermann, A.; Krausslich, H.G.; Mizokami, M.; Bartenschlager, R.; Liang, T.J. Production of infectious hepatitis C virus in tissue culture from a cloned viral genome. Nat. Med. 2005, 11, 791–796. [Google Scholar] [CrossRef]
- Sir, D.; Chen, W.L.; Choi, J.; Wakita, T.; Yen, T.S.; Ou, J.H. Induction of incomplete autophagic response by hepatitis C virus via the unfolded protein response. Hepatology 2008, 48, 1054–1061. [Google Scholar] [CrossRef]
- Ke, P.Y.; Chen, S.S. Activation of the unfolded protein response and autophagy after hepatitis C virus infection suppresses innate antiviral immunity in vitro. J. Clin. Invest. 2011, 121, 37–56. [Google Scholar] [CrossRef]
- Saeed, M.; Suzuki, R.; Watanabe, N.; Masaki, T.; Tomonaga, M.; Muhammad, A.; Kato, T.; Matsuura, Y.; Watanabe, H.; Wakita, T.; Suzuki, T. Role of the endoplasmic reticulum-associated degradation (ERAD) pathway in degradation of hepatitis C virus envelope proteins and production of virus particles. J. Biol. Chem. 2011, 286, 37264–37273. [Google Scholar]
- Joyce, M.A.; Walters, K.A.; Lamb, S.E.; Yeh, M.M.; Zhu, L.F.; Kneteman, N.; Doyle, J.S.; Katze, M.G.; Tyrrell, D.L. HCV induces oxidative and ER stress, and sensitizes infected cells to apoptosis in SCID/Alb-uPA mice. PLoS Pathog. 2009, 5, e1000291. [Google Scholar]
- Merquiol, E.; Uzi, D.; Mueller, T.; Goldenberg, D.; Nahmias, Y.; Xavier, R.J.; Tirosh, B.; Shibolet, O. HCV causes chronic endoplasmic reticulum stress leading to adaptation and interference with the unfolded protein response. PLoS One 2011, 6, e24660. [Google Scholar]
- Asselah, T.; Bieche, I.; Mansouri, A.; Laurendeau, I.; Cazals-Hatem, D.; Feldmann, G.; Bedossa, P.; Paradis, V.; Martinot-Peignoux, M.; Lebrec, D.; Guichard, C.; Ogier-Denis, E.; Vidaud, M.; Tellier, Z.; Soumelis, V.; Marcellin, P.; Moreau, R. In vivo hepatic endoplasmic reticulum stress in patients with chronic hepatitis C. J. Pathol. 2010, 221, 264–274. [Google Scholar]
- Deretic, V.; Levine, B. Autophagy, immunity, and microbial adaptations. Cell. Host Microbe 2009, 5, 527–549. [Google Scholar] [CrossRef]
- Mizushima, N.; Levine, B.; Cuervo, A.M.; Klionsky, D.J. Autophagy fights disease through cellular self-digestion. Nature 2008, 451, 1069–1075. [Google Scholar]
- Kroemer, G.; Marino, G.; Levine, B. Autophagy and the integrated stress response. Mol. Cell. 2010, 40, 280–293. [Google Scholar] [CrossRef]
- Matsunaga, K.; Morita, E.; Saitoh, T.; Akira, S.; Ktistakis, N.T.; Izumi, T.; Noda, T.; Yoshimori, T. Autophagy requires endoplasmic reticulum targeting of the PI3-kinase complex via Atg14L. J. Cell. Biol. 2010, 190, 511–521. [Google Scholar]
- Axe, E.L.; Walker, S.A.; Manifava, M.; Chandra, P.; Roderick, H.L.; Habermann, A.; Griffiths, G.; Ktistakis, N.T. Autophagosome formation from membrane compartments enriched in phosphatidylinositol 3-phosphate and dynamically connected to the endoplasmic reticulum. J. Cell. Biol. 2008, 182, 685–701. [Google Scholar] [CrossRef]
- Fujita, N.; Itoh, T.; Omori, H.; Fukuda, M.; Noda, T.; Yoshimori, T. The Atg16L complex specifies the site of LC3 lipidation for membrane biogenesis in autophagy. Mol. Biol. Cell. 2008, 19, 2092–2100. [Google Scholar]
- Mizushima, N.; Levine, B. Autophagy in mammalian development and differentiation. Nat. Cell. Biol. 2010, 12, 823–830. [Google Scholar] [CrossRef]
- Levine, B.; Kroemer, G. Autophagy in the pathogenesis of disease. Cell 2008, 132, 27–42. [Google Scholar]
- Suhy, D.A.; Giddings, T.H., Jr.; Kirkegaard, K. Remodeling the endoplasmic reticulum by poliovirus infection and by individual viral proteins: an autophagy-like origin for virus-induced vesicles. J. Virol. 2000, 74, 8953–8965. [Google Scholar] [CrossRef]
- Prentice, E.; Jerome, W.G.; Yoshimori, T.; Mizushima, N.; Denison, M.R. Coronavirus replication complex formation utilizes components of cellular autophagy. J. Biol. Chem. 2004, 279, 10136–10141. [Google Scholar]
- Jackson, W.T.; Giddings, T.H., Jr.; Taylor, M.P.; Mulinyawe, S.; Rabinovitch, M.; Kopito, R.R.; Kirkegaard, K. Subversion of cellular autophagosomal machinery by RNA viruses. PLoS Biol. 2005, 3, e156. [Google Scholar] [CrossRef]
- Orvedahl, A.; MacPherson, S.; Sumpter, R., Jr.; Talloczy, Z.; Zou, Z.; Levine, B. Autophagy protects against Sindbis virus infection of the central nervous system. Cell. Host. Microbe 2010, 7, 115–127. [Google Scholar]
- Liu, Y.; Schiff, M.; Czymmek, K.; Talloczy, Z.; Levine, B.; Dinesh-Kumar, S.P. Autophagy regulates programmed cell death during the plant innate immune response. Cell 2005, 121, 567–577. [Google Scholar] [CrossRef]
- Liang, X.H.; Kleeman, L.K.; Jiang, H.H.; Gordon, G.; Goldman, J.E.; Berry, G.; Herman, B.; Levine, B. Protection against fatal Sindbis virus encephalitis by beclin, a novel Bcl-2-interacting protein. J. Virol. 1998, 72, 8586–8596. [Google Scholar]
- Orvedahl, A.; Levine, B. Eating the enemy within: autophagy in infectious diseases. Cell Death Differ. 2009, 16, 57–69. [Google Scholar] [CrossRef]
- Lee, H.K.; Lund, J.M.; Ramanathan, B.; Mizushima, N.; Iwasaki, A. Autophagy-dependent viral recognition by plasmacytoid dendritic cells. Science 2007, 315, 1398–1401. [Google Scholar]
- Paludan, C.; Schmid, D.; Landthaler, M.; Vockerodt, M.; Kube, D.; Tuschl, T.; Munz, C. Endogenous MHC class II processing of a viral nuclear antigen after autophagy. Science 2005, 307, 593–596. [Google Scholar]
- Ait-Goughoulte, M.; Kanda, T.; Meyer, K.; Ryerse, J.S.; Ray, R.B.; Ray, R. Hepatitis C virus genotype 1a growth and induction of autophagy. J. Virol. 2008, 82, 2241–2249. [Google Scholar]
- Dreux, M.; Gastaminza, P.; Wieland, S.F.; Chisari, F.V. The autophagy machinery is required to initiate hepatitis C virus replication. Proc. Natl. Acad. Sci. USA 2009, 106, 14046–14051. [Google Scholar]
- Heaton, N.S.; Randall, G. Dengue virus-induced autophagy regulates lipid metabolism. Cell Host Microbe 2010, 8, 422–432. [Google Scholar] [CrossRef]
- Lee, Y.R.; Lei, H.Y.; Liu, M.T.; Wang, J.R.; Chen, S.H.; Jiang-Shieh, Y.F.; Lin, Y.S.; Yeh, T.M.; Liu, C.C.; Liu, H.S. Autophagic machinery activated by dengue virus enhances virus replication. Virology 2008, 374, 240–248. [Google Scholar]
- Li, J.K.; Liang, J.J.; Liao, C.L.; Lin, Y.L. Autophagy is involved in the early step of Japanese encephalitis virus infection. Microbes Infect. 2012, 14, 159–168. [Google Scholar] [CrossRef]
- Panyasrivanit, M.; Greenwood, M.P.; Murphy, D.; Isidoro, C.; Auewarakul, P.; Smith, D.R. Induced autophagy reduces virus output in dengue infected monocytic cells. Virology 2011, 418, 74–84. [Google Scholar]
- Sir, D.; Kuo, C.F.; Tian, Y.; Liu, H.M.; Huang, E.J.; Jung, J.U.; Machida, K.; Ou, J.H. Replication of hepatitis C virus RNA on autophagosomal membranes. J. Biol. Chem. 2012, 287, 18036–18043. [Google Scholar]
- Shrivastava, S.; Raychoudhuri, A.; Steele, R.; Ray, R.; Ray, R.B. Knockdown of autophagy enhances the innate immune response in hepatitis C virus-infected hepatocytes. Hepatology 2011, 53, 406–414. [Google Scholar] [CrossRef]
- Taguwa, S.; Kambara, H.; Fujita, N.; Noda, T.; Yoshimori, T.; Koike, K.; Moriishi, K.; Matsuura, Y. Dysfunction of autophagy participates in vacuole formation and cell death in cells replicating hepatitis C virus. J. Virol. 2011, 85, 13185–13194. [Google Scholar] [CrossRef]
- Tanida, I.; Fukasawa, M.; Ueno, T.; Kominami, E.; Wakita, T.; Hanada, K. Knockdown of autophagy-related gene decreases the production of infectious hepatitis C virus particles. Autophagy 2009, 5, 937–945. [Google Scholar]
- Kim, I.; Xu, W.; Reed, J.C. Cell death and endoplasmic reticulum stress: disease relevance and therapeutic opportunities. Nat. Rev. Drug Discov. 2008, 7, 1013–1030. [Google Scholar] [CrossRef]
- Ogata, M.; Hino, S.; Saito, A.; Morikawa, K.; Kondo, S.; Kanemoto, S.; Murakami, T.; Taniguchi, M.; Tanii, I.; Yoshinaga, K.; Shiosaka, S.; Hammarback, J.A.; Urano, F.; Imaizumi, K. Autophagy is activated for cell survival after endoplasmic reticulum stress. Mol. Cell. Biol. 2006, 26, 9220–9231. [Google Scholar]
- Schroder, M. Endoplasmic reticulum stress responses. Cell Mol. Life Sci. 2008, 65, 862–894. [Google Scholar] [CrossRef]
- Mohl, B.P.; Tedbury, P.R.; Griffin, S.; Harris, M. Hepatitis C virus-induced autophagy is independent of the unfolded protein response. J. Virol. 2012, 86, 10724–10732. [Google Scholar]
- Su, W.C.; Chao, T.C.; Huang, Y.L.; Weng, S.C.; Jeng, K.S.; Lai, M.M. Rab5 and class III phosphoinositide 3-kinase Vps34 are involved in hepatitis C virus NS4B-induced autophagy. J. Virol. 2011, 85, 10561–10571. [Google Scholar] [CrossRef]
- Shrivastava, S.; Bhanja Chowdhury, J.; Steele, R.; Ray, R.; Ray, R.B. Hepatitis C Virus Upregulates Beclin1 for Induction of Autophagy and Activates mTOR Signaling. J. Virol. 2012, 86, 8705–8712. [Google Scholar]
- Gregoire, I.P.; Richetta, C.; Meyniel-Schicklin, L.; Borel, S.; Pradezynski, F.; Diaz, O.; Deloire, A.; Azocar, O.; Baguet, J.; Le Breton, M.; Mangeot, P.E.; Navratil, V.; Joubert, P.E.; Flacher, M.; Vidalain, P.O.; Andre, P.; Lotteau, V.; Biard-Piechaczyk, M.; Rabourdin-Combe, C.; Faure, M. IRGM is a common target of RNA viruses that subvert the autophagy network. PLoS Pathog. 2011, 7, e1002422. [Google Scholar]
- Guevin, C.; Manna, D.; Belanger, C.; Konan, K.V.; Mak, P.; Labonte, P. Autophagy protein ATG5 interacts transiently with the hepatitis C virus RNA polymerase (NS5B) early during infection. Virology 2010, 405, 1–7. [Google Scholar] [CrossRef]
- Chu, V.C.; Bhattacharya, S.; Nomoto, A.; Lin, J.; Zaidi, S.K.; Oberley, T.D.; Weinman, S.A.; Azhar, S.; Huang, T.T. Persistent expression of hepatitis C virus non-structural proteins leads to increased autophagy and mitochondrial injury in human hepatoma cells. PLoS One 2011, 6, e28551. [Google Scholar]
- Vescovo, T.; Romagnoli, A.; Perdomo, A.B.; Corazzari, M.; Ciccosanti, F.; Alonzi, T.; Nardacci, R.; Ippolito, G.; Tripodi, M.; Garcia-Monzon, C.; Lo Iacono, O.; Piacentini, M.; Fimia, G.M. Autophagy protects cells from HCV-induced defects in lipid metabolism. Gastroenterology 2012, 142, 644–653. [Google Scholar]
- Das, G.C.; Hollinger, F.B. Molecular pathways for glucose homeostasis, insulin signaling and autophagy in hepatitis C virus induced insulin resistance in a cellular model. Virology 2012.
- Best, S.M. Viral subversion of apoptotic enzymes: escape from death row. Annu. Rev. Microbiol. 2008, 62, 171–192. [Google Scholar] [CrossRef]
- Richard, A.; Tulasne, D. Caspase cleavage of viral proteins, another way for viruses to make the best of apoptosis. Cell Death Dis. 2012, 3, e277. [Google Scholar]
- Strasser, A.; O'Connor, L.; Dixit, V.M. Apoptosis signaling. Annu. Rev. Biochem. 2000, 69, 217–245. [Google Scholar] [CrossRef]
- Varfolomeev, E.E.; Ashkenazi, A. Tumor necrosis factor: an apoptosis JuNKie? Cell 2004, 116, 491–497. [Google Scholar] [CrossRef]
- Ashkenazi, A.; Dixit, V.M. Apoptosis control by death and decoy receptors. Curr. Opin. Cell. Biol. 1999, 11, 255–260. [Google Scholar]
- Wang, C.; Youle, R.J. The role of mitochondria in apoptosis. Annu.Rev. Genet. 2009, 43, 95–118. [Google Scholar] [CrossRef]
- Rong, Y.; Distelhorst, C.W. Bcl-2 protein family members: versatile regulators of calcium signaling in cell survival and apoptosis. Annu. Rev. Physiol. 2008, 70, 73–91. [Google Scholar]
- Wallach, D.; Varfolomeev, E.E.; Malinin, N.L.; Goltsev, Y.V.; Kovalenko, A.V.; Boldin, M.P. Tumor necrosis factor receptor and Fas signaling mechanisms. Annu. Rev. Immunol. 1999, 17, 331–367. [Google Scholar] [CrossRef]
- Bodmer, J.L.; Schneider, P.; Tschopp, J. The molecular architecture of the TNF superfamily. Trends Biochem.Sci. 2002, 27, 19–26. [Google Scholar] [CrossRef]
- Ashkenazi, A.; Dixit, V.M. Death receptors: signaling and modulation. Science 1998, 281, 1305–1308. [Google Scholar]
- Benali-Furet, N.L.; Chami, M.; Houel, L.; De Giorgi, F.; Vernejoul, F.; Lagorce, D.; Buscail, L.; Bartenschlager, R.; Ichas, F.; Rizzuto, R.; Paterlini-Brechot, P. Hepatitis C virus core triggers apoptosis in liver cells by inducing ER stress and ER calcium depletion. Oncogene 2005, 24, 4921–4933. [Google Scholar]
- Chiou, H.L.; Hsieh, Y.S.; Hsieh, M.R.; Chen, T.Y. HCV E2 may induce apoptosis of Huh-7 cells via a mitochondrial-related caspase pathway. Biochem. Biophys. Res. Commun. 2006, 345, 453–458. [Google Scholar] [CrossRef]
- Ciccaglione, A.R.; Marcantonio, C.; Costantino, A.; Equestre, M.; Rapicetta, M. Expression of HCV E1 protein in baculovirus-infected cells: effects on cell viability and apoptosis induction. Intervirology 2003, 46, 121–126. [Google Scholar]
- Su, Y.P.; Shien, J.H.; Liu, H.J.; Yin, H.S.; Lee, L.H. Avian reovirus core protein muA expressed in Escherichia coli possesses both NTPase and RTPase activities. J. Gen. Virol. 2007, 88, 1797–1805. [Google Scholar] [CrossRef]
- Nomura-Takigawa, Y.; Nagano-Fujii, M.; Deng, L.; Kitazawa, S.; Ishido, S.; Sada, K.; Hotta, H. Non-structural protein 4A of Hepatitis C virus accumulates on mitochondria and renders the cells prone to undergoing mitochondria-mediated apoptosis. J. Gen. Virol. 2006, 87, 1935–1945. [Google Scholar]
- Prikhod'ko, E.A.; Prikhod'ko, G.G.; Siegel, R.M.; Thompson, P.; Major, M.E.; Cohen, J.I. The NS3 protein of hepatitis C virus induces caspase-8-mediated apoptosis independent of its protease or helicase activities. Virology 2004, 329, 53–67. [Google Scholar] [CrossRef]
- Zhu, N.; Khoshnan, A.; Schneider, R.; Matsumoto, M.; Dennert, G.; Ware, C.; Lai, M.M. Hepatitis C virus core protein binds to the cytoplasmic domain of tumor necrosis factor (TNF) receptor 1 and enhances TNF-induced apoptosis. J. Virol. 1998, 72, 3691–3697. [Google Scholar]
- Ray, R.B.; Meyer, K.; Steele, R.; Shrivastava, A.; Aggarwal, B.B.; Ray, R. Inhibition of tumor necrosis factor (TNF-alpha)-mediated apoptosis by hepatitis C virus core protein. J. Biol. Chem. 1998, 273, 2256–2259. [Google Scholar]
- Lee, S.H.; Kim, Y.K.; Kim, C.S.; Seol, S.K.; Kim, J.; Cho, S.; Song, Y.L.; Bartenschlager, R.; Jang, S.K. E2 of hepatitis C virus inhibits apoptosis. J. Immunol. 2005, 175, 8226–8235. [Google Scholar]
- Tanaka, M.; Nagano-Fujii, M.; Deng, L.; Ishido, S.; Sada, K.; Hotta, H. Single-point mutations of hepatitis C virus NS3 that impair p53 interaction and anti-apoptotic activity of NS3. Biochem. Biophys. Res. Commun. 2006, 340, 792–799. [Google Scholar] [CrossRef]
- Lan, K.H.; Sheu, M.L.; Hwang, S.J.; Yen, S.H.; Chen, S.Y.; Wu, J.C.; Wang, Y.J.; Kato, N.; Omata, M.; Chang, F.Y.; Lee, S.D. HCV NS5A interacts with p53 and inhibits p53-mediated apoptosis. Oncogene 2002, 21, 4801–4811. [Google Scholar]
- Deng, L.; Adachi, T.; Kitayama, K.; Bungyoku, Y.; Kitazawa, S.; Ishido, S.; Shoji, I.; Hotta, H. Hepatitis C virus infection induces apoptosis through a Bax-triggered, mitochondrion-mediated, caspase 3-dependent pathway. J. Virol. 2008, 82, 10375–10385. [Google Scholar] [CrossRef]
- Deng, Z.; Yan, H.; Hu, J.; Zhang, S.; Peng, P.; Liu, Q.; Guo, D. Hepatitis C virus sensitizes host cells to TRAIL-induced apoptosis by up-regulating DR4 and DR5 via a MEK1-dependent pathway. PLoS One 2012, 7, e37700. [Google Scholar]
- Zhu, H.; Dong, H.; Eksioglu, E.; Hemming, A.; Cao, M.; Crawford, J.M.; Nelson, D.R.; Liu, C. Hepatitis C virus triggers apoptosis of a newly developed hepatoma cell line through antiviral defense system. Gastroenterology 2007, 133, 1649–1659. [Google Scholar] [CrossRef]
- Johnson, D.G.; Walker, C.L. Cyclins and cell cycle checkpoints. Annu. Rev. Pharmacol. Toxicol. 1999, 39, 295–312. [Google Scholar]
- Zhao, R.Y.; Elder, R.T. Viral infections and cell cycle G2/M regulation. Cell Res. 2005, 15, 143–149. [Google Scholar] [CrossRef]
- Sanchez, V.; Spector, D.H. Subversion of cell cycle regulatory pathways. Curr. Top. Microbiol. Immunol. 2008, 325, 243–262. [Google Scholar]
- Izumi, T.; Io, K.; Matsui, M.; Shirakawa, K.; Shinohara, M.; Nagai, Y.; Kawahara, M.; Kobayashi, M.; Kondoh, H.; Misawa, N.; Koyanagi, Y.; Uchiyama, T.; Takaori-Kondo, A. HIV-1 viral infectivity factor interacts with TP53 to induce G2 cell cycle arrest and positively regulate viral replication. Proc. Natl. Acad. Sci. USA 2010, 107, 20798–20803. [Google Scholar]
- Hassan, M.; Ghozlan, H.; Abdel-Kader, O. Activation of RB/E2F signaling pathway is required for the modulation of hepatitis C virus core protein-induced cell growth in liver and non-liver cells. Cell Signal. 2004, 16, 1375–1385. [Google Scholar] [CrossRef]
- Munakata, T.; Nakamura, M.; Liang, Y.; Li, K.; Lemon, S.M. Down-regulation of the retinoblastoma tumor suppressor by the hepatitis C virus NS5B RNA-dependent RNA polymerase. Proc. Natl. Acad. Sci. USA 2005, 102, 18159–18164. [Google Scholar]
- Munakata, T.; Liang, Y.; Kim, S.; McGivern, D.R.; Huibregtse, J.; Nomoto, A.; Lemon, S.M. Hepatitis C virus induces E6AP-dependent degradation of the retinoblastoma protein. PLoS Pathog. 2007, 3, 1335–1347. [Google Scholar]
- McGivern, D.R.; Villanueva, R.A.; Chinnaswamy, S.; Kao, C.C.; Lemon, S.M. Impaired replication of hepatitis C virus containing mutations in a conserved NS5B retinoblastoma protein-binding motif. J. Virol. 2009, 83, 7422–7433. [Google Scholar]
- Machida, K.; Liu, J.C.; McNamara, G.; Levine, A.; Duan, L.; Lai, M.M. Hepatitis C virus causes uncoupling of mitotic checkpoint and chromosomal polyploidy through the Rb pathway. J. Virol. 2009, 83, 12590–12600. [Google Scholar]
- Kannan, R.P.; Hensley, L.L.; Evers, L.E.; Lemon, S.M.; McGivern, D.R. Hepatitis C virus infection causes cell cycle arrest at the level of initiation of mitosis. J. Virol. 2011, 85, 7989–8001. [Google Scholar]
- Lai, C.K.; Jeng, K.S.; Machida, K.; Cheng, Y.S.; Lai, M.M. Hepatitis C virus NS3/4A protein interacts with ATM, impairs DNA repair and enhances sensitivity to ionizing radiation. Virology 2008, 370, 295–309. [Google Scholar]
- Ariumi, Y.; Kuroki, M.; Dansako, H.; Abe, K.; Ikeda, M.; Wakita, T.; Kato, N. The DNA damage sensors ataxia-telangiectasia mutated kinase and checkpoint kinase 2 are required for hepatitis C virus RNA replication. J. Virol. 2008, 82, 9639–9646. [Google Scholar]
- Machida, K.; McNamara, G.; Cheng, K.T.; Huang, J.; Wang, C.H.; Comai, L.; Ou, J.H.; Lai, M.M. Hepatitis C virus inhibits DNA damage repair through reactive oxygen and nitrogen species and by interfering with the ATM-NBS1/Mre11/Rad50 DNA repair pathway in monocytes and hepatocytes. J. Immunol. 2010, 185, 6985–6998. [Google Scholar] [CrossRef]
- Dhillon, A.S.; Hagan, S.; Rath, O.; Kolch, W. MAP kinase signalling pathways in cancer. Oncogene 2007, 26, 3279–3290. [Google Scholar] [CrossRef]
- Tan, S.L.; Nakao, H.; He, Y.; Vijaysri, S.; Neddermann, P.; Jacobs, B.L.; Mayer, B.J.; Katze, M.G. NS5A, a nonstructural protein of hepatitis C virus, binds growth factor receptor-bound protein 2 adaptor protein in a Src homology 3 domain/ligand-dependent manner and perturbs mitogenic signaling. Proc. Natl. Acad. Sci. USA 1999, 96, 5533–5538. [Google Scholar]
- Georgopoulou, U.; Caravokiri, K.; Mavromara, P. Suppression of the ERK1/2 signaling pathway from HCV NS5A protein expressed by herpes simplex recombinant viruses. Arch. Virol. 2003, 148, 237–251. [Google Scholar] [CrossRef]
- Macdonald, A.; Crowder, K.; Street, A.; McCormick, C.; Saksela, K.; Harris, M. The hepatitis C virus non-structural NS5A protein inhibits activating protein-1 function by perturbing ras-ERK pathway signaling. J. Biol. Chem. 2003, 278, 17775–17784. [Google Scholar]
- He, Y.; Tan, S.L.; Tareen, S.U.; Vijaysri, S.; Langland, J.O.; Jacobs, B.L.; Katze, M.G. Regulation of mRNA translation and cellular signaling by hepatitis C virus nonstructural protein NS5A. J. Virol. 2001, 75, 5090–5098. [Google Scholar]
- He, Y.; Yan, W.; Coito, C.; Li, Y.; Gale, M., Jr.; Katze, M.G. The regulation of hepatitis C virus (HCV) internal ribosome-entry site-mediated translation by HCV replicons and nonstructural proteins. J. Gen. Virol. 2003, 84, 535–543. [Google Scholar] [CrossRef]
- Liu, Q.; Bhat, R.A.; Prince, A.M.; Zhang, P. The hepatitis C virus NS2 protein generated by NS2-3 autocleavage is required for NS5A phosphorylation. Biochem. Biophys. Res. Commun. 1999, 254, 572–577. [Google Scholar] [CrossRef]
- Park, K.J.; Choi, S.H.; Choi, D.H.; Park, J.M.; Yie, S.W.; Lee, S.Y.; Hwang, S.B. 1Hepatitis C virus NS5A protein modulates c-Jun N-terminal kinase through interaction with tumor necrosis factor receptor-associated factor 2. J. Biol. Chem. 2003, 278, 30711–30718. [Google Scholar]
- He, Y.; Nakao, H.; Tan, S.L.; Polyak, S.J.; Neddermann, P.; Vijaysri, S.; Jacobs, B.L.; Katze, M.G. Subversion of cell signaling pathways by hepatitis C virus nonstructural 5A protein via interaction with Grb2 and P85 phosphatidylinositol 3-kinase. J. Virol. 2002, 76, 9207–9217. [Google Scholar] [CrossRef]
- Street, A.; Macdonald, A.; Crowder, K.; Harris, M. The Hepatitis C virus NS5A protein activates a phosphoinositide 3-kinase-dependent survival signaling cascade. J. Biol. Chem. 2004, 279, 12232–12241. [Google Scholar]
- Oyadomari, S.; Harding, H.P.; Zhang, Y.; Oyadomari, M.; Ron, D. Dephosphorylation of translation initiation factor 2alpha enhances glucose tolerance and attenuates hepatosteatosis in mice. Cell Metab. 2008, 7, 520–532. [Google Scholar]
- Sun, L.P.; Li, L.; Goldstein, J.L.; Brown, M.S. Insig required for sterol-mediated inhibition of Scap/SREBP binding to COPII proteins in vitro. J. Biol. Chem. 2005, 280, 26483–26490. [Google Scholar] [CrossRef]
- Sun, L.P.; Seemann, J.; Goldstein, J.L.; Brown, M.S. Sterol-regulated transport of SREBPs from endoplasmic reticulum to Golgi: Insig renders sorting signal in Scap inaccessible to COPII proteins. Proc. Natl. Acad. Sci. USA 2007, 104, 6519–6526. [Google Scholar]
- Brodsky, J.L.; Fisher, E.A. The many intersecting pathways underlying apolipoprotein B secretion and degradation. Trends Endocrinol. Metab. 2008, 19, 254–259. [Google Scholar] [CrossRef]
- Ota, T.; Gayet, C.; Ginsberg, H.N. Inhibition of apolipoprotein B100 secretion by lipid-induced hepatic endoplasmic reticulum stress in rodents. J. Clin. Invest. 2008, 118, 316–332. [Google Scholar]
- Kodama, Y.; Brenner, D.A. c-Jun N-terminal kinase signaling in the pathogenesis of nonalcoholic fatty liver disease: Multiple roles in multiple steps. Hepatology 2009, 49, 6–8. [Google Scholar] [CrossRef]
- Kodama, Y.; Kisseleva, T.; Iwaisako, K.; Miura, K.; Taura, K.; De Minicis, S.; Osterreicher, C.H.; Schnabl, B.; Seki, E.; Brenner, D.A. c-Jun N-terminal kinase-1 from hematopoietic cells mediates progression from hepatic steatosis to steatohepatitis and fibrosis in mice. Gastroenterology 2009, 137, 1467–1477, e1465.. [Google Scholar]
- Ozcan, U.; Cao, Q.; Yilmaz, E.; Lee, A.H.; Iwakoshi, N.N.; Ozdelen, E.; Tuncman, G.; Gorgun, C.; Glimcher, L.H.; Hotamisligil, G.S. Endoplasmic reticulum stress links obesity, insulin action, and type 2 diabetes. Science 2004, 306, 457–461. [Google Scholar]
- Fernandez, P.M.; Tabbara, S.O.; Jacobs, L.K.; Manning, F.C.; Tsangaris, T.N.; Schwartz, A.M.; Kennedy, K.A.; Patierno, S.R. Overexpression of the glucose-regulated stress gene GRP78 in malignant but not benign human breast lesions. Breast Cancer Res. Treat. 2000, 59, 15–26. [Google Scholar] [CrossRef]
- Shuda, M.; Kondoh, N.; Imazeki, N.; Tanaka, K.; Okada, T.; Mori, K.; Hada, A.; Arai, M.; Wakatsuki, T.; Matsubara, O.; Yamamoto, N.; Yamamoto, M. Activation of the ATF6, XBP1 and grp78 genes in human hepatocellular carcinoma: a possible involvement of the ER stress pathway in hepatocarcinogenesis. J. Hepatol. 2003, 38, 605–614. [Google Scholar]
- Lee, A.S. GRP78 induction in cancer: therapeutic and prognostic implications. Cancer Res. 2007, 67, 3496–3499. [Google Scholar] [CrossRef]
- Carrasco, D.R.; Sukhdeo, K.; Protopopova, M.; Sinha, R.; Enos, M.; Carrasco, D.E.; Zheng, M.; Mani, M.; Henderson, J.; Pinkus, G.S.; Munshi, N.; Horner, J.; Ivanova, E.V.; Protopopov, A.; Anderson, K.C.; Tonon, G.; DePinho, R.A. The differentiation and stress response factor XBP-1 drives multiple myeloma pathogenesis. Cancer Cell 2007, 11, 349–360. [Google Scholar] [CrossRef]
- Halliwell, B. Oxidative stress and cancer: have we moved forward? Biochem. J. 2007, 401, 1–11. [Google Scholar] [CrossRef]
- Jaeschke, H. Reactive oxygen and mechanisms of inflammatory liver injury: Present concepts. J. Gastroenterol. Hepatol. 2011, 26 Suppl 1, 173–179. [Google Scholar] [CrossRef]
- Singh, R.; Kaushik, S.; Wang, Y.; Xiang, Y.; Novak, I.; Komatsu, M.; Tanaka, K.; Cuervo, A.M.; Czaja, M.J. Autophagy regulates lipid metabolism. Nature 2009, 458, 1131–1135. [Google Scholar]
- Singh, R.; Xiang, Y.; Wang, Y.; Baikati, K.; Cuervo, A.M.; Luu, Y.K.; Tang, Y.; Pessin, J.E.; Schwartz, G.J.; Czaja, M.J. Autophagy regulates adipose mass and differentiation in mice. J. Clin. Invest. 2009, 119, 3329–3339. [Google Scholar]
- Kimmelman, A.C. The dynamic nature of autophagy in cancer. Genes Dev. 2011, 25, 1999–2010. [Google Scholar] [CrossRef]
- Balaburski, G.M.; Hontz, R.D.; Murphy, M.E. p53 and ARF: unexpected players in autophagy. Trends Cell Biol. 2010, 20, 363–369. [Google Scholar] [CrossRef]
- Degtyarev, M.; De Maziere, A.; Orr, C.; Lin, J.; Lee, B.B.; Tien, J.Y.; Prior, W.W.; van Dijk, S.; Wu, H.; Gray, D.C.; Davis, D.P.; Stern, H.M.; Murray, L.J.; Hoeflich, K.P.; Klumperman, J.; Friedman, L.S.; Lin, K. Akt inhibition promotes autophagy and sensitizes PTEN-null tumors to lysosomotropic agents. J. Cell. Biol. 2008, 183, 101–116. [Google Scholar] [CrossRef]
- Qu, X.; Yu, J.; Bhagat, G.; Furuya, N.; Hibshoosh, H.; Troxel, A.; Rosen, J.; Eskelinen, E.L.; Mizushima, N.; Ohsumi, Y.; Cattoretti, G.; Levine, B. Promotion of tumorigenesis by heterozygous disruption of the beclin 1 autophagy gene. J. Clin. Invest. 2003, 112, 1809–1820. [Google Scholar]
- Yue, Z.; Jin, S.; Yang, C.; Levine, A.J.; Heintz, N. Beclin 1, an autophagy gene essential for early embryonic development, is a haploinsufficient tumor suppressor. Pro. Natl. Acad. Sci. USA 2003, 100, 15077–15082. [Google Scholar]
- Takamura, A.; Komatsu, M.; Hara, T.; Sakamoto, A.; Kishi, C.; Waguri, S.; Eishi, Y.; Hino, O.; Tanaka, K.; Mizushima, N. Autophagy-deficient mice develop multiple liver tumors. Genes Dev 2011, 25, 795–800. [Google Scholar] [CrossRef]
- Komatsu, M.; Kurokawa, H.; Waguri, S.; Taguchi, K.; Kobayashi, A.; Ichimura, Y.; Sou, Y.S.; Ueno, I.; Sakamoto, A.; Tong, K.I.; Kim, M.; Nishito, Y.; Iemura, S.; Natsume, T.; Ueno, T.; Kominami, E.; Motohashi, H.; Tanaka, K.; Yamamoto, M. The selective autophagy substrate p62 activates the stress responsive transcription factor Nrf2 through inactivation of Keap1. Nat. Cell Biol. 2010, 12, 213–223. [Google Scholar]
- Inami, Y.; Waguri, S.; Sakamoto, A.; Kouno, T.; Nakada, K.; Hino, O.; Watanabe, S.; Ando, J.; Iwadate, M.; Yamamoto, M.; Lee, M.S.; Tanaka, K.; Komatsu, M. Persistent activation of Nrf2 through p62 in hepatocellular carcinoma cells. J. Cell Biol 2011, 193, 275–284. [Google Scholar] [CrossRef]
- Degenhardt, K.; Mathew, R.; Beaudoin, B.; Bray, K.; Anderson, D.; Chen, G.; Mukherjee, C.; Shi, Y.; Gelinas, C.; Fan, Y.; Nelson, D.A.; Jin, S.; White, E. Autophagy promotes tumor cell survival and restricts necrosis, inflammation, and tumorigenesis. Cancer Cell 2006, 10, 51–64. [Google Scholar] [CrossRef]
- Amaravadi, R.K.; Thompson, C.B. The roles of therapy-induced autophagy and necrosis in cancer treatment. Clin. Cancer Res. 2007, 13, 7271–7279. [Google Scholar] [CrossRef]
- Amaravadi, R.K.; Yu, D.; Lum, J.J.; Bui, T.; Christophorou, M.A.; Evan, G.I.; Thomas-Tikhonenko, A.; Thompson, C.B. Autophagy inhibition enhances therapy-induced apoptosis in a Myc-induced model of lymphoma. J. Clin. Invest. 2007, 117, 326–336. [Google Scholar] [CrossRef]
- Maher, J.J.; Scott, M.K.; Saito, J.M.; Burton, M.C. Adenovirus-mediated expression of cytokine-induced neutrophil chemoattractant in rat liver induces a neutrophilic hepatitis. Hepatology 1997, 25, 624–630. [Google Scholar] [CrossRef]
- Lawson, J.A.; Fisher, M.A.; Simmons, C.A.; Farhood, A.; Jaeschke, H. Parenchymal cell apoptosis as a signal for sinusoidal sequestration and transendothelial migration of neutrophils in murine models of endotoxin and Fas-antibody-induced liver injury. Hepatology 1998, 28, 761–767. [Google Scholar] [CrossRef]
- Faouzi, S.; Burckhardt, B.E.; Hanson, J.C.; Campe, C.B.; Schrum, L.W.; Rippe, R.A.; Maher, J.J. Anti-Fas induces hepatic chemokines and promotes inflammation by an NF-kappa B-independent, caspase-3-dependent pathway. J. Biol. Chem. 2001, 276, 49077–49082. [Google Scholar]
- Ogasawara, J.; Watanabe-Fukunaga, R.; Adachi, M.; Matsuzawa, A.; Kasugai, T.; Kitamura, Y.; Itoh, N.; Suda, T.; Nagata, S. Lethal effect of the anti-Fas antibody in mice. Nature 1993, 364, 806–809. [Google Scholar] [CrossRef]
- Canbay, A.; Taimr, P.; Torok, N.; Higuchi, H.; Friedman, S.; Gores, G.J. Apoptotic body engulfment by a human stellate cell line is profibrogenic. Lab. Invest. 2003, 83, 655–663. [Google Scholar]
- Kurosaka, K.; Watanabe, N.; Kobayashi, Y. Production of proinflammatory cytokines by resident tissue macrophages after phagocytosis of apoptotic cells. Cell. Immunol. 2001, 211, 1–7. [Google Scholar] [CrossRef]
- Takehara, T.; Liu, X.; Fujimoto, J.; Friedman, S.L.; Takahashi, H. Expression and role of Bcl-xL in human hepatocellular carcinomas. Hepatology 2001, 34, 55–61. [Google Scholar]
- Sieghart, W.; Losert, D.; Strommer, S.; Cejka, D.; Schmid, K.; Rasoul-Rockenschaub, S.; Bodingbauer, M.; Crevenna, R.; Monia, B.P.; Peck-Radosavljevic, M.; Wacheck, V. Mcl-1 overexpression in hepatocellular carcinoma: a potential target for antisense therapy. J. Hepatol. 2006, 44, 151–157. [Google Scholar]
- Beerheide, W.; Tan, Y.J.; Teng, E.; Ting, A.E.; Jedpiyawongse, A.; Srivatanakul, P. Downregulation of proapoptotic proteins Bax and Bcl-X(S) in p53 overexpressing hepatocellular carcinomas. Biochem. Biophys. Res. Commun. 2000, 273, 54–61. [Google Scholar] [CrossRef]
- Duan, X.X.; Ou, J.S.; Li, Y.; Su, J.J.; Ou, C.; Yang, C.; Yue, H.F.; Ban, K.C. Dynamic expression of apoptosis-related genes during development of laboratory hepatocellular carcinoma and its relation to apoptosis. World J. Gastroenterol. 2005, 11, 4740–4744. [Google Scholar]
- Ito, T.; Shiraki, K.; Sugimoto, K.; Yamanaka, T.; Fujikawa, K.; Ito, M.; Takase, K.; Moriyama, M.; Kawano, H.; Hayashida, M.; Nakano, T.; Suzuki, A. Survivin promotes cell proliferation in human hepatocellular carcinoma. Hepatology 2000, 31, 1080–1085. [Google Scholar] [CrossRef]
- Ye, C.P.; Qiu, C.Z.; Huang, Z.X.; Su, Q.C.; Zhuang, W.; Wu, R.L.; Li, X.F. Relationship between survivin expression and recurrence, and prognosis in hepatocellular carcinoma. World J. Gastroenterol. 2007, 13, 6264–6268. [Google Scholar] [CrossRef]
- Breuhahn, K.; Longerich, T.; Schirmacher, P. Dysregulation of growth factor signaling in human hepatocellular carcinoma. Oncogene 2006, 25, 3787–3800. [Google Scholar] [CrossRef]
- Bunney, T.D.; Katan, M. Phosphoinositide signalling in cancer: beyond PI3K and PTEN. Nat. Rev. Cancer 2010, 10, 342–352. [Google Scholar] [CrossRef]
- Farazi, P.A.; DePinho, R.A. Hepatocellular carcinoma pathogenesis: from genes to environment. Nat. Rev. Cancer 2006, 6, 674–687. [Google Scholar] [CrossRef]
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Ke, P.-Y.; Chen, S.S.-L. Hepatitis C Virus and Cellular Stress Response: Implications to Molecular Pathogenesis of Liver Diseases. Viruses 2012, 4, 2251-2290. https://doi.org/10.3390/v4102251
Ke P-Y, Chen SS-L. Hepatitis C Virus and Cellular Stress Response: Implications to Molecular Pathogenesis of Liver Diseases. Viruses. 2012; 4(10):2251-2290. https://doi.org/10.3390/v4102251
Chicago/Turabian StyleKe, Po-Yuan, and Steve S.-L. Chen. 2012. "Hepatitis C Virus and Cellular Stress Response: Implications to Molecular Pathogenesis of Liver Diseases" Viruses 4, no. 10: 2251-2290. https://doi.org/10.3390/v4102251
APA StyleKe, P.-Y., & Chen, S. S.-L. (2012). Hepatitis C Virus and Cellular Stress Response: Implications to Molecular Pathogenesis of Liver Diseases. Viruses, 4(10), 2251-2290. https://doi.org/10.3390/v4102251