
Evolution of Phage Tail Sheath Protein



Abstract
:1. Introduction
2. Materials and Methods
2.1. Data Acquisition and Annotation
2.2. Protein Sequence Alignment and Phylogeny
2.3. Protein Tertiary Structure Modelling, Visualisation and In Silico Analysis
3. Results
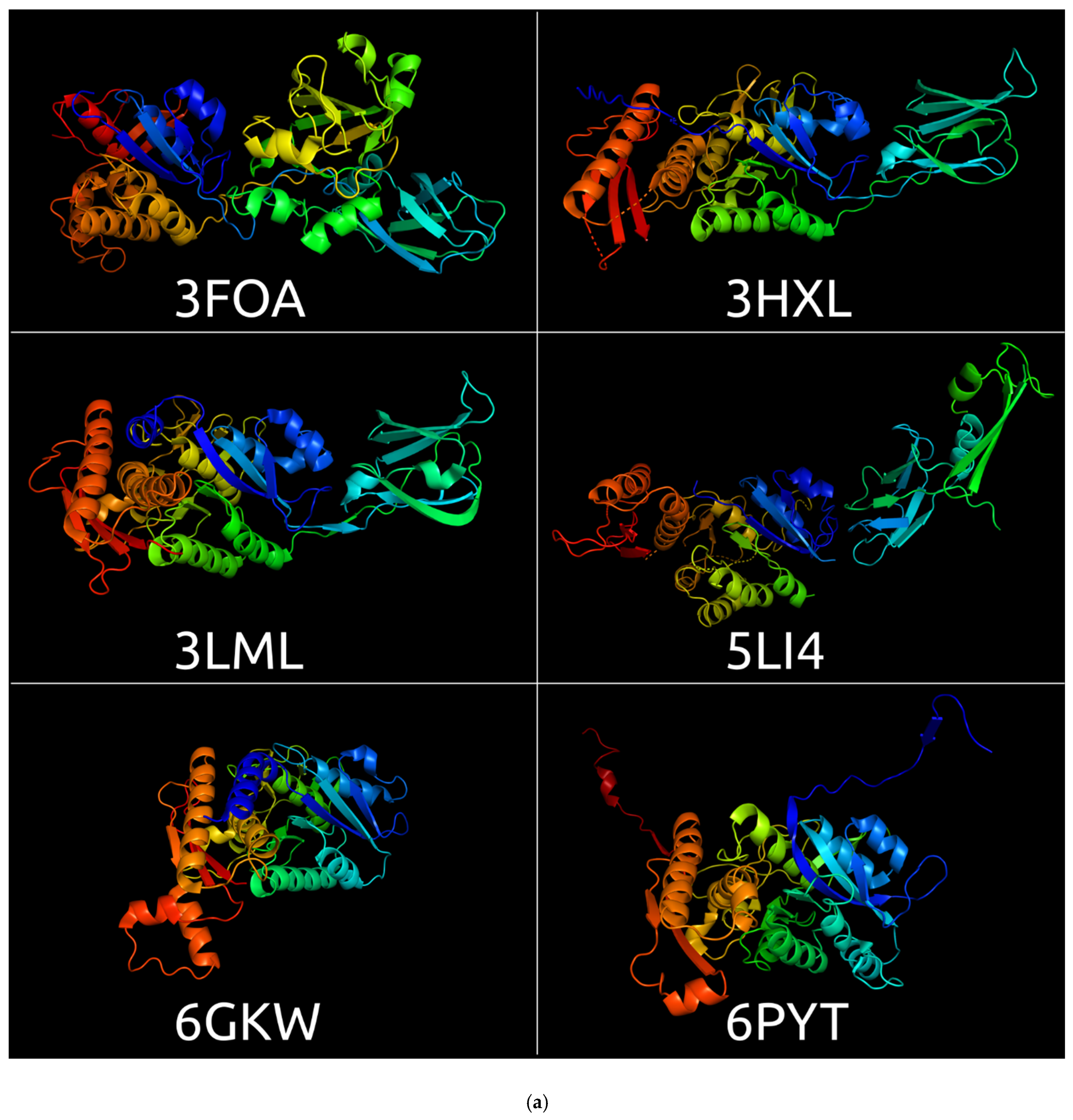
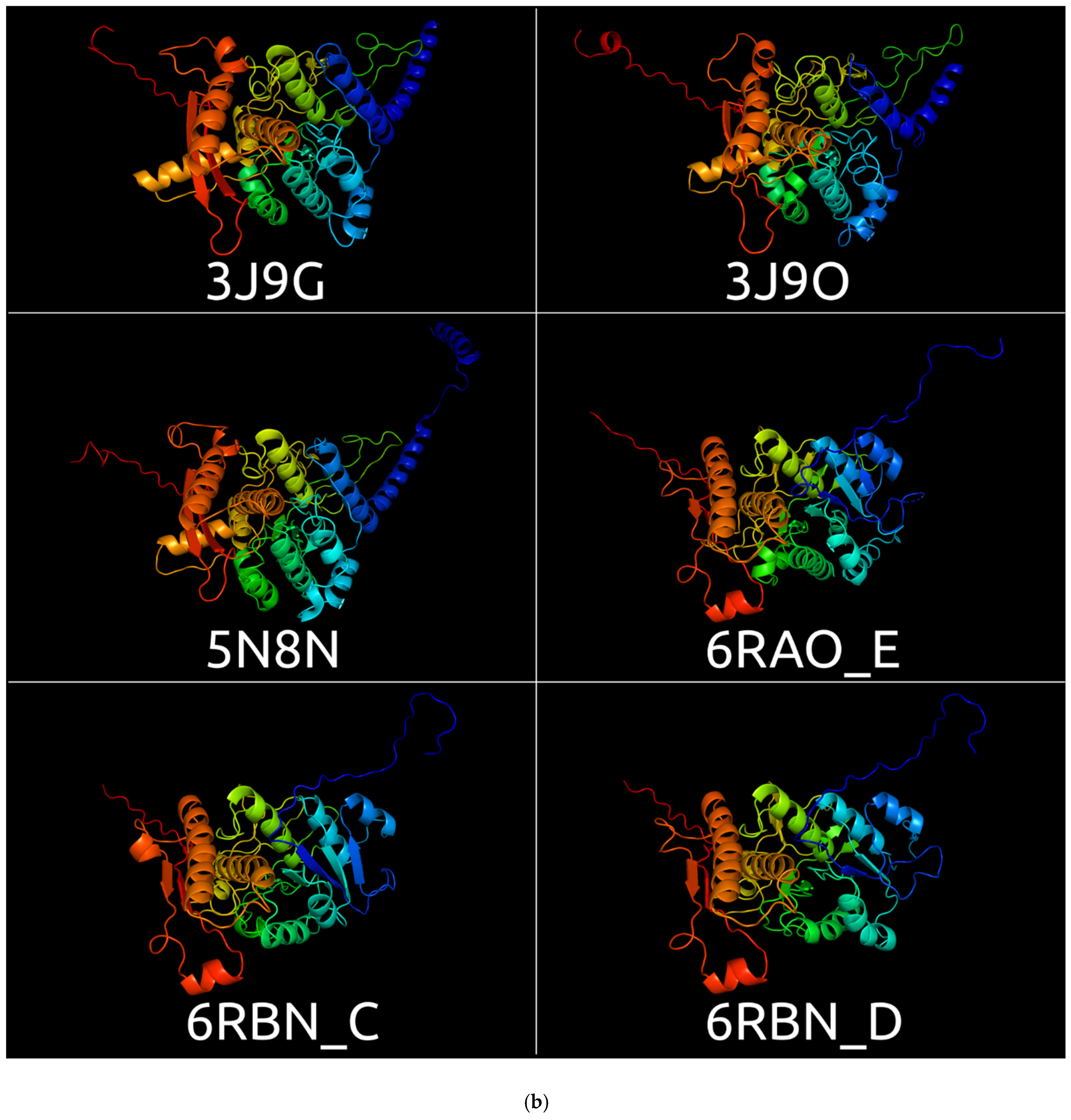
3.1. Sheath Proteins in the RCSB Protein Bank Database
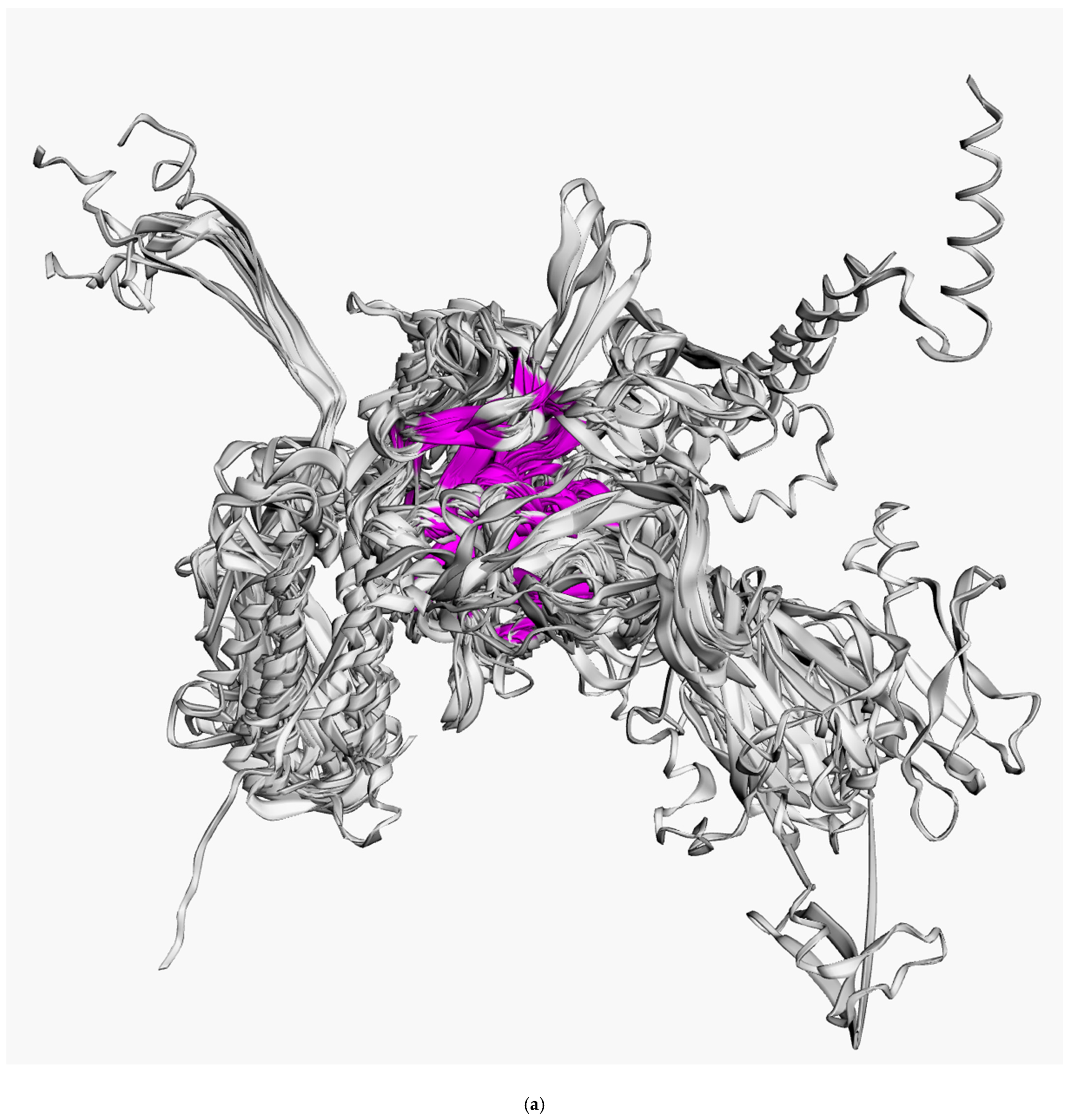
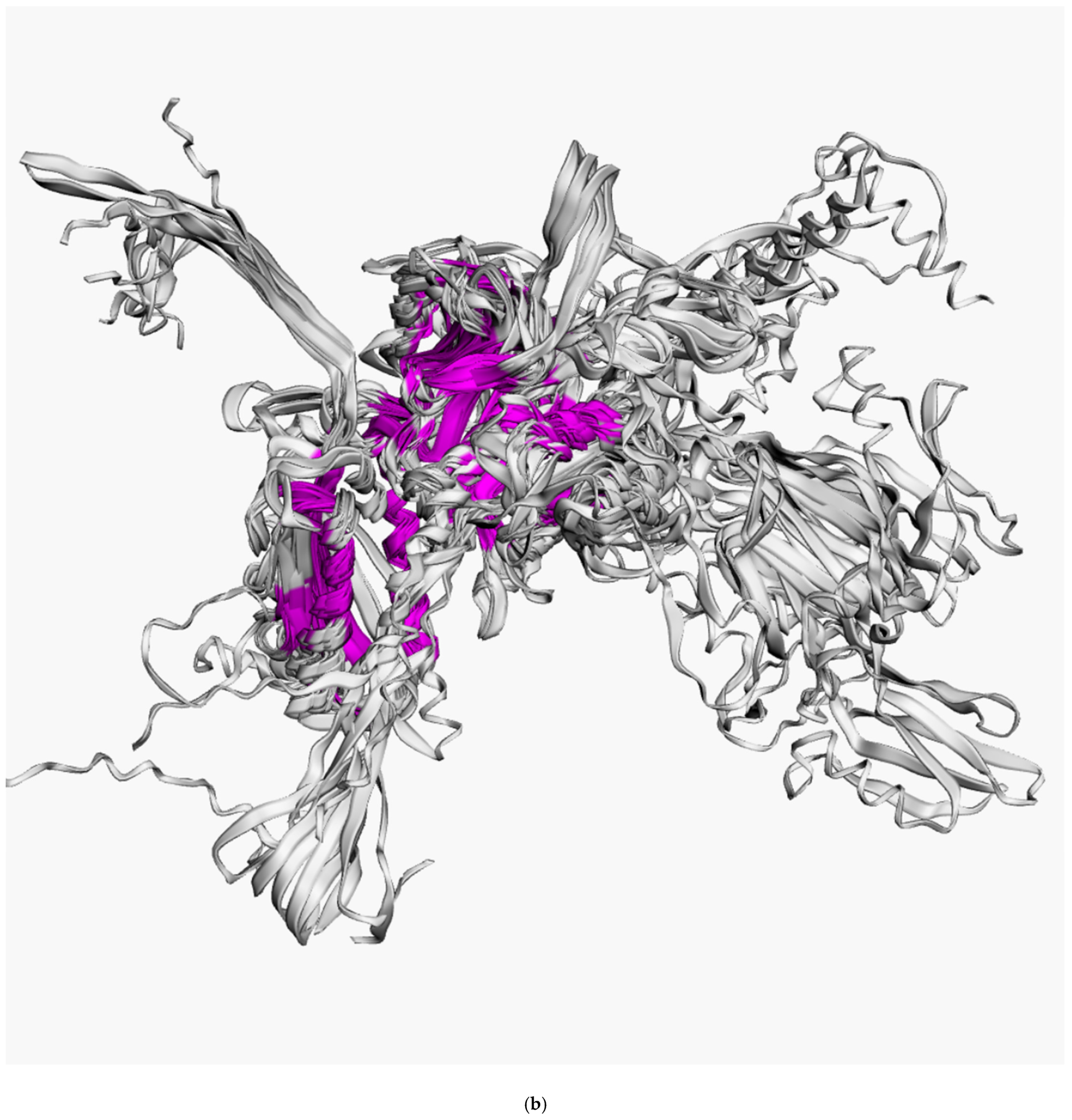
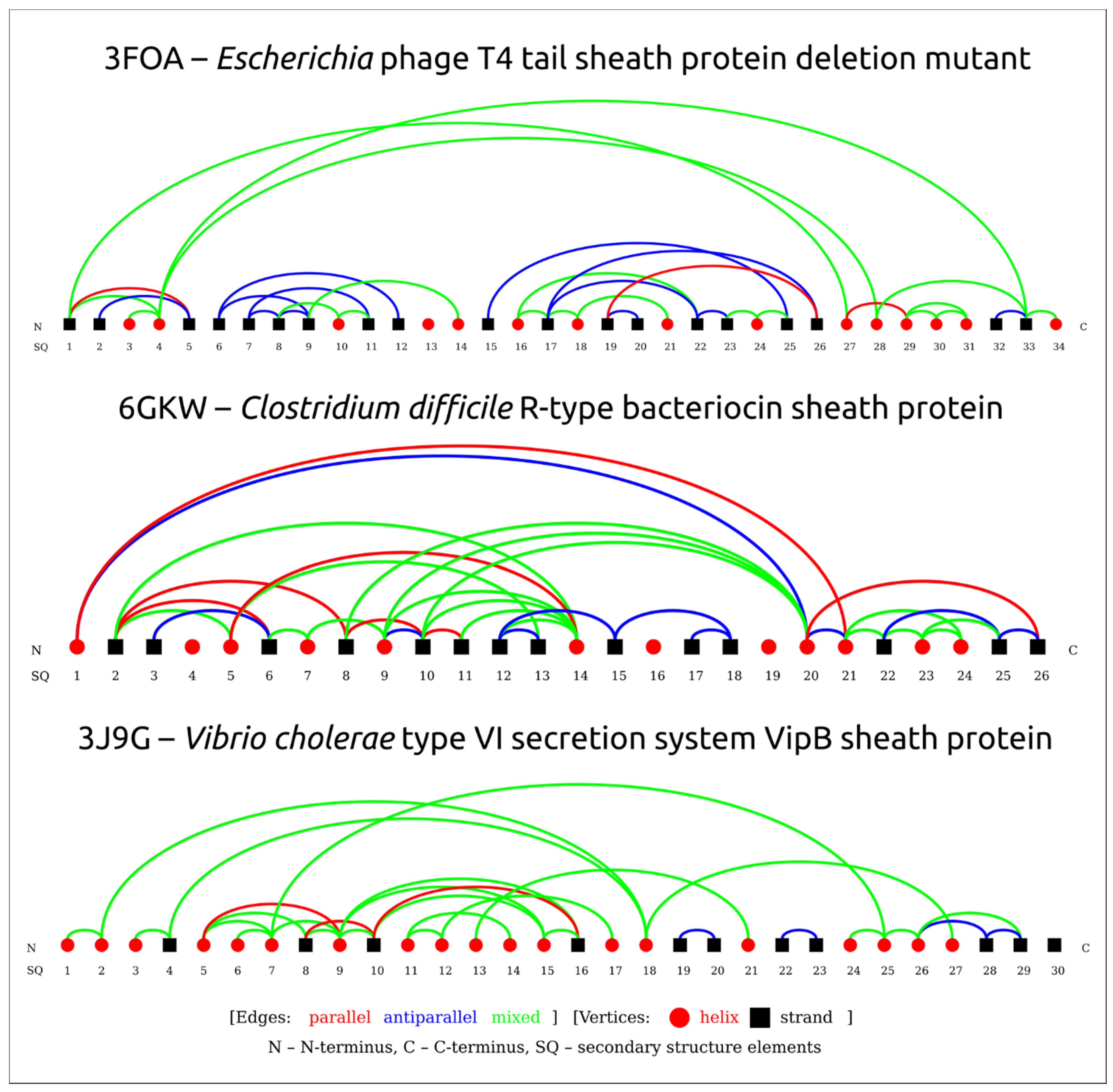
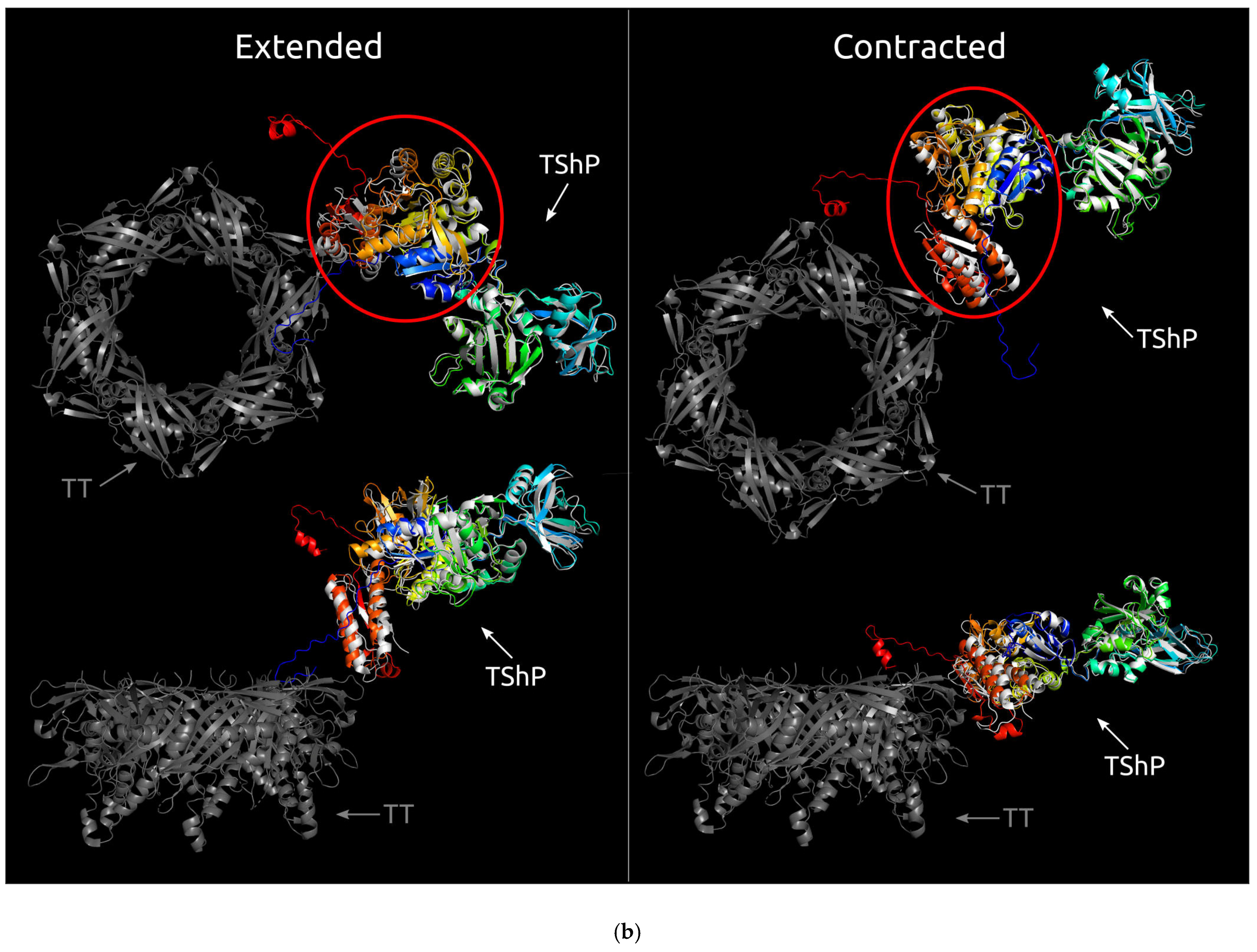
3.2. Positioning of the Conserved Core in Experimentally Determined TShPs
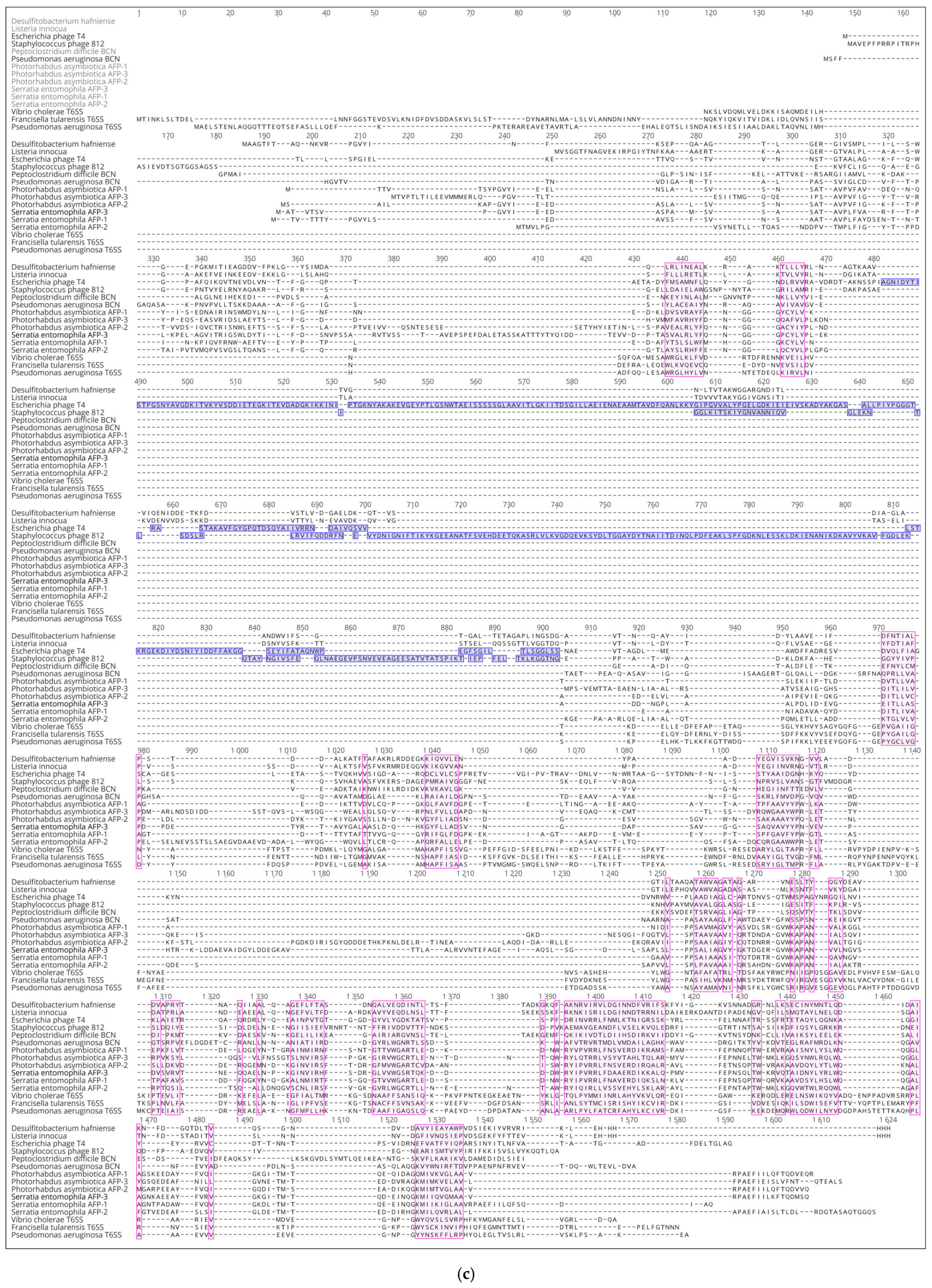
3.3. Choosing Representative Sequences for Modelling
- Haloarcula phages of Haloferacalesvirus genus: Ten complete genomes (HCTV-6, -7, -8, -9, -10, -11, -15 and HJTV-1, -2, -3) possess similar genome organisation and length. The predicted TShPs possess 431 to 438 amino acid (aa) residues. HCTV-6 and HCTV-15 are identical and differ in their primary sequence from eight other TSPs (% identity is about 45–47%). The latter proteins are very similar, or identical, showing a pairwise identity of 84% and higher.
- Halobacterium phages of Myohalovirus genus: two complete genomes (phiH and ChaoS9) contain two TSPs of about 430 aa length that have 52% identity and identical HHM-HHM motif comparison results obtained with HHpred [28].
- Haloferax phage HF1 of Haloferacalesvirus genus: the HF1 tail sheath protein amino acid sequence is similar to the TShPs of Haloarcula phages HCTV-7, -8, -9, -10, -11, and HJTV 1, -2, -3, and has an identity to Haloarcula phages of about 90%.
- Halorubrum phages of Haloferacalesvirus genus: 28 genomes encode the TShPs of about 430 aa lengths. Of these, 23 TShP sequences are very similar to one another (83–99%). They belong to Halobrum phage HF2; Halorubrum Tailed Viruses 5 and 8; phages Hardycor2; Halorubrum viruses HRTV-9, -10, -13, -14, -15, -16, -17, -18, -19, -20, -21, -22, -23, -24, -26; Halorubrum virus HSTV-4; Serpecor1; and VOLN27B. Tail sheath proteins from Halovirus HSTV-2, Halorubrum Tailed Virus 7, and Halorubrum viruses HRTV-2 and HRTV-11 have 97–98% identity with one another and less than 50% identity with all other Halobrum phages, and constitute another group of Halorubrum phage TShPs. The TShPs of phages HRTV-25 and HRTV-27 show less than 40–50% primary sequence identity with one another and all other Halobrum TShPs.
- Natrialba phage φCh1 of Myohalovirus genus: a TShP was revealed by a BLAST and HMM search. The protein is 426 aa in length and shows about 40% identity with Halobacterium phages.
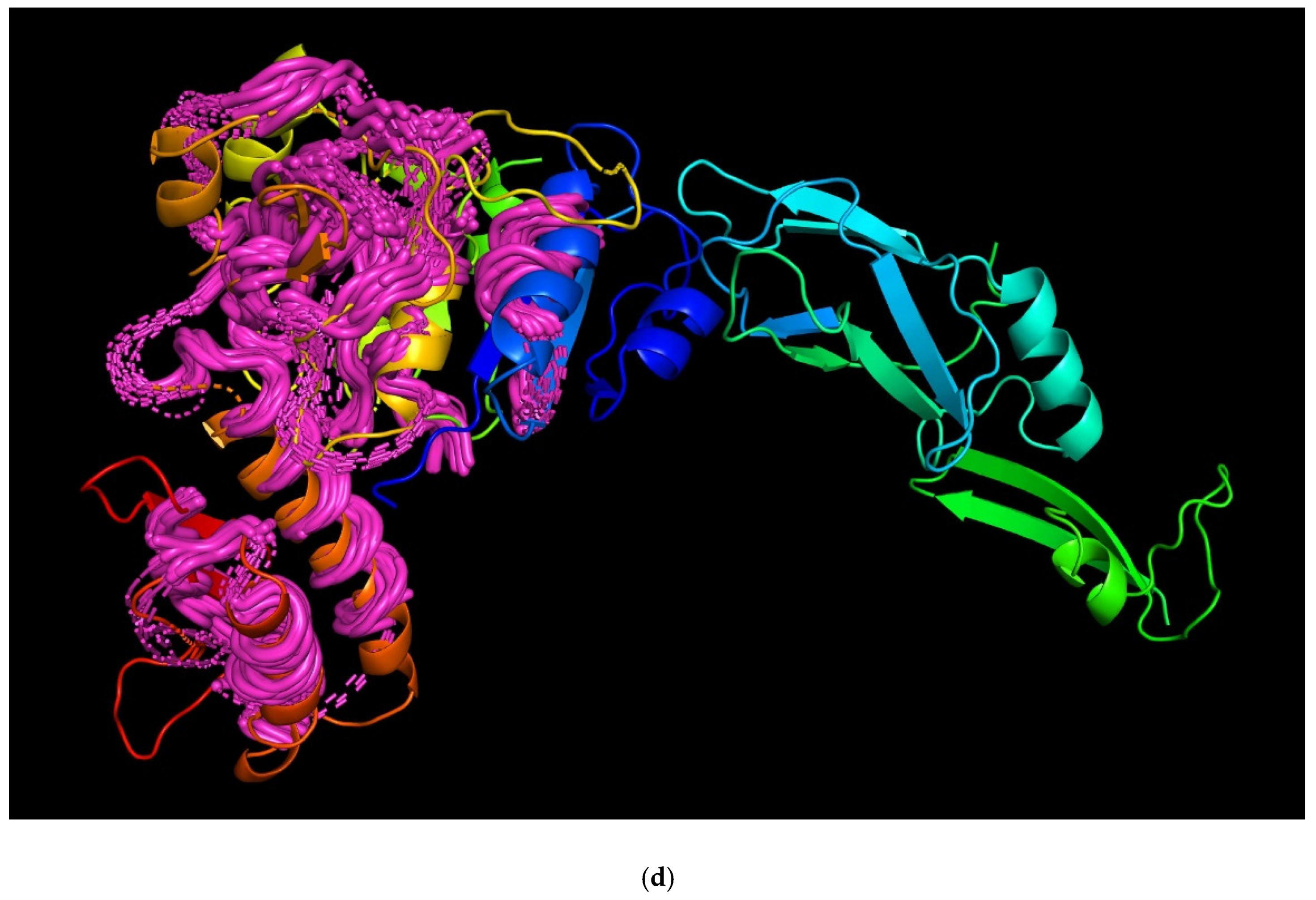
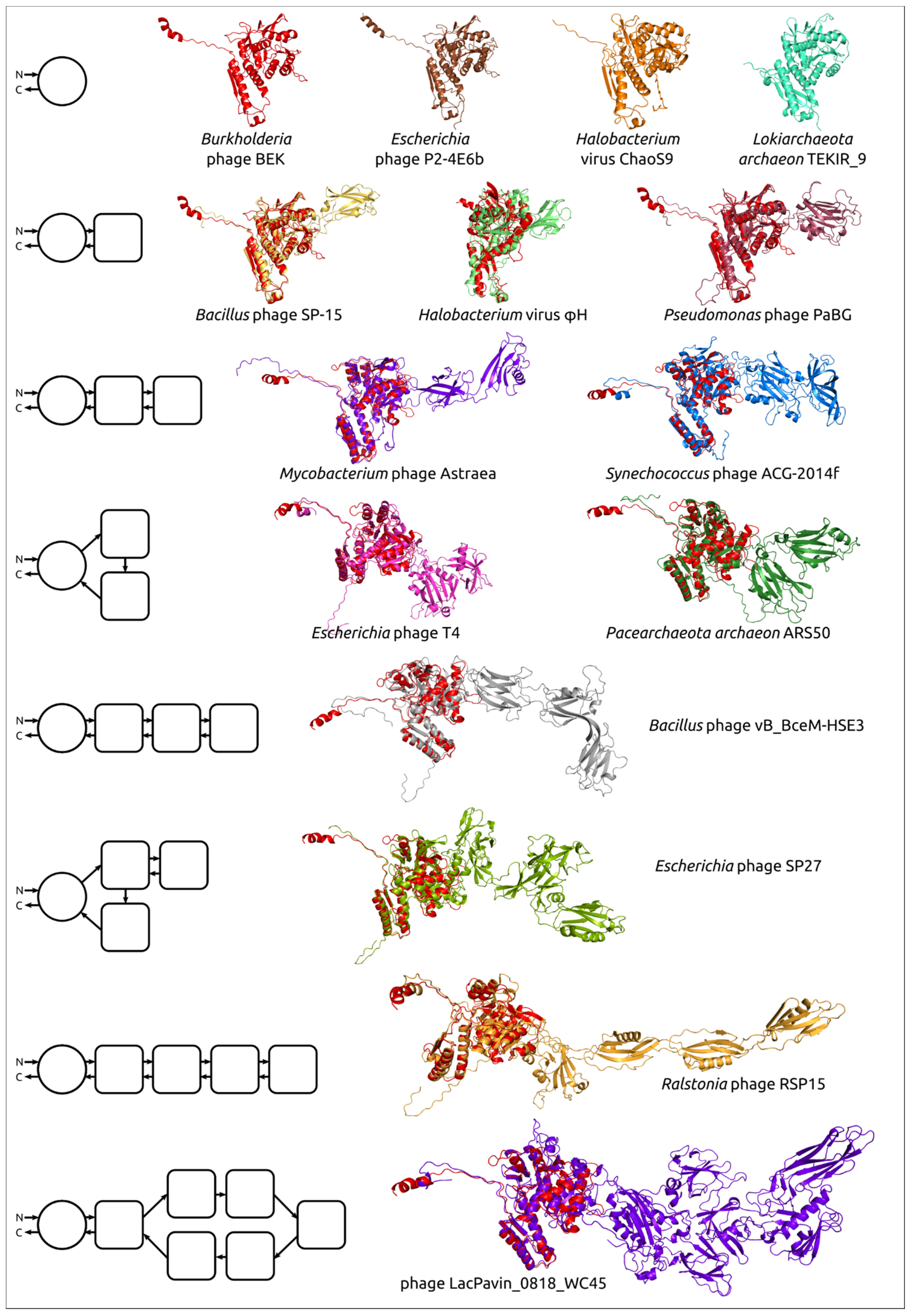
3.4. Modelling and General Structural Analysis of Representative Sheath Proteins
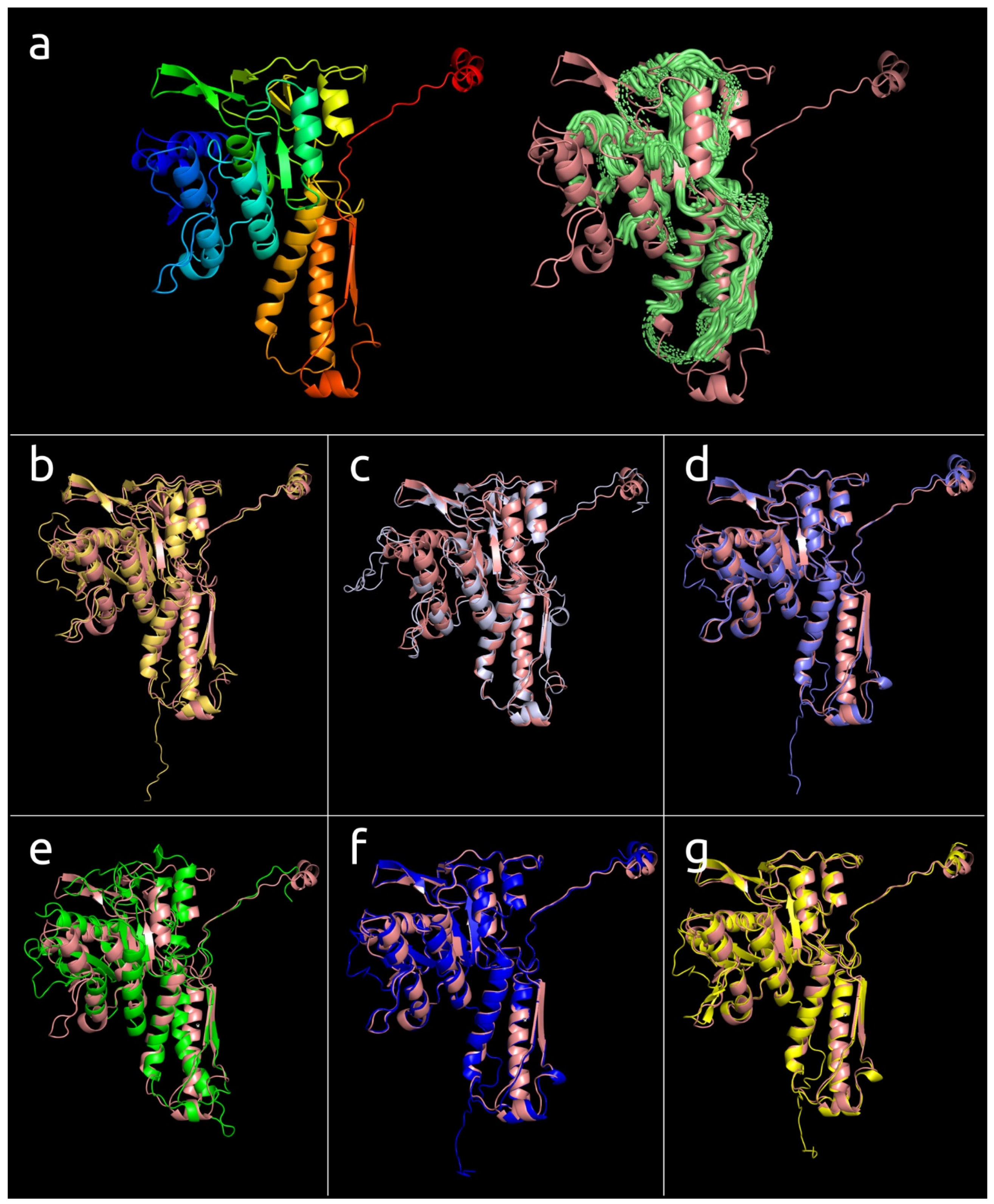
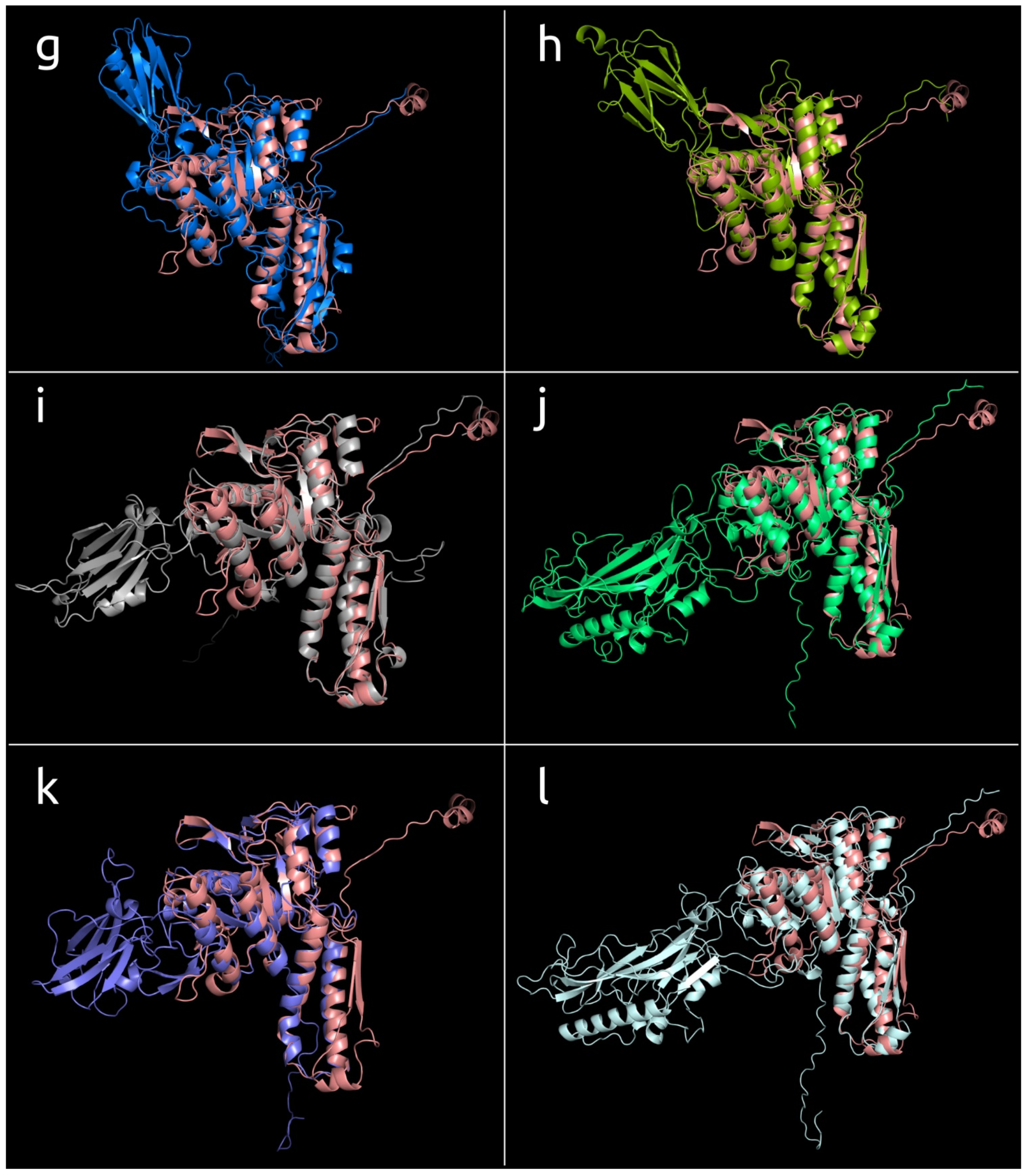
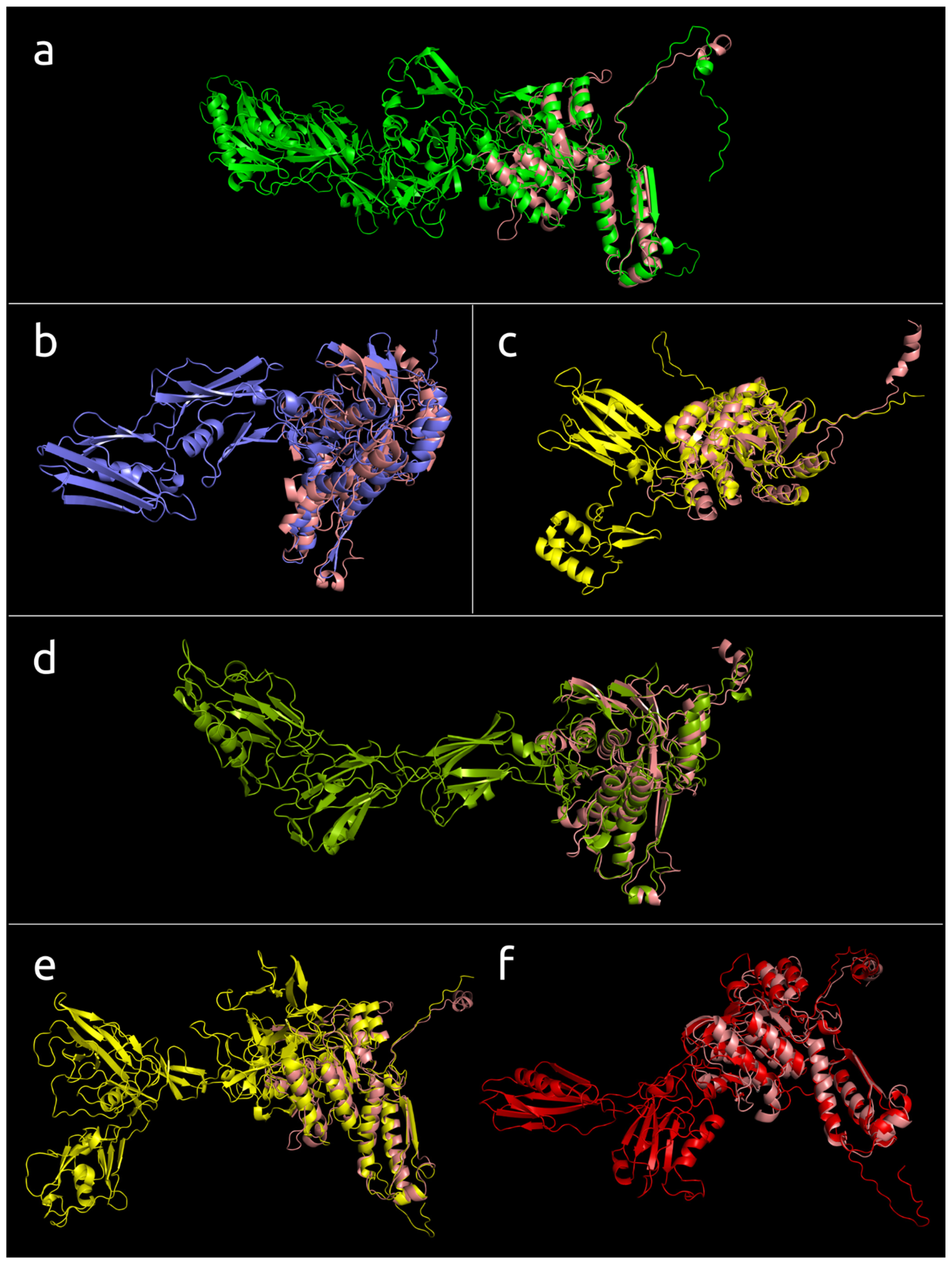
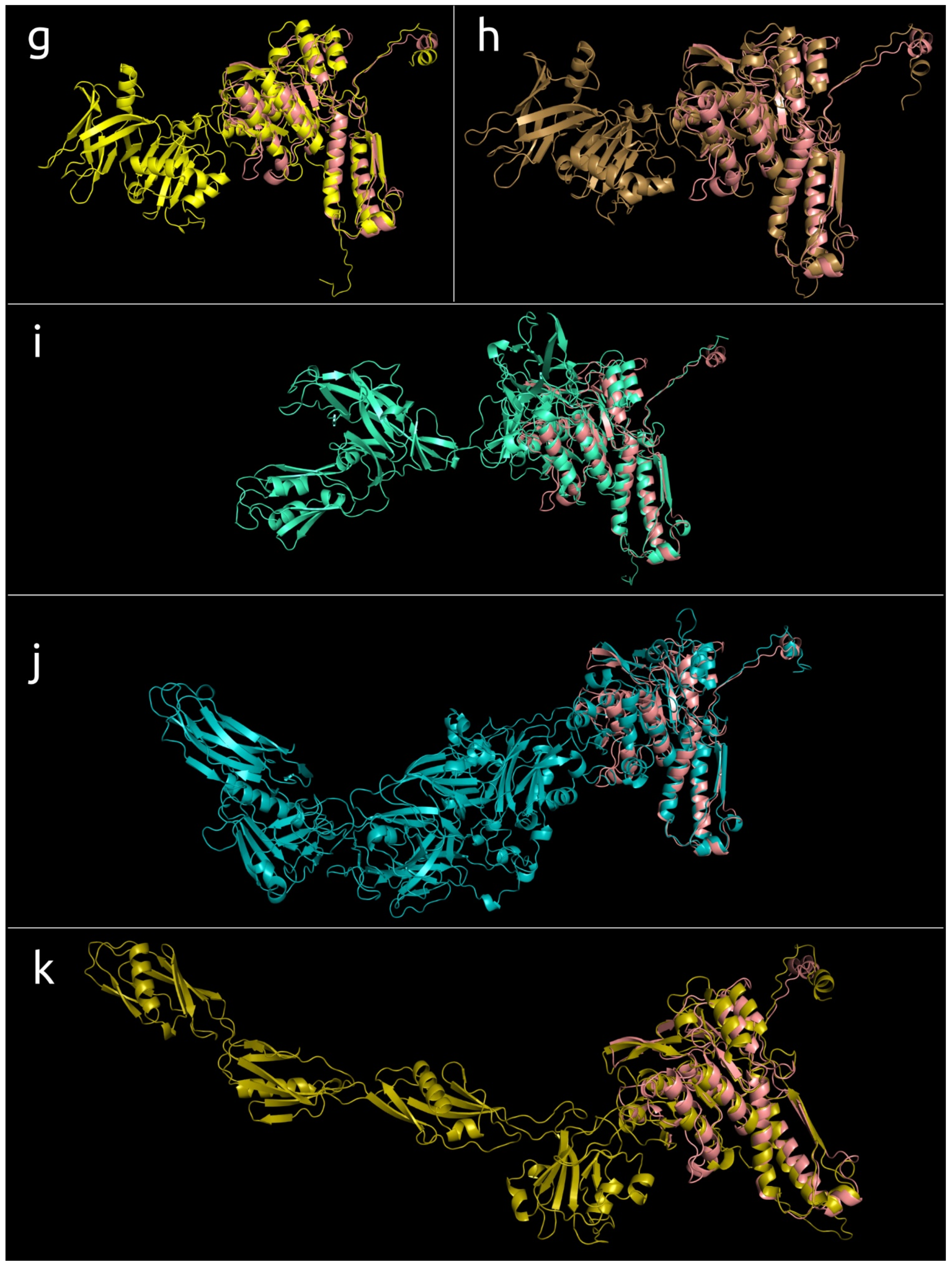
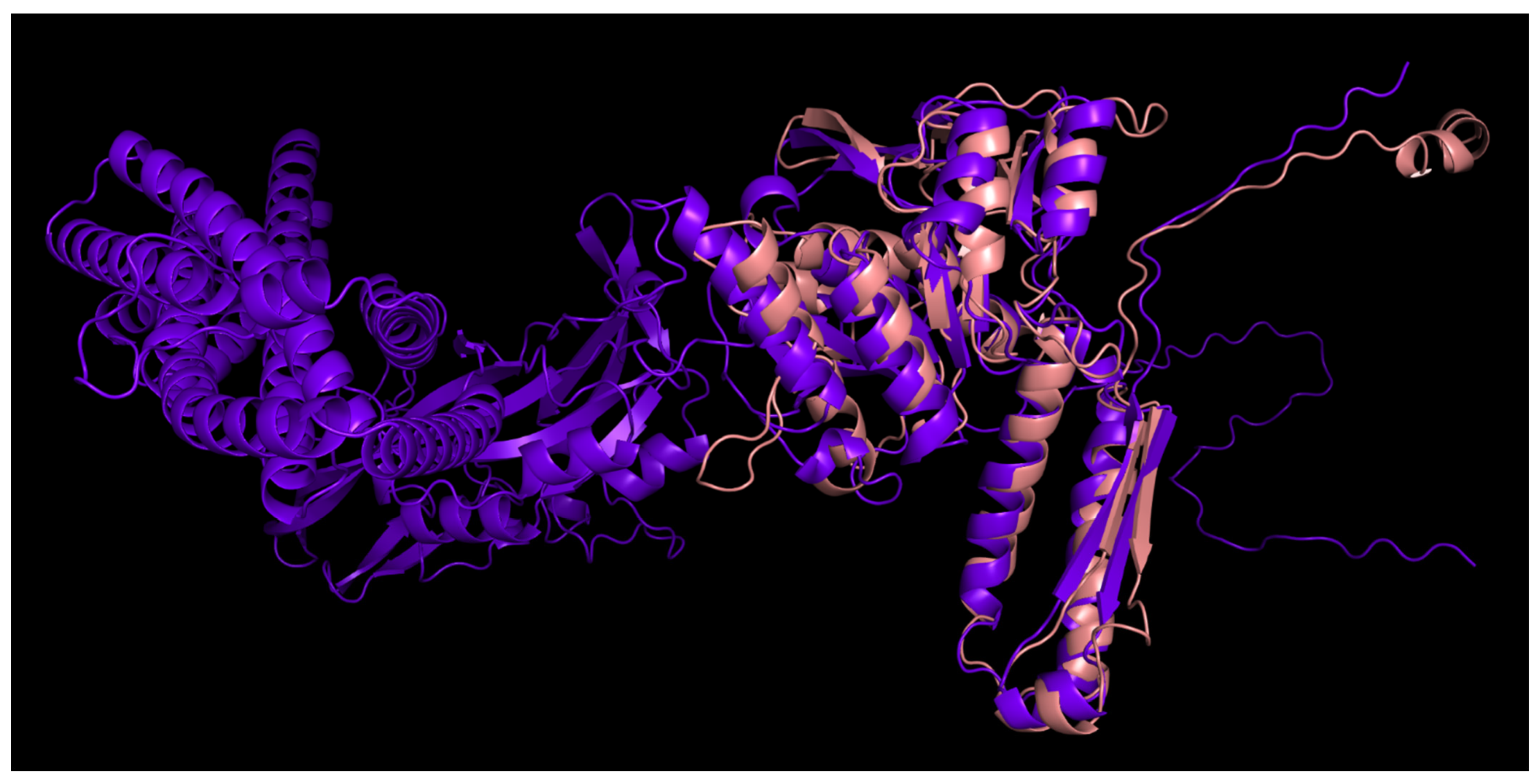
3.4.1. One-Domain Contractile Sheath Proteins (Type 1)
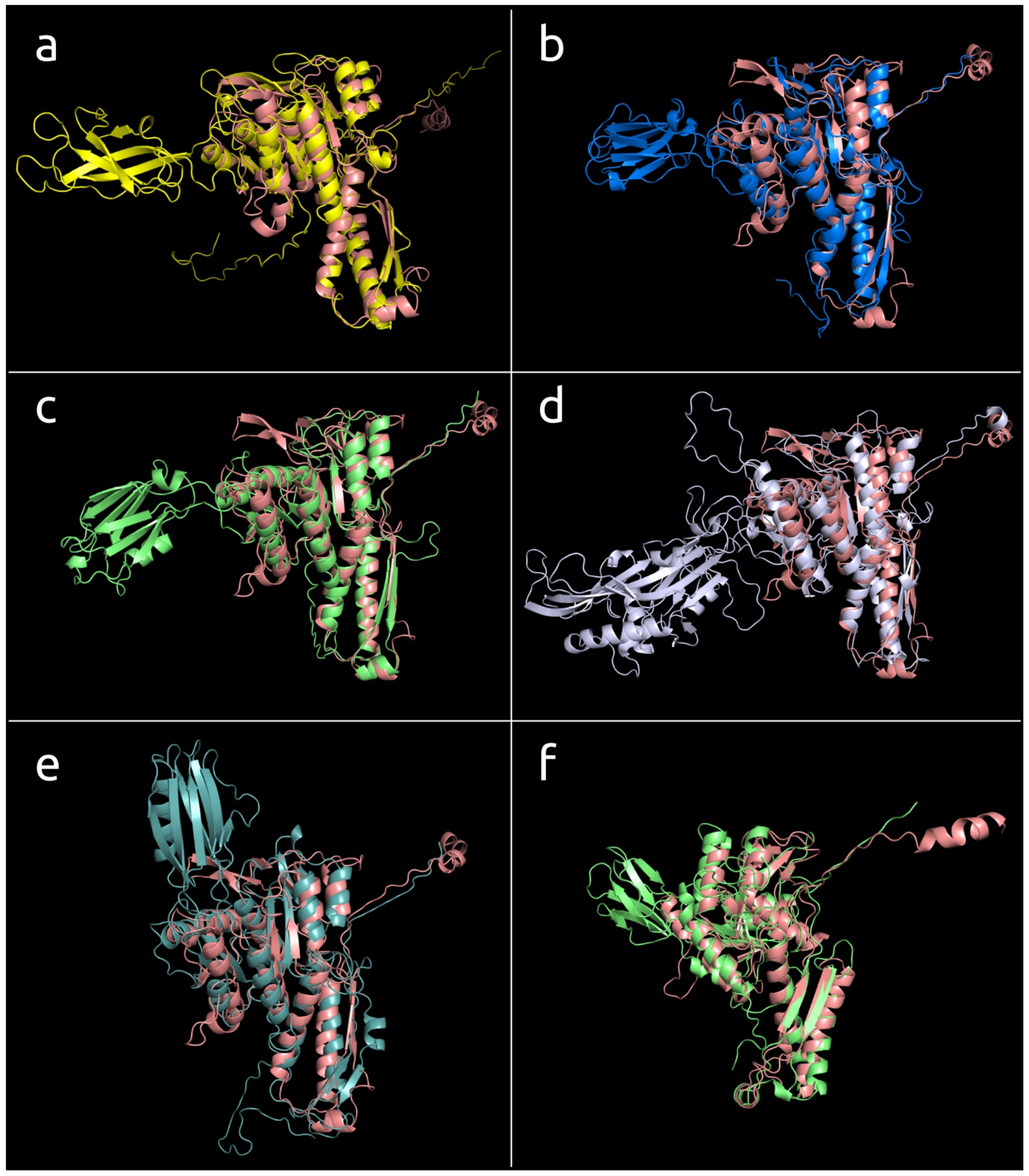
3.4.2. Two-Domains Contractile Sheath Proteins (Type 2)
3.4.3. Multiple Domain Contractile Sheath Proteins (Type 3)
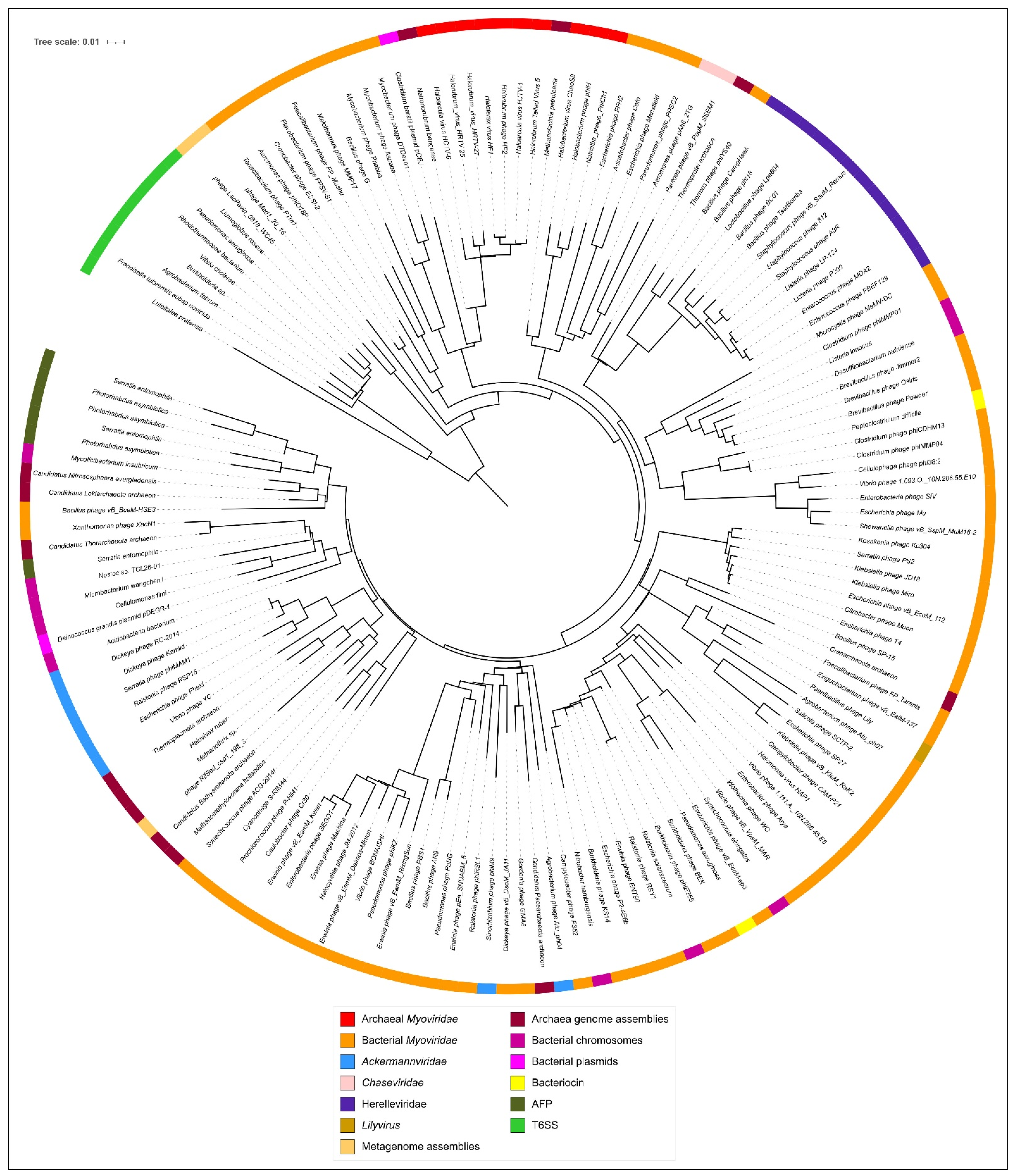
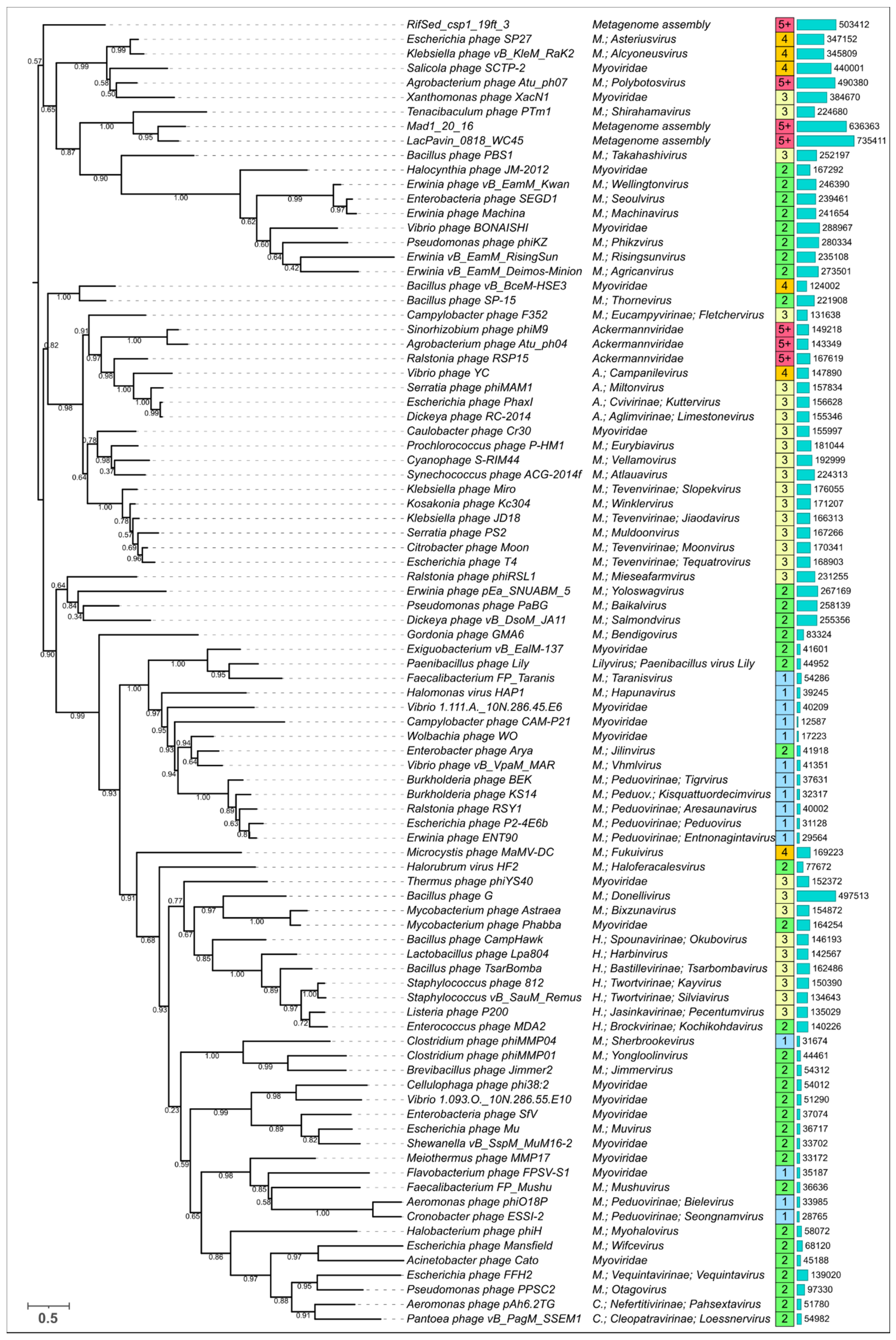
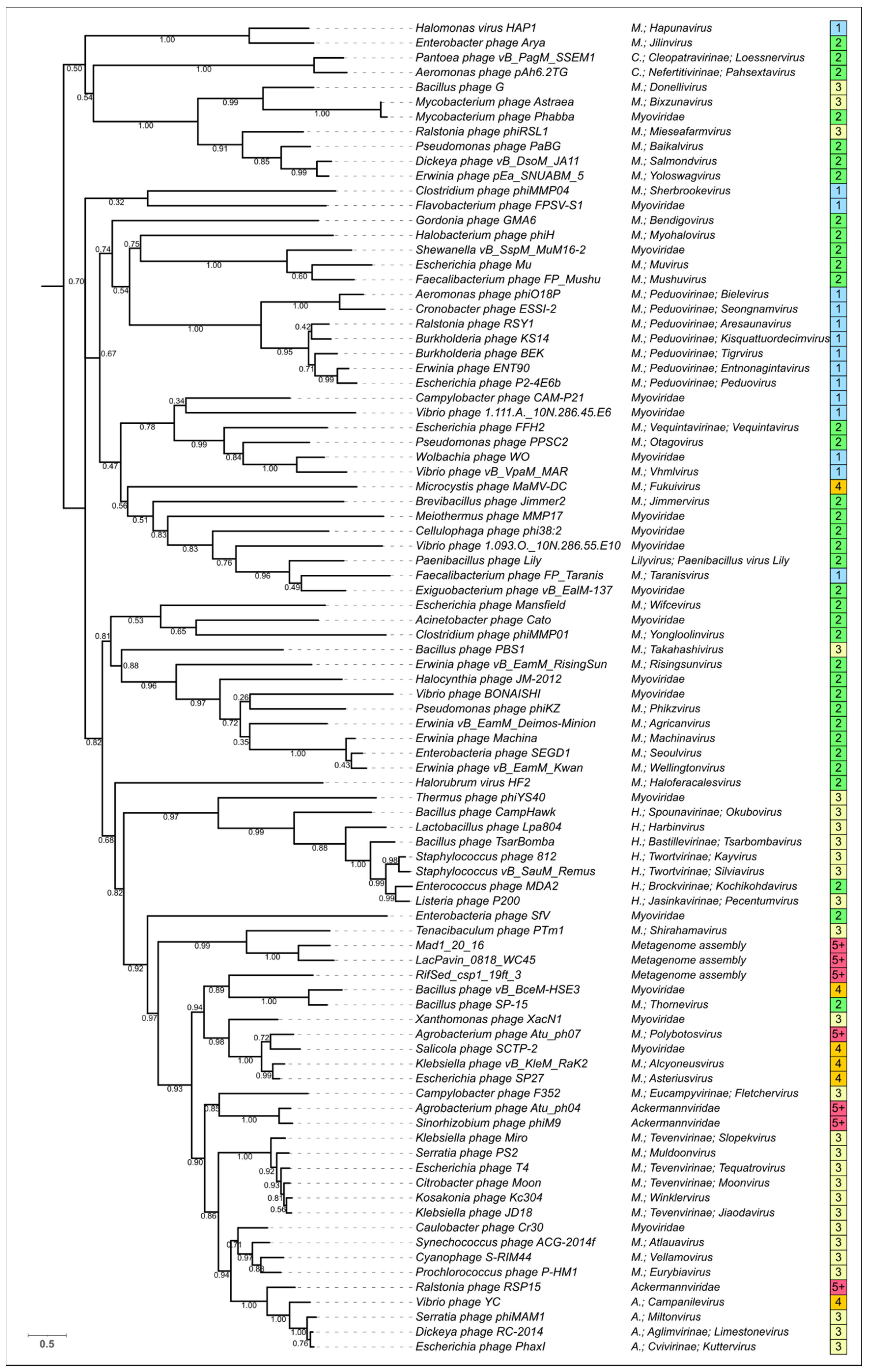
3.5. Phylogenetic Analysis of Modelled Sheath Proteins
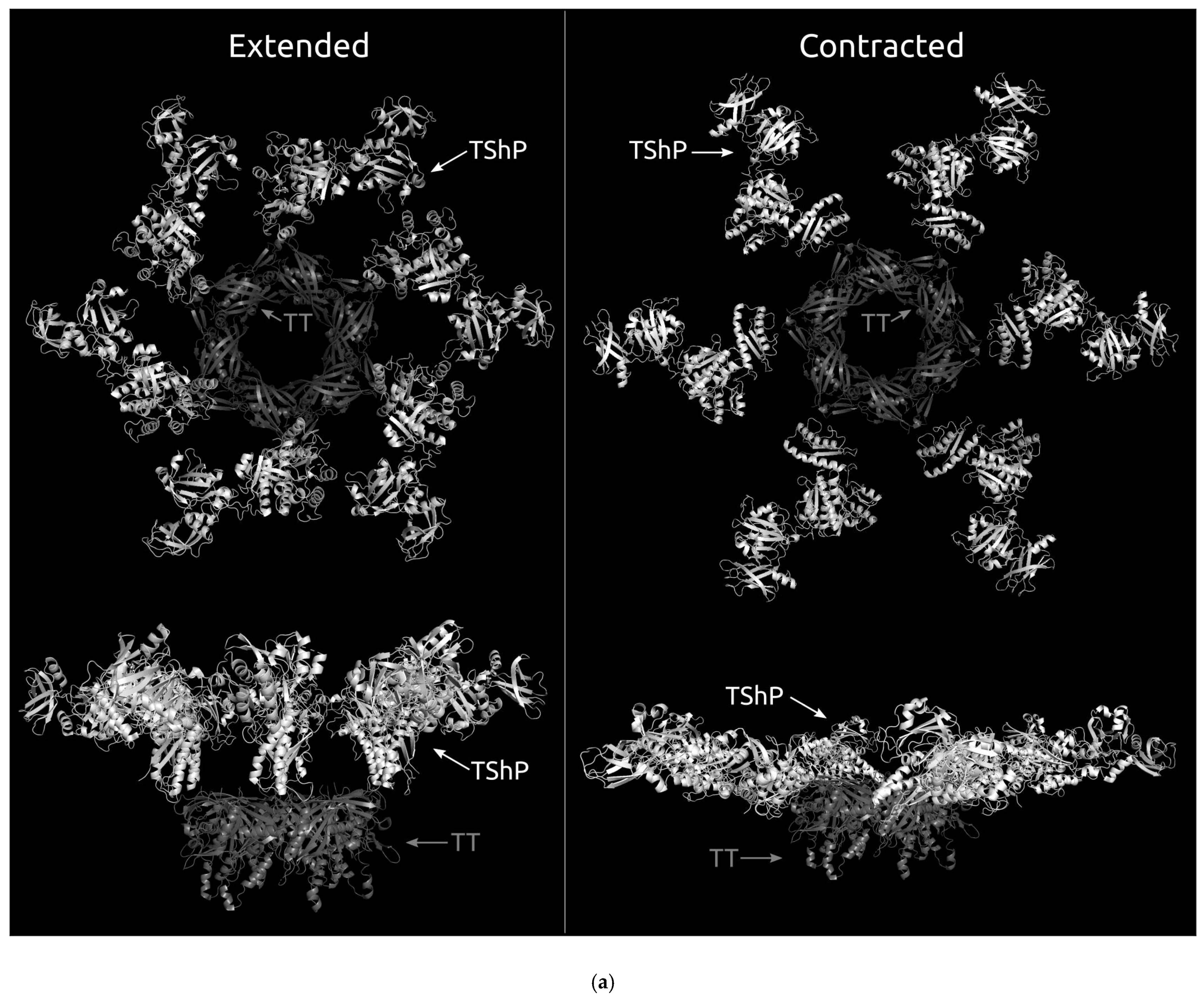
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Aksyuk, A.A.; Leiman, P.G.; Kurochkina, L.P.; Shneider, M.M.; Kostyuchenko, V.A.; Mesyanzhinov, V.V.; Rossmann, M.G. The tail sheath structure of bacteriophage T4: A molecular machine for infecting bacteria. EMBO J. 2009, 28, 821–829. [Google Scholar] [CrossRef] [PubMed]
- Aksyuk, A.A.; Kurochkina, L.P.; Fokine, A.; Forouhar, F.; Mesyanzhinov, V.V.; Tong, L.; Rossmann, M.G. Structural conservation of the myoviridae phage tail sheath protein fold. Structure 2011, 19, 1885–1894. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, F.; Li, N.; Wang, X.; Cheng, J.; Huang, Y.; Yang, Y.; Yang, J.; Cai, B.; Wang, Y.-P.; Jin, Q.; et al. Cryo-EM structure and assembly of an extracellular contractile injection system. Cell 2019, 177, 370–383.e15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Desfosses, A.; Venugopal, H.; Joshi, T.; Felix, J.; Jessop, M.; Jeong, H.; Hyun, J.; Heymann, J.B.; Hurst, M.R.H.; Gutsche, I.; et al. Atomic structures of an entire contractile injection system in both the extended and contracted states. Nat. Microbiol. 2019, 4, 1885–1894. [Google Scholar] [CrossRef] [PubMed]
- Belyaeva, N.N.; Azizbekyan, R.R. Fine structure of new Bacillus subtilis phage AR9 with complex morphology. Virology 1968, 34, 176–179. [Google Scholar] [CrossRef]
- Bönemann, G.; Pietrosiuk, A.; Diemand, A.; Zentgraf, H.; Mogk, A. remodelling of VipA/VipB tubules by ClpV-mediated threading is crucial for type VI protein secretion. EMBO J. 2009, 28, 315–325. [Google Scholar] [CrossRef] [Green Version]
- Brackmann, M.; Wang, J.; Basler, M. Type VI secretion system sheath inter-subunit interactions modulate its contraction. EMBO Rep. 2018, 19, 225–233. [Google Scholar] [CrossRef]
- Chen, L.; Song, N.; Liu, B.; Zhang, N.; Alikhan, N.-F.; Zhou, Z.; Zhou, Y.; Zhou, S.; Zheng, D.; Chen, M.; et al. Genome-wide identification and characterization of a superfamily of bacterial extracellular contractile injection systems. Cell Rep. 2019, 29, 511–521.e2. [Google Scholar] [CrossRef] [Green Version]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Žídek, A.; Potapenko, A.; et al. Highly accurate protein structure prediction with alphafold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef]
- Zheng, W.; Zhang, C.; Li, Y.; Pearce, R.; Bell, E.W.; Zhang, Y. Folding non-homologous proteins by coupling deep-learning contact maps with I-TASSER assembly simulations. Cell Rep. Methods 2021, 1, 100014. [Google Scholar] [CrossRef]
- Baek, M.; DiMaio, F.; Anishchenko, I.; Dauparas, J.; Ovchinnikov, S.; Lee, G.R.; Wang, J.; Cong, Q.; Kinch, L.N.; Schaeffer, R.D.; et al. Accurate prediction of protein structures and interactions using a three-track neural network. Science 2021, 373, 871–876. [Google Scholar] [CrossRef] [PubMed]
- Komarevtsev, S.K.; Evseev, P.V.; Shneider, M.M.; Popova, E.A.; Tupikin, A.E.; Stepanenko, V.N.; Kabilov, M.R.; Shabunin, S.V.; Osmolovskiy, A.A.; Miroshnikov, K.A. Gene analysis, cloning, and heterologous expression of protease from a micromycete Aspergillus ochraceus capable of activating protein C of blood plasma. Microorganisms 2021, 9, 1936. [Google Scholar] [CrossRef] [PubMed]
- Evseev, P.; Lukianova, A.; Tarakanov, R.; Tokmakova, A.; Shneider, M.; Ignatov, A.; Miroshnikov, K. Curtobacterium spp. and Curtobacterium flaccumfaciens: Phylogeny, genomics-based taxonomy, pathogenicity, and diagnostics. Curr. Issues Mol. Biol. 2022, 44, 889–927. [Google Scholar] [CrossRef]
- UniProt. Available online: https://www.uniprot.org/ (accessed on 25 May 2022).
- AlphaFold Protein Structure Database. Available online: https://alphafold.ebi.ac.uk/ (accessed on 9 April 2022).
- Johnson, J.E.; Olson, A.J. Icosahedral virus structures and the protein data bank. J. Biol. Chem. 2021, 296, 100554. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.; Xiang, Y. Structures of the tailed bacteriophages that infect gram-positive bacteria. Curr. Opin. Virol. 2020, 45, 65–74. [Google Scholar] [CrossRef]
- Dedeo, C.L.; Teschke, C.M.; Alexandrescu, A.T. Keeping it together: Structures, functions, and applications of viral decoration proteins. Viruses 2020, 12, 1163. [Google Scholar] [CrossRef]
- Linares, R.; Arnaud, C.-A.; Degroux, S.; Schoehn, G.; Breyton, C. Structure, function and assembly of the long, flexible tail of siphophages. Curr. Opin. Virol. 2020, 45, 34–42. [Google Scholar] [CrossRef]
- Dedeo, C.L.; Cingolani, G.; Teschke, C.M. Portal protein: The orchestrator of capsid assembly for the DsDNA tailed bacteriophages and herpesviruses. Annu. Rev. Virol. 2019, 6, 141–160. [Google Scholar] [CrossRef]
- Zinke, M.; Schröder, G.F.; Lange, A. Major tail proteins of bacteriophages of the order caudovirales. J. Biol. Chem. 2022, 298, 101472. [Google Scholar] [CrossRef]
- Taylor, N.M.I.; van Raaij, M.J.; Leiman, P.G. Contractile injection systems of bacteriophages and related systems. Mol. Microbiol. 2018, 108, 6–15. [Google Scholar] [CrossRef] [Green Version]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The protein data bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Home—Genome—NCBI. Available online: https://www.ncbi.nlm.nih.gov/genome (accessed on 11 November 2021).
- Delcher, A.L.; Bratke, K.A.; Powers, E.C.; Salzberg, S.L. Identifying bacterial genes and endosymbiont DNA with glimmer. Bioinformatics 2007, 23, 673–679. [Google Scholar] [CrossRef] [PubMed]
- Seemann, T. Prokka: Rapid prokaryotic genome annotation. Bioinformatics 2014, 30, 2068–2069. [Google Scholar] [CrossRef] [PubMed]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Gabler, F.; Nam, S.-Z.; Till, S.; Mirdita, M.; Steinegger, M.; Söding, J.; Lupas, A.N.; Alva, V. Protein sequence analysis using the MPI bioinformatics toolkit. Curr. Protoc. Bioinform. 2020, 72, e108. [Google Scholar] [CrossRef]
- Jones, P.; Binns, D.; Chang, H.-Y.; Fraser, M.; Li, W.; McAnulla, C.; McWilliam, H.; Maslen, J.; Mitchell, A.; Nuka, G.; et al. InterProScan 5: Genome-scale protein function classification. Bioinformatics 2014, 30, 1236–1240. [Google Scholar] [CrossRef] [Green Version]
- Kelley, L.A.; Mezulis, S.; Yates, C.M.; Wass, M.N.; Sternberg, M.J.E. The Phyre2 web portal for protein modeling, prediction and analysis. Nat. Protoc. 2015, 10, 845–858. [Google Scholar] [CrossRef] [Green Version]
- PyMOL | Pymol.Org. Available online: https://pymol.org/2/ (accessed on 11 November 2021).
- Sievers, F.; Wilm, A.; Dineen, D.; Gibson, T.J.; Karplus, K.; Li, W.; Lopez, R.; McWilliam, H.; Remmert, M.; Söding, J.; et al. Fast, scalable generation of high-quality protein multiple sequence alignments using clustal omega. Mol. Syst. Biol. 2011, 7, 539. [Google Scholar] [CrossRef]
- Katoh, K.; Misawa, K.; Kuma, K.; Miyata, T. MAFFT: A Novel Method for rapid multiple sequence alignment based on fast fourier transform. Nucleic Acids Res. 2002, 30, 3059–3066. [Google Scholar] [CrossRef] [Green Version]
- Price, M.N.; Dehal, P.S.; Arkin, A.P. FastTree 2—Approximately maximum-likelihood trees for large alignments. PLoS ONE 2010, 5, e9490. [Google Scholar] [CrossRef]
- Kozlov, A.M.; Darriba, D.; Flouri, T.; Morel, B.; Stamatakis, A. RAxML-NG: A fast, scalable and user-friendly tool for maximum likelihood phylogenetic inference. Bioinformatics 2019, 35, 4453–4455. [Google Scholar] [CrossRef] [Green Version]
- Edler, D.; Klein, J.; Antonelli, A.; Silvestro, D. RaxmlGUI 2.0: A graphical interface and toolkit for phylogenetic analyses using RAxML. Methods Ecol. Evol. 2021, 12, 373–377. [Google Scholar] [CrossRef]
- Darriba, D.; Posada, D.; Kozlov, A.M.; Stamatakis, A.; Morel, B.; Flouri, T. ModelTest-NG: A new and scalable tool for the selection of DNA and protein evolutionary models. Mol. Biol. Evol. 2020, 37, 291–294. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Letunic, I.; Bork, P. Interactive tree of life (ITOL) v5: An online tool for phylogenetic tree display and annotation. Nucleic Acids Res. 2021, 49, W293–W296. [Google Scholar] [CrossRef]
- Dong, R.; Pan, S.; Peng, Z.; Zhang, Y.; Yang, J. MTM-Align: A server for fast protein structure database search and multiple protein structure alignment. Nucleic Acids Res. 2018, 46, W380–W386. [Google Scholar] [CrossRef] [PubMed]
- Dong, R.; Peng, Z.; Zhang, Y.; Yang, J. MTM-Align: An algorithm for fast and accurate multiple protein structure alignment. Bioinformatics 2018, 34, 1719–1725. [Google Scholar] [CrossRef] [Green Version]
- PHYLIP Home Page. Available online: https://evolution.genetics.washington.edu/phylip/ (accessed on 13 March 2022).
- Wolf, J.N.; Keßler, M.; Ackermann, J.; Koch, I. PTGL: Extension to graph-based topologies of cryo-EM data for large protein structures. Bioinformatics 2021, 37, 1032–1034. [Google Scholar] [CrossRef] [PubMed]
- Bank, R.P.D. RCSB PDB: Homepage. Available online: https://www.rcsb.org/ (accessed on 19 March 2022).
- Pukatzki, S.; Ma, A.T.; Revel, A.T.; Sturtevant, D.; Mekalanos, J.J. Type VI secretion system translocates a phage tail spike-like protein into target cells where it cross-links actin. Proc. Natl. Acad. Sci. USA 2007, 104, 15508–15513. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leiman, P.G.; Basler, M.; Ramagopal, U.A.; Bonanno, J.B.; Sauder, J.M.; Pukatzki, S.; Burley, S.K.; Almo, S.C.; Mekalanos, J.J. Type VI secretion apparatus and phage tail-associated protein complexes share a common evolutionary origin. Proc. Natl. Acad. Sci. USA 2009, 106, 4154–4159. [Google Scholar] [CrossRef] [Green Version]
- Bank, R.P.D. RCSB PDB-3HXL: Crystal Structure of the Sheath Tail Protein (DSY3957) from Desulfitobacterium hafniense, Northeast Structural Genomics Consortium Target DhR18. Available online: https://www.rcsb.org/structure/3HXL (accessed on 20 March 2022).
- Bank, R.P.D. RCSB PDB-3LML: Crystal Structure of the Sheath Tail Protein Lin1278 from Listeria innocua, Northeast Structural Genomics Consortium Target LkR115. Available online: https://www.rcsb.org/structure/3LML (accessed on 20 March 2022).
- Nováček, J.; Šiborová, M.; Benešík, M.; Pantůček, R.; Doškař, J.; Plevka, P. Structure and genome release of twort-like myoviridae phage with a double-layered baseplate. Proc. Natl. Acad. Sci. USA 2016, 113, 9351–9356. [Google Scholar] [CrossRef] [Green Version]
- Schwemmlein, N.; Pippel, J.; Gazdag, E.-M.; Blankenfeldt, W. Crystal structures of R-type bacteriocin sheath and tube proteins CD1363 and CD1364 From Clostridium difficile in the pre-assembled state. Front. Microbiol. 2018, 9, 1750. [Google Scholar] [CrossRef] [PubMed]
- Ge, P.; Scholl, D.; Prokhorov, N.S.; Avaylon, J.; Shneider, M.M.; Browning, C.; Buth, S.A.; Plattner, M.; Chakraborty, U.; Ding, K.; et al. Action of a minimal contractile bactericidal nanomachine. Nature 2020, 580, 658–662. [Google Scholar] [CrossRef] [PubMed]
- Clemens, D.L.; Ge, P.; Lee, B.-Y.; Horwitz, M.A.; Zhou, Z.H. Atomic structure and mutagenesis of T6SS reveals interlaced array essential to function. Cell 2015, 160, 940–951. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salih, O.; He, S.; Planamente, S.; Stach, L.; MacDonald, J.T.; Manoli, E.; Scheres, S.H.W.; Filloux, A.; Freemont, P.S. Atomic structure of type VI contractile sheath from Pseudomonas aeruginosa. Structure 2018, 26, 329–336.e3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kudryashev, M.; Wang, R.Y.-R.; Brackmann, M.; Scherer, S.; Maier, T.; Baker, D.; DiMaio, F.; Stahlberg, H.; Egelman, E.H.; Basler, M. Structure of the type VI secretion system contractile sheath. Cell 2015, 160, 952–962. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, J.; Ericson, C.F.; Lien, Y.-W.; Rutaganira, F.U.N.; Eisenstein, F.; Feldmüller, M.; King, N.; Pilhofer, M. Identification and structure of an extracellular contractile injection system from the marine bacterium Algoriphagus machipongonensis. Nat. Microbiol. 2022, 7, 397–410. [Google Scholar] [CrossRef]
- Weiss, G.L.; Eisenstein, F.; Kieninger, A.-K.; Xu, J.; Minas, H.A.; Gerber, M.; Feldmüller, M.; Maldener, I.; Forchhammer, K.; Pilhofer, M. Structure of a thylakoid-anchored contractile injection system in multicellular cyanobacteria. Nat. Microbiol. 2022, 7, 386–396. [Google Scholar] [CrossRef]
- Fokine, A.; Zhang, Z.; Kanamaru, S.; Bowman, V.D.; Aksyuk, A.A.; Arisaka, F.; Rao, V.B.; Rossmann, M.G. The molecular architecture of the bacteriophage T4 neck. J. Mol. Biol. 2013, 425, 1731–1744. [Google Scholar] [CrossRef] [Green Version]
- Taxonomy. Available online: https://talk.ictvonline.org/taxonomy/ (accessed on 15 March 2022).
- Carson, S.; Bruff, E.; DeFoor, W.; Dums, J.; Groth, A.; Hatfield, T.; Iyer, A.; Joshi, K.; McAdams, S.; Miles, D.; et al. Genome sequences of six paenibacillus larvae siphoviridae phages. Genome Announc. 2015, 3, e00101-15. [Google Scholar] [CrossRef] [Green Version]
- Beims, H.; Bunk, B.; Erler, S.; Mohr, K.I.; Spröer, C.; Pradella, S.; Günther, G.; Rohde, M.; von der Ohe, W.; Steinert, M. Discovery of paenibacillus larvae ERIC V: Phenotypic and genomic comparison to genotypes ERIC I-IV reveal different inventories of virulence factors which correlate with epidemiological prevalences of american foulbrood. Int. J. Med. Microbiol. 2020, 310, 151394. [Google Scholar] [CrossRef]
- Prangishvili, D.; Bamford, D.H.; Forterre, P.; Iranzo, J.; Koonin, E.V.; Krupovic, M. The enigmatic archaeal virosphere. Nat. Rev. Microbiol. 2017, 15, 724–739. [Google Scholar] [CrossRef] [PubMed]
- Koonin, E.V.; Dolja, V.V.; Krupovic, M.; Varsani, A.; Wolf, Y.I.; Yutin, N.; Zerbini, F.M.; Kuhn, J.H. Global organization and proposed megataxonomy of the virus world. Microbiol. Mol. Biol. Rev. 2020, 84, e00061-19. [Google Scholar] [CrossRef] [PubMed]
- Evseev, P.; Sykilinda, N.; Gorshkova, A.; Kurochkina, L.; Ziganshin, R.; Drucker, V.; Miroshnikov, K. Pseudomonas phage PaBG—A jumbo member of an old parasite family. Viruses 2020, 12, 721. [Google Scholar] [CrossRef] [PubMed]
- Al-Shayeb, B.; Sachdeva, R.; Chen, L.-X.; Ward, F.; Munk, P.; Devoto, A.; Castelle, C.J.; Olm, M.R.; Bouma-Gregson, K.; Amano, Y.; et al. Clades of huge phages from across earth’s ecosystems. Nature 2020, 578, 425–431. [Google Scholar] [CrossRef] [Green Version]
- Wittmann, J.; Turner, D.; Millard, A.D.; Mahadevan, P.; Kropinski, A.M.; Adriaenssens, E.M. From orphan phage to a proposed new family-the diversity of N4-like viruses. Antibiotics 2020, 9, 663. [Google Scholar] [CrossRef]
- McPartland, J.; Rothman-Denes, L.B. The tail sheath of bacteriophage N4 Interacts with the Escherichia coli receptor. J. Bacteriol. 2009, 191, 525–532. [Google Scholar] [CrossRef] [Green Version]
- Richardson, J.S. The anatomy and taxonomy of protein structure. In Advances in Protein Chemistry; Anfinsen, C.B., Edsall, J.T., Richards, F.M., Eds.; Academic Press: Cambridge, MA, USA, 1981; Volume 34, pp. 167–339. [Google Scholar]
- Zhang, Y.; Skolnick, J. Scoring function for automated assessment of protein structure template quality. Proteins 2004, 57, 702–710. [Google Scholar] [CrossRef]
- Botstein, D. A theory of modular evolution for bacteriophages. Ann. N. Y. Acad. Sci. 1980, 354, 484–490. [Google Scholar] [CrossRef]
- Murthy, K.G.K.; Deb, A.; Goonesekera, S.; Szabó, C.; Salzman, A.L. Identification of conserved domains in Salmonella muenchen flagellin that are essential for its ability to activate TLR5 and to induce an inflammatory response in vitro. J. Biol. Chem. 2004, 279, 5667–5675. [Google Scholar] [CrossRef] [Green Version]
- Forstnerič, V.; Ivičak-Kocjan, K.; Plaper, T.; Jerala, R.; Benčina, M. The Role of the C-terminal D0 domain of flagellin in activation of toll like receptor 5. PLoS Pathog. 2017, 13, e1006574. [Google Scholar] [CrossRef]
- Yuan, Y.; Gao, M. Jumbo bacteriophages: An overview. Front. Microbiol. 2017, 8, 403. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fraser, J.S.; Yu, Z.; Maxwell, K.L.; Davidson, A.R. Ig-Like domains on bacteriophages: A tale of promiscuity and deceit. J. Mol. Biol. 2006, 359, 496–507. [Google Scholar] [CrossRef] [PubMed]
- Buttimer, C.; Hendrix, H.; Oliveira, H.; Casey, A.; Neve, H.; McAuliffe, O.; Ross, R.P.; Hill, C.; Noben, J.-P.; O’Mahony, J.; et al. Jumbo bacteriophages: An overview. Front. Microbiol. 2017, 8, 403. [Google Scholar]
- Williams, A.F.; Barclay, A.N. The immunoglobulin superfamily—Domains for cell surface recognition. Annu. Rev. Immunol. 1988, 6, 381–405. [Google Scholar] [CrossRef]
- Stone, N.P.; Demo, G.; Agnello, E.; Kelch, B.A. Principles for enhancing virus capsid capacity and stability from a thermophilic virus capsid structure. Nat. Commun. 2019, 10, 4471. [Google Scholar] [CrossRef] [Green Version]
- Mateu, M.G. Assembly, stability and dynamics of virus capsids. Arch. Biochem. Biophys. 2013, 531, 65–79. [Google Scholar] [CrossRef] [Green Version]
- Leiman, P.G.; Chipman, P.R.; Kostyuchenko, V.A.; Mesyanzhinov, V.V.; Rossmann, M.G. Three-dimensional rearrangement of proteins in the tail of bacteriophage T4 on infection of its host. Cell 2004, 118, 419–429. [Google Scholar] [CrossRef] [Green Version]
- Leiman, P.G.; Kanamaru, S.; Mesyanzhinov, V.V.; Arisaka, F.; Rossmann, M.G. Structure and morphogenesis of bacteriophage T4. Cell. Mol. Life Sci. 2003, 60, 2356–2370. [Google Scholar] [CrossRef]
- Tétart, F.; Desplats, C.; Kutateladze, M.; Monod, C.; Ackermann, H.-W.; Krisch, H.M. Phylogeny of the major head and tail genes of the wide-ranging T4-type bacteriophages. J. Bacteriol. 2001, 183, 358–366. [Google Scholar] [CrossRef] [Green Version]
- Evseev, P.; Lukianova, A.; Sykilinda, N.; Gorshkova, A.; Bondar, A.; Shneider, M.; Kabilov, M.; Drucker, V.; Miroshnikov, K. Pseudomonas phage MD8: Genetic mosaicism and challenges of taxonomic classification of lambdoid bacteriophages. Int. J. Mol. Sci. 2021, 22, 10350. [Google Scholar] [CrossRef]
- Baquero, D.P.; Liu, Y.; Wang, F.; Egelman, E.H.; Prangishvili, D.; Krupovic, M. Structure and assembly of archaeal viruses. Adv. Virus Res. 2020, 108, 127–164. [Google Scholar] [CrossRef] [PubMed]




















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| PDB Code | Description | Organism | Resolution | Method | References |
|---|---|---|---|---|---|
| 3FO8 | Crystal structure of the bacteriophage T4 tail sheath protein, protease-resistant fragment gp18PR | Escherichia phage T4 | 1.8 Å | X-ray diffraction | [1] |
| 3FOA | Crystal structure of the bacteriophage T4 tail sheath protein, deletion mutant gp18M | Escherichia phage T4 | 3.5 Å | X-ray diffraction | [1] |
| 3HXL | Crystal structure of the sheath tail protein (DSY3957) from Desulfitobacterium hafniense | Desulfitobacterium hafniense | 1.90 Å | X-ray diffraction | [46] |
| 3LML | Crystal structure of the sheath tail protein Lin1278 from Listeria innocua, Northeast Structural Genomics Consortium Target LkR115 | Listeria innocua | 3.3 Å | X-ray diffraction | [47] |
| 3SPE | Crystal structure of the tail sheath protein protease-resistant fragment from bacteriophage phiKZ | Pseudomonas phage phiKZ | 2.4 Å | X-ray diffraction | [2] |
| 5LI4 | Bacteriophage phi812K1-420 (Staphylococcus phage 812) tail sheath protein after contraction. This structure is related to 5LI2, 5LII, 5LIJ | Staphylococcus phage 812 | 4.2 Å | Electron microscopy | [48] |
| 6GKW | Crystal structure of the R-type bacteriocin (diffocin) sheath protein CD1363 from Clostridium difficile 630 in the pre-assembled state | Clostridium difficile | 1.9 Å | X-ray diffraction | [49] |
| 6PYT | CryoEM structure of precontracted pyocin R2 trunk from Pseudomonas aeruginosa | Pseudomonas aeruginosa | 2.9 Å | Electron microscopy | [50] |
| 3J9O | CryoEM structure of a type VI secretion system from Francisella tularensis subsp. novicida U112 | Francisella tularensis subsp. novicida | 3.70 Å | Electron microscopy | [51] |
| 5N8N | CryoEM structure of contracted sheath of a Pseudomonas aeruginosa type VI secretion system consisting of TssB1 and TssC | Pseudomonas aeruginosa | 3.28 Å | Electron microscopy | [52] |
| 3J9G | Atomic model of the VipA/VipB, the type VI secretion system contractile sheath of Vibrio cholerae | Vibrio cholerae | 3.5 Å | Electron microscopy | [53] |
| 6RAO | Cryo-EM structure of the anti-feeding prophage (AFP) baseplate for Serratia entomophila. This structure is related to 6RAP, 6RBK, 6RBN, 6RC8, 6RGL | Serratia entomophila | 3.1 Å | Electron microscopy | [4] |
| 6J0B | Cryo-EM structure of an extracellular contractile injection system (CIS), PVC sheath-tube complex in extended state from Photorhabdus asymbiotica subsp. asymbiotica ATCC 43949 | Photorhabdus asymbiotica subsp. asymbiotica ATCC 43949 | 2.9 Å | Electron microscopy | [3] |
| 7AE0 | Cryo-EM structure of an extracellular contractile injection system from the marine bacterium Algoriphagus machipongonensis with the sheath-tube module in its extended state. This structure is related to 7ADZ, 7AE0, 7AEB, 7AEF, 7AEK | Algoriphagus machipongonensis | 2.4 Å | Electron microscopy | [54] |
| 7B5I | Cryo-EM structure of the contractile injection system cap complex from Anabaena PCC7120 | Nostoc sp. | 2.8 Å | Electron microscopy | [55] |
| # | Organism Name (AFP, Anti-Feeding Prophage; BCN, Bacteriocin; CHR, Chromosome or Genome Assembly; PMD, Plasmid; T6SS, Type VI Secretion System) | NCBI Taxonomy | Length of Sheath Protein, Amino Acid Residues | Number of Domains in the Modelled Structure |
|---|---|---|---|---|
| 1 | Acinetobacter phage Cato | Myoviridae | 487 | 2 |
| 2 | Aeromonas phage pAh6_2TG | Chaseviridae; Nefertitivirinae; Pahsextavirus | 472 | 2 |
| 3 | Aeromonas phage phiO18P | Myoviridae; Peduovirinae; Bielevirus | 370 | 1 |
| 4 | AFP-6J0B-SP1 Photorhabdus asymbiotica ATCC43949 | Gammaproteobacteria; Enterobacterales; Morganellaceae | 355 | 1 |
| 5 | AFP-6J0N-SP2 Photorhabdus asymbiotica ATCC43949 | Gammaproteobacteria; Enterobacterales; Morganellaceae | 440 | 1 |
| 6 | AFP-6J0N-SP3 Photorhabdus asymbiotica ATCC43949 | Gammaproteobacteria; Enterobacterales; Morganellaceae | 424 | 1 |
| 7 | AFP-6RAO-SP3 Serratia entomophila | Gammaproteobacteria; Enterobacterales; Yersiniaceae | 417 | 1 |
| 8 | AFP-6RBN-SP1 Serratia entomophila | Gammaproteobacteria; Enterobacterales; Yersiniaceae; Serratia | 354 | 1 |
| 9 | AFP-6RBN-SP2 Serratia entomophila | Gammaproteobacteria; Enterobacterales; Yersiniaceae; Serratia | 451 | 1 |
| 10 | Agrobacterium phage Atu_ph04 | Ackermannviridae | 838 | 5+ |
| 11 | Agrobacterium phage Atu_ph07 | Myoviridae; Polybotosvirus | 1086 | 5+ |
| 12 | Bacillus phage AR9 | Myoviridae | 987 | 3 |
| 13 | Bacillus phage BC01 | Herelleviridae; Bastillevirinae; Tsarbombavirus | 568 | 3 |
| 14 | Bacillus phage CampHawk | Herelleviridae; Spounavirinae; Okubovirus | 571 | 3 |
| 15 | Bacillus phage G | Myoviridae; Donellivirus | 579 | 3 |
| 16 | Bacillus phage PBS1 | Myoviridae; Takahashivirus | 987 | 3 |
| 17 | Bacillus phage phi18 | Herelleviridae; Spounavirinae; Okubovirus | 571 | 3 |
| 18 | Bacillus phage SP-15 | Myoviridae; Thornevirus | 494 | 2 |
| 19 | Bacillus phage TsarBomba | Herelleviridae; Bastillevirinae; Tsarbombavirus | 568 | 3 |
| 20 | Bacillus phage vB_BceM-HSE3 | Myoviridae | 727 | 4 |
| 21 | BCN-6GKW-Peptoclostridium difficile | Firmicutes; Clostridia; Clostridiales; Peptostreptococcaceae; Clostridioides | 356 | 1 |
| 22 | BCN-6PYT-Pseudomonas aeruginosa PAO1 | Gammaproteobacteria; Pseudomonadales; Pseudomonadaceae; Pseudomonas | 386 | 1 |
| 23 | Brevibacillus phage Jimmer2 | Myoviridae; Jimmervirus | 437 | 2 |
| 24 | Brevibacillus phage Osiris | Myoviridae; Jimmervirus | 437 | 2 |
| 25 | Brevibacillus phage Powder | Myoviridae; Jimmervirus | 437 | 2 |
| 26 | Burkholderia phage BEK | Myoviridae; Peduovirinae; Tigrvirus | 342 | 1 |
| 27 | Burkholderia phage KS14 | Myoviridae; Peduovirinae; Kisquattuordecimvirus | 391 | 1 |
| 28 | Burkholderia phage phiE255 | Myoviridae; Bcepmuvirus | 477 | 2 |
| 29 | Campylobacter phage CAM-P21 | Myoviridae | 397 | 1 |
| 30 | Campylobacter phage F352 | Myoviridae; Eucampyvirinae; Fletchervirus | 636 | 3 |
| 31 | Caulobacter phage Cr30 | Myoviridae | 688 | 3 |
| 32 | Cellulophaga phage phi38:2 | Myoviridae | 508 | 2 |
| 33 | CHR-3HXL-Desulfitobacterium hafniense | Firmicutes; Clostridia; Clostridiales; Peptococcaceae; Desulfitobacterium | 446 | 2 |
| 34 | CHR-3LML-Listeria innocua | Firmicutes; Bacilli; Bacillales; Listeriaceae; Listeria | 460 | 2 |
| 35 | CHR-Acidobacteria bacterium Mor1 | Acidobacteria | 410 | 1 |
| 36 | CHR-Candidatus Bathyarchaeota archaeon isolate Bin-L-2 | Candidatus Bathyarchaeota | 321 | 1 |
| 37 | CHR-Candidatus Lokiarchaeota archaeon isolate TEKIR_9 | Asgard group; Candidatus Lokiarchaeota | 369 | 1 |
| 38 | CHR-Candidatus Nitrososphaera evergladensis SR1 | Thaumarchaeota; Nitrososphaeria; Nitrososphaerales; Nitrososphaeraceae | 521 | 2 |
| 39 | CHR-Candidatus Pacearchaeota archaeon isolate ARS50 | Candidatus Pacearchaeota | 634 | 3 |
| 40 | CHR-Candidatus Thorarchaeota archaeon isolate 2_13 | Asgard group; Candidatus Thorarchaeota | 577 | 2 |
| 41 | CHR-Cellulomonas fimi ATCC 484 | Actinobacteria; Micrococcales; Cellulomonadaceae | 523 | 2 |
| 42 | CHR-Crenarchaeota archaeon isolate LB_CRA_1 | Crenarchaeota | 805 | 4 |
| 43 | CHR-Halovivax ruber XH-70 | Euryarchaeota; Stenosarchaea group; HaloNatrialbales; Natrialbaceae | 574 | 3 |
| 44 | CHR-Methanolacinia_petrolearia_DSM_11571 | Euryarchaeota; Methanomicrobia; Methanomicrobiales; Methanomicrobiaceae; Methanolacinia | 343 | 1 |
| 45 | CHR-Methanomethylovorans hollandica DSM 15978 | Euryarchaeota; Stenosarchaea group; Methanomicrobia; Methanosarcinales; Methanosarcinaceae | 540 | 2 |
| 46 | CHR-Methanothrix sp. isolate bin.308 Contig_420493 | Euryarchaeota; Stenosarchaea group; Methanomicrobia; Methanosarcinales; Methanosaetaceae | 509 | 2 |
| 47 | CHR-Microbacterium wangchenii strain dk512 | Actinobacteria; Micrococcales; Microbacteriaceae | 520 | 2 |
| 48 | CHR-Mycolicibacterium insubricum JCM 16366 | Actinobacteria; Corynebacteriales; Mycobacteriaceae | 508 | 2 |
| 49 | CHR-Natronorubrum bangense strain JCM 10635 | Euryarchaeota; Stenosarchaea group; HaloNatrialbales; Natrialbaceae | 348 | 1 |
| 50 | CHR-Nitrobacter hamburgensis X14 | Alphaproteobacteria; Rhizobiales; Bradyrhizobiaceae | 478 | 2 |
| 51 | CHR-Nostoc sp. TCL26-01 | Cyanobacteria; Nostocales; Nostocaceae; Nostoc | 474 | 2 |
| 52 | CHR-Ralstonia solanacearum strain UW774 | Betaproteobacteria; Burkholderiales; Burkholderiaceae | 476 | 2 |
| 53 | CHR-Synechococcus elongatus PCC 6301 | Cyanobacteria; Synechococcales; Synechococcaceae | 474 | 2 |
| 54 | CHR-Thermoplasmata archaeon isolate B28_G1 | Euryarchaeota; Diaforarchaea group; Thermoplasmata | 436 | 2 |
| 55 | CHR-Thermoprotei archaeon B19_G17 | Archaea; Crenarchaeota; Thermoprotei | 452 | 2 |
| 56 | Citrobacter phage Moon | Myoviridae; Tevenvirinae; Moonvirus | 658 | 3 |
| 57 | Clostridium phage phiCDHM13 | Myoviridae; Sherbrookevirus | 355 | 1 |
| 58 | Clostridium phage phiMMP01 | Myoviridae; Yongloolinvirus | 436 | 2 |
| 59 | Clostridium phage phiMMP04 | Myoviridae; Sherbrookevirus | 355 | 1 |
| 60 | Cronobacter phage ESSI-2 | Myoviridae; Peduovirinae; Seongnamvirus | 375 | 1 |
| 61 | Cyanophage S-RIM44 | Myoviridae; Vellamovirus | 635 | 3 |
| 62 | Dickeya phage Kamild | Ackermannviridae; Aglimvirinae; Limestonevirus | 632 | 3 |
| 63 | Dickeya phage RC-2014 | Ackermannviridae; Aglimvirinae; Limestonevirus | 632 | 3 |
| 64 | Dickeya phage vB_DsoM_JA11 | Myoviridae; Salmondvirus | 558 | 2 |
| 65 | Enterobacter phage Arya | Myoviridae; Jilinvirus | 477 | 2 |
| 66 | Enterobacteria phage SEGD1 | Myoviridae; Seoulvirus | 681 | 2 |
| 67 | Enterobacteria phage SfV | Myoviridae | 498 | 2 |
| 68 | Enterococcus phage MDA2 | Herelleviridae; Brockvirinae; Kochikohdavirus | 569 | 2 |
| 69 | Enterococcus phage PBEF129 | Herelleviridae; Brockvirinae; Kochikohdavirus | 569 | 3 |
| 70 | Erwinia phage ENT90 | Myoviridae; Peduovirinae; Entnonagintavirus | 389 | 1 |
| 71 | Erwinia phage Machina | Myoviridae; Machinavirus | 680 | 2 |
| 72 | Erwinia phage pEa_SNUABM_5 | Myoviridae; Yoloswagvirus | 563 | 2 |
| 73 | Erwinia phage vB_EamM_Deimos-Minion | Myoviridae; Agricanvirus | 695 | 2 |
| 74 | Erwinia phage vB_EamM_Kwan | Myoviridae; Wellingtonvirus | 681 | 2 |
| 75 | Erwinia phage vB_EamM_RisingSun | Myoviridae; Risingsunvirus | 713 | 2 |
| 76 | Escherichia phage FFH2 | Myoviridae; Vequintavirinae | 458 | 2 |
| 77 | Escherichia phage Mansfield | Myoviridae; Wifcevirus | 512 | 2 |
| 78 | Escherichia phage Mu | Myoviridae; Muvirus | 495 | 2 |
| 79 | Escherichia phage P2-4E6b | Myoviridae; Peduovirinae; Peduovirus | 396 | 1 |
| 80 | Escherichia phage PhaxI | Ackermannviridae; Cvivirinae; Kuttervirus | 631 | 3 |
| 81 | Escherichia phage SP27 | Myoviridae; Asteriusvirus | 887 | 4 |
| 82 | Escherichia phage T4 | Myoviridae; Tevenvirinae; Tequatrovirus | 659 | 3 |
| 83 | Escherichia phage vB_EcoM_112 | Myoviridae; Tevenvirinae; Tequatrovirus | 659 | 3 |
| 84 | Escherichia phage vB_EcoM-ep3 | Myoviridae; Jilinvirus | 475 | 2 |
| 85 | Exiguobacterium phage vB_EalM-137 | Myoviridae | 482 | 2 |
| 86 | Faecalibacterium phage FP_Mushu | Myoviridae; Mushuvirus | 481 | 2 |
| 87 | Faecalibacterium phage FP_Taranis | Myoviridae; Taranisvirus | 384 | 1 |
| 88 | Flavobacterium phage FPSV-S1 | Myoviridae | 390 | 1 |
| 89 | Gordonia phage GMA6 | Myoviridae; Bendigovirus | 482 | 2 |
| 90 | Haloarcula virus HCTV-6 | Myoviridae; Haloferacalesvirus | 437 | 2 |
| 91 | Haloarcula virus HJTV-1 | Myoviridae; Haloferacalesvirus | 430 | 2 |
| 92 | Halobacterium phage phiH | Myoviridae; Myohalovirus | 432 | 2 |
| 93 | Halobacterium virus ChaoS9 | Myoviridae; Myohalovirus | 434 | 2 |
| 94 | Halocynthia phage JM-2012 | Myoviridae | 681 | 2 |
| 95 | Haloferax virus HF1 | Myoviridae; Haloferacalesvirus | 430 | 2 |
| 96 | Halomonas virus HAP1 | Myoviridae; Hapunavirus | 388 | 1 |
| 97 | Halorubrum phage HF2 | Myoviridae; Haloferacalesvirus | 430 | 2 |
| 98 | Halorubrum Tailed Virus 5 | Myoviridae; Haloferacalesvirus | 430 | 2 |
| 99 | Halorubrum_virus_HRTV-25 | Myoviridae; Haloferacalesvirus | 431 | 2 |
| 100 | Halorubrum_virus_HRTV-27 | Myoviridae; Haloferacalesvirus | 430 | 2 |
| 101 | Klebsiella phage JD18 | Myoviridae; Tevenvirinae; Jiaodavirus | 657 | 3 |
| 102 | Klebsiella phage Miro | Myoviridae; Tevenvirinae; Slopekvirus | 663 | 3 |
| 103 | Klebsiella phage vB_KleM_RaK2 | Myoviridae; Alcyoneusvirus | 888 | 4 |
| 104 | Kosakonia phage Kc304 | Myoviridae; Winklervirus | 660 | 3 |
| 105 | Lactobacillus phage Lpa804 | Herelleviridae; Harbinvirus | 612 | 3 |
| 106 | Listeria phage LP-124 | Herelleviridae; Jasinkavirinae; Pecentumvirus | 562 | 3 |
| 107 | Listeria phage P200 | Herelleviridae; Jasinkavirinae; Pecentumvirus | 562 | 3 |
| 108 | Meiothermus phage MMP17 | Myoviridae | 472 | 2 |
| 109 | Microcystis phage MaMV-DC | Myoviridae; Fukuivirus | 774 | 4 |
| 110 | Mycobacterium phage Astraea | Myoviridae; Bixzunavirus | 581 | 3 |
| 111 | Mycobacterium phage DTDevon | Myoviridae; Bixzunavirus | 581 | 3 |
| 112 | Mycobacterium phage Phabba | Myoviridae | 482 | 2 |
| 113 | Natrialba_phage_PhiCh1 | Myoviridae; Myohalovirus | 426 | 2 |
| 114 | Paenibacillus phage Lily | Lilyvirus; Paenibacillus virus Lily | 478 | 2 |
| 115 | Pantoea phage vB_PagM_SSEM1 | Chaseviridae; Cleopatravirinae; Loessnervirus | 483 | 2 |
| 116 | phage LacPavin_0818_WC45 | metagenome assembly | 1283 | 5+ |
| 117 | phage Mad1_20_16 | metagenome assembly | 1248 | 5+ |
| 118 | phage RifSed_csp1_19ft_3 | metagenome assembly | 881 | 5+ |
| 119 | PMD-Clostridium baratii str Sullivan plasmid pCBJ | Firmicutes; Clostridia; Clostridiales; Clostridiaceae | 814 | 4 |
| 120 | PMD-Deinococcus grandis ATCC 43672 plasmid pDEGR-1 | Deinococcus-Thermus; Deinococci; Deinococcales; Deinococcaceae | 539 | 2 |
| 121 | Prochlorococcus phage P-HM1 | Myoviridae; Eurybiavirus | 669 | 3 |
| 122 | Pseudomonas phage PaBG | Myoviridae; Baikalvirus | 547 | 2 |
| 123 | Pseudomonas phage phiKZ | Myoviridae; Phikzvirus | 695 | 2 |
| 124 | Pseudomonas_phage_PPSC2 | Myoviridae; Otagovirus | 427 | 2 |
| 125 | Ralstonia phage phiRSL1 | Myoviridae; Mieseafarmvirus | 648 | 3 |
| 126 | Ralstonia phage RSP15 | Ackermannviridae | 826 | 5+ |
| 127 | Ralstonia phage RSY1 | Myoviridae; Peduovirinae; Aresaunavirus | 391 | 1 |
| 128 | Salicola phage SCTP-2 | Myoviridae | 955 | 4 |
| 129 | Serratia phage phiMAM1 | Ackermannviridae; Miltonvirus | 636 | 3 |
| 130 | Serratia phage PS2 | Myoviridae; Muldoonvirus | 663 | 3 |
| 131 | Shewanella phage vB_SspM_MuM16-2 | Myoviridae | 493 | 2 |
| 132 | Sinorhizobium phage phiM9 | Ackermannviridae | 838 | 5+ |
| 133 | Staphylococcus phage 812 | Herelleviridae; Twortvirinae; Kayvirus | 587 | 3 |
| 134 | Staphylococcus phage A3R | Herelleviridae; Twortvirinae; Kayvirus | 587 | 3 |
| 135 | Staphylococcus phage vB_SauM_Remus | Herelleviridae; Twortvirinae; Silviavirus | 586 | 3 |
| 136 | Synechococcus phage ACG-2014f | Myoviridae; Atlauavirus | 731 | 3 |
| 137 | T6SS-3J9G-Vibrio cholerae | Gammaproteobacteria; Vibrionales; Vibrionaceae | 432 | 1 |
| 138 | T6SS-3J9O-Francisella tularensis subsp novicida | Gammaproteobacteria; Thiotrichales; Francisellaceae | 506 | 1 |
| 139 | T6SS-5N8N-Pseudomonas aeruginosa | Gammaproteobacteria; Pseudomonadales; Pseudomonadaceae | 498 | 1 |
| 140 | T6SS-Agrobacterium fabrum C58 | Alphaproteobacteria; Hyphomicrobiales; Rhizobiaceae | 493 | 1 |
| 141 | T6SS-Burkholderia sp MSMB0852 | Betaproteobacteria; Burkholderiales; Burkholderiaceae; Burkholderia | 499 | 1 |
| 142 | T6SS-Limnoglobus roseus strain PX52 | Planctomycetes; Planctomycetia; Gemmatales; Gemmataceae | 491 | 1 |
| 143 | T6SS-Luteitalea pratensis DSM 100886 | Acidobacteria; Vicinamibacteria; Vicinamibacteraceae | 493 | 1 |
| 144 | T6SS-Rhodothermaceae bacterium RA | Bacteroidetes; Bacteroidetes Order II. Incertae sedis; Rhodothermaceae | 509 | 1 |
| 145 | Tenacibaculum phage PTm1 | Myoviridae; Shirahamavirus | 1032 | 3 |
| 146 | Thermus phage phiYS40 | Myoviridae | 648 | 3 |
| 147 | Vibrio phage 1.093.O._10N.286.55.E10 | Myoviridae | 486 | 2 |
| 148 | Vibrio phage 1.111.A._10N.286.45.E6 | Myoviridae | 378 | 1 |
| 149 | Vibrio phage BONAISHI | Myoviridae | 682 | 2 |
| 150 | Vibrio phage vB_VpaM_MAR | Myoviridae; Vhmlvirus | 386 | 1 |
| 151 | Vibrio phage YC | Ackermannviridae; Campanilevirus | 756 | 4 |
| 152 | Wolbachia phage WO | Myoviridae | 383 | 1 |
| 153 | Xanthomonas phage XacN1 | Myoviridae | 714 | 3 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Evseev, P.; Shneider, M.; Miroshnikov, K. Evolution of Phage Tail Sheath Protein. Viruses 2022, 14, 1148. https://doi.org/10.3390/v14061148
Evseev P, Shneider M, Miroshnikov K. Evolution of Phage Tail Sheath Protein. Viruses. 2022; 14(6):1148. https://doi.org/10.3390/v14061148
Chicago/Turabian StyleEvseev, Peter, Mikhail Shneider, and Konstantin Miroshnikov. 2022. "Evolution of Phage Tail Sheath Protein" Viruses 14, no. 6: 1148. https://doi.org/10.3390/v14061148
APA StyleEvseev, P., Shneider, M., & Miroshnikov, K. (2022). Evolution of Phage Tail Sheath Protein. Viruses, 14(6), 1148. https://doi.org/10.3390/v14061148