
An Unwanted Association: The Threat to Papaya Crops by a Novel Potexvirus in Northwest Argentina

 ,
,  , , , , , ,
, , , , , ,  ,
,
Abstract
1. Introduction
2. Materials and Methods
2.1. Sample Collections and Virus Maintenance
2.2. Electron Microscopy
2.3. Purification of Virus-like Particles, Library Preparation and Next-Generation Sequencing
2.4. Sequence Processing and De Novo Assembly
2.5. Phylogenetic Analysis
2.6. Host Range Assay
2.7. Virus Detection
2.7.1. RT-PCR Detection
2.7.2. Sequence Read Archive (SRA) Searches
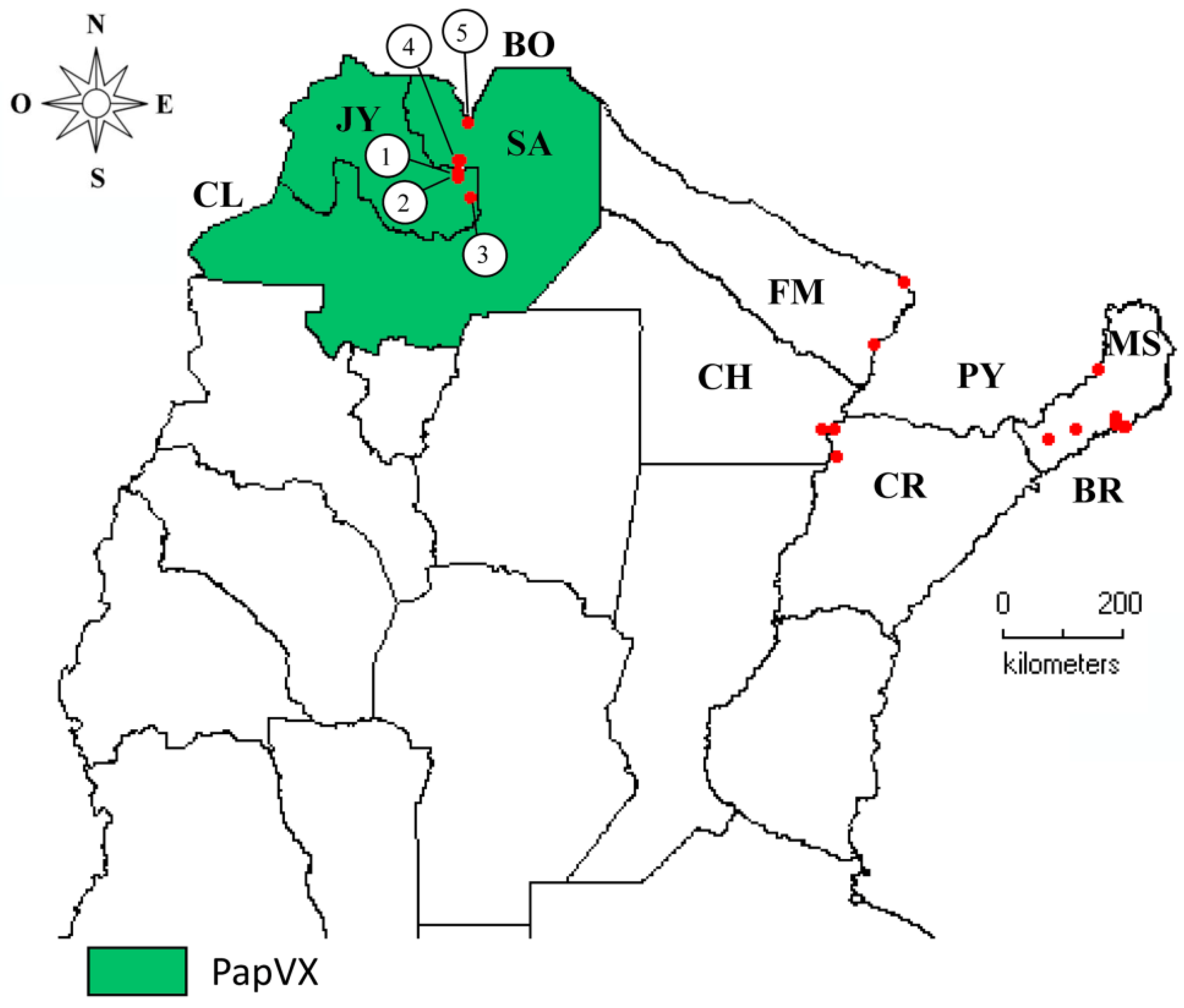
2.8. Virus Distribution and Genetic Analyses
3. Results
3.1. Papaya Symptoms and Particle Observations
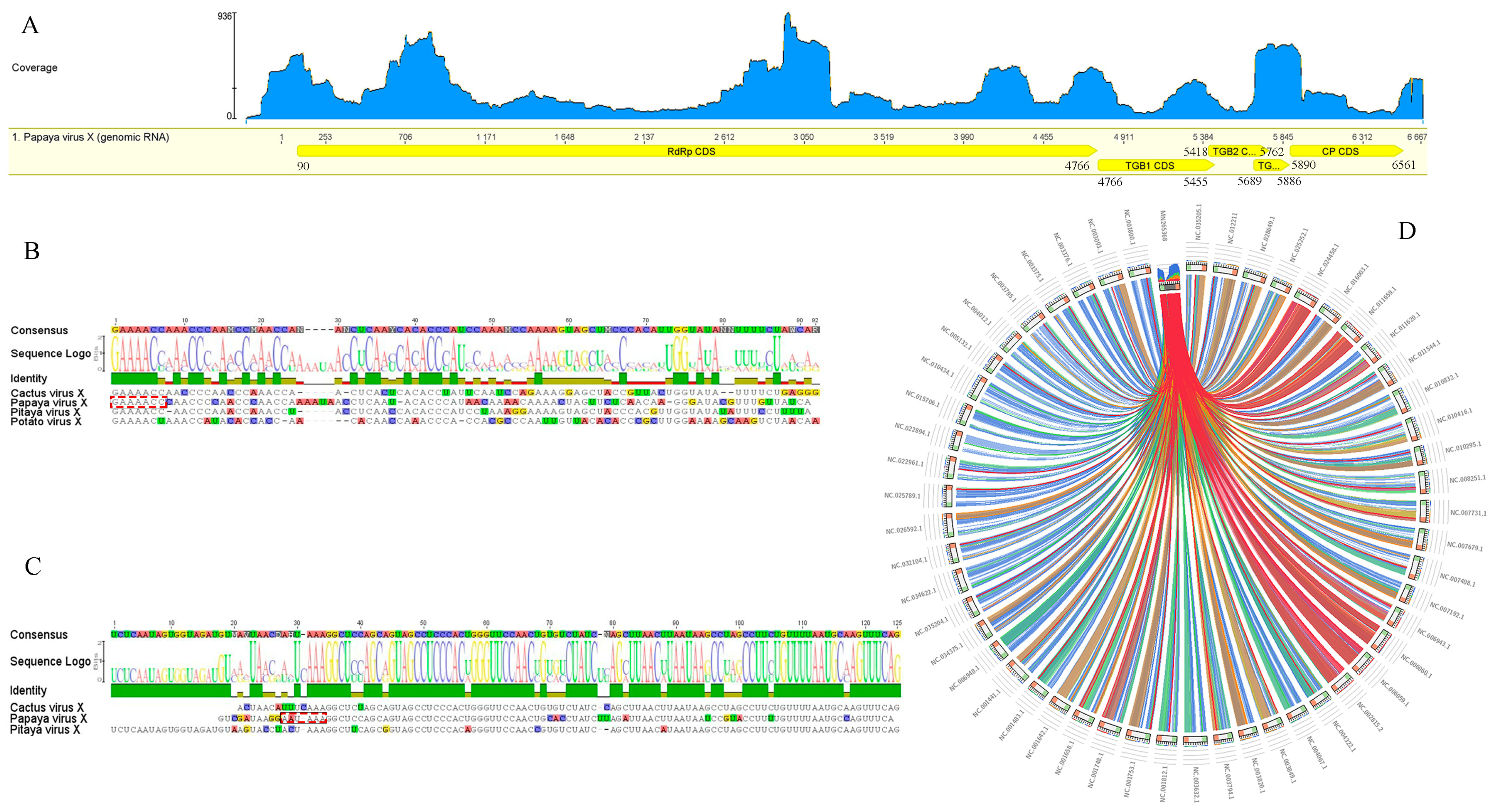
3.2. Sequence Assembly
3.3. Genome Organization and Identification
3.4. Exploring Publicly Available Transcriptome Datasets
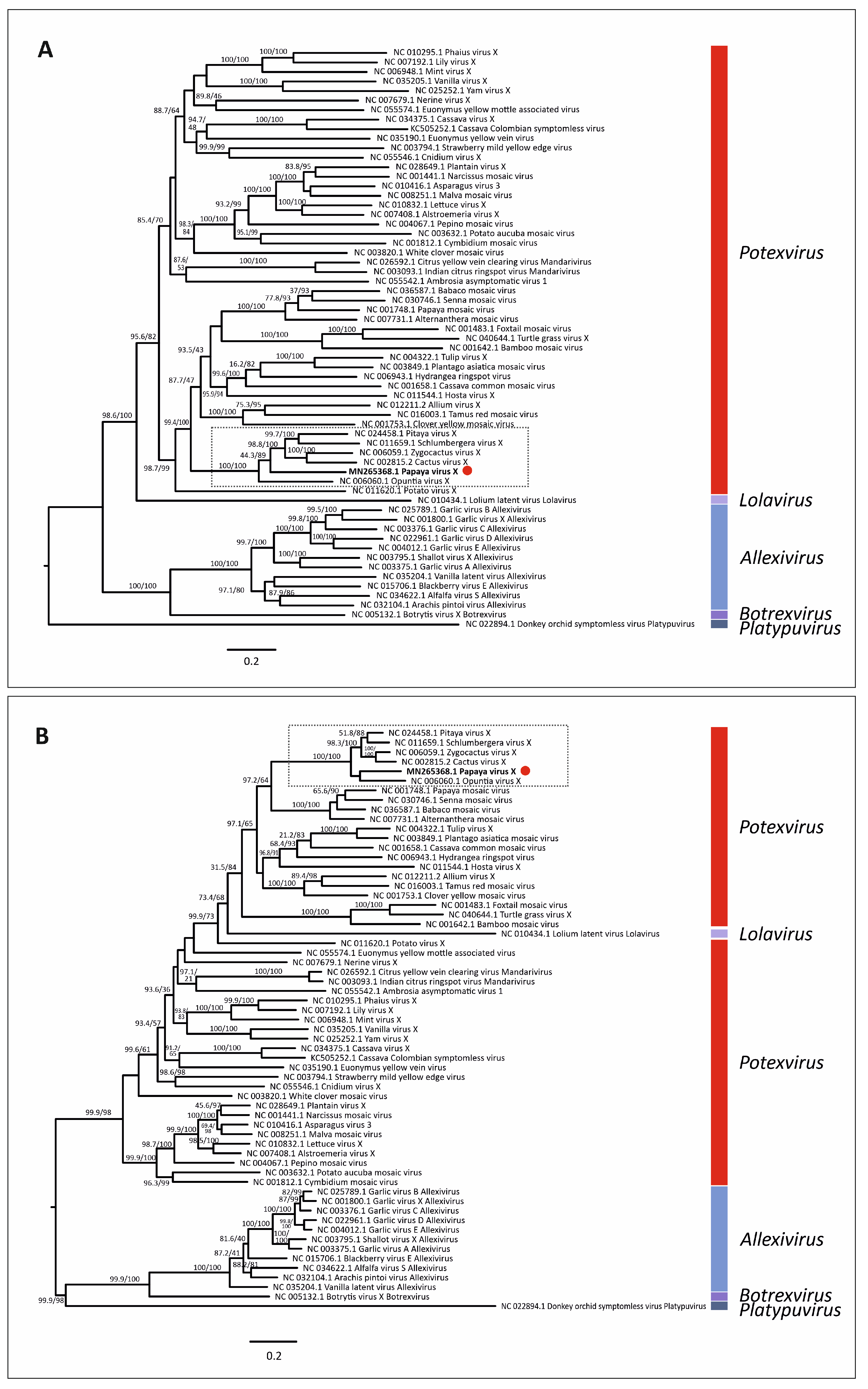
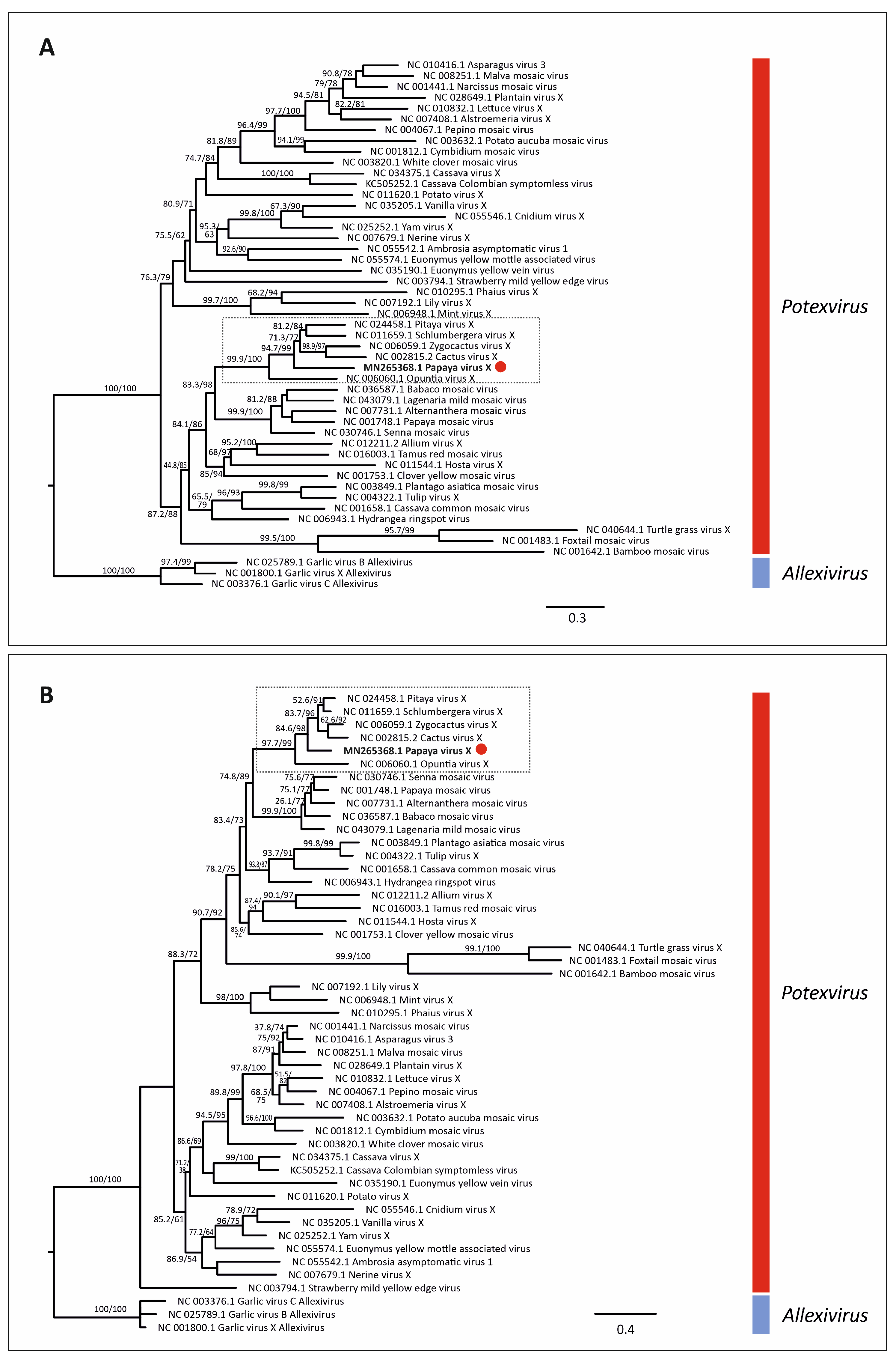
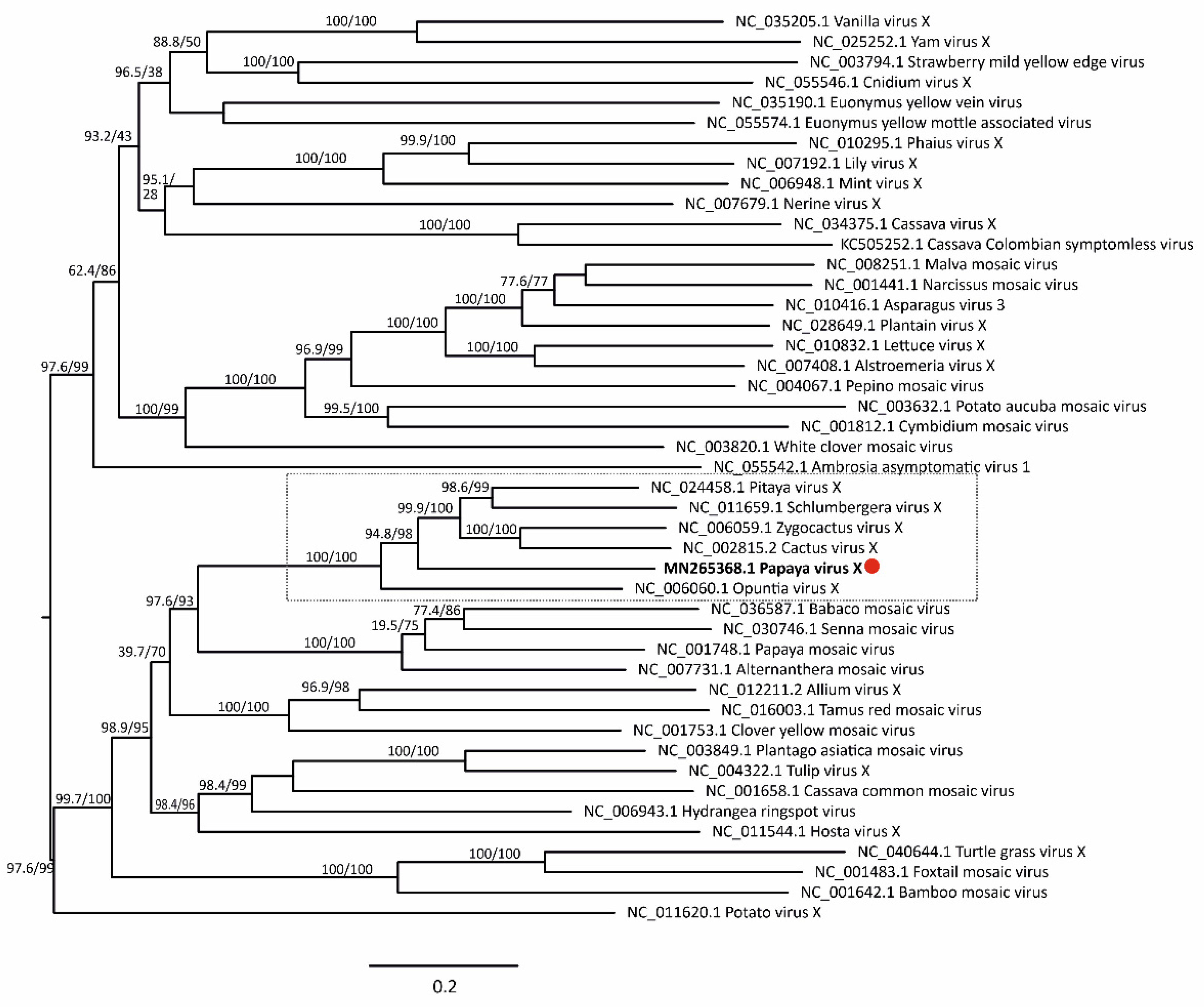
3.5. Virus Phylogenetic Analysis
3.6. Host Range Assay and Virus Detection
3.7. Virus Distribution and Genetic Analyses
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Yeh, S.D.; Bau, H.J.; Kung, Y.J.; Yu, T.; Papaya, A. Biotechnology in Agriculture and Forestry. In Transgenic Crops V; Pua, E.C., Davey, M.R., Eds.; Springer: Berlin, Germany, 2007; Volume 60, pp. 73–96. [Google Scholar]
- FAO. FAO Food and Agriculture Data. 2022. Available online: http://www.fao.org/faostat/ (accessed on 12 April 2022).
- Tennant, P.F.; Fermin, G.A.; Roye, M.E. Viruses infecting papaya (Carica papaya L.): Etiology, pathogenesis, and molecular biology. Plant Viruses 2007, 1, 178–188. [Google Scholar]
- Tripathi, S.; Suzuki, J.Y.; Ferreira, S.A.; Gonsalves, D. Papaya ringspot virus-P: Characteristics, pathogenicity, sequence variability and control. Mol. Plant Pathol. 2008, 9, 269–280. [Google Scholar] [CrossRef] [PubMed]
- Sa Antunes, T.F.; Amaral, R.J.V.; Ventura, J.A.; Godinho, M.T.; Amaral, J.G.; Souza, F.O.; Zerbini, P.A.; Zerbini, F.M.; Fernandes, P.M.B. The dsRNA Virus Papaya Meleira Virus and an ssRNA Virus Are Associated with Papaya Sticky Disease. PLoS ONE 2016, 11, e0155240. [Google Scholar] [CrossRef] [PubMed]
- Cabrera Mederos, D.; Giolitti, F.; Torres, C.; Portal, O. Distribution and phylodynamics of papaya ringspot virus on Carica papaya in Cuba. Plant Pathol. 2019, 68, 239–250. [Google Scholar] [CrossRef]
- Reyes-Proaño, E.; Cornejo-Franco, J.F.; Alvarez-Quinto, R.; Mollov, D.; Karasev, A.V.; Quito-Avila, D.F. ‘Sticky’ disease of papaya in Ecuador: Same disease as in Brazil but caused by two different viruses? Phytopathology 2022, 112, P446. [Google Scholar]
- Laurance, W.F.; Sayer, J.; Cassman, K.G. Agricultural expansion and its impacts on tropical nature. Trends Ecol. Evol. 2014, 29, 107–116. [Google Scholar] [CrossRef]
- Jones, R.A. Plant virus emergence and evolution: Origins, new encounter scenarios, factors driving emergence, effects of changing world conditions, and prospects for control. Virus Res. 2009, 141, 113–130. [Google Scholar] [CrossRef]
- Ghini, R.; Bettiol, W.; Hamada, E. Diseases in tropical and plantation crops as affected by climate changes: Current knowledge and perspectives. Plant Pathol. 2011, 60, 122–132. [Google Scholar] [CrossRef]
- Cabrera Mederos, D.; Zotto, A.D.; Galdeano, E.; Portal, O.; Giolitti, F. First report of papaya ringspot virus infecting Carica papaya in Argentina. J. Plant Pathol. 2016, 98, 687. [Google Scholar] [CrossRef]
- Sit, T.; AbouHaidar, M.G.; Holý, S. Nucleotide Sequence of Papaya Mosaic Virus RNA. J. Gen. Virol. 1989, 70, 2325–2331. [Google Scholar] [CrossRef]
- Kreuze, J.F.; Vaira, A.M.; Menzel, W.; Candresse, T.; Zavriev, S.K.; Hammond, J.; Ryu, K.H. ICTV Report Consortium ICTV Virus Taxonomy Profile: Alphaflexiviridae. J. Gen. Virol. 2020, 101, 699–700. [Google Scholar] [CrossRef] [PubMed]
- Alvarez-Quinto, R.; Cornejo-Franco, J.F.; Quito-Avila, D.F. Characterization of a not so new potexvirus from babaco (Vasconcellea × heilbornii). PLoS ONE 2017, 12, e0189519. [Google Scholar] [CrossRef] [PubMed]
- Blawid, R.; Silva, J.; Nagata, T. Discovering and sequencing new plant viral genomes by next-generation sequencing: Description of a practical pipeline. Ann. Appl. Biol. 2017, 170, 301–314. [Google Scholar] [CrossRef]
- Medina-Salguero, A.X.; Cornejo-Franco, J.F.; Grinstead, S.; Mollov, D.; Mowery, J.D.; Flores, F.; Quito-Avila, D.F. Sequencing, genome analysis and prevalence of a cytorhabdovirus discovered in Carica papaya. PLoS ONE 2019, 14, e0215798. [Google Scholar] [CrossRef] [PubMed]
- Alcalá-Briseño, R.I.; Casarrubias-Castillo, K.; López-Ley, D.; Garrett, K.A.; Silva-Rosales, L. Network Analysis of the Papaya Orchard Virome from Two Agroecological Regions of Chiapas, Mexico. mSystems 2020, 5, e00423-19. [Google Scholar] [CrossRef] [PubMed]
- Mumo, N.N.; Mamati, G.E.; Ateka, E.M.; Rimberia, F.K.; Asudi, G.O.; Boykin, L.; Machuka, E.M.; Njuguna, J.N.; Pelle, R.; Stomeo, F. Metagenomic Analysis of Plant Viruses Associated With Papaya Ringspot Disease in Carica papaya L. in Kenya. Front. Microbiol. 2020, 11, 205. [Google Scholar] [CrossRef]
- Rumbou, A.; Candresse, T.; Von Bargen, S.; Büttner, C. Next-Generation Sequencing Reveals a Novel Emaravirus in Diseased Maple Trees From a German Urban Forest. Front. Microbiol. 2021, 11, 621179. [Google Scholar] [CrossRef]
- Bejerman, N.; Debat, H.; Dietzgen, R.G. The Plant Negative-Sense RNA Virosphere: Virus Discovery Through New Eyes. Front. Microbiol. 2020, 11, 588427. [Google Scholar] [CrossRef]
- Lauber, C.; Seitz, S. Opportunities and Challenges of Data-Driven Virus Discovery. Biomolecules 2022, 12, 1073. [Google Scholar] [CrossRef]
- Hull, R. Plant Virology, 5th ed.; Academic Press: Waltham, MA, USA, 2014. [Google Scholar]
- Kitajima, E.W. Electron Microscopy of Plant Viruses; University of Sao Paulo: Sao Paulo, Brazil, 1997. [Google Scholar]
- Filloux, D.; Dallot, S.; Delaunay, A.; Galzi, S.; Jacquot, E.; Roumagnac, P. Metagenomics Approaches Based on Virion-Associated Nucleic Acids (VANA): An Innovative Tool for Assessing Without A Priori Viral Diversity of Plants. Plant Pathol. 2015, 1302, 249–257. [Google Scholar] [CrossRef]
- Lockhart, B.E.L. Purification and Serology of a Bacilliform Virus Associated with Banana Streak Disease. Phytopathology 1986, 76, 995–999. [Google Scholar] [CrossRef]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [PubMed]
- Haas, B.J.; Papanicolaou, A.; Yassour, M.; Grabherr, M.; Blood, P.D.; Bowden, J.; Couger, M.B.; Eccles, D.; Li, B.; Lieber, M.; et al. De novo transcript sequence recon-struction from RNA-seq using the Trinity platform for reference generation and analysis. Nat. Protoc. 2013, 8, 1494–1512. [Google Scholar] [CrossRef] [PubMed]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef] [PubMed]
- Edgar, R.C. MUSCLE: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004, 32, 1792–1797. [Google Scholar] [CrossRef]
- Gouy, M.; Guindon, S.; Gascuel, O. SeaView Version 4: A Multiplatform Graphical User Interface for Sequence Alignment and Phylogenetic Tree Building. Mol. Biol. Evol. 2010, 27, 221–224. [Google Scholar] [CrossRef]
- Darzentas, N. Circoletto: Visualizing sequence similarity with Circos. Bioinformatics 2010, 26, 2620–2621. [Google Scholar] [CrossRef]
- Muhire, B.M.; Varsani, A.; Martin, D.P. SDT: A Virus Classification Tool Based on Pairwise Sequence Alignment and Identity Calculation. PLoS ONE 2014, 9, e108277. [Google Scholar] [CrossRef]
- Talavera, G.; Castresana, J. Improvement of Phylogenies after Removing Divergent and Ambiguously Aligned Blocks from Protein Sequence Alignments. Syst. Biol. 2007, 56, 564–577. [Google Scholar] [CrossRef]
- Kalyaanamoorthy, S.; Minh, B.Q.; Wong, T.K.F.; Von Haeseler, A.; Jermiin, L.S. ModelFinder: Fast model selection for accurate phylogenetic estimates. Nat. Methods 2017, 14, 587–589. [Google Scholar] [CrossRef]
- Nguyen, L.-T.; Schmidt, H.A.; Von Haeseler, A.; Minh, B.Q. IQ-TREE: A Fast and Effective Stochastic Algorithm for Estimating Maximum-Likelihood Phylogenies. Mol. Biol. Evol. 2015, 32, 268–274. [Google Scholar] [CrossRef] [PubMed]
- Trifinopoulos, J.; Nguyen, L.-T.; Von Haeseler, A.; Minh, B.Q. W-IQ-TREE: A fast online phylogenetic tool for maximum likelihood analysis. Nucleic Acids Res. 2016, 44, W232–W235. [Google Scholar] [CrossRef] [PubMed]
- Guindon, S.; Dufayard, J.-F.; Lefort, V.; Anisimova, M.; Hordijk, W.; Gascuel, O. New Algorithms and Methods to Estimate Maximum-Likelihood Phylogenies: Assessing the Performance of PhyML 3.0. Syst. Biol. 2010, 59, 307–321. [Google Scholar] [CrossRef] [PubMed]
- Hoang, D.T.; Chernomor, O.; Von Haeseler, A.; Minh, B.Q.; Vinh, L.S. UFBoot2: Improving the Ultrafast Bootstrap Approximation. Mol. Biol. Evol. 2018, 35, 518–522. [Google Scholar] [CrossRef] [PubMed]
- Martin, D.P.; Murrell, B.; Golden, M.; Khoosal, A.; Muhire, B. RDP4: Detection and analysis of recombination patterns in virus genomes. Virus Evol. 2015, 1, vev003. [Google Scholar] [CrossRef] [PubMed]
- Van der Vlugt, R.; Berendsen, M. Development of a General Potexvirus Detection Method. Eur. J. Plant Pathol. 2002, 108, 367–371. [Google Scholar] [CrossRef]
- Edgar, R.C.; Taylor, J.; Lin, V.; Altman, T.; Barbera, P.; Meleshko, D.; Lohr, D.; Novakovsky, G.; Buchfink, B.; Al-Shayeb, B.; et al. Petabase-scale sequence alignment catalyses viral discovery. Nature 2022, 602, 142–147. [Google Scholar] [CrossRef]
- Babaian, A.; Edgar, R. Ribovirus classification by a polymerase barcode sequence. PeerJ 2022, 10, e14055. [Google Scholar] [CrossRef]
- Debat, H.; Bejerman, N. A glimpse into the DNA virome of the unique “living fossil” Welwitschia mirabilis. Gene 2022, 843, 146806. [Google Scholar] [CrossRef]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Tuo, D.; Shen, W.; Yang, Y.; Yan, P.; Li, X.; Zhou, P. Development and Validation of a Multiplex Reverse Transcription PCR Assay for Simultaneous Detection of Three Papaya Viruses. Viruses 2014, 6, 3893–3906. [Google Scholar] [CrossRef]
- Martelli, G.P.; Adams, M.J.; Kreuze, J.F.; Dolja, V.V. Family Flexiviridae: A Case Study in Virion and Genome Plasticity. Annu. Rev. Phytopathol. 2007, 45, 73–100. [Google Scholar] [CrossRef]
- Rozanov, M.N.; Koonin, E.V.; Gorbalenya, A. Conservation of the putative methyltransferase domain: A hallmark of the ‘Sindbis-like’ supergroup of positive-strand RNA viruses. J. Gen. Virol. 1992, 73, 2129–2134. [Google Scholar] [CrossRef]
- Koonin, E.V. The phylogeny of RNA-dependent RNA polymerases of positive-strand RNA viruses. J. Gen. Virol. 1991, 72, 2197–2206. [Google Scholar] [CrossRef]
- Chen, I.-H.; Chou, W.-J.; Lee, P.-Y.; Hsu, Y.-H.; Tsai, C.-H. The AAUAAA Motif of Bamboo Mosaic Virus RNA Is Involved in Minus-Strand RNA Synthesis and Plus-Strand RNA Polyadenylation. J. Virol. 2005, 79, 14555–14561. [Google Scholar] [CrossRef]
- Adams, M.J.; Antoniw, J.F.; Bar-Joseph, M.; Brunt, A.A.; Candresse, T.; Foster, G.; Martelli, G.P.; Milne, R.G.; Fauquet, C.M. Virology Division News: The new plant virus family Flexiviridae and assessment of molecular criteria for species demarcation. Arch. Virol. 2004, 149, 1045–1060. [Google Scholar] [CrossRef]
- Chávez-Calvillo, G.; Contreras-Paredes, C.A.; Mora-Macias, J.; Noa-Carrazana, J.C.; Serrano-Rubio, A.A.; Dinkova, T.D.; Carrillo-Tripp, M.; Silva-Rosales, L. Antagonism or synergism between papaya ringspot virus and papaya mosaic virus in Carica papaya is determined by their order of infection. Virology 2016, 489, 179–191. [Google Scholar] [CrossRef]
- Fiallo-Olivé, E.; Trenado, H.P.; Louro, D.; Navas-Castillo, J. Recurrent speciation of a tomato yellow leaf curl geminivirus in Portugal by recombination. Sci. Rep. 2019, 9, 1332. [Google Scholar] [CrossRef]
- Rubio, L.; Galipienso, L.; Ferriol, I. Detection of Plant Viruses and Disease Management: Relevance of Genetic Diversity and Evolution. Front. Plant Sci. 2020, 11, 1092. [Google Scholar] [CrossRef]
- Jones, R.A.; Naidu, R.A. Global Dimensions of Plant Virus Diseases: Current Status and Future Perspectives. Annu. Rev. Virol. 2019, 6, 387–409. [Google Scholar] [CrossRef]
- Kutnjak, D.; Tamisier, L.; Adams, I.; Boonham, N.; Candresse, T.; Chiumenti, M.; De Jonghe, K.; Kreuze, J.; Lefebvre, M.; Silva, G.; et al. A Primer on the Analysis of High-Throughput Sequencing Data for Detection of Plant Viruses. Microorganisms 2021, 9, 841. [Google Scholar] [CrossRef]
- Cornejo-Franco, J.F.; Alvarez-Quinto, R.A.; Quito-Avila, D.F. Transmission of the umbra-like Papaya virus Q in Ecuador and its association with meleira-related viruses from Brazil. Crop Prot. 2018, 110, 99–102. [Google Scholar] [CrossRef]
- Roossinck, M.J.; García-Arenal, F. Ecosystem simplification, biodiversity loss and plant virus emergence. Curr. Opin. Virol. 2015, 10, 56–62. [Google Scholar] [CrossRef]
- Medina, C.G.V.; Teppa, E.; Bornancini, V.A.; Flores, C.R.; Marino-Buslje, C.; Lambertini, P.M.L. Tomato Apical Leaf Curl Virus: A Novel, Monopartite Geminivirus Detected in Tomatoes in Argentina. Front. Microbiol. 2018, 8, 2665. [Google Scholar] [CrossRef]
- Reyna, P.G.; Bejerman, N.; Laguna, I.G.; Pardina, P.R. Biological and molecular characterization of bean bushy stunt virus, a novel bipartite begomovirus infecting common bean in northwestern Argentina. J. Behav. Health Serv. Res. 2021, 166, 1409–1414. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Virus | Genome Region | Position | Amino Acid Motif | Sequence | Amplicon Size (bp) |
|---|---|---|---|---|---|
| PapVX | RdRp | 1167-1173 | HQQAKDE | CACCARCARGCNARRGATGA | 737 |
| 1412-1406 | TFDANTE | TCDGTGTTKGCRTCRAADGT |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cabrera Mederos, D.; Debat, H.; Torres, C.; Portal, O.; Jaramillo Zapata, M.; Trucco, V.; Flores, C.; Ortiz, C.; Badaracco, A.; Acuña, L.; et al. An Unwanted Association: The Threat to Papaya Crops by a Novel Potexvirus in Northwest Argentina. Viruses 2022, 14, 2297. https://doi.org/10.3390/v14102297
Cabrera Mederos D, Debat H, Torres C, Portal O, Jaramillo Zapata M, Trucco V, Flores C, Ortiz C, Badaracco A, Acuña L, et al. An Unwanted Association: The Threat to Papaya Crops by a Novel Potexvirus in Northwest Argentina. Viruses. 2022; 14(10):2297. https://doi.org/10.3390/v14102297
Chicago/Turabian StyleCabrera Mederos, Dariel, Humberto Debat, Carolina Torres, Orelvis Portal, Margarita Jaramillo Zapata, Verónica Trucco, Ceferino Flores, Claudio Ortiz, Alejandra Badaracco, Luis Acuña, and et al. 2022. "An Unwanted Association: The Threat to Papaya Crops by a Novel Potexvirus in Northwest Argentina" Viruses 14, no. 10: 2297. https://doi.org/10.3390/v14102297
APA StyleCabrera Mederos, D., Debat, H., Torres, C., Portal, O., Jaramillo Zapata, M., Trucco, V., Flores, C., Ortiz, C., Badaracco, A., Acuña, L., Nome, C., Quito-Avila, D., Bejerman, N., Castellanos Collazo, O., Sánchez-Rodríguez, A., & Giolitti, F. (2022). An Unwanted Association: The Threat to Papaya Crops by a Novel Potexvirus in Northwest Argentina. Viruses, 14(10), 2297. https://doi.org/10.3390/v14102297