Antiviral Therapy for Hepatitis C Virus: Beyond the Standard of Care

{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Small Molecule Inhibitors of HCV Replication in Development
2.1. Virus-Specific Strategies
2.1.1. Entry Inhibitors

2.1.2. P7 Inhibitors
2.1.3. NS3 Protease Inhibitors
2.1.4. NS3 Helicase Inhibitors
2.1.5. Presumed Inhibitors ofNS3/NS4A Interaction
2.1.6. Inhibitors of NS4B-HCV RNA Binding
2.1.7. NS5A Inhibitors
2.1.8. NS5B (RNA-Dependent RNA Polymerase) Inhibitors
a. Nucleoside polymerase inhibitors
b. Nucleotide analogues
c. Non-nucleoside polymerase inhibitors
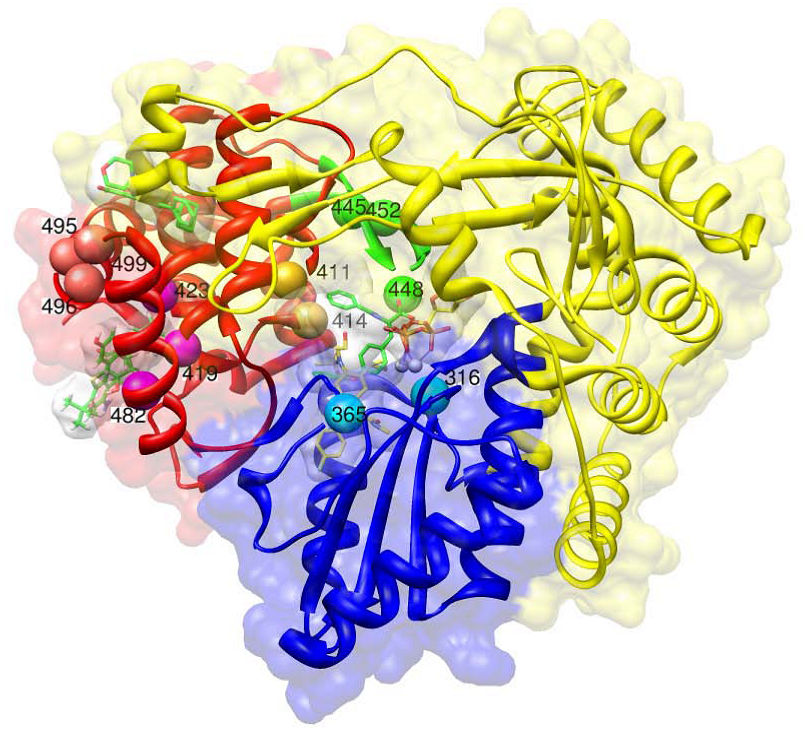
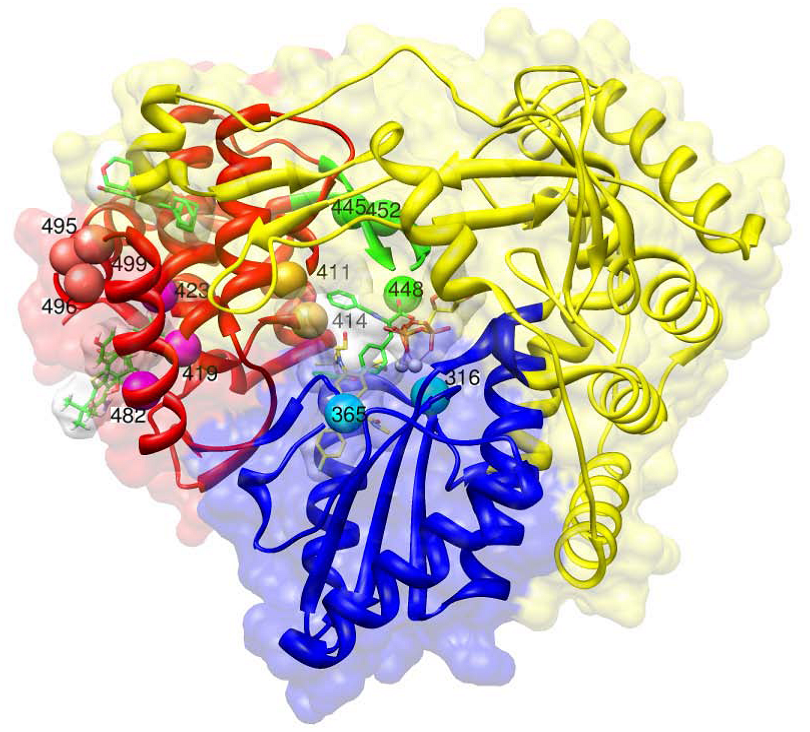
Thumb domain 1 (site A)
Thumb domain 2 (site B)
Palm domain 1 (site C)
Palm domain 2 (site D)
2.2. Host-Specific Strategies
2.2.1. Cyclophilin Binding Molecules
2.2.2. Glycosylation Inhibitors
2.2.3. Statins
2.2.4. Nitazoxanide (Alinia®, Romark laboratories)
2.2.5. Silymarin Extracts
3. Combination Therapy
4. Discussion
Acknowledgments
References
- Tang, H.; Grise, H. Cellular and molecular biology of HCV infection and hepatitis. Clin. Sci. (Lond.) 2009, 117, 49–65. [Google Scholar] [CrossRef] [PubMed]
- Shepard, C.W.; Finelli, L.; Alter, M.J. Global epidemiology of hepatitis C virus infection. Lancet Infect. Dis. 2005, 5, 558–567. [Google Scholar] [CrossRef] [PubMed]
- Dienstag, J.L.; McHutchison, J.G. American Gastroenterological Association technical review on the management of hepatitis C. Gastroenterology 2006, 130, 231–264. [Google Scholar] [CrossRef] [PubMed]
- Manns, M.P.; Wedemeyer, H.; Cornberg, M. Treating viral hepatitis C: Efficacy, side effects, and complications. Gut. 2006, 55, 1350–1359. [Google Scholar] [CrossRef] [PubMed]
- Rustgi, V.K. Albinterferon alfa-2b, a novel fusion protein of human albumin and human interferon alfa-2b, for chronic hepatitis C. Curr. Med. Res. Opin. 2009, 25, 991–1002. [Google Scholar] [CrossRef] [PubMed]
- Benhamou, Y.; Afdhal, N.H.; Nelson, D.R.; Shiffman, M.L.; Halliman, D.G.; Heise, J.; Chun, E.; Pockros, P.J. A phase III study of the safety and efficacy of viramidine versus ribavirin in treatment-naive patients with chronic hepatitis C: ViSER1 results. Hepatology. 2009, 50, 717–726. [Google Scholar] [CrossRef] [PubMed]
- Brass, V.; Moradpour, D.; Blum, H.E. Hepatitis C virus infection: In vivo and in vitro models. J. Viral Hepat. 2007, 14, 64–67. [Google Scholar] [CrossRef] [PubMed]
- Perrault, M.; Pecheur, E.I. The hepatitis C virus and its hepatic environment: A toxic but finely tuned partnership. Biochem. J. 2009, 423, 303–314. [Google Scholar] [CrossRef] [PubMed]
- Masson, D.; Koseki, M.; Ishibashi, M.; Larson, C.J.; Miller, S.G.; King, B.D.; Tall, A.R. Increased HDL cholesterol and apoA-I in humans and mice treated with a novel SR-BI inhibitor. Arterioscler. Thromb. Vasc. Biol. 2009, 29, 2054–2060. [Google Scholar] [CrossRef] [PubMed]
- iTherX homepage. Available online: http://www.itherx.com/press.html (accessed 3 February 2009).
- Qian, D.; Coburn, G.; Han, A.Q.; de Muys, J.M.; Gauss, C.; Provoncha, K.; Canfield, M.; Paul, D.; Mohamed, S.; Moorji, S.; Fisch, D.; Murga, J.; Rotshteyn, Y.; Maddon, P.J.; Olson, W.C. Preclinical characterization of Pro 206, an orally active small-molecule Hepatitis C (HCV) entry inhibitor . Copenhagen, Denmark, 2009; pp. S5–S6. In 44th Annual Meeting of the European Association for the Study of the Liver ; 4. [Google Scholar]
- de Muys, J.M.; Coburn, G.; Han, A.; Provoncha, K.; Paul, D.; Moorji, S.; Fisch, D.; Murga, J.; Qian, D.; Maddon, P.J.; Olson, W.C. Discovery of potent and selective inhibitors of Hepatitis C virus (HCV) entry . Nice, France, 2009; p. P52. In 16th International Symposium on Hepatitis C Virus & Related Viruses; 10. [Google Scholar]
- de Bruijne, J.; Bergmann, J.; Weegink, C.; van Nieuwkerk, K.; de Knegt, R.; van de Wetering de Rooij, J.; Van Vliet, A.; Molenkamp, R.; Schinkel, J.; Reesink, H.; Janssen, H. JTK-652 is a novel HCV entry inhibitor: Results of a Phase I study evaluating safety, tolerability and antiviral activity in chronic Hepatitis C patients. Copenhagen, Denmark; In 44th Annual Meeting of the European Association for the Study of the Liver ; 4 2009; p. S342. [Google Scholar]
- Boyd, M.R.; Gustafson, K.R.; McMahon, J.B.; Shoemaker, R.H.; O'Keefe, B.R.; Mori, T.; Gulakowski, R.J.; Wu, L.; Rivera, M.I.; Laurencot, C.M.; Currens, M.J.; Cardellina, J.H.; Buckheit, R.W.; Nara, P.L.; Pannell, L.K.; Sowder, R.C.; Henderson, L.E. Discovery of cyanovirin-N, a novel human immunodeficiency virus-inactivating protein that binds viral surface envelope glycoprotein gp120: potential applications to microbicide development . Antimicrob. Agents Chemother. 1997, 41, 1521–1530. [Google Scholar] [PubMed]
- Helle, F.; Wychowski, C.; Vu-Dac, N.; Gustafson, K.R.; Voisset, C.; Dubuisson, J. Cyanovirin-N inhibits hepatitis C virus entry by binding to envelope protein glycans. J. Biol. Chem. 2006, 281, 25177–25183. [Google Scholar] [CrossRef] [PubMed]
- Steinmann, E.; Penin, F.; Kallis, S.; Patel, A.H.; Bartenschlager, R.; Pietschmann, T. Hepatitis C virus p7 protein is crucial for assembly and release of infectious virions. Plos Pathogens 2007, 3, 962–971. [Google Scholar] [CrossRef]
- Sakai, A.; St Claire, M.S.; Faulk, K.; Govindarajan, S.; Emerson, S.U.; Purcell, R.H.; Bukh, J. The p7 polypeptide of hepatitis C virus is critical for infectivity and contains functionally important genotype-specific sequences. Proc. Natl. Acad. Sci. USA 2003, 100, 11646–11651. [Google Scholar] [CrossRef]
- Steinmann, E.; Whitfield, T.; Kallis, S.; Dwek, R.A.; Zitzmann, N.; Pietschmann, T.; Bartenschlager, R. Antiviral effects of amantadine and iminosugar derivatives against hepatitis C virus. Hepatology 2007, 46, 330–338. [Google Scholar] [CrossRef] [PubMed]
- Luscombe, C.A.; Huang, Z.; Murray, M.G.; Miller, M.; Wilkinson, J.; Ewart, G.D. A novel Hepatitis C virus p7 ion channel inhibitor, BIT225, inhibits bovine viral diarrhea virus in vitro and shows synergism with recombinant interferon-alpha-2b and nucleoside analogues . Antiviral Res. 2010, in press. [Google Scholar]
- Riordan, S.; Luscombe, C.A.; Ewart, G.; Wilkinson, J.; Quan, K.; Marjason, J.; Legget, B.; Dore, G.J.; Fenn, C.; Miller, M. A Phase Ib placebo-controlled, randomized study of the safety, pharmacokinetics and antiviral activity of p7 inhibitor BIT225 in patients with Hepatitis C Virus (HCV) infection . Hep. Dart. 2009. Available online: http://www.ihlpress.com/pdf%20files/hepdart09_presentations/late_breaker/11_Luscombe%20HepDART%202009%20Oral%20presentation.pdf. [Google Scholar]
- Pawlotsky, J.M.; Chevaliez, S.; McHutchison, J.G. The hepatitis C virus life cycle as a target for new antiviral therapies. Gastroenterology 2007, 132, 1979–1998. [Google Scholar] [CrossRef] [PubMed]
- Cheng, W.; Dumont, S.; Tinoco Jr., I.; Bustamante, C. NS3 helicase actively separates RNA strands and senses sequence barriers ahead of the opening fork . Proc. Natl. Acad. Sci. USA. 2007, 104, 13954–13959. [Google Scholar] [CrossRef]
- Lamarre, D.; Anderson, P.C.; Bailey, M.; Beaulieu, P.; Bolger, G.; Bonneau, P.; Bos, M.; Cameron, D.R.; Cartier, M.; Cordingley, M.G.; Faucher, A.M.; Goudreau, N.; Kawai, S.H.; Kukolj, G.; Lagace, L.; LaPlante, S.R.; Narjes, H.; Poupart, M.A.; Rancourt, J.; Sentjens, R.E.; St George, R.; Simoneau, B.; Steinmann, G.; Thibeault, D.; Tsantrizos, Y.S.; Weldon, S.M.; Yong, C.L.; Llinas-Brunet, M. An NS3 protease inhibitor with antiviral effects in humans infected with hepatitis C virus. Nature. 2003, 426, 186–189. [Google Scholar] [CrossRef] [PubMed]
- Hinrichsen, H.; Benhamou, Y.; Wedemeyer, H.; Reiser, M.; Sentjens, R.E.; Calleja, J.L.; Forns, X.; Erhardt, A.; Cronlein, J.; Chaves, R.L.; Yong, C.L.; Nehmiz, G.; Steinmann, G.G. Short-term antiviral efficacy of BILN 2061, a hepatitis C virus serine protease inhibitor, in hepatitis C genotype 1 patients. Gastroenterology. 2004, 127, 1347–1355. [Google Scholar] [CrossRef] [PubMed]
- Reiser, M.; Hinrichsen, H.; Benhamou, Y.; Reesink, H.W.; Wedemeyer, H.; Avendano, C.; Riba, N.; Yong, C.L.; Nehmiz, G.; Steinmann, G.G. Antiviral efficacy of NS3-serine protease inhibitor BILN-2061 in patients with chronic genotype 2 and 3 hepatitis C. Hepatology. 2005, 41, 832–835. [Google Scholar] [CrossRef] [PubMed]
- Lu, L.; Pilot-Matias, T.J.; Stewart, K.D.; Randolph, J.T.; Pithawalla, R.; He, W.; Huang, P.P.; Klein, L.L.; Mo, H.; Molla, A. Mutations conferring resistance to a potent hepatitis C virus serine protease inhibitor in vitro. Antimicrob. Agents Chemother. 2004, 48, 2260–2266. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.; Kwong, A.D.; Perni, R.B. Discovery and development of VX-950, a novel, covalent, and reversible inhibitor of hepatitis C virus NS3 4A serine protease. Infect. Disord. Drug Targets 2006, 6, 3–16. [Google Scholar] [CrossRef] [PubMed]
- Perni, R.B.; Almquist, S.J.; Byrn, R.A.; Chandorkar, G.; Chaturvedi, P.R.; Courtney, L.F.; Decker, C.J.; Dinehart, K.; Gates, C.A.; Harbeson, S.L.; Heiser, A.; Kalkeri, G.; Kolaczkowski, E.; Lin, K.; Luong, Y.P.; Rao, B.G.; Taylor, W.P.; Thomson, J.A.; Tung, R.D.; Wei, Y.; Kwong, A.D.; Lin, C. Preclinical profile of VX-950, a potent, selective, and orally bioavailable inhibitor of hepatitis C virus NS3-4A serine protease. Antimicrob. Agents Chemother. 2006, 50, 899–909. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.; Lin, K.; Luong, Y.P.; Rao, B.G.; Wei, Y.Y.; Brennan, D.L.; Fulghum, J.R.; Hsiao, H.M.; Ma, S.; Maxwell, J.P.; Cottrell, K.M.; Perni, R.B.; Gates, C.A.; Kwong, A.D. In vitro resistance studies of hepatitis C virus serine protease inhibitors, VX-950 and BILN 2061: structural analysis indicates different resistance mechanisms. J. Biol. Chem. 2004, 279, 17508–17514. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.; Gates, C.A.; Rao, B.G.; Brennan, D.L.; Fulghum, J.R.; Luong, Y.P.; Frantz, J.D.; Lin, K.; Ma, S.; Wei, Y.Y.; Perni, R.B.; Kwong, A.D. In vitro studies of cross-resistance mutations against two hepatitis C virus serine protease inhibitors, VX-950 and BILN 2061. J. Biol. Chem. 2005, 280, 36784–36791. [Google Scholar] [CrossRef] [PubMed]
- Reesink, H.W.; Zeuzem, S.; Weegink, C.J.; Forestier, N.; van Vliet, A. Rapid decline of viral RNA in hepatitis C patients treated with VX-950: A phase Ib, placebo-controlled, randomized study. Gastroenterology. 2006, 131, 997–1002. [Google Scholar] [CrossRef] [PubMed]
- Sarrazin, C.; Kieffer, T.L.; Bartels, D.; Hanzelka, B.; Muh, U.; Welker, M.; Wincheringer, D.; Zhou, Y.; Chu, H.M.; Lin, C.; Weegink, C.; Reesink, H.; Zeuzem, S.; Kwong, A.D. Dynamic hepatitis C virus genotypic and phenotypic changes in patients treated with the protease inhibitor telaprevir. Gastroenterology. 2007, 132, 1767–1777. [Google Scholar] [CrossRef] [PubMed]
- Forestier, N.; Reesink, H.W.; Weegink, C.J.; McNair, L.; Kieffer, T.L.; Chu, H.M.; Purdy, S.; Jansen, P.L.; Zeuzem, S. Antiviral activity of telaprevir (VX-950) and peginterferon alfa-2a in patients with hepatitis C. Hepatology. 2007, 46, 640–648. [Google Scholar] [CrossRef] [PubMed]
- Zeuzem, S. Telaprevir, peginterferon alfa-2a, and ribavirin for 28 days in chronic hepatitis C patients. J. Hepatol. 2008, 49, 157–159. [Google Scholar] [CrossRef] [PubMed]
- McHutchison, J.G.; Everson, G.T.; Gordon, S.C.; Jacobson, I.M.; Sulkowski, M.; Kauffman, R.; McNair, L.; Alam, J.; Muir, A.J. Telaprevir with peginterferon and ribavirin for chronic HCV genotype 1 infection. N. Engl. J. Med. 2009, 360, 1827–1838. [Google Scholar] [CrossRef] [PubMed]
- Hezode, C.; Forestier, N.; Dusheiko, G.; Ferenci, P.; Pol, S.; Goeser, T.; Bronowicki, J.P.; Bourliere, M.; Gharakhanian, S.; Bengtsson, L.; McNair, L.; George, S.; Kieffer, T.; Kwong, A.; Kauffman, R.S.; Alam, J.; Pawlotsky, J.M.; Zeuzem, S. Telaprevir and peginterferon with or without ribavirin for chronic HCV infection. N. Engl. J. Med. 2009, 360, 1839–1850. [Google Scholar] [CrossRef] [PubMed]
- McHutchison, J.G.; Manns, M.P.; Muir, A.; Terrault, N.; Jacobson, I.M.; Afdhal, N.H.; Heathcote, E.; Zeuzem, S.; Reesink, H.W.; Bsharat, M.; George, S.; Adda, N.; Di Bisceglie, A.M. PROVE3 final results and 1-year durability of SVR with telaprevir-based regimen in hepatitis C genotype 1 - infected patients with prior non-response, viral breakthrough or relapse to peginterferon-alpha-2a/b and ribavirin therapy . 2009; p. 66. In 60th Annual Meeting of the American Association for the Study of Liver Diseases (AASLD); 10 30. [Google Scholar]
- Foster, G.R.; Hezode, C.; Bronowicki, J.P.; Carosi, G.; Weiland, O.; Verlinden, L.; van Heeswijk, R.; Vangeneugden, T.; Picchio, G.; Beumont-Mauviel, M. Activity of Telaprevir Alone Or in Combination with Peginterferon Alfa-2A and Ribavirin in Treatment-Naive Genotype 2 and 3 Hepatitis-C Patients: Interim Results of Study C209 . J. Hepatol. 2009, 50, S22–S22. [Google Scholar] [CrossRef]
- Benhamou, Y.; Moussalli, J.; Ratziu, V.; Lebray, P.; Gysen, V.; de Backer, K.; Ghys, A.; van Heeswijk, R.; Vangeneugden, I.; Picchio, G.; Beumont-Mauviel, M. Results of A Proof of Concept Study (C210) of Telaprevir Monotherapy and in Combination with Peginterferon Alfa-2A and Ribavirin in Treatment-Naive Genotype 4 Hcv Patients . J. Hepatol. 2009, 50, S6–S6. [Google Scholar] [CrossRef]
- Malcolm, B.A.; Liu, R.; Lahser, F.; Agrawal, S.; Belanger, B.; Butkiewicz, N.; Chase, R.; Gheyas, F.; Hart, A.; Hesk, D.; Ingravallo, P.; Jiang, C.; Kong, R.; Lu, J.; Pichardo, J.; Prongay, A.; Skelton, A.; Tong, X.; Venkatraman, S.; Xia, E.; Girijavallabhan, V.; Njoroge, F.G. SCH 503034, a mechanism-based inhibitor of hepatitis C virus NS3 protease, suppresses polyprotein maturation and enhances the antiviral activity of alpha interferon in replicon cells. Antimicrob. Agents Chemother. 2006, 50, 1013–1020. [Google Scholar] [CrossRef] [PubMed]
- Tong, X.; Chase, R.; Skelton, A.; Chen, T.; Wright-Minogue, J.; Malcolm, B.A. Identification and analysis of fitness of resistance mutations against the HCV protease inhibitor SCH 503034. Antiviral Res. 2006, 70, 28–38. [Google Scholar] [CrossRef] [PubMed]
- Sarrazin, C.; Rouzier, R.; Wagner, F.; Forestier, N.; Larrey, D.; Gupta, S.K.; Hussain, M.; Shah, A.; Cutler, D.; Zhang, J.; Zeuzem, S. SCH 503034, a novel hepatitis C virus protease inhibitor, plus pegylated interferon alpha-2b for genotype 1 nonresponders. Gastroenterology 2007, 132, 1270–1278. [Google Scholar] [CrossRef] [PubMed]
- Susser, S.; Welsch, C.; Wang, Y.; Zettler, M.; Domingues, F.S.; Karey, U.; Hughes, E.; Ralston, R.; Tong, X.; Herrmann, E.; Zeuzem, S.; Sarrazin, C. Characterization of resistance to the protease inhibitor boceprevir in hepatitis C virus-infected patients. Hepatology. 2009, 50, 1709–1718. [Google Scholar] [CrossRef] [PubMed]
- Kwo, P.; Lawitz, E.; McCone, J.; Schiff, E.; Vierling, J.; Pound, D.; Davis, M.; Galati, J.; Gordon, S.; Ravendhran, N.; Rossaro, L.; Anderson, F.; Jacobson, I.; Rubin, R.; Koury, K.; Brass, C.; Chaudri, E.; Albrecht, J. HCV SPRINT-1 final results: SVR24 from a phase 2 study of boceprevir plus pegintron (peginterferon alpha-2b)/ribavirin in treatment-naive subjects with genotype-1 chronic hepatitis C . In J. Hepatol. 44th Annual Meeting of the European Association for the Study of the Liver ; 4 22-26; Volume 50, Copenhagen, Denmark., 2009; p. S4. [Google Scholar]
- Kwo, P.Y.; Lawitz, E.; McCone, J.; Schiff, E.R.; Vierling, J.M.; Pound, D.; Davis, M.; Galati, J.S.; Gordon, S.C.; Ravendhran, N.; Rossaro, L.; Anderson, F.H.; Jacobson, I.M.; Rubin, R.; Koury, K.; Boparai, N.; Chaudhri, E.I.; Brass, C.A.; Albrecht, J.K. Response-guided therapy for boceprevir combination treatment? - results from HCV SPRINT-1 . Boston, MA, USA, 2009; p. 1582. In 60th Annual Meeting of the American Association for the Study of Liver Diseases (AASLD); 10 30. [Google Scholar]
- Lin, T.I.; Lenz, O.; Fanning, G.; Verbinnen, T.; Delouvroy, F.; Scholliers, A.; Vermeiren, K.; Rosenquist, A.; Edlund, M.; Samuelsson, B.; Vrang, L.; de Kock, H.; Wigerinck, P.; Raboisson, P.; Simmen, K. In vitro activity and preclinical profile of TMC435350, a potent hepatitis C virus protease inhibitor. Antimicrob. Agents Chemother. 2009, 53, 1377–1385. [Google Scholar] [CrossRef] [PubMed]
- Reesink, H.W.; Fanning, G.C.; Abou, F.K.; Weegink, C.; van Vliet, A.; van 't, K.G.; Lenz, O.; Aharchi, F.; Marien, K.; Van Remoortere, P.; de Kock, H.; Broeckaert, F.; Meyvisch, P.; Van Beirendonck, E.; Simmen, K.; Verloes, R. Rapid HCV-RNA decline with once-daily TMC435: A Phase I study in healthy volunteers and hepatitis C patients. Gastroenterology. 2010, 138, 913–921. [Google Scholar] [CrossRef] [PubMed]
- Marcellin, P.; Reesink, H.; Berg, T.; Cramp, M.; Flisiak, R.; Van Vlierberghe, H.; Verloes, R.; Lenz, O.; Peeters, M.; Sekar, V.; De Smedt, G. Antiviral activity and safety of TMC435 combined with peginterferon alpha-2a and ribavirin in patients with genotype 1 hepatitis C infection who failed previous IFN-based therapy . Copenhagen, Denmark, 2009; p. S385. In J. Hepatol. 44th Annual Meeting of the European Association for the Study of the Liver ; 4 22-26; Volume 50. [Google Scholar]
- Seiwert, S.D.; Andrews, S.W.; Jiang, Y.; Serebryany, V.; Tan, H.; Kossen, K.; Rajagopalan, P.T.; Misialek, S.; Stevens, S.K.; Stoycheva, A.; Hong, J.; Lim, S.R.; Qin, X.; Rieger, R.; Condroski, K.R.; Zhang, H.; Do, M.G.; Lemieux, C.; Hingorani, G.P.; Hartley, D.P.; Josey, J.A.; Pan, L.; Beigelman, L.; Blatt, L.M. Preclinical characteristics of the hepatitis C virus NS3/4A protease inhibitor ITMN-191 (R7227). Antimicrob. Agents Chemother. 2008, 52, 4432–4441. [Google Scholar] [CrossRef] [PubMed]
- Tan, H.; Rajyaguru, S.; Wu, T.; McCown, M.; Ali, S.; Jiang, W.R.; Otto, M.; Furman, P.; Najera, I.; Klumpp, K.; Symons, J.; Cammack, N.; Blatt, L.; Seiwert, S. Combination of the NS3/4A protease inhibitor ITMN-191 (R7227) with the active moiety of the NS5B inhibitors R1626 or R7128 enhances replicon clearance and reduces the emergence of drug resistant variants . San Francisco, CA, USA, 2008; In 59th Annual Meeting of the American Association for the Study of Liver Disease; 10 31. [Google Scholar]
- Forestier, N.; Larrey, D.; Guyader, D. Treatment of chronic hepatitis C virus (HCV) Genotype 1 patients with the NS3/4A protease inhibitor ITMN-191 leads to rapid reductions in plasma HCV RNA: Results of a phase 1b multiple ascending dose (MAD) study . San Francisco, CA, USA, 2008; In 59th Annual Meeting of the American Association for the Study of Liver Disease; 10 31. [Google Scholar]
- Forestier, N.; Larrey, D.; Marcellin, P.; Benhamou, Y.; Guyader, D.; Bradford, W.; Porter, S.; Patat, A.; Rouzier, R.; Zeuzem, S. Antiviral activity and safety of ITMN-191 in combination with peginterferon alpha-2a and ribavirin in patients with chronic hepatitis C virus (HCV) . Copenhagen, Denmark, 2009; In J. Hepatol. 44th Annual Meeting of the European Association for the Study of the Liver ; 4 22-26. [Google Scholar]
- Liverton, N.J.; Carroll, S.S.; Dimuzio, J.; Fandozzi, C.; Graham, D.J.; Hazuda, D.; Holloway, M.K.; Ludmerer, S.W.; McCauley, J.A.; McIntyre, C.J.; Olsen, D.B.; Rudd, M.T.; Stahlhut, M.; Vacca, J.P. MK-7009: A Potent and Selective Inhibitor of Hepatitis C Virus NS3/4A Protease. Antimicrob. Agents Chemother. 2009, 54, 305–311. [Google Scholar] [CrossRef] [PubMed]
- Manns, M.P.; Gane, E.J.; Rodriguez-Torres, M.; Stoehr, A.D.; Yeh, C.; Marcellin, P.; Wiedmann, R.T.; Hwang, P.; Barnard, R.J.Q.E.; Kartsonis, N.A.; Lee, A.W. Early Viral Response ( EVR ) Rates in treatment - naïve patients with Chronic Hepatitis C ( CHC ) genotype 1 infection treated with MK - 7009, a novel NS3/4a protease inhibitor, in combination with pegylated Interferon alpha - 2a and ribavirin for 28 days . Boston, MA, USA, 2009; In 60th Annual Meeting of the American Association for the Study of Liver Diseases (AASLD); 10 30. [Google Scholar]
- Manns, M.P.; Bourliere, M.; Benhamou, Y.; Pol, S.; Bonacini, M.; Berg, T.; Trepo, C.; Wright, D.; Calleja, J.L.; Steinmann, G.; Huang, D.B.; Mikl, J.; Kukolj, G.; Stern, J. Safety and antiviral activity of BI 201335, a new HCV NS3 protease inhibitor, in treatment-naïve patients with chronic hepatitis C genotype 1 infection given as monotherapy and in combination with peginterferon alfa-2a and ribavirin . San Francisco, CA, USA, 2008; p. 1849. In 59th Annual Meeting of the American Association for the Study of Liver Disease; 10 31. [Google Scholar]
- Manns, M.P.; Bourliere, M.; Benhamou, Y.; Pol, S.; Bonacini, M.; Berg, T.; Trepo, C.; Wright, D.; Calleja, J.L.; Steinmann, G.; Huang, D.B.; Mikl, J.; Kukolj, G.; Stern, J. Safety and antiviral activity of BI201335, a new HCV NS3 protease inhibitor, in combination therapy with peginterferon-alpha-2a and ribavirin for 28 days in P + R treatment-experienced patients with chronic hepatitis C genotype 1 infection . San Francisco, CA, USA, 2008; p. 1882. In 59th Annual Meeting of the American Association for the Study of Liver Disease; 10 31. [Google Scholar]
- Kukolj, G.; Benhamou, Y.; Manns, M.P.; Bourliere, M.; Pol, S.; Schuchmann, M.; Cartier, M.; Huang, D.; Lagace, L.; Steinmann, G.; Stern, J.O. BI 201335, a potent HCV NS3 protease inhibitor, in treatment-naÃÂïve and -experienced chronic HCV genotype-1 infection: Genotypic and phenotypic analysis of the NS3 protease domain . Copenhagen, Denmark, 2009; In J. Hepatol. 44th Annual Meeting of the European Association for the Study of the Liver ; 4 22-26. [Google Scholar]
- Pol, S.; Berg, T.; Bonacini, M.; Schuchmann, M.; Lalezari, J.; Erhardt, A.; Bourliere, M.; Manns, M.P.; Yong, C.L.; Steinmann, G.; Stern, J.O.; Scherer, J.; Boecher, W.O. Virological response and safety of BI 201335 protease inhibitor, peginterferon alfa 2a and ribavirin treatment of HCV genotype - 1 patients with compensated liver cirrhosis and non - response to previous peginterferon / ribavirin . Boston, MA, USA; LB162009; In 60th Annual Meeting of the American Association for the Study of Liver Diseases (AASLD); 10 30. [Google Scholar]
- Reesink, H.; Bergmann, J.; de Bruijne, J.; Weegink, C.; van Lier, J.; van Vliet, A.; Keung, A.; Li, J.; O'Mara, E.; Treitel, M.; Hughes, E.; Janssen, H.; de Knegt, R. Safety and antiviral activity of SCH 900518 administered as monotherapy and in combination with peginterferon afa-2b to naive and treatment-experienced HCV infected patients . Copenhagen, Denmark, 2009; p. S35. In J. Hepatol. 44th Annual Meeting of the European Association for the Study of the Liver ; 4 22-26; Volume 50. [Google Scholar]
- de Bruijne, J.; Bergmann, J.F.; Weegink, C.J.; Molenkamp, R.; Schinkel, J.; Treitel, M.A.; Hughes, E.A.; van Vliet, A.A.; de Knegt, R.J.; Reesink, H.W.; Janssen, H.L. SVR results in chronic Hepatitis C genotype 1 patients dosed with SCH 900518 and peginterferon alfa - 2b for 2 weeks, followed by peginterferon alfa - 2b and ribavirin for 24/48 weeks: An interim analysis . Boston, MA, USA, 2009; p. 1555. In 60th Annual Meeting of the American Association for the Study of Liver Diseases (AASLD); 10 30. [Google Scholar]
- Vierling, J.M.; Poordad, F.; Lawitz, E.; Ghalib, R.H.; Lee, W.M.; Ravendhran, N.; Galati, J.S.; Bacon, B.R.; Flamm, S.L.; Balart, L.A.; Freilich, B.; Schiff, E.R.; Jacobson, I.M.; Kwo, P.Y.; Gordon, S.C.; Sulkowski, M.S.; Boparai, N.; Chaudri, E.I.; Brass, C.; Hughes, E.A.; Albrecht, J.K. Once daily Narlaprevir ( SCH 900518 ) in combination with PEGINTRONTM (Peginterferon alfa - 2b ) /Ribavirin for treatment - naïve subjects with genotype - 1 CHC: Interim results from NEXT - 1, a phase 2a study. Boston, US; LB42009; In 60th Annual Meeting of the American Association for the Study of Liver Diseases (AASLD); 10 30. [Google Scholar]
- Bretner, M.; Baier, A.; Kopanska, K.; Najda, A.; Schoof, A.; Reinholz, M.; Lipniacki, A.; Piasek, A.; Kulikowski, T.; Borowski, P. Synthesis and biological activity of 1H-benzotriazole and 1H-benzimidazole analogues--inhibitors of the NTpase/helicase of HCV and of some related Flaviviridae. Antivir. Chem. Chemother. 2005, 16, 315–326. [Google Scholar] [PubMed]
- Maga, G.; Gemma, S.; Fattorusso, C.; Locatelli, G.A.; Butini, S.; Persico, M.; Kukreja, G.; Romano, M.P.; Chiasserini, L.; Savini, L.; Novellino, E.; Nacci, V.; Spadari, S.; Campiani, G. Specific targeting of hepatitis C virus NS3 RNA helicase. Discovery of the potent and selective competitive nucleotide-mimicking inhibitor QU663. Biochemistry. 2005, 44, 9637–9644. [Google Scholar] [CrossRef] [PubMed]
- Boguszewska-Chachulska, A.M.; Krawczyk, M.; Najda, A.; Kopanska, K.; Stankiewicz-Drogon, A.; Zagorski-Ostoja, W.; Bretner, M. Searching for a new anti-HCV therapy: Synthesis and properties of tropolone derivatives. Biochem. Biophys. Res. Commun. 2006, 341, 641–647. [Google Scholar] [CrossRef] [PubMed]
- Ujjinamatada, R.K.; Baier, A.; Borowski, P.; Hosmane, R.S. An analogue of AICAR with dual inhibitory activity against WNV and HCV NTPase/helicase: Synthesis and in vitro screening of 4-carbamoyl-5-(4,6-diamino-2,5-dihydro-1,3,5-triazin-2-yl)imidazole-1-beta-D-ribo furanoside. Bioorg. Med. Chem. Lett. 2007, 17, 2285–2288. [Google Scholar] [CrossRef] [PubMed]
- Manfroni, G.; Paeshuyse, J.; Massari, S.; Zanoli, S.; Gatto, B.; Maga, G.; Tabarrini, O.; Cecchetti, V.; Fravolini, A.; Neyts, J. Inhibition of subgenomic hepatitis C virus RNA replication by acridone derivatives: Identification of an NS3 helicase inhibitor. J. Med. Chem. 2009, 52, 3354–3365. [Google Scholar] [CrossRef] [PubMed]
- Gozdek, A.; Zhukov, I.; Polkowska, A.; Poznanski, J.; Stankiewicz-Drogon, A.; Pawlowicz, J.M.; Zagorski-Ostoja, W.; Borowski, P.; Boguszewska-Chachulska, A.M. NS3 Peptide, a novel potent hepatitis C virus NS3 helicase inhibitor: Its mechanism of action and antiviral activity in the replicon system. Antimicrob. Agents Chemother. 2008, 52, 393–401. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.S.; Chiou, C.T.; Chen, G.S.; Chen, S.C.; Hu, C.Y.; Chi, W.K.; Chu, Y.D.; Hwang, L.H.; Chen, P.J.; Chen, D.S.; Liaw, S.H.; Chern, J.W. Structure-based discovery of triphenylmethane derivatives as inhibitors of hepatitis C virus helicase. J. Med. Chem. 2009, 52, 2716–2723. [Google Scholar] [CrossRef] [PubMed]
- Penin, F.; Dubuisson, J.; Rey, F.A.; Moradpour, D.; Pawlotsky, J.M. Structural biology of hepatitis C virus. Hepatology 2004, 39, 5–19. [Google Scholar] [CrossRef] [PubMed]
- Wyles, D.L.; Kaihara, K.A.; Schooley, R.T. Synergy of a hepatitis C virus (HCV) NS4A antagonist in combination with HCV protease and polymerase inhibitors. Antimicrob. Agents Chemother. 2008, 52, 1862–1864. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.; Zhao, Y.; Fabrycki, J.; Hou, X.; Nie, X.; Sanchez, A.; Phadke, A.; Deshpande, M.; Agarwal, A.; Huang, M. Selection of replicon variants resistant to ACH-806, a novel hepatitis C virus inhibitor with no cross-resistance to NS3 protease and NS5B polymerase inhibitors. Antimicrob. Agents Chemother. 2008, 52, 2043–2052. [Google Scholar] [CrossRef] [PubMed]
- Pottage, J.C.; Lawitz, E.; Mazur, D.; Wyles, D.L.; Vargas, H.E.; Ghalib, R.; Gugliotti, R.; Donohue, M.; Robison, H. Short-term antiviral activity and safety of ACH-806 (GS-9132), an NS4A antagonist, in HCV genotyp 1 infected individuals . Barcelona, Spain, 2007; pp. S294–S295. In 42th Annual Meeting of the European Association for the Study of the Liver ; 4. [Google Scholar]
- El Hage, N. and Luo, G. Replication of hepatitis C virus RNA occurs in a membrane-bound replication complex containing nonstructural viral proteins and RNA. J. Gen. Virol. 2003, 84, 2761–2769. [Google Scholar] [CrossRef] [PubMed]
- Einav, S.; Gerber, D.; Bryson, P.D.; Sklan, E.H.; Elazar, M.; Maerkl, S.J.; Glenn, J.S.; Quake, S.R. Discovery of a hepatitis C target and its pharmacological inhibitors by microfluidic affinity analysis. Nat. Biotechnol. 2008, 26, 1019–1027. [Google Scholar] [CrossRef] [PubMed]
- Macdonald, A. and Harris, M. Hepatitis C virus NS5A: Tales of a promiscuous protein. J. Gen. Virol. 2004, 85, 2485–2502. [Google Scholar] [CrossRef] [PubMed]
- Schmitz, U.; Tan, S.L. NS5A--from obscurity to new target for HCV therapy. Recent Pat Antiinfect. Drug Discov. 2008, 3, 77–92. [Google Scholar]
- Evans, M.J.; Rice, C.M.; Goff, S.P. Phosphorylation of hepatitis C virus nonstructural protein 5A modulates its protein interactions and viral RNA replication. Proc. Natl. Acad. Sci. U. S. A. 2004, 101, 13038–13043. [Google Scholar] [CrossRef] [PubMed]
- Appel, N.; Zayas, M.; Miller, S.; Krijnse-Locker, J.; Schaller, T.; Friebe, P.; Kallis, S.; Engel, U.; Bartenschlager, R. Essential role of domain III of nonstructural protein 5A for hepatitis C virus infectious particle assembly . PLoS. Pathog. 2008, 4, e1000035. [Google Scholar] [CrossRef] [PubMed]
- Moradpour, D.; Brass, V.; Penin, F. Function follows form: The structure of the N-terminal domain of HCV NS5A. Hepatology. 2005, 42, 732–735. [Google Scholar] [CrossRef] [PubMed]
- Arrow therapeutics Home Page . Available online: http://www.arrowt.co.uk/product-hcv.asp (accessed 27 November 2006 ). [CrossRef] [PubMed]
- Quinkert, D.; Deneka, M.; Najarro, P.; Chapman, J.; Bushnell, D.; Mathews, N.; Cockerill, S.; Powell, K. HCV inhibitors targeting NS5A. San Antonio, Texas, 2008; In 15th International Symposium on Hepatitis C Virus and Related Viruses; 10 5. [Google Scholar]
- Deneka, M.; Quinkert, D.; Rupassara, D.; Thomas, E.; Bone, P.; Granycome, C.; Bell, T.; Chapman, J.; Carter, M.; Mathews, N.; Najarro, P. Characterisation of a small molecule inhibitor of HCV replication that targets NS5A . Nice, France, 2009; In 16th International Symposium on Hepatitis C Virus and Related Viruses; 10 3. [Google Scholar]
- Gao, M.; Fridell, R.; O'Boyle, D.; Qiu, D.; Sun, J.; Lemm, J.; Nower, P.; Valera, L.; Voss, S.; Liu, M.; Belema, M.; Nguyen, V.; Romine, J.; Martin, S.; StLaurent, D.; Serrano-Wu, M.; Snyder, L.; Colonno, R.; Hamann, L.; Meanwell, N. HCV NS5A inhibitor: From screen hit to clinic. San Antonio, TX, USA, 2008. [Google Scholar]
- Lemm, J.A.; O'Boyle, D.; Liu, M.; Nower, P.T.; Colonno, R.; Deshpande, M.S.; Snyder, L.B.; Martin, S.W.; St Laurent, D.R.; Serrano-Wu, M.H.; Romine, J.L.; Meanwell, N.A.; Gao, M. Identification of Hepatitis C Virus NS5A Inhibitors. J. Virol. 2009, 84, 482–491. [Google Scholar] [CrossRef]
- Nettles, R.; Chien, C.; Persson, A.; Min Gao Belema, M.; Meanwell, N.; DeMicco, M.; Marbury, T.; Goldwater, R.; Northup, P.; Coumbis, J.; Kraft, W.; Charlton, M.; Lopez-Talavera, J.; Grasela, D. BMS-790052 is a first-in-class potent hepatitis C virus (HCV) NS5A inhibitor for patients with chronic HCV infection: Results from a proof-of-concept study . San Francisco, CA, USA, 2008; In 59th Annual Meeting of the American Association for the Study of Liver Disease; 10 31. [Google Scholar]
- Bressanelli, S.; Tomei, L.; Roussel, A.; Incitti, I.; Vitale, R.L.; Mathieu, M.; De Francesco, R.; Rey, F.A. Crystal structure of the RNA-dependent RNA polymerase of hepatitis C virus. Proc. Natl. Acad. Sci. U. S. A 1999, 96, 13034–13039. [Google Scholar] [CrossRef] [PubMed]
- Pierra, C.; Amador, A.; Benzaria, S.; Cretton-Scott, E.; D'Amours, M.; Mao, J.; Mathieu, S.; Moussa, A.; Bridges, E.G.; Standring, D.N.; Sommadossi, J.P.; Storer, R.; Gosselin, G. Synthesis and pharmacokinetics of valopicitabine (NM283), an efficient prodrug of the potent anti-HCV agent 2'-C-methylcytidine. J Med. Chem. 2006, 49, 6614–6620. [Google Scholar] [CrossRef] [PubMed]
- Migliaccio, G.; Tomassini, J.E.; Carroll, S.S.; Tomei, L.; Altamura, S.; Bhat, B.; Bartholomew, L.; Bosserman, M.R.; Ceccacci, A.; Colwell, L.F.; Cortese, R.; De Francesco, R.; Eldrup, A.B.; Getty, K.L.; Hou, X.S.; LaFemina, R.L.; Ludmerer, S.W.; MacCoss, M.; McMasters, D.R.; Stahlhut, M.W.; Olsen, D.B.; Hazuda, D.J.; Flores, O.A. Characterization of resistance to non-obligate chain-terminating ribonucleoside analogs that inhibit hepatitis C virus replication in vitro. J Biol. Chem. 2003, 278, 49164–49170. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.J.; Afdhal, N.; Godofsky, E.; Dienstag, J.; Rustgi, V.; Schick, L.; McInery, D.; Fielman, B.A.; Brown, N.A. Pharmacokinetics and pharmacodynamics of valopicitabine (NM283), a new nucleoside HCV polymerase inhibitor: Results from a phase I/II dose-escalation trial in patients with HCV-1 infection . Paris, France, 2005; p. 229. Volume 626, In 40th Annual Meeting of the European Association for the Study of the Liver; 4. [Google Scholar]
- Afdhal, N.; O'Brien, C.; Godofsky, E.; Rodriguez-Torres, M.; Pappas, S.C.; Lawitz, E.; Pockros, P.; Sulkowski, M.; Jacobson, I.; Chao, G.; Knox, S.; Pietropaolo, K.; Brown, N.A. Valopicitabine (NM283), alone or with peginterferon, compared to peginterferon/ribavirine (pegIFN/RBV) retreatment in patients with HCV-1 infection and prior non-response to pegIFN/RBV: One-year results . 42th Annual Meeting of the European Association for the Study of the Liver 2007, S5. [Google Scholar]
- Klumpp, K.; Leveque, V.; Le Pogam, S.; Ma, H.; Jiang, W.R.; Kang, H.; Granycome, C.; Singer, M.; Laxton, C.; Hang, J.Q.; Sarma, K.; Smith, D.B.; Heindl, D.; Hobbs, C.J.; Merrett, J.H.; Symons, J.; Cammack, N.; Martin, J.A.; Devos, R.; Najera, I. The novel nucleoside analog R1479 (4'-azidocytidine) is a potent inhibitor of NS5B-dependent RNA synthesis and hepatitis C virus replication in cell culture. J Biol. Chem. 2006, 281, 3793–3799. [Google Scholar] [CrossRef] [PubMed]
- Le Pogam, S.; Jiang, W.R.; Leveque, V.; Rajyaguru, S.; Ma, H.; Kang, H.; Jiang, S.; Singer, M.; Ali, S.; Klumpp, K.; Smith, D.; Symons, J.; Cammack, N.; Najera, I. In vitro selected Con1 subgenomic replicons resistant to 2'-C-methyl-cytidine or to R1479 show lack of cross resistance. Virology 2006, 351, 349–359. [Google Scholar] [CrossRef] [PubMed]
- Roberts, S.K.; Cooksley, G.; Dore, G.J.; Robson, R.; Shaw, D.; Berns, H.; Hill, G.; Klumpp, K.; Najera, I.; Washington, C. Robust antiviral activity of R1626, a novel nucleoside analog: A randomized, placebo-controlled study in patients with chronic hepatitis C. Hepatology 2008, 48, 398–406. [Google Scholar] [CrossRef] [PubMed]
- Pockros, P.; Nelson, D.; Godofsky, E.; Rodriguez-Torres, M.; Everson, G.T.; Fried, M.W.; Ghalib, R.; Harrison, S.; Nyberg, L.; Shiffman, M.L.; Chan, A.; Hill, G. High Relapse Rate Seen at Week 72 for Patients Treated with R1626 Combination Therapy. Hepatology 2008, 48, 1349–1350. [Google Scholar] [CrossRef] [PubMed]
- Pockros, P.J.; Nelson, D.; Godofsky, E.; Rodriguez-Torres, M.; Everson, G.T.; Fried, M.W.; Ghalib, R.; Harrison, S.; Nyberg, L.; Shiffman, M.L.; Najera, I.; Chan, A.; Hill, G. R1626 plus peginterferon Alfa-2a provides potent suppression of hepatitis C virus RNA and significant antiviral synergy in combination with ribavirin. Hepatology 2008, 48, 385–397. [Google Scholar] [CrossRef] [PubMed]
- Stuyver, L.J.; McBrayer, T.R.; Tharnish, P.M.; Clark, J.; Hollecker, L.; Lostia, S.; Nachman, T.; Grier, J.; Bennett, M.A.; Xie, M.Y.; Schinazi, R.F.; Morrey, J.D.; Julander, J.L.; Furman, P.A.; Otto, M.J. Inhibition of hepatitis C replicon RNA synthesis by beta-D-2'-deoxy-2'-fluoro-2'-C-methylcytidine: A specific inhibitor of hepatitis C virus replication. Antivir. Chem. Chemother. 2006, 17, 79–87. [Google Scholar] [PubMed]
- Ali, S.; Leveque, V.; Le Pogam, S.; Ma, H.; Philipp, F.; Inocencio, N.; Smith, M.; Alker, A.; Kang, H.; Najera, I.; Klumpp, K.; Symons, J.; Cammack, N.; Jiang, W.R. Selected replicon variants with low-level in vitro resistance to the hepatitis C virus NS5B polymerase inhibitor PSI-6130 lack cross-resistance with R1479 . Antimicrob. Agents Chemother. 2008, 52, 4356–4369. [Google Scholar] [CrossRef] [PubMed]
- Reddy, R.; Rodriguez-Torres, M.; Gane, E.; Robson, R.; Lalezari, J.; Everson, G.T.; DeJesus, E.; Mchutchison, J.G.; Vargas, H.E.; Beard, A.; Rodriguez, A.V.; Hill, G.; Symonds, W.; Berrey, M. Antiviral activity, pharmacokinetics, safety, and tolerability of R7128, a novel nucleoside HCV RNA polymerase inhibitor, following multiple, ascending, oral doses in patients with HCV genotype 1 infection who have failed prior interferon therapy . Boston, MA, USA, 2007; pp. 862A–863A. In 58th Annual Meeting of the American Association for the Study of Liver Disease; 10; LB9. [Google Scholar]
- Lalezari, J.; Gane, E.; Rodriguez-Torres, M.; DeJesus, E.; Nelson, D.; Everson, G.T.; Jacobson, I.; Reddy, R.; Hill, G.; Beard, A.; Symonds, W.; Berrey, M.; Mchutchison, J.G. Potent antiviral activity of the HCV nucleoside polymerase inhibitor R7128 with peg-IFN and ribavirin: Interim results of R7128 500mg BID for 28 days . Milan, Italy, 2008; p. S29. In 43th Annual Meeting of the European Association for the Study of the Liver ; 4; Volume 66. [Google Scholar]
- Gane, E.; Rodriguez-Torres, M.; Nelson, D.; Jacobson, I.; Mchutchison, J.G.; Jeffers, L.; Beard, A.; Walker, S.; Shulman, N.; Symonds, W.; Albanis, E.; Berrey, M. Antiviral activity of the HCV nucleoside polymerae inhibitor R7128 in HCV genotype 2 and 3 prior non-responders: Interim results of R7128 1500mg BID with peg-IFN and ribavirin for 28 days . San Francisco, CA, USA, 2008; p. 1024A. In 59th Annual Meeting of the American Association for the Study of Liver Disease; 10 31. [Google Scholar]
- Zhou, X.J.; Pietropaolo, K.; Sullivan-Bolyai, J.; Kuca, B.; Liu, W.; Xu, L.; Belanger, B.; Khan, S.; Mayers, D. IDX184, a liver-targeted nucleotide HCV polymerase inhibitor: Results of a first-in-man safety and pharmacokinetic study . Copenhagen, Denmark; Volume 966, 2009; p. S351. In 44th Annual Meeting of the European Association for the Study of the Liver ; 4. [Google Scholar]
- Lalezari, J.; Ashmut, D.; Casiro, A.; Vargas, H.E.; Dubuc, P.; Liu, W.; Pietropaolo, K.; Zhou, X.; Sullivan-Bolyai, J.; Mayers, D. Antiviral Activity, Safety and Pharmacokinetics of IDX184, A Liver - Targeted Nucleotide HCV Polymerase Inhibitor, in Patients with Chronic Hepatitis C . Boston, MA, USA, 2009; In 60th Annual Meeting of the American Association for the Study of Liver Diseases (AASLD); 10 30. [Google Scholar]
- Furman, P.A.; Wang, P.; Niu, C.; Bao, D.; Symonds, W.; Nagarathnam, D.; Steuer, H.M.; Rachakonda, S.; Bruce, S.R.; Otto, M.J.; Sofia, M. PSI-7851: A novel liver-targeting nucleotide prodrug for the treatment of Hepatitis C. 59th Annual Meeting of the American Association for the Study of Liver Disease 2008. [Google Scholar]
- Rodriguez-Torres, M.; Lawitz, E.; Flach, S.; Denning, J.M.; Albanis, E.; Symonds, W.; Berrey, M. Antiviral Activity, Pharmacokinetics, Safety, and Tolerability of PSI - 7851, a Novel Nucleotide Polymerase Inhibitor for HCV, Following Single and 3 Day Multiple Ascending Oral Doses in Healthy Volunteers and Patients with Chronic HCV Infection . Boston, MA, USA, 2009; LB17In 60th Annual Meeting of the American Association for the Study of Liver Diseases (AASLD); 10 30. [Google Scholar]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF chimera - A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef] [PubMed]
- Holm, L. and Park, J. DaliLite workbench for protein structure comparison. Bioinformatics 2000, 16, 566–567. [Google Scholar] [CrossRef] [PubMed]
- Kukolj, G.; McGibbon, G.A.; McKercher, G.; Marquis, M.; Lefebvre, S.; Thauvette, L.; Gauthier, J.; Goulet, S.; Poupart, M.A.; Beaulieu, P.L. Binding site characterization and resistance to a class of non-nucleoside inhibitors of the hepatitis C virus NS5B polymerase. J. Biol. Chem. 2005, 280, 39260–39267. [Google Scholar] [CrossRef] [PubMed]
- De Francesco, R.; Paonessa, G.; Olsen, D.B.; Rowley, M.; Crescenzi, B.; Habermann, J.; Narjes, F.; Laufer, R. Robust antiviral efficacy of a "finger-loop" allosteric inhibitor of the HCV polymerase in HCV infected chimpanzees . Hawai, USA, 2007; In the HepDart meeting; 12 24. [Google Scholar]
- Brainard, D.M.; Anderson, M.S.; Petry, A.; Van Dyck, K.; De Lepeleire, I.; Sneddon, K.; Cummings, C.E.; Nachbar, R.B.; Barnard, R.J.; Sun, P.; Panorchan, P.; Sanderson, J.B.; Udezue, E.; Wagner, F.; Iwamoto, M.; Chodakewitz, J.; Wagner, J.A. Safety and Antiviral Activity of NS5B Polymerase Inhibitor MK-3281, in Treatment-Naïve Genotype 1A, 1B AND 3 HCV-Infected Patients . Boston, MA, USA, 2009; p. 1568. In 60th Annual Meeting of the American Association for the Study of Liver Diseases (AASLD); 10. [Google Scholar]
- Erhardt, A.; Deterding, K.; Benhamou, Y.; Reiser, M.; Forns, X.; Pol, S.; Calleja, J.L.; Ross, S.; Spangenberg, H.C.; Garcia-Samaniego, J.; Fuchs, M.; Enriquez, J.; Wiegand, J.; Stern, J.; Wu, K.; Kukolj, G.; Marquis, M.; Beaulieu, P.; Nehmiz, G.; Steffgen, J. Safety, pharmacokinetics and antiviral effect of BILB 1941, a novel hepatitis C virus RNA polymerase inhibitor, after 5 days oral treatment. Antivir. Ther. 2009, 14, 23–32. [Google Scholar] [PubMed]
- Chan, L.; Pereira, O.; Reddy, T.J.; Das, S.K.; Poisson, C.; Courchesne, M.; Proulx, M.; Siddiqui, A.; Yannopoulos, C.G.; Nguyen-Ba, N.; Roy, C.; Nasturica, D.; Moinet, C.; Bethell, R.; Hamel, M.; L'heureux, L.; David, M.; Nicolas, O.; Courtemanche-Asselin, P.; Brunette, S.; Bilimoria, D.; Bedard, J. Discovery of thiophene-2-carboxylic acids as potent inhibitors of HCV NS5B polymerase and HCV subgenomic RNA replication. Part 2: Tertiary amides. Bioorg. Med. Chem. Lett. 2004, 14, 797–800. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Ng, K.K.; Cherney, M.M.; Chan, L.; Yannopoulos, C.G.; Bedard, J.; Morin, N.; Nguyen-Ba, N.; Alaoui-Ismaili, M.H.; Bethell, R.C.; James, M.N. Non-nucleoside analogue inhibitors bind to an allosteric site on HCV NS5B polymerase. Crystal structures and mechanism of inhibition. J. Biol. Chem. 2003, 278, 9489–9495. [Google Scholar] [CrossRef] [PubMed]
- Biswal, B.K.; Wang, M.; Cherney, M.M.; Chan, L.; Yannopoulos, C.G.; Bilimoria, D.; Bedard, J.; James, M.N. Non-nucleoside inhibitors binding to hepatitis C virus NS5B polymerase reveal a novel mechanism of inhibition. J Mol. Biol. 2006, 361, 33–45. [Google Scholar] [CrossRef] [PubMed]
- Le Pogam, S.; Kang, H.; Harris, S.F.; Leveque, V.; Giannetti, A.M.; Ali, S.; Jiang, W.R.; Rajyaguru, S.; Tavares, G.; Oshiro, C.; Hendricks, T.; Klumpp, K.; Symons, J.; Browner, M.F.; Cammack, N.; Najera, I. Selection and characterization of replicon variants dually resistant to thumb- and palm-binding nonnucleoside polymerase inhibitors of the hepatitis C virus. J. Virol. 2006, 80, 6146–6154. [Google Scholar] [CrossRef] [PubMed]
- Cooper, C.; Lawitz, E.; Ghali, P.; Rodriguez-Torres, M.; Anderson, F.; Lee, S.; Proulx, L. Antiviral activity of the non-nucleoside polymerase inhibitor, VCH-759, in chronic Hepatitis C patients: Results from a randomized, double-blind, placebo-controlled, ascending multiple dose study . Boston, MA, USA, 2007; p. 864A. In 58th Annual Meeting of the American Association for the Study of Liver Disease; 10; LB9. [Google Scholar]
- Nicolas, O.; Boivin, I.; St-Denis, C.; Bedard, J. Genotypic analysis of HCV NS5B variants selected from patients treated with VCH-759 . Milan, Italy, 2008; p. S317. In 43th Annual Meeting of the European Association for the Study of the Liver; 4; 844. [Google Scholar]
- Proulx, L.; Bourgault, B.; Chauret, N.; Larouche, R.; Tanguay, M.; Thibert, R. Results of a safety, tolerability and pharmacokinetic phase I study of VCH-916, a novel polymerase inhibitor for HCV, following single ascending doses in healthy volunteers . Milan, Italy, 2008; pp. S320–S321. In 43th Annual Meeting of the European Association for the Study of the Liver; 4. [Google Scholar]
- Lawitz, E.; Cooper, C.; Rodriguez-Torres, M.; Ghalib, R.; Lalonde, R.; Sheikh, A.; Bourgault, B.; Chauret, N.; Proulx, L.; Mchutchison, J.G. Safety, tolerability and antiviral activity of VCH-916, a novel non-nucleoside HCV polymerase inhibitor in patients with chronic HCV genotype-1 infection . Copenhagen, Denmark; Volume 92, p. S37. 2009; In 44th Annual Meeting of the European Association for the Study of the Liver ; 4. [Google Scholar]
- Howe, A.Y.; Cheng, H.; Thompson, I.; Chunduru, S.K.; Herrmann, S.; O'Connell, J.; Agarwal, A.; Chopra, R.; Del Vecchio, A.M. Molecular mechanism of a thumb domain hepatitis C virus nonnucleoside RNA-dependent RNA polymerase inhibitor. Antimicrob. Agents Chemother. 2006, 50, 4103–4113. [Google Scholar] [CrossRef] [PubMed]
- Howe, A.Y.; Bloom, J.; Baldick, C.J.; Benetatos, C.A.; Cheng, H.; Christensen, J.S.; Chunduru, S.K.; Coburn, G.A.; Feld, B.; Gopalsamy, A.; Gorczyca, W.P.; Herrmann, S.; Johann, S.; Jiang, X.; Kimberland, M.L.; Krisnamurthy, G.; Olson, M.; Orlowski, M.; Swanberg, S.; Thompson, I.; Thorn, M.; Del Vecchio, A.; Young, D.C.; van Zeijl, M.; Ellingboe, J.W.; Upeslacis, J.; Collett, M.; Mansour, T.S.; O'Connell, J.F. Novel nonnucleoside inhibitor of hepatitis C virus RNA-dependent RNA polymerase. Antimicrob. Agents Chemother. 2004, 48, 4813–4821. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Tatlock, J.; Linton, A.; Gonzalez, J.; Jewell, T.; Patel, L.; Ludlum, S.; Drowns, M.; Rahavendran, S.V.; Skor, H.; Hunter, R.; Shi, S.T.; Herlihy, K.J.; Parge, H.; Hickey, M.; Yu, X.; Chau, F.; Nonomiya, J.; Lewis, C. Discovery of (R)-6-Cyclopentyl-6-(2-(2,6-diethylpyridin-4-yl)ethyl)-3-((5,7-dimethyl-[1 ,2,4]triazolo[1,5-a]pyrimidin-2-yl)methyl)-4-hydroxy-5,6-dihydro-pyran-2-on e (PF-00868554) as a Potent and Orally Available Hepatitis C Virus Polymerase Inhibitor. J Med. Chem. 2009, 52, 1255–1258. [Google Scholar] [CrossRef]
- Shi, S.T.; Herlihy, K.J.; Graham, J.P.; Nonomiya, J.; Rahavendran, S.V.; Skor, H.; Irvine, R.; Binford, S.; Tatlock, J.; Li, H.; Gonzalez, J.; Linton, A.; Patick, A.K.; Lewis, C. Preclinical characterization of PF-00868554, a potent nonnucleoside inhibitor of the hepatitis C virus RNA-dependent RNA polymerase. Antimicrob. Agents Chemother. 2009, 53, 2544–2552. [Google Scholar] [CrossRef] [PubMed]
- Hammond, J.L.; Rosario, M.C.; Wagner, F.; Mazur, D.; Kantaridis, C.; Purohit, V.S.; Durham, K.; Jagannatha, S.; DeBruin, M.F. Antiviral Activity of the Hcv Polymerase Inhibitor Pf-00868554 Administered As Monotherapy in Hcv Genotype 1 Infected Subjects . San Francisco, CA, USA, 2008; pp. 1024A–1025A. In 59th Annual Meeting of the American Association for the Study of Liver Disease; 10 31. [Google Scholar]
- Jacobson, I.; Pockros, P.; Lalezari, J.; Lawitz, E.; Rodriguez-Torres, M.; DeJesus, E.; Haas, F.; Martorell, C.; Pruitt, R.; Durham, K.; Srinivasan, S.; Rosario, M.; Jagannatha, S.; Hammond, J. Antiviral Activity of Filibuvir in Combination with Pegylated Interferon Alfa-2A and Ribavirin for 28 Days in Treatment Naive Patients Chronically Infected with Hcv Genotype 1 . Copenhagen, Denmark, 2009; Volume 50, pp. S382–S383. In 44th Annual Meeting of the European Association for the Study of the Liver ; 4. [Google Scholar]
- Gu, B.H.; Johnston, V.K.; Gutshall, L.L.; Nguyen, T.T.; Gontarek, R.R.; Darcy, M.G.; Tedesco, R.; Dhanak, D.; Duffy, K.J.; Kao, C.C.; Sarisky, R.T. Arresting initiation of hepatitis C virus RNA synthesis using heterocyclic derivatives. Journal of Biological Chemistry 2003, 278, 16602–16607. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, T.T.; Gates, A.T.; Gutshall, L.L.; Johnston, V.K.; Gu, B.; Duffy, K.J.; Sarisky, R.T. Resistance profile of a hepatitis C virus RNA-dependent RNA polymerase benzothiadiazine inhibitor. Antimicrob. Agents Chemother. 2003, 47, 3525–3530. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Torres, M.; Lawitz, E.; Cohen, D.; Larsen, L.M.; Menon, R.; Collins, C.; Marsh, T.; Gibbs, S.; Bernstein, B. Treatment-naïve, HCV genotype 1-infected subjects show significantly greater HCV RNA decreases when treated with 28 days of ABT-333 plus peginterferon and ribavirin compared to peginterferon and ribavirin alone . Boston, MA, USA, 2009; LB16In 60th Annual Meeting of the American Association for the Study of Liver Diseases (AASLD); 10 30. [Google Scholar]
- Chen, C.M.; He, Y.; Lu, L.; Lim, H.B.; Tripathi, R.L.; Middleton, T.; Hernandez, L.E.; Beno, D.W.; Long, M.A.; Kati, W.M.; Bosse, T.D.; Larson, D.P.; Wagner, R.; Lanford, R.E.; Kohlbrenner, W.E.; Kempf, D.J.; Pilot-Matias, T.J.; Molla, A. Activity of a potent hepatitis C virus polymerase inhibitor in the chimpanzee model. Antimicrob. Agents Chemother. 2007, 51, 4290–4296. [Google Scholar] [CrossRef] [PubMed]
- Lawitz, E.; Rodriguez-Torres, M.; DeMico, M.; Nguyen, T.; Godofsky, E.; Appleman, J.; Rahimy, M.; Crowley, C.; Freddo, J. Antiviral Activity of Ana598, A Potent Non-Nucleoside Polymerase Inhibitor, in Chronic Hepatitis C Patients . J. Hepatol. 2009, 50, S384. [Google Scholar] [CrossRef]
- Anadys Pharmaceuticals Inc. Press releases. Available online: http://www.anadyspharma.com/investor/press_releases.asp?year=2009 (accessed 23 April 2009).
- Slater, M.J.; Amphlett, E.M.; Andrews, D.M.; Bravi, G.; Burton, G.; Cheasty, A.G.; Corfield, J.A.; Ellis, M.R.; Fenwick, R.H.; Fernandes, S.; Guidetti, R.; Haigh, D.; Hartley, C.D.; Howes, P.D.; Jackson, D.L.; Jarvest, R.L.; Lovegrove, V.L.; Medhurst, K.J.; Parry, N.R.; Price, H.; Shah, P.; Singh, O.M.; Stocker, R.; Thommes, P.; Wilkinson, C.; Wonacott, A. Optimization of novel acyl pyrrolidine inhibitors of hepatitis C virus RNA-dependent RNA polymerase leading to a development candidate. J Med. Chem. 2007, 50, 897–900. [Google Scholar] [CrossRef] [PubMed]
- Gray, F.; Amphlett, E.; Bright, H.; Chambers, L.; Cheasty, A.; Fenwick, R.; Haigh, D.; Hartley, D.; Howes, P.; Jarvest, R.; Mirzai, F.; Nerozzi, F.; Parry, N.; Slater, M.; Smith, S.; Thommes, P.; Wilkinson, C.; Williams, E. GSK625433; A novel and highly potent inhibitor of the HCVNS5B polymerase . Barcelona, Spain, 2007; p. S225. In 42th Annual Meeting of the European Association for the Study of the Liver ; 4. [Google Scholar]
- Gopalsamy, A.; Aplasca, A.; Ciszewski, G.; Park, K.; Ellingboe, J.W.; Orlowski, M.; Feld, B.; Howe, A.Y. Design and synthesis of 3,4-dihydro-1H-[1]-benzothieno[2,3-c]pyran and 3,4-dihydro-1H-pyrano[3,4-b]benzofuran derivatives as non-nucleoside inhibitors of HCV NS5B RNA dependent RNA polymerase. Bioorg. Med. Chem. Lett. 2006, 16, 457–460. [Google Scholar] [CrossRef] [PubMed]
- Howe, A.Y.; Cheng, H.; Johann, S.; Mullen, S.; Chunduru, S.K.; Young, D.C.; Bard, J.; Chopra, R.; Krishnamurthy, G.; Mansour, T.; O'Connell, J. Molecular mechanism of hepatitis C virus replicon variants with reduced susceptibility to a benzofuran inhibitor, HCV-796. Antimicrob. Agents Chemother. 2008, 52, 3327–3338. [Google Scholar] [CrossRef] [PubMed]
- Chandra, P.; Raible, D.; Harper, D.; Speth, J.; Villano, S.; Bichier, G. Antiviral activity of the non-nucleoside polymerase inhibitor, HCV-796, in patients with chronic hepatitis C virus: Preliminary results from a randomized, double-blind, placebo-controlled, ascending multiple dose study . Gastroenterology 2006, A748. [Google Scholar]
- Villano, S.A.; Raible, D.; Harper, D.; Speth, J.; Chandra, P.; Shaw, P.; Bichier, G. Antiviral activity of the non-nucleoside polymerase inhibitor, HCV-796, in combination with pegylated interferon alfa-2b in treatment-naive patients with chronic HCV . J. Hepatol. 2007, 46, S24. [Google Scholar] [CrossRef]
- Vliegen, I.; Paeshuyse, J.; De Burghgraeve, T.; Lehman, L.S.; Paulson, M.; Shih, I.H.; Mabery, E.; Boddeker, N.; De Clercq, E.; Reiser, H.; Oare, D.; Lee, W.A.; Zhong, W.; Bondy, S.; Purstinger, G.; Neyts, J. Substituted imidazopyridines as potent inhibitors of HCV replication. J Hepatol 2009, 50, 999–1009. [Google Scholar] [CrossRef] [PubMed]
- Edlich, F. and Fischer. Pharmacological targeting of catalyzed protein folding: The example of peptide bond cis/trans isomerases . Handb. Exp. Pharmacol. 2006, 359–404. [Google Scholar]
- Watashi, K.; Ishii, N.; Hijikata, M.; Inoue, D.; Murata, T.; Miyanari, Y.; Shimotohno, K. Cyclophilin B is a functional regulator of hepatitis C virus RNA polymerase. Mol. Cell. 2005, 19, 111–122. [Google Scholar] [CrossRef] [PubMed]
- Chatterji, U.; Bobardt, M.; Selvarajah, S.; Yang, F.; Tang, H.; Sakamoto, N.; Vuagniaux, G.; Parkinson, T.; Gallay, P. The isomerase active site of cyclophilin A is critical for hepatitis C virus replication. J. Biol. Chem. 2009, 284, 16998–17005. [Google Scholar] [CrossRef] [PubMed]
- Kaul, A.; Stauffer, S.; Berger, C.; Pertel, T.; Schmitt, J.; Kallis, S.; Zayas, M.; Lohmann, V.; Luban, J.; Bartenschlager, R. Essential role of cyclophilin A for hepatitis C virus replication and virus production and possible link to polyprotein cleavage kinetics . PLoS. Pathog. 2009, 5, e1000546. [Google Scholar] [CrossRef] [PubMed]
- Yang, F.; Robotham, J.M.; Nelson, H.B.; Irsigler, A.; Kenworthy, R.; Tang, H. Cyclophilin A is an essential cofactor for hepatitis C virus infection and the principal mediator of cyclosporine resistance in vitro. J. Virol. 2008, 82, 5269–5278. [Google Scholar] [CrossRef] [PubMed]
- Nakagawa, M.; Sakamoto, N.; Enomoto, N.; Tanabe, Y.; Kanazawa, N.; Koyama, T.; Kurosaki, M.; Maekawa, S.; Yamashiro, T.; Chen, C.H.; Itsui, Y.; Kakinuma, S.; Watanabe, M. Specific inhibition of hepatitis C virus replication by cyclosporin A. Biochem. Biophys. Res. Commun. 2004, 313, 42–47. [Google Scholar] [CrossRef] [PubMed]
- Watashi, K.; Hijikata, M.; Hosaka, M.; Yamaji, M.; Shimotohno, K. Cyclosporin A suppresses replication of hepatitis C virus genome in cultured hepatocytes. Hepatology 2003, 38, 1282–1288. [Google Scholar] [CrossRef] [PubMed]
- Nakagawa, M.; Sakamoto, N.; Tanabe, Y.; Koyama, T.; Itsui, Y.; Takeda, Y.; Chen, C.H.; Kakinuma, S.; Oooka, S.; Maekawa, S.; Enomoto, N.; Watanabe, M. Suppression of hepatitis C virus replication by cyclosporin a is mediated by blockade of cyclophilins. Gastroenterology 2005, 129, 1031–1041. [Google Scholar] [CrossRef] [PubMed]
- Paeshuyse, J.; Kaul, A.; De Clercq, E.; Rosenwirth, B.; Dumont, J.M.; Scalfaro, P.; Bartenschlager, R.; Neyts, J. The non-immunosuppressive cyclosporin DEBIO-025 is a potent inhibitor of hepatitis C virus replication in vitro. Hepatology 2006, 43, 761–770. [Google Scholar] [CrossRef] [PubMed]
- Coelmont, L.; Kaptein, S.; Paeshuyse, J.; Vliegen, I.; Dumont, J.M.; Vuagniaux, G.; Neyts, J. Debio 025, a cyclophilin binding molecule, is highly efficient in clearing hepatitis C virus (HCV) replicon-containing cells when used alone or in combination with specifically targeted antiviral therapy for HCV (STAT-C) inhibitors. Antimicrob. Agents Chemother. 2009, 53, 967–976. [Google Scholar] [CrossRef] [PubMed]
- Coelmont, L.; Gallay, P.; Bobardt, M.; Kaptein, S.; Paeshuyse, J.; Vliegen, I.; Vuagniaux, G.; Neyts, J. Particular in vitro anti-HCV activities and resistance profile of the cyclophilin inhibitor Debio 025 . 2009, S36. [Google Scholar]
- Kaul, A.; Stauffer, S.; Berger, C.; Pertel, T.; Schmitt, J.; Kallis, S.; Zayas, M.; Lohmann, V.; Luban, J.; Bartenschlager, R. Essential role of cyclophilin A for hepatitis C virus replication and virus production and possible link to polyprotein cleavage kinetics . PLoS. Pathog. 2009, 5, e1000546. [Google Scholar] [CrossRef] [PubMed]
- Flisiak, R.; Horban, A.; Gallay, P.; Bobardt, M.; Selvarajah, S.; Wiercinska-Drapalo, A.; Siwak, E.; Cielniak, I.; Higersberger, J.; Kierkus, J.; Aeschlimann, C.; Grosgurin, P.; Nicolas-Metral, V.; Dumont, J.M.; Porchet, H.; Crabbe, R.; Scalfaro, P. The cyclophilin inhibitor Debio-025 shows potent anti-hepatitis C effect in patients coinfected with hepatitis C and human immunodeficiency virus. Hepatology. 2008, 47, 817–826. [Google Scholar] [CrossRef] [PubMed]
- Flisiak, R.; Feinman, S.V.; Jablkowski, M.; Horban, A.; Kryczka, W.; Pawlowska, M.; Heathcote, J.E.; Mazzella, G.; Vandelli, C.; Nicolas-Metral, V.; Grosgurin, P.; Liz, J.S.; Scalfaro, P.; Porchet, H.; Crabbe, R. The cyclophilin inhibitor Debio 025 combined with PEG IFNalpha2a significantly reduces viral load in treatment-naive hepatitis C patients. Hepatology. 2009, 49, 1460–1468. [Google Scholar] [CrossRef] [PubMed]
- Nelson, D.R.; Ghalib, R.H.; Sulkowski, M.; Schiff, E.; Rustgi, V.; Pockros, P.J.; Wang, C.; Decosterd Kerhuel, G.; Grosgurin, P.; Porchet, H.; Crabbe, R. Efficacy and safety of the cyclophilin inhibitor Debio 025 in combination with pegylated interferon alpha-2a and ribavirin in previously null-responder genotype 1 HCV patients . 2009, S40. [Google Scholar]
- Mathy, J.E.; Ma, S.; Compton, T.; Lin, K. Combinations of cyclophilin inhibitor NIM811 with hepatitis C Virus NS3-4A Protease or NS5B polymerase inhibitors enhance antiviral activity and suppress the emergence of resistance. Antimicrob. Agents Chemother. 2008, 52, 3267–3275. [Google Scholar] [CrossRef] [PubMed]
- Lawitz, E.; Rouzier, R.; Nguyen, T.; Huang, M.; Ke, J.; Praestgaard, J.; Serra, D.; Koziel, M.; Evans, T. Safety and antiviral efficacy of 14 days of the cyclophilin inhibitor NIM811 in combination with pegylated interferon alpha 2a in relapsed genotype 1 HCV infected patients . 2009, S379. [Google Scholar]
- Hopkins, S.; Scorneaux, B.; Huang, Z.; Murray, M.G.; Wring, S.; Smitley, C.; Harris, R.; Erdmann, F.; Fisher, G.; Ribeill, Y. SCY-635: A Novel Non-Immunosuppressive Analog of Cyclosporin A that Exhibits Potent Inhibition of Hepatitis C Virus RNA Replication in vitro. Antimicrob. Agents Chemother. 2009, 54, 660–672. [Google Scholar] [CrossRef] [PubMed]
- Hopkins, S.; Heuman, D.; Gavis, E.; Lalezari, J.; Glutzer, E.; DiMassimo, B.; Rusnak, P.; Wring, S.; Smitley, C.; Ribeill, Y. Safety, plasma pharmacokinetics, and anti-viral activity of SCY-635 in adult patients with chronic hepatitis C virus infection . 2009, 50, S36. [Google Scholar] [PubMed]
- Ellgaard, L. and Helenius, A. ER quality control: Towards an understanding at the molecular level. Curr. Opin. Cell Biol. 2001, 13, 431–437. [Google Scholar] [CrossRef] [PubMed]
- Herscovics, A. Importance of glycosidases in mammalian glycoprotein biosynthesis. Biochim. Biophys. Acta 1999, 1473, 96–107. [Google Scholar] [PubMed]
- Goffard, A.; Callens, N.; Bartosch, B.; Wychowski, C.; Cosset, F.L.; Montpellier, C.; Dubuisson, J. Role of N-linked glycans in the functions of hepatitis C virus envelope glycoproteins. J Virol. 2005, 79, 8400–8409. [Google Scholar] [CrossRef] [PubMed]
- Durantel, D.; Alotte, C.; Zoulim, F. Glucosidase inhibitors as antiviral agents for hepatitis B and C. Curr. Opin. Investig. Drugs 2007, 8, 125–129. [Google Scholar] [PubMed]
- Chapel, C.; Garcia, C.; Bartosch, B.; Roingeard, P.; Zitzmann, N.; Cosset, F.L.; Dubuisson, J.; Dwek, R.A.; Trepo, C.; Zoulim, F.; Durantel, D. Reduction of the infectivity of hepatitis C virus pseudoparticles by incorporation of misfolded glycoproteins induced by glucosidase inhibitors. J Gen. Virol. 2007, 88, 1133–1143. [Google Scholar] [CrossRef] [PubMed]
- Durantel, D.; Chapel, C.; Samuel, E.; Compain, P.; Blu, J.; Martin, O.; Alotte, C.; Bartosch, B.; Zoulim, F. Novel glucosidase inhibitors with anti-HBV and anti-HCV activity . Nice, France, 2009; In 16th International Symposium on Hepatitis C Virus & Related Viruses; 10; pp. 198, 248. [Google Scholar]
- Whitby, K.; Taylor, D.; Patel, D.; Ahmed, P.; Tyms, A.S. Action of celgosivir (6 O-butanoyl castanospermine) against the pestivirus BVDV: Implications for the treatment of hepatitis C. Antivir. Chem. Chemother. 2004, 15, 141–151. [Google Scholar] [PubMed]
- Katia, K.; Yoshida, E.; Kunimoto, D.; Anderson, F.; Sherman, M.; Marotta, P.; Scully, L.; Peltekian, K.; Enns, R.; Diaz-Mitoma, F.; Lee, S.; Worobetz, L.; Pankovich, J.; Petersen, A. Phase II study of celgosivir in combination with peginterferon alfa-2b and ribavirin in chronic hepatitis C genotyp-1 non-responder patitents . Washington, DC, USA, 2007; p. 324227. In Digestive Disease Week; 5. [Google Scholar]
- Ye, J. Reliance of host cholesterol metabolic pathways for the life cycle of hepatitis C virus. PLoS. Pathog. 2007, 3, 1017–1022. [Google Scholar] [CrossRef]
- Burlone, M.E. and Budkowska, A. Hepatitis C virus cell entry: Role of lipoproteins and cellular receptors. J Gen. Virol. 2009, 90, 1055–1070. [Google Scholar] [CrossRef] [PubMed]
- Andre, P.; Komurian-Pradel, F.; Deforges, S.; Perret, M.; Berland, J.L.; Sodoyer, M.; Pol, S.; Brechot, C.; Paranhos-Baccala, G.; Lotteau, V. Characterization of low- and very-low-density hepatitis C virus RNA-containing particles. J. Virol 2002, 76, 6919–6928. [Google Scholar] [CrossRef] [PubMed]
- Amemiya, F.; Maekawa, S.; Itakura, Y.; Kanayama, A.; Matsui, A.; Takano, S.; Yamaguchi, T.; Itakura, J.; Kitamura, T.; Inoue, T.; Sakamoto, M.; Yamauchi, K.; Okada, S.; Yamashita, A.; Sakamoto, N.; Itoh, M.; Enomoto, N. Targeting lipid metabolism in the treatment of hepatitis C virus infection. J. Infect. Dis. 2008, 197, 361–370. [Google Scholar] [CrossRef] [PubMed]
- Schachter, M. Chemical, pharmacokinetic and pharmacodynamic properties of statins: An update. Fundam. Clin. Pharmacol. 2005, 19, 117–125. [Google Scholar] [CrossRef] [PubMed]
- Ikeda, M.; Abe, K.; Yamada, M.; Dansako, H.; Naka, K.; Kato, N. Different anti-HCV profiles of statins and their potential for combination therapy with interferon. Hepatology 2006, 44, 117–125. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.S.; Peng, L.F.; Lin, W.; Choe, W.-H.; Sakamoto, N.; Schreiber, S.L.; Chung, R.T. A cell-based, high-throughput screen for small molecule regulators of hepatitis C virus replication. Gastroenterology 2007, 132, 311–320. [Google Scholar] [CrossRef] [PubMed]
- Delang, L.; Paeshuyse, J.; Vliegen, I.; Leyssen, P.; Obeid, S.; Durantel, D.; Zoulim, F.; Op, d.B.; Neyts, J. Statins potentiate the in vitro anti-hepatitis C virus activity of selective hepatitis C virus inhibitors and delay or prevent resistance development. Hepatology 2009, 50, 6–16. [Google Scholar] [CrossRef] [PubMed]
- Kapadia, S.B.; Chisari, F.V. Hepatitis C virus RNA replication is regulated by host geranylgeranylation and fatty acids. Proc. Natl. Acad. Sci. U. S. A 2005, 102, 2561–2566. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Gale, M.; Keller, B.C.; Huang, H.; Brown, M.S.; Goldstein, J.L.; Ye, J. Identification of FBL2 as a geranylgeranylated cellular protein required for hepatitis C virus RNA replication . Molecular Cell 2005, 18, 425–434. [Google Scholar] [CrossRef] [PubMed]
- O'Leary, J.G.; Chan, J.L.; McMahon, C.M.; Chung, R.T. Atorvastatin does not exhibit antiviral activity against HCV at conventional doses: A pilot clinical trial. Hepatology 2007, 45, 895–898. [Google Scholar] [CrossRef] [PubMed]
- George, J.O.; Kenedi, C.; Brown, K.; Zekry, A.; Jhaveri, R.; Kilaru, R.; Conrad, A.; Mchutchison, J.G.; Patel, K. A pilot study to assess the impact of rosuvastatin therapy on HCV RNA and lipid fractions in chronic hepatitis c patients . Gastroenterology 2007, 132, A741–A741. [Google Scholar]
- Bader, T.; Fazili, J.; Madhoun, M.; Aston, C.; Hughes, D.; Rizvi, S.; Seres, K.; Hasan, M. Fluvastatin Inhibits Hepatitis C Replication in Humans. Am J Gastroenterol 2008, 103, 1383–1389. [Google Scholar] [CrossRef] [PubMed]
- Harrison, S.; Rossaro, L.; Hu, K.; Patel, K.; Tillman, H.L.; Dhaliwal, S.; Torres, D.M.; Koury, K.; Goteti, V.; Brass, C.A.; Noviello, S.; Albrecht, J.K.; Mchutchison, J.G.; Sulkowski, M. Relationship of the Use of Statins and Elevated Low - Density Lipoprotein ( LDL ) or Total Cholesterol ( TC ) to Virologic Response in Patients Treated for Hepatitis C Virus ( HCV ) in the IDEAL Study . Boston, MA, USA, 2009; p. 120. In 60th Annual Meeting of the American Association for the Study of Liver Diseases (AASLD); 10 30. [Google Scholar]
- Sezaki, H.; Suzuki, F.; Akuta, N.; Yatsuji, H.; Hosaka, T.; Kobayashi, M.; Suzuki, Y.; Arase, Y.; Ikeda, K.; Miyakawa, Y.; Kumada, H. An Open Pilot Study Exploring the Efficacy of Fluvastatin, Pegylated Interferon and Ribavirin in Patients with Hepatitis C Virus Genotype 1b in High Viral Loads. Intervirology 2009, 52, 43–48. [Google Scholar] [CrossRef] [PubMed]
- Rossignol, J.F.; Keeffe, E.B. Thiazolides: A new class of drugs for the treatment of chronic hepatitis B and C. Future Microbiol. 2008, 3, 539–545. [Google Scholar] [CrossRef] [PubMed]
- Korba, B.E.; Elazar, M.; Lui, P.; Rossignol, J.F.; Glenn, J.S. Potential for hepatitis C virus resistance to nitazoxanide or tizoxanide. Antimicrob. Agents Chemother. 2008, 52, 4069–4071. [Google Scholar] [CrossRef] [PubMed]
- Korba, B.E.; Montero, A.B.; Farrar, K.; Gaye, K.; Mukerjee, S.; Ayers, M.S.; Rossignol, J.F. Nitazoxanide, tizoxanide and other thiazolides are potent inhibitors of hepatitis B virus and hepatitis C virus replication. Antiviral Res. 2008, 77, 56–63. [Google Scholar] [CrossRef] [PubMed]
- Rossignol, J.F.; Kabil, S.M.; El Gohary, Y.; Elfert, A.; Keeffe, E.B. Clinical trial: Randomized, double-blind, placebo-controlled study of nitazoxanide monotherapy for the treatment of patients with chronic hepatitis C genotype 4. Aliment. Pharmacol. Ther. 2008, 28, 574–580. [Google Scholar] [CrossRef] [PubMed]
- Rossignol, J.F.; Elfert, A.; El Gohary, Y.; Keeffe, E.B. Improved virologic response in chronic hepatitis C genotype 4 treated with nitazoxanide, peginterferon, and ribavirin. Gastroenterology 2009, 136, 856–862. [Google Scholar] [CrossRef] [PubMed]
- Basu, P.; Rayapudi, K.; Shah, N.; Pacana, T.; Brown, R.S. Effects of High Dose Ribavirin (RBV), Alinia (Nitazoxanide) and Pegylated Interferon (PEG)alfa-2a in Attaining Sustained Viral Response (SVR) in Treatment of Chronic Hepatitis C (ERAIS-C trial) - Interim Results in Naïve Genotype 1 Patients . Boston, MA, USA, 2009; p. 904. In 60th Annual Meeting of the American Association for the Study of Liver Diseases (AASLD); 10 30. [Google Scholar]
- Yoffe, B.; Gasitashvili, K.; Khaoustov, V. Pilot study of lead-in nitazoxanide plus pegylated alpha-2a interferon and ribavirin in HCV-genotype 1 nonresponders with cirrhosis: Interim results . Boston, MA, USA, 2009; p. 1580. In 60th Annual Meeting of the American Association for the Study of Liver Diseases (AASLD); 10 30. [Google Scholar]
- Ahmed-Belkocem, A.; Ahnou, N.; Barbotte, L.; Wychowski, C.; Brillet, R.; Pohl, R.T.; Pawlotsky, J.M. Silibinin and Related Compounds Are Direct Inhibitors of Hepatitis C Virus Rna-Dependent Rna Polymerase . Hepatology 2009, 50, 412A–412A. [Google Scholar] [CrossRef]
- Polyak, S.J.; Morishima, C.; Shuhart, M.C.; Wang, C.C.; Liu, Y.Z.; Lee, D.Y.W. Inhibition of T-cell inflammatory cytokines, hepatocyte NF-kappa B signaling, and HCV infection by standardized silymarin. Gastroenterology 2007, 132, 1925–1936. [Google Scholar] [CrossRef] [PubMed]
- Ferenci, P.; Scherzer, T.M.; Kerschner, H.; Rutter, K.; Beinhardt, S.; Hofer, H.; Schoniger-Hekele, M.; Holzmann, H.; Steindl-Munda, P. Silibinin is a potent antiviral agent in patients with chronic hepatitis C not responding to pegylated interferon/ribavirin therapy. Gastroenterology 2008, 135, 1561–1567. [Google Scholar] [CrossRef] [PubMed]
- Gordon, A.; Hobbs, D.A.; Bowden, D.S.; Bailey, M.J.; Mitchell, J.; Francis, A.J.; Roberts, S.K. Effects of Silybum marianum on serum hepatitis C virus RNA, alanine aminotransferase levels and well-being in patients with chronic hepatitis C. J Gastroenterol. Hepatol 2006, 21, 275–280. [Google Scholar] [CrossRef] [PubMed]
- Hawke, R.L.; Schrieber, S.J.; Soule, T.A.; Wen, Z.; Smith, P.C.; Reddy, K.R.; Wahed, A.S.; Belle, S.H.; Afdhal, N.H.; Navarro, V.J.; Berman, J.; Liu, Q.Y.; Doo, E.; Fried, M.W. Silymarin Ascending Multiple Oral Dosing Phase I Study in Noncirrhotic Patients With Chronic Hepatitis C . J Clin. Pharmacol. 2009. [Google Scholar]
- McCown, M.F.; Rajyaguru, S.; Le Pogam, S.; Ali, S.; Jiang, W.R.; Kang, H.; Symons, J.; Cammack, N.; Najera, I. The hepatitis C virus replicon presents a higher barrier to resistance to nucleoside analogs than to nonnucleoside polymerase or protease inhibitors. Antimicrob. Agents Chemother. 2008, 52, 1604–1612. [Google Scholar] [CrossRef] [PubMed]
- Coelmont, L.; Kaptein, S.; Paeshuyse, J.; Vliegen, I.; Dumont, J.M.; Vuagniaux, G.; Neyts, J. Debio 025, a Cyclophilin Binding Molecule, Is Highly Efficient in Clearing Hepatitis C Virus (HCV) Replicon-Containing Cells When Used Alone or in Combination with Specifically Targeted Antiviral Therapy for HCV (STAT-C) Inhibitors. Antimicrobial Agents and Chemotherapy 2009, 53, 967–976. [Google Scholar] [CrossRef] [PubMed]
- Gane, E.; Roberts, S.K.; Stedman, C.; Angus, P.W.; Ritchie, B.; Elston, R.; Ipe, D.; Morcos, P.; Najera, I.; Chu, T.; Berrey, M.; Bradford, W.; Laughlin, M.; Shulman, N.; Smith, P.F. Combination Therapy With A Nucleoside Polymerase (R7128) And Protease (R7227/ITMN-191) Inhibitor In HCV: Safety, Pharmacokinetics, And Virologic Results From INFORM-1 . Boston, MA, USA, 2009; p. 193. In 60th Annual Meeting of the American Association for the Study of Liver Diseases (AASLD); 10 30. [Google Scholar]
- Le Pogam, S.; Chhabra, M.; Ali, S.; Yan, J.; Ilnicka, M.J.; Kang, H.; Wong, J.; Kosaka, A.; Ewing, A.; Seshaadri, A.; De La Rosa, A.; Bradford, W.; Klumpp, K.; Shulman, N.; Smith, P.F.; Cammack, N.; Najera, I. Combination therapy with nucleoside polymerase R7128 and protease R7227/ITMN-191 inhibitors in genotype 1 HCV infected patients: Interim resistance analysis of INFORM-1 cohorts A-D . Boston, MA, USA, 2009; p. 1580. In 60th Annual Meeting of the American Association for the Study of Liver Diseases (AASLD); 10 30. [Google Scholar]
© 2010 by the authors; licensee MDPI, Basel, Switzerland This is an Open Access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Share and Cite
Delang, L.; Coelmont, L.; Neyts, J. Antiviral Therapy for Hepatitis C Virus: Beyond the Standard of Care. Viruses 2010, 2, 826-866. https://doi.org/10.3390/v2040826
Delang L, Coelmont L, Neyts J. Antiviral Therapy for Hepatitis C Virus: Beyond the Standard of Care. Viruses. 2010; 2(4):826-866. https://doi.org/10.3390/v2040826
Chicago/Turabian StyleDelang, Leen, Lotte Coelmont, and Johan Neyts. 2010. "Antiviral Therapy for Hepatitis C Virus: Beyond the Standard of Care" Viruses 2, no. 4: 826-866. https://doi.org/10.3390/v2040826
APA StyleDelang, L., Coelmont, L., & Neyts, J. (2010). Antiviral Therapy for Hepatitis C Virus: Beyond the Standard of Care. Viruses, 2(4), 826-866. https://doi.org/10.3390/v2040826