Hepatitis C Virus P7—A Viroporin Crucial for Virus Assembly and an Emerging Target for Antiviral Therapy

Abstract
: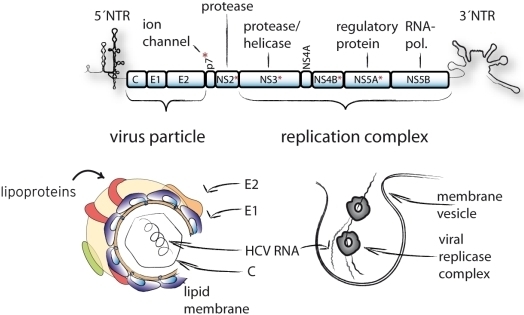
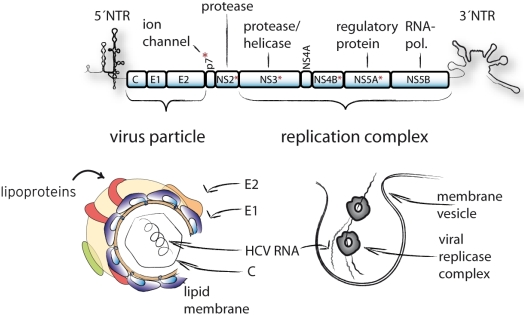{kind=link}
{kind=link}
{kind=link}
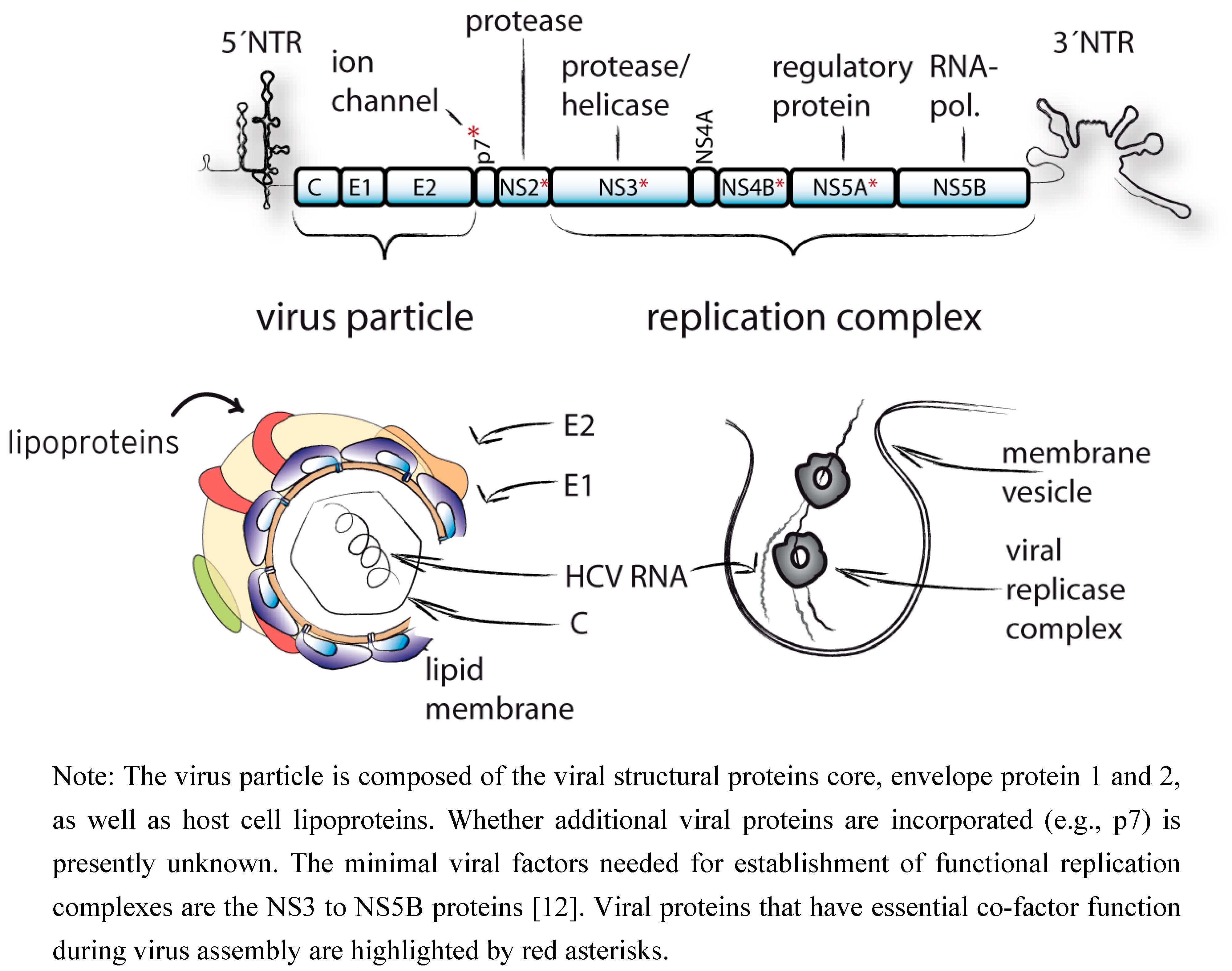{kind=link}
{kind=link}
{kind=link}
1. Introduction
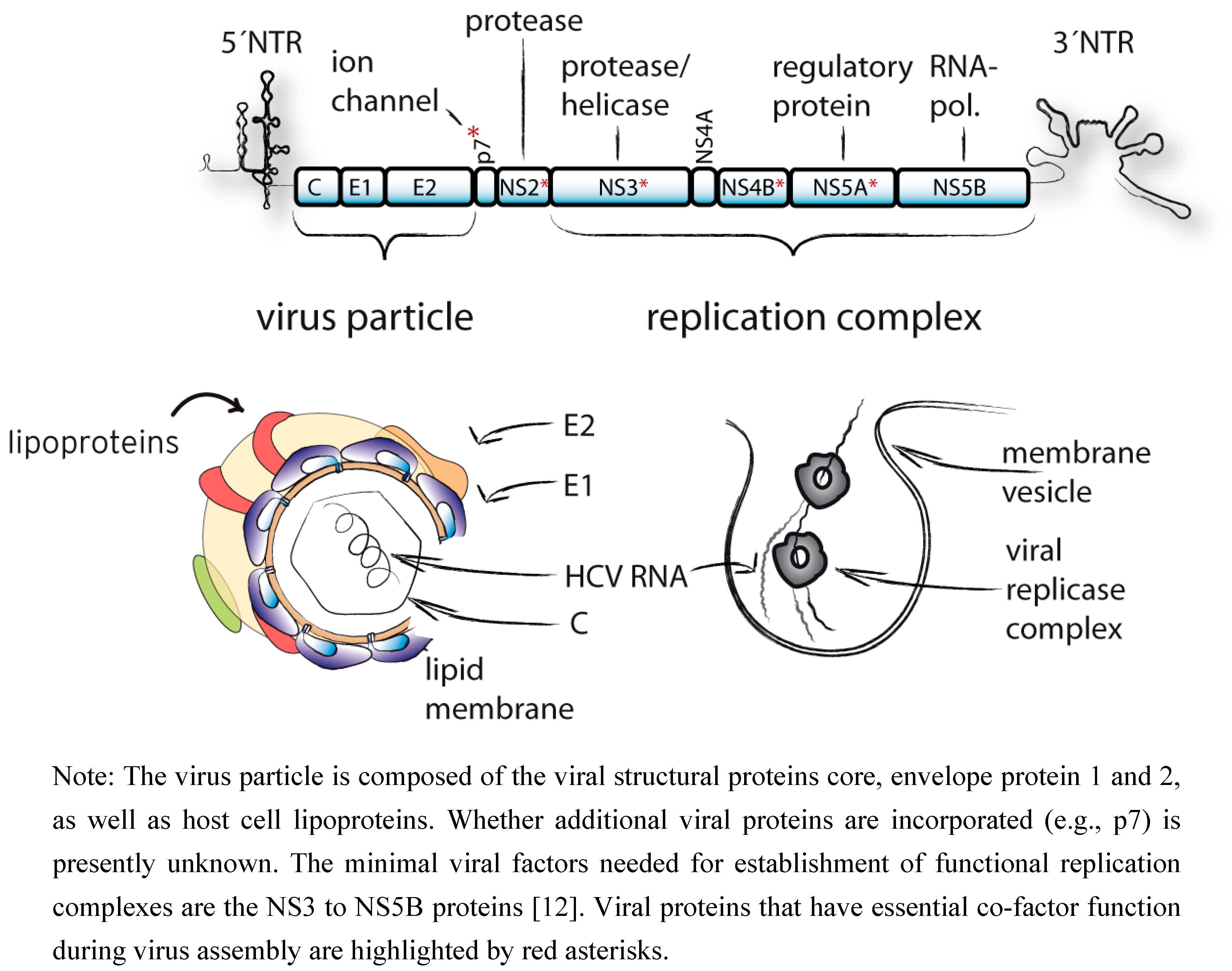
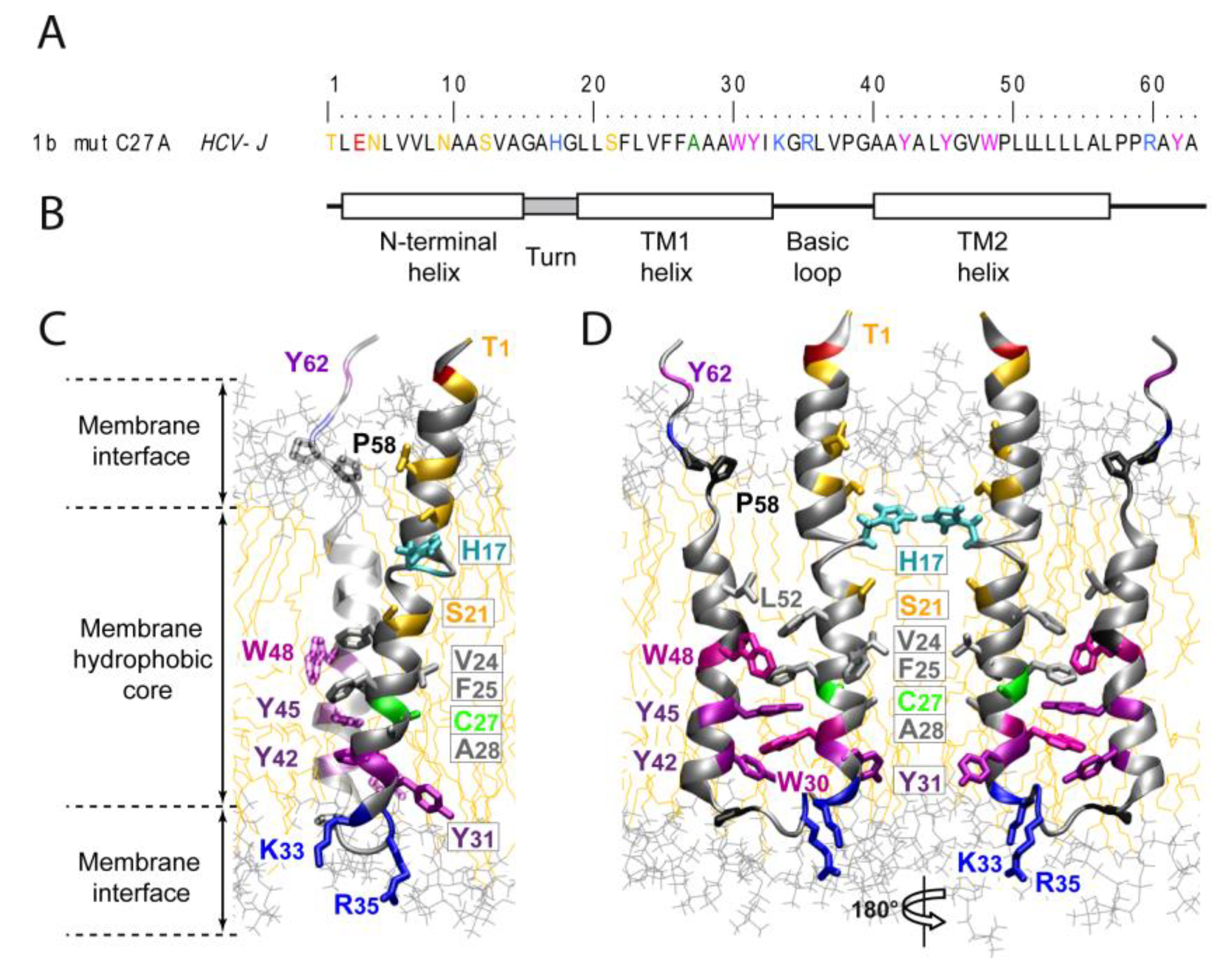
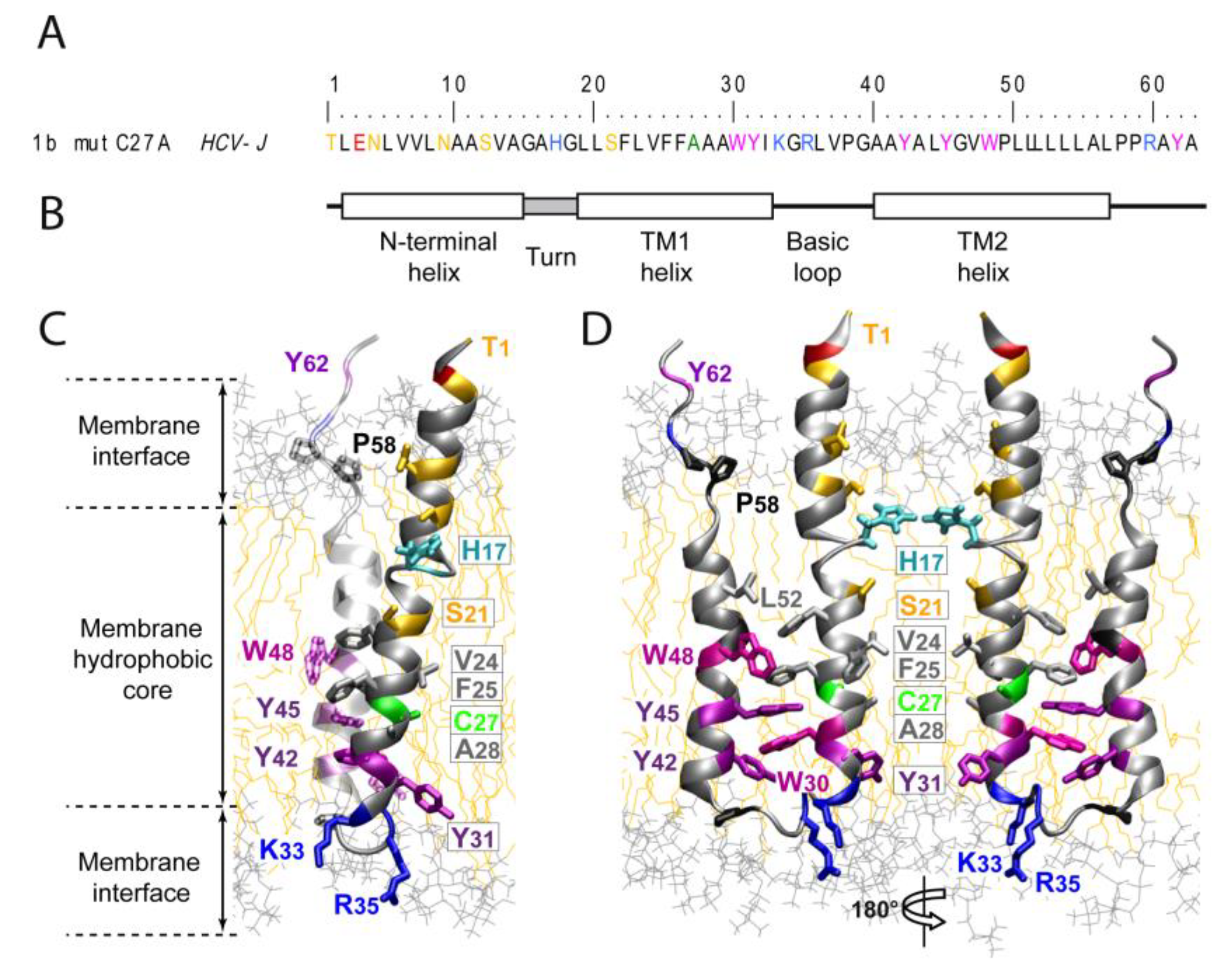
2. Structure and Function of HCV P7
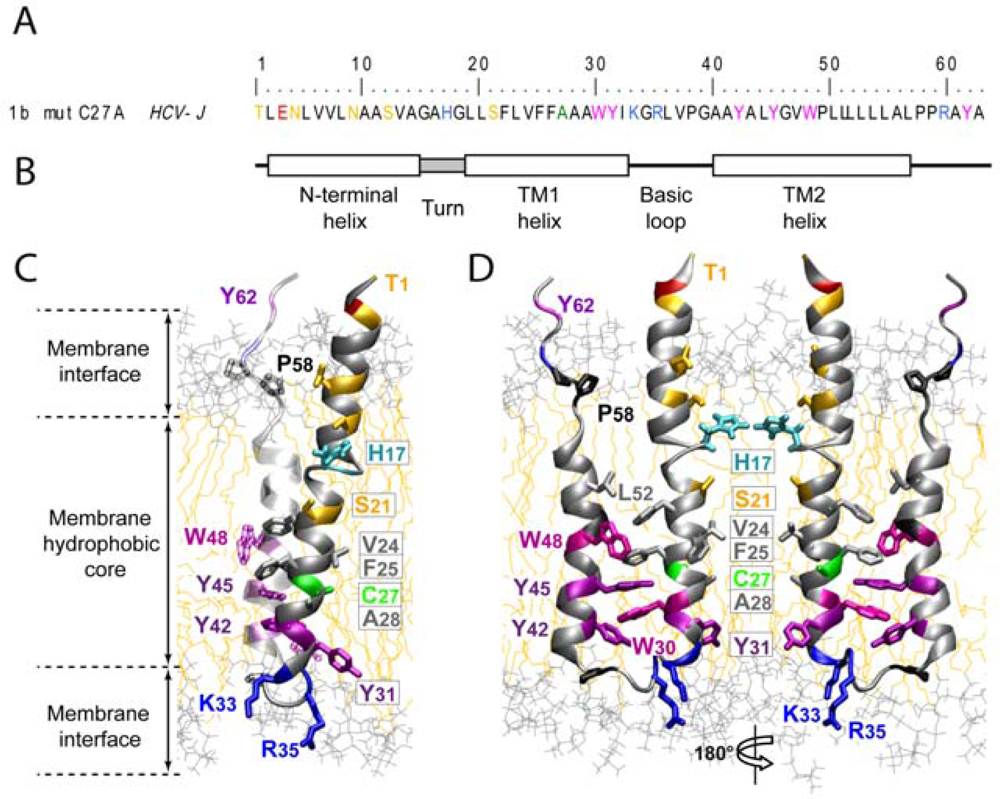
2.1. P7 Topology and Structure

2.2. P7—A Viroporin and True Ion Channel?
2.3. Subcellular Localization of P7
2.4. Clues of P7 Function Based on Animal and Tissue Culture Experiments
3. P7-Based Antiviral Strategies
3.1. Amantadine
3.2. Amilorides
3.3. Iminosugar Derivatives
3.4. BIT225
4. Conclusion and Outlook
Acknowledgements
References and Notes
- Alter, M.J. Epidemiology of hepatitis C virus infection. World J. Gastroenterol. 2007, 13, 2436–2441. [Google Scholar] [CrossRef] [PubMed]
- Lauer, G.M.; Walker, B.D. Hepatitis C virus infection. N. Engl. J. Med. 2001, 345, 41–52. [Google Scholar] [CrossRef] [PubMed]
- Brown, R.S. Hepatitis C and liver transplantation. Nature 2005, 436, 973–978. [Google Scholar] [CrossRef] [PubMed]
- Simmonds, P.; Bukh, J.; Combet, C.; Deleage, G.; Enomoto, N.; Feinstone, S.; Halfon, P.; Inchauspe, G.; Kuiken, C.; Maertens, G.; Mizokami, M.; Murphy, D.G.; Okamoto, H.; Pawlotsky, J.M.; Penin, F.; Sablon, E.; Shin, I.T.; Stuyver, L.J.; Thiel, H.J.; Viazov, S.; Weiner, A.J.; Widell, A. Consensus proposals for a unified system of nomenclature of hepatitis C virus genotypes. Hepatology 2005, 42, 962–973. [Google Scholar] [CrossRef] [PubMed]
- Manns, M.P.; Wedemeyer, H.; Cornberg, M. Treating viral hepatitis C: Efficacy, side effects, and complications. Gut 2006, 55, 1350–1359. [Google Scholar] [CrossRef]
- Sarrazin, C.; Zeuzem, S. Resistance to direct antiviral agents in patients with hepatitis C virus infection. Gastroenterology 2010, 138, 447–462. [Google Scholar] [CrossRef]
- Bartenschlager, R.; Frese, M.; Pietschmann, T. Novel insights into hepatitis C virus replication and persistence. Advan. Virus Res. 2004, 63, 71–180. [Google Scholar]
- Grakoui, A.; Wychowski, C.; Lin, C.; Feinstone, S.M.; Rice, C.M. Expression and identification of hepatitis C virus polyprotein cleavage products. J. Virol. 1993, 67, 1385–1395. [Google Scholar] [CrossRef]
- Hijikata, M.; Kato, N.; Ootsuyama, Y.; Nakagawa, M.; Shimotohno, K. Gene mapping of the putative structural region of the hepatitis C virus genome by in vitro processing analysis. Proc. Natl. Acad. Sci. U. S. A. 1991, 88, 5547–5551. [Google Scholar] [CrossRef]
- McLauchlan, J.; Lemberg, M.K.; Hope, G.; Martoglio, B. Intramembrane proteolysis promotes trafficking of hepatitis C virus core protein to lipid droplets. Embo. J. 2002, 21, 3980–3988. [Google Scholar] [CrossRef]
- Gosert, R.; Egger, D.; Lohmann, V.; Bartenschlager, R.; Blum, H.E.; Bienz, K.; Moradpour, D. Identification of the hepatitis C virus RNA replication complex in Huh-7 cells harboring subgenomic replicons. J. Virol. 2003, 77, 5487–5492. [Google Scholar] [CrossRef] [PubMed]
- Lohmann, V.; Korner, F.; Koch, J.; Herian, U.; Theilmann, L.; Bartenschlager, R. Replication of subgenomic hepatitis C virus RNAs in a hepatoma cell line. Science 1999, 285, 110–113. [Google Scholar] [CrossRef] [PubMed]
- Bartosch, B.; Dubuisson, J.; Cosset, F.L. Infectious hepatitis C virus pseudo-particles containing functional E1-E2 envelope protein complexes. J. Exp. Med. 2003, 197, 633–642. [Google Scholar] [CrossRef] [PubMed]
- Hsu, M.; Zhang, J.; Flint, M.; Logvinoff, C.; Cheng-Mayer, C.; Rice, C.M.; McKeating, J.A. Hepatitis C virus glycoproteins mediate pH-dependent cell entry of pseudotyped retroviral particles. Proc. Natl. Acad. Sci. U. S. A. 2003, 100, 7271–7276. [Google Scholar] [CrossRef]
- Lindenbach, B.D.; Evans, M.J.; Syder, A.J.; Wolk, B.; Tellinghuisen, T.L.; Liu, C.C.; Maruyama, T.; Hynes, R.O.; Burton, D.R.; McKeating, J.A.; Rice, C.M. Complete replication of hepatitis C virus in cell culture. Science 2005, 309, 623–626. [Google Scholar] [CrossRef]
- Wakita, T.; Pietschmann, T.; Kato, T.; Date, T.; Miyamoto, M.; Zhao, Z.; Murthy, K.; Habermann, A.; Krausslich, H.G.; Mizokami, M.; Bartenschlager, R.; Liang, T.J. Production of infectious hepatitis C virus in tissue culture from a cloned viral genome. Nat. Med. 2005, 11, 791–796. [Google Scholar] [CrossRef]
- Zhong, J.; Gastaminza, P.; Cheng, G.; Kapadia, S.; Kato, T.; Burton, D.R.; Wieland, S.F.; Uprichard, S.L.; Wakita, T.; Chisari, F.V. Robust hepatitis C virus infection in vitro. Proc. Natl. Acad. Sci. U. S. A. 2005, 102, 9294–9299. [Google Scholar] [CrossRef]
- Bartenschlager, R.; Sparacio, S. Hepatitis C virus molecular clones and their replication capacity in vivo and in cell culture. Virus Res. 2007, 127, 195–207. [Google Scholar] [CrossRef]
- Chang, K.S.; Jiang, J.; Cai, Z.; Luo, G. Human apolipoprotein e is required for infectivity and production of hepatitis C virus in cell culture. J. Virol. 2007, 81, 13783–13793. [Google Scholar] [CrossRef]
- Gastaminza, P.; Cheng, G.; Wieland, S.; Zhong, J.; Liao, W.; Chisari, F.V. Cellular determinants of hepatitis C virus assembly, maturation, degradation, and secretion. J. Virol. 2008, 82, 2120–2129. [Google Scholar] [CrossRef]
- Huang, H.; Sun, F.; Owen, D.M.; Li, W.; Chen, Y.; Gale, M., Jr.; Ye, J. Hepatitis C virus production by human hepatocytes dependent on assembly and secretion of very low-density lipoproteins. Proc. Natl. Acad. Sci. U. S. A. 2007, 104, 5848–5853. [Google Scholar] [CrossRef] [PubMed]
- Appel, N.; Zayas, M.; Miller, S.; Krijnse-Locker, J.; Schaller, T.; Friebe, P.; Kallis, S.; Engel, U.; Bartenschlager, R. Essential role of domain III of nonstructural protein 5A for hepatitis C virus infectious particle assembly. PLoS Pathog. 2008, 4, e1000035. [Google Scholar] [CrossRef] [PubMed]
- Jones, D.M.; Patel, A.H.; Targett-Adams, P.; McLauchlan, J. The hepatitis C virus NS4B protein can trans-complement viral RNA replication and modulates production of infectious virus. J. Virol. 2009, 83, 2163–2177. [Google Scholar] [CrossRef] [PubMed]
- Phan, T.; Beran, R.K.; Peters, C.; Lorenz, I.C.; Lindenbach, B.D. Hepatitis C virus NS2 protein contributes to virus particle assembly via opposing epistatic interactions with the E1-E2 glycoprotein and NS3-NS4A enzyme complexes. J. Virol. 2009, 83, 8379–8395. [Google Scholar] [CrossRef] [PubMed]
- Tellinghuisen, T.L.; Foss, K.L.; Treadaway, J. Regulation of hepatitis C virion production via phosphorylation of the NS5A protein. PLoS Pathog. 2008, 4, e1000032. [Google Scholar] [CrossRef] [PubMed]
- Yi, M.; Ma, Y.; Yates, J.; Lemon, S.M. Compensatory mutations in E1, p7, NS2, and NS3 enhance yields of cell culture-infectious intergenotypic chimeric hepatitis C virus. J. Virol. 2007, 81, 629–638. [Google Scholar] [CrossRef] [PubMed]
- Dubuisson, J.; Hsu, H.H.; Cheung, R.C.; Greenberg, H.B.; Russell, D.G.; Rice, C.M. Formation and intracellular localization of hepatitis C virus envelope glycoprotein complexes expressed by recombinant vaccinia and Sindbis viruses. J. Virol. 1994, 68, 6147–6160. [Google Scholar] [CrossRef]
- Lin, C.; Lindenbach, B.D.; Pragai, B.M.; McCourt, D.W.; Rice, C.M. Processing in the hepatitis C virus E2-NS2 region: Identification of p7 and two distinct E2-specific products with different C termini. J. Virol. 1994, 68, 5063–5073. [Google Scholar] [CrossRef]
- Mizushima, H.; Hijikata, M.; Asabe, S.; Hirota, M.; Kimura, K.; Shimotohno, K. Two hepatitis C virus glycoprotein E2 products with different C termini. J. Virol. 1994, 68, 6215–6222. [Google Scholar] [CrossRef]
- Carrere-Kremer, S.; Montpellier-Pala, C.; Cocquerel, L.; Wychowski, C.; Penin, F.; Dubuisson, J. Subcellular localization and topology of the p7 polypeptide of hepatitis C virus. J. Virol. 2002, 76, 3720–3730. [Google Scholar] [CrossRef]
- Isherwood, B.J.; Patel, A.H. Analysis of the processing and transmembrane topology of the E2p7 protein of hepatitis C virus. J. Gen. Virol. 2005, 86, 667–676. [Google Scholar] [CrossRef] [PubMed]
- Yamaga, A.K.; Ou, J.H. Membrane topology of the hepatitis C virus NS2 protein. J. Biol. Chem. 2002, 277, 33228–33234. [Google Scholar] [CrossRef] [PubMed]
- Griffin, S.D.; Beales, L.P.; Clarke, D.S.; Worsfold, O.; Evans, S.D.; Jaeger, J.; Harris, M.P.; Rowlands, D.J. The p7 protein of hepatitis C virus forms an ion channel that is blocked by the antiviral drug, Amantadine. FEBS Lett. 2003, 535, 34–38. [Google Scholar] [CrossRef] [PubMed]
- Clarke, D.; Griffin, S.; Beales, L.; Gelais, C.S.; Burgess, S.; Harris, M.; Rowlands, D. Evidence for the formation of a heptameric ion channel complex by the hepatitis C virus p7 protein in vitro. J. Biol. Chem. 2006, 281, 37057–37068. [Google Scholar] [CrossRef]
- Patargias, G.; Zitzmann, N.; Dwek, R.; Fischer, W.B. Protein-protein interactions: Modeling the hepatitis C virus ion channel p7. J. Med. Chem. 2006, 49, 648–655. [Google Scholar] [CrossRef]
- Luik, P.; Chew, C.; Aittoniemi, J.; Chang, J.; Wentworth, P., Jr.; Dwek, R.A.; Biggin, P.C.; Venien-Bryan, C.; Zitzmann, N. The 3-dimensional structure of a hepatitis C virus p7 ion channel by electron microscopy. Proc. Natl. Acad. Sci. U. S. A. 2009, 106, 12712–12716. [Google Scholar] [CrossRef]
- Montserret, R.; Saint, N.; Vanbelle, C.; Salvay, A.G.; Simorre, J.P.; Ebel, C.; Sapay, N.; Renisio, J.G.; Bockmann, A.; Steinmann, E.; Pietschmann, T.; Dubuisson, J.; Chipot, C.; Penin, F. NMR structure and ion channel activity of the p7 protein from hepatitis C virus. J. Biol. Chem. 2010. Available online: http://www.ncbi.nlm.nih.gov/pubmed/20667830 (accessed on 17 September 2010). [CrossRef]
- Pavlovic, D.; Neville, D.C.; Argaud, O.; Blumberg, B.; Dwek, R.A.; Fischer, W.B.; Zitzmann, N. The hepatitis C virus p7 protein forms an ion channel that is inhibited by long-alkyl-chain iminosugar derivatives. Proc. Natl. Acad. Sci. U. S. A. 2003, 100, 6104–6108. [Google Scholar] [CrossRef]
- Premkumar, A.; Wilson, L.; Ewart, G.D.; Gage, P.W. Cation-selective ion channels formed by p7 of hepatitis C virus are blocked by hexamethylene amiloride. FEBS Lett. 2004, 557, 99–103. [Google Scholar] [CrossRef]
- Chew, C.F.; Vijayan, R.; Chang, J.; Zitzmann, N.; Biggin, P.C. Determination of pore-lining residues in the hepatitis C virus p7 protein. Biophys. J. 2009, 96, L10–L12. [Google Scholar] [CrossRef]
- Haqshenas, G.; Dong, X.; Ewart, G.; Bowden, S.; Gowans, E.J. A 2a/1b full-length p7 inter-genotypic chimeric genome of hepatitis C virus is infectious in vitro. Virology 2007, 360, 17–26. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez, M.E.; Carrasco, L. Viroporins. FEBS Lett. 2003, 552, 28–34. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, T.; Orba, Y.; Okada, Y.; Sunden, Y.; Kimura, T.; Tanaka, S.; Nagashima, K.; Hall, W.W.; Sawa, H. The human polyoma JC virus agnoprotein acts as a viroporin. PLoS Pathog. 2010, 6, e1000801. [Google Scholar] [CrossRef] [PubMed]
- Chizhmakov, I.V.; Geraghty, F.M.; Ogden, D.C.; Hayhurst, A.; Antoniou, M.; Hay, A.J. Selective proton permeability and pH regulation of the influenza virus M2 channel expressed in mouse erythroleukaemia cells. J. Physiol. 1996, 494, 329–336. [Google Scholar] [CrossRef]
- Pinto, L.H.; Holsinger, L.J.; Lamb, R.A. Influenza virus M2 protein has ion channel activity. Cell 1992, 69, 517–528. [Google Scholar] [CrossRef]
- Ciampor, F.; Bayley, P.M.; Nermut, M.V.; Hirst, E.M.; Sugrue, R.J.; Hay, A.J. Evidence that the amantadine-induced, M2-mediated conversion of influenza A virus hemagglutinin to the low pH conformation occurs in an acidic trans Golgi compartment. Virology 1992, 188, 14–24. [Google Scholar] [CrossRef]
- StGelais, C.; Tuthill, T.J.; Clarke, D.S.; Rowlands, D.J.; Harris, M.; Griffin, S. Inhibition of hepatitis C virus p7 membrane channels in a liposome-based assay system. Antivir. Res. 2007, 76, 48–58. [Google Scholar] [CrossRef]
- Griffin, S.D.; Harvey, R.; Clarke, D.S.; Barclay, W.S.; Harris, M.; Rowlands, D.J. A conserved basic loop in hepatitis C virus p7 protein is required for amantadine-sensitive ion channel activity in mammalian cells but is dispensable for localization to mitochondria. J. Gen. Virol. 2004, 85, 451–461. [Google Scholar] [CrossRef]
- Koutsoudakis, G.; Kaul, A.; Steinmann, E.; Kallis, S.; Lohmann, V.; Pietschmann, T.; Bartenschlager, R. Characterization of the early steps of hepatitis C virus infection by using luciferase reporter viruses. J. Virol. 2006, 80, 5308–5320. [Google Scholar] [CrossRef]
- Tscherne, D.M.; Jones, C.T.; Evans, M.J.; Lindenbach, B.D.; McKeating, J.A.; Rice, C.M. Time- and temperature-dependent activation of hepatitis C virus for low-pH-triggered entry. J. Virol. 2006, 80, 1734–1741. [Google Scholar] [CrossRef]
- Pinto, L.H.; Dieckmann, G.R.; Gandhi, C.S.; Papworth, C.G.; Braman, J.; Shaughnessy, M.A.; Lear, J.D.; Lamb, R.A.; DeGrado, W.F. A functionally defined model for the M2 proton channel of influenza A virus suggests a mechanism for its ion selectivity. Proc. Natl. Acad. Sci. U. S. A. 1997, 94, 11301–11306. [Google Scholar] [CrossRef]
- StGelais, C.; Foster, T.L.; Verow, M.; Atkins, E.; Fishwick, C.W.; Rowlands, D.; Harris, M.; Griffin, S. Determinants of hepatitis C virus p7 ion channel function and drug sensitivity identified in vitro. J. Virol. 2009, 83, 7970–7981. [Google Scholar] [CrossRef]
- Steinmann, E.; Penin, F.; Kallis, S.; Patel, A.H.; Bartenschlager, R.; Pietschmann, T. Hepatitis C virus p7 protein is crucial for assembly and release of infectious virions. PLoS Pathog. 2007, 3, e103. [Google Scholar] [CrossRef] [PubMed]
- Meshkat, Z.; Audsley, M.; Beyer, C.; Gowans, E.J.; Haqshenas, G. Reverse genetic analysis of a putative, influenza virus M2 HXXXW-like motif in the p7 protein of hepatitis C virus. J. Viral Hepatitis 2009, 16, 187–194. [Google Scholar] [CrossRef] [PubMed]
- Unwin, N. Acetylcholine receptor channel imaged in the open state. Nature 1995, 373, 37–43. [Google Scholar] [CrossRef] [PubMed]
- Biggin, P.C.; Sansom, M.S. Channel gating: Twist to open. Curr. Biol. 2001, 11, R364–R366. [Google Scholar] [CrossRef]
- Perozo, E.; Cortes, D.M.; Cuello, L.G. Structural rearrangements underlying K+-channel activation gating. Science 1999, 285, 73–78. [Google Scholar] [CrossRef]
- Griffin, S.; Clarke, D.; McCormick, C.; Rowlands, D.; Harris, M. Signal peptide cleavage and internal targeting signals direct the hepatitis C virus p7 protein to distinct intracellular membranes. J. Virol. 2005, 79, 15525–15536. [Google Scholar] [CrossRef]
- Haqshenas, G.; Mackenzie, J.M.; Dong, X.; Gowans, E.J. Hepatitis C virus p7 protein is localized in the endoplasmic reticulum when it is encoded by a replication-competent genome. J. Gen. Virol. 2007, 88, 134–142. [Google Scholar] [CrossRef]
- Paroutis, P.; Touret, N.; Grinstein, S. The pH of the secretory pathway: Measurement, determinants, and regulation. Physiology (Bethesda) 2004, 19, 207–215. [Google Scholar] [CrossRef]
- Sakai, A.; Claire, M.S.; Faulk, K.; Govindarajan, S.; Emerson, S.U.; Purcell, R.H.; Bukh, J. The p7 polypeptide of hepatitis C virus is critical for infectivity and contains functionally important genotype-specific sequences. Proc. Natl. Acad. Sci. U. S. A. 2003, 100, 11646–11651. [Google Scholar] [CrossRef] [PubMed]
- Jones, C.T.; Murray, C.L.; Eastman, D.K.; Tassello, J.; Rice, C.M. Hepatitis C virus p7 and NS2 proteins are essential for production of infectious virus. J. Virol. 2007, 81, 8374–8383. [Google Scholar] [CrossRef] [PubMed]
- Harada, T.; Tautz, N.; Thiel, H.J. E2-p7 region of the bovine viral diarrhea virus polyprotein: Processing and functional studies. J. Virol. 2000, 74, 9498–9506. [Google Scholar] [CrossRef] [PubMed]
- Elbers, K.; Tautz, N.; Becher, P.; Stoll, D.; Rumenapf, T.; Thiel, H.J. Processing in the pestivirus E2-NS2 region: Identification of proteins p7 and E2p7. J. Virol. 1996, 70, 4131–4135. [Google Scholar] [CrossRef] [PubMed]
- Brohm, C.; Steinmann, E.; Friesland, M.; Lorenz, I.C.; Patel, A.; Penin, F.; Bartenschlager, R.; Pietschmann, T. Characterization of determinants important for hepatitis C virus p7 function in morphogenesis by using trans-complementation. J. Virol. 2009, 83, 11682–11693. [Google Scholar] [CrossRef] [PubMed]
- Carrere-Kremer, S.; Montpellier, C.; Lorenzo, L.; Brulin, B.; Cocquerel, L.; Belouzard, S.; Penin, F.; Dubuisson, J. Regulation of hepatitis C virus polyprotein processing by signal peptidase involves structural determinants at the p7 sequence junctions. J. Biol. Chem. 2004, 279, 41384–41392. [Google Scholar] [CrossRef]
- Pietschmann, T.; Kaul, A.; Koutsoudakis, G.; Shavinskaya, A.; Kallis, S.; Steinmann, E.; Abid, K.; Negro, F.; Dreux, M.; Cosset, F.L.; Bartenschlager, R. Construction and characterization of infectious intragenotypic and intergenotypic hepatitis C virus chimeras. Proc. Natl. Acad. Sci. U. S. A. 2006, 103, 7408–7413. [Google Scholar] [CrossRef]
- Murray, C.L.; Jones, C.T.; Tassello, J.; Rice, C.M. Alanine scanning of the hepatitis C virus core protein reveals numerous residues essential for production of infectious virus. J. Virol. 2007, 81, 10220–10231. [Google Scholar] [CrossRef]
- Mould, J.A.; Drury, J.E.; Frings, S.M.; Kaupp, U.B.; Pekosz, A.; Lamb, R.A.; Pinto, L.H. Permeation and activation of the M2 ion channel of influenza A virus. J. Biol. Chem. 2000, 275, 31038–31050. [Google Scholar] [CrossRef]
- Pinto, L.H.; Lamb, R.A. Understanding the mechanism of action of the anti-influenza virus drug amantadine. Trends Microbiol. 1995, 3, 271. [Google Scholar] [CrossRef]
- Tosteson, M.T.; Pinto, L.H.; Holsinger, L.J.; Lamb, R.A. Reconstitution of the influenza virus M2 ion channel in lipid bilayers. J. Membr. Biol. 1994, 142, 117–126. [Google Scholar] [CrossRef]
- Ewart, G.D.; Mills, K.; Cox, G.B.; Gage, P.W. Amiloride derivatives block ion channel activity and enhancement of virus-like particle budding caused by HIV-1 protein Vpu. Eur. Biophys. J. 2002, 31, 26–35. [Google Scholar] [CrossRef] [PubMed]
- Cady, S.D.; Schmidt-Rohr, K.; Wang, J.; Soto, C.S.; Degrado, W.F.; Hong, M. Structure of the amantadine binding site of influenza M2 proton channels in lipid bilayers. Nature 2010, 463, 689–692. [Google Scholar] [CrossRef] [PubMed]
- Smith, J.P. Treatment of chronic hepatitis C with amantadine. Dig. Dis. Sci. 1997, 42, 1681–1687. [Google Scholar] [CrossRef]
- Van Soest, H.; Van der Schaar, P.J.; Koek, G.H.; De Vries, R.A.; Van Ooteghem, N.A.; Van Hoek, B.; Drenth, J.P.; Vrolijk, J.M.; Lieverse, R.J.; Houben, P.; Van der Sluys Veer, A.; Siersema, P.D.; Schipper, M.E.; Van Erpecum, K.J.; Boland, G.J. No beneficial effects of amantadine in treatment of chronic hepatitis C patients. Dig. Liver Dis. 2009, 42, 496–502. [Google Scholar] [CrossRef] [PubMed]
- Von Wagner, M.; Hofmann, W.P.; Teuber, G.; Berg, T.; Goeser, T.; Spengler, U.; Hinrichsen, H.; Weidenbach, H.; Gerken, G.; Manns, M.; Buggisch, P.; Herrmann, E.; Zeuzem, S. Placebo-controlled trial of 400 mg amantadine combined with peginterferon alfa-2a and ribavirin for 48 weeks in chronic hepatitis C virus-1 infection. Hepatology 2008, 48, 1404–1411. [Google Scholar] [CrossRef]
- Cook, G.A.; Opella, S.J. NMR studies of p7 protein from hepatitis C virus. Eur. Biophys. J. 2010, 39, 1097–1104. [Google Scholar] [CrossRef]
- Steinmann, E.; Whitfield, T.; Kallis, S.; Dwek, R.A.; Zitzmann, N.; Pietschmann, T.; Bartenschlager, R. Antiviral effects of amantadine and iminosugar derivatives against hepatitis C virus. Hepatology 2007, 46, 330–338. [Google Scholar] [CrossRef]
- Griffin, S.; Stgelais, C.; Owsianka, A.M.; Patel, A.H.; Rowlands, D.; Harris, M. Genotype-dependent sensitivity of hepatitis C virus to inhibitors of the p7 ion channel. Hepatology 2008, 48, 1779–1790. [Google Scholar] [CrossRef]
- Ewart, G.D.; Sutherland, T.; Gage, P.W.; Cox, G.B. The Vpu protein of human immunodeficiency virus type 1 forms cation-selective ion channels. J. Virol. 1996, 70, 7108–7115. [Google Scholar] [CrossRef]
- Lemaitre, V.; Ali, R.; Kim, C.G.; Watts, A.; Fischer, W.B. Interaction of amiloride and one of its derivatives with Vpu from HIV-1: A molecular dynamics simulation. FEBS Lett. 2004, 563, 75–81. [Google Scholar] [CrossRef] [PubMed]
- Dwek, R.A.; Butters, T.D.; Platt, F.M.; Zitzmann, N. Targeting glycosylation as a therapeutic approach. Nat. Rev. Drug Discov. 2002, 1, 65–75. [Google Scholar] [CrossRef] [PubMed]
- Durantel, D.; Branza-Nichita, N.; Carrouee-Durantel, S.; Butters, T.D.; Dwek, R.A.; Zitzmann, N. Study of the mechanism of antiviral action of iminosugar derivatives against bovine viral diarrhea virus. J. Virol. 2001, 75, 8987–8998. [Google Scholar] [CrossRef] [PubMed]
- Helenius, A.; Aebi, M. Roles of N-linked glycans in the endoplasmic reticulum. Annu. Rev. Biochem. 2004, 73, 1019–1049. [Google Scholar] [CrossRef]
- Pawlotsky, J.M.; Chevaliez, S.; McHutchison, J.G. The hepatitis C virus life cycle as a target for new antiviral therapies. Gastroenterology 2007, 132, 1979–1998. [Google Scholar] [CrossRef]
- Luscombe, C.A.; Huang, Z.; Murray, M.G.; Miller, M.; Wilkinson, J.; Ewart, G.D. A novel Hepatitis C virus p7 ion channel inhibitor, BIT225, inhibits bovine viral diarrhea virus in vitro and shows synergism with recombinant interferon-alpha-2b and nucleoside analogues. Antivir. Res. 2010, 86, 144–153. [Google Scholar] [CrossRef]
© 2010 by the authors. licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Steinmann, E.; Pietschmann, T. Hepatitis C Virus P7—A Viroporin Crucial for Virus Assembly and an Emerging Target for Antiviral Therapy. Viruses 2010, 2, 2078-2095. https://doi.org/10.3390/v2092078
Steinmann E, Pietschmann T. Hepatitis C Virus P7—A Viroporin Crucial for Virus Assembly and an Emerging Target for Antiviral Therapy. Viruses. 2010; 2(9):2078-2095. https://doi.org/10.3390/v2092078
Chicago/Turabian StyleSteinmann, Eike, and Thomas Pietschmann. 2010. "Hepatitis C Virus P7—A Viroporin Crucial for Virus Assembly and an Emerging Target for Antiviral Therapy" Viruses 2, no. 9: 2078-2095. https://doi.org/10.3390/v2092078
APA StyleSteinmann, E., & Pietschmann, T. (2010). Hepatitis C Virus P7—A Viroporin Crucial for Virus Assembly and an Emerging Target for Antiviral Therapy. Viruses, 2(9), 2078-2095. https://doi.org/10.3390/v2092078