
Regulation of the Macroautophagic Machinery, Cellular Differentiation, and Immune Responses by Human Oncogenic γ-Herpesviruses


{kind=link}
{kind=link}
Abstract
1. Introduction on Human Oncogenic γ-Herpesviruses
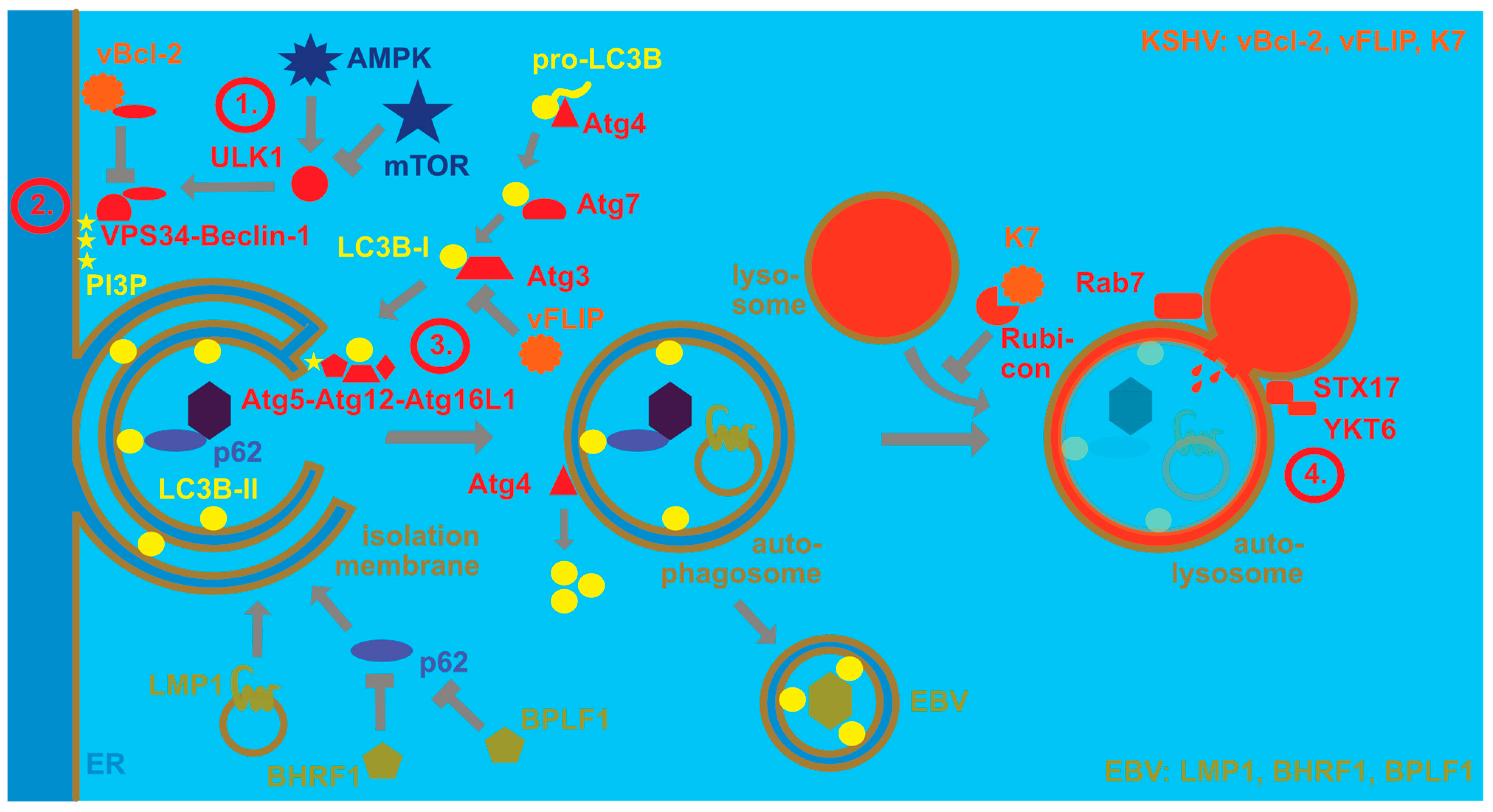
2. Regulation of Macroautophagy Proteins by EBV and KSHV
3. Influence of Human γ-Herpesviruses on B Cell Differentiation and Lymphomagenesis
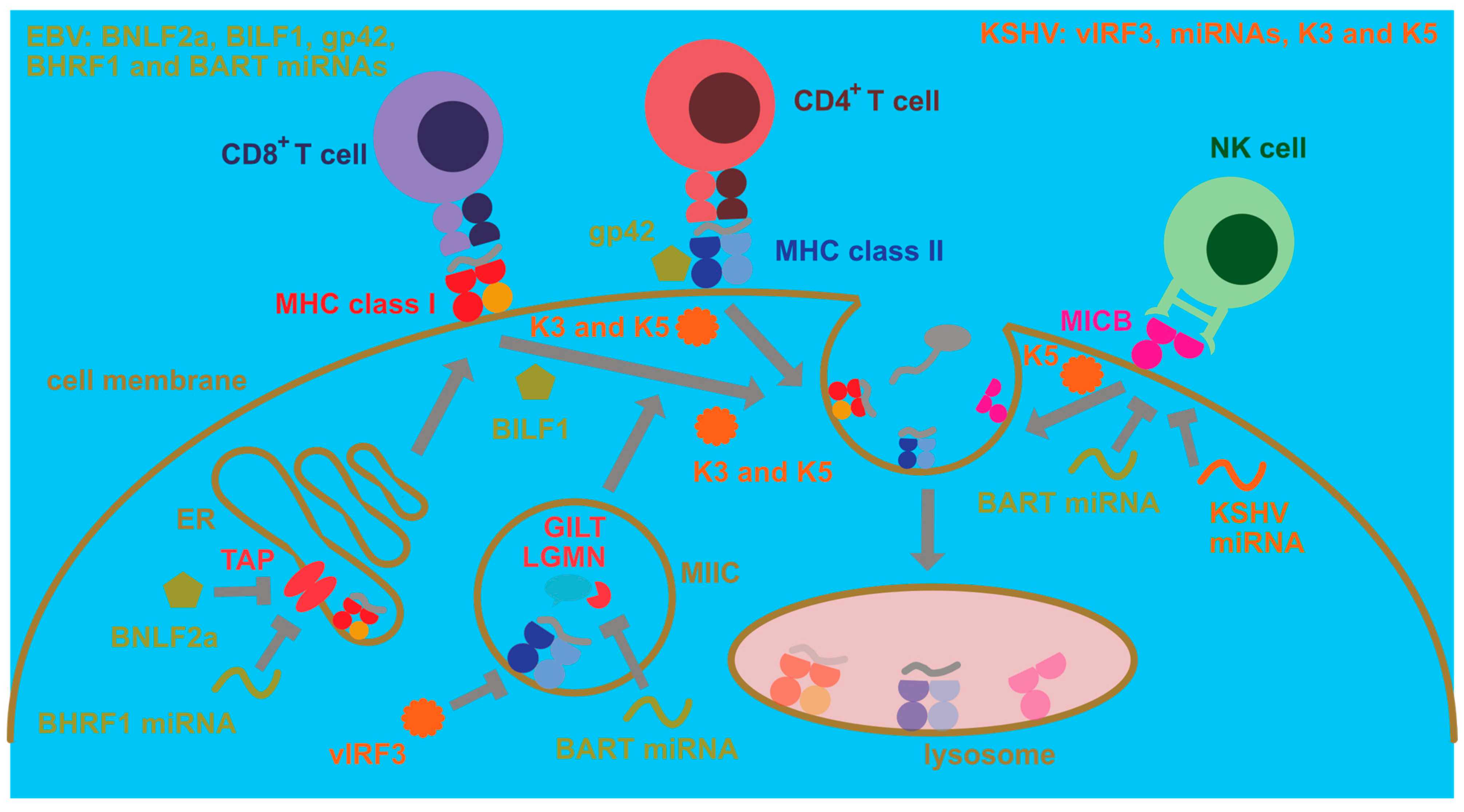
4. Manipulation of Antigen Presentation and Immune Recognition by EBV and KSHV
5. Conclusions and Outlook
Funding
Acknowledgments
Conflicts of Interest
References
- Azab, W.; Dayaram, A.; Greenwood, A.D.; Osterrieder, N. How host specific are herpesviruses? Lessons from herpesviruses infecting wild and endangered mammals. Annu. Rev. Virol. 2018, 5, 53–68. [Google Scholar] [CrossRef] [PubMed]
- Farrell, P.J. Epstein-Barr virus and cancer. Annu. Rev. Pathol. 2019, 14, 29–53. [Google Scholar] [CrossRef]
- Münz, C. Latency and lytic replication in the oncogenesis of the Epstein Barr virus. Nat. Rev. Micobiol. 2019, 17, 691–700. [Google Scholar] [CrossRef]
- Cesarman, E.; Damania, B.; Krown, S.E.; Martin, J.; Bower, M.; Whitby, D. Kaposi sarcoma. Nat. Rev. Dis. Prim. 2019, 5, 1–21. [Google Scholar] [CrossRef]
- Dunmire, S.K.; Verghese, P.S.; Balfour, H.H., Jr. Primary Epstein-Barr virus infection. J. Clin. Virol. 2018, 102, 84–92. [Google Scholar] [CrossRef] [PubMed]
- Cesarman, E. Gammaherpesviruses and Lymphoproliferative Disorders. Annu. Rev. Pathol. Mech. Dis. 2014, 9, 349–372. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Sathiyamoorthy, K.; Zhang, X.; Schaller, S.; White, B.E.P.; Jardetzky, T.S.; Longnecker, R. Ephrin receptor A2 is a functional entry receptor for Epstein-Barr virus. Nat. Microbiol. 2018, 3, 172–180. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Li, Y.; Wang, H.-B.; Zhang, A.; Chen, M.-L.; Fang, Z.-X.; Dong, X.-D.; Li, S.-B.; Du, Y.; Xiong, D.; et al. Ephrin receptor A2 is an epithelial cell receptor for Epstein-Barr virus entry. Nat. Microbiol. 2018, 3, 164–171. [Google Scholar] [CrossRef]
- Hahn, A.S.; Kaufmann, J.K.; Wies, E.; Naschberger, E.; Panteleev-Ivlev, J.; Schmidt, K.; Holzer, A.; Schmidt, M.; Chen, J.; Konig, S.; et al. The ephrin receptor tyrosine kinase A2 is a cellular receptor for Kaposi’s sarcoma-associated herpesvirus. Nat. Med. 2012, 18, 961–966. [Google Scholar] [CrossRef]
- Tugizov, S.M.; Berline, J.W.; Palefsky, J.M. Epstein-Barr virus infection of polarized tongue and nasopharyngeal epithelial cells. Nat. Med. 2003, 9, 307–314. [Google Scholar] [CrossRef]
- Tugizov, S.M.; Herrera, R.; Palefsky, J.M. Epstein-Barr Virus Transcytosis through Polarized Oral Epithelial Cells. J. Virol. 2013, 87, 8179–8194. [Google Scholar] [CrossRef] [PubMed]
- Thorley-Lawson, D.A. EBV persistence-introducing the virus. Curr. Top. Microbiol. Immunol. 2015, 390, 151–209. [Google Scholar] [PubMed]
- Babcock, J.G.; Hochberg, D.; Thorley-Lawson, A.D. The expression pattern of Epstein-Barr virus latent genes in vivo is dependent upon the differentiation stage of the infected B cell. Immunity 2000, 13, 497–506. [Google Scholar] [CrossRef]
- Skalsky, R.L.; Cullen, B.R. EBV noncoding RNAs. Curr. Top. Microbiol. Immunol. 2015, 391, 181–217. [Google Scholar] [PubMed]
- Babcock, G.J.; Decker, L.L.; Volk, M.; Thorley-Lawson, D.A. EBV persistence in memory B cells in vivo. Immunity 1998, 9, 395–404. [Google Scholar] [CrossRef]
- Hochberg, D.; Middeldorp, J.M.; Catalina, M.; Sullivan, J.L.; Luzuriaga, K.; Thorley-Lawson, D.A. Demonstration of the Burkitt’s lymphoma Epstein-Barr virus phenotype in dividing latently infected memory cells in vivo. Proc. Natl. Acad. Sci. USA 2004, 101, 239–244. [Google Scholar] [CrossRef] [PubMed]
- Woellmer, A.; Arteaga-Salas, J.M.; Hammerschmidt, W. BYLF1 governs CpG-methylated chromatin of Epstein-Barr virus reversing epigenetic repression. PLoS Pathog. 2012, 8, e1002902. [Google Scholar] [CrossRef]
- Laichalk, L.L.; Thorley-Lawson, D.A. Terminal differentiation into plasma cells initiates the replicative cycle of Epstein-Barr virus in vivo. J. Virol. 2005, 79, 1296–1307. [Google Scholar] [CrossRef]
- McHugh, D.; Caduff, N.; Barros, M.H.M.; Rämer, P.; Raykova, A.; Murer, A.; Landtwing, V.; Quast, I.; Styles, C.T.; Spohn, M.; et al. Persistent KSHV infection increases EBV-associated tumor formation in vivo via enhanced EBV lytic gene expression. Cell Host Microbe 2017, 22, 61–73. [Google Scholar] [CrossRef]
- Faure, A.; Hayes, M.; Sugden, B. How Kaposi’s sarcoma-associated herpesvirus stably transforms peripheral B cells towards lymphomagenesis. Proc. Natl. Acad. Sci. USA 2019, 116, 16519–16528. [Google Scholar] [CrossRef]
- Bigi, R.; Landis, J.T.; An, H.; Caro-Vegas, C.; Raab-Traub, N.; Dittmer, D.P. Epstein-Barr virus enhances genome maintenance of Kaposi sarcoma-associated herpesvirus. Proc. Natl. Acad. Sci. USA 2018, 115, E11379–E11387. [Google Scholar] [CrossRef]
- Blackbourn, D.J.; Lennette, E.; Klencke, B.; Moses, A.; Chandran, B.; Weinstein, M.; Glogau, R.G.; Witte, M.H.; Way, D.L.; Kutzkey, T.; et al. The restricted cellular host range of human herpesvirus 8. AIDS 2000, 14, 1123–1133. [Google Scholar] [CrossRef] [PubMed]
- Labo, N.; Marshall, V.; Miley, W.; Davis, E.; McCann, B.; Stolka, K.B.; Ndom, P.; Hemingway-Foday, J.J.; Abassora, M.; Newton, R.; et al. Mutual detection of Kaposi’s sarcoma-associated herpesvirus and Epstein-Barr virus in blood and saliva of cameroonians with and without Kaposi’s sarcoma. Int. J. Cancer 2019, 145, 2468–2477. [Google Scholar] [CrossRef]
- Sallah, N.; Miley, W.; Labo, N.; Carstensen, T.; Fatumo, S.; Gurdasani, D.; Pollard, M.O.; Dilthey, A.T.; Mentzer, A.J.; Marshall, V.; et al. Distinct genetic architectures and environmental factors associate with host response to the gamma2-herpesvirus infections. Nat. Commun. 2020, 11, 3849. [Google Scholar] [CrossRef]
- Hutt-Fletcher, L.M. The long and complicated relationship between Epstein-Barr virus and epithelial cells. J. Virol. 2017, 91, e01677-16. [Google Scholar] [CrossRef] [PubMed]
- Borza, C.M.; Hutt-Fletcher, L.M. Alternate replication in B cells and epithelial cells switches tropism of Epstein-Barr virus. Nat. Med. 2002, 8, 594–599. [Google Scholar] [CrossRef] [PubMed]
- Shannon-Lowe, C.; Rowe, M. Epstein-Barr virus infection of polarized epithelial cells via the basolateral surface by memory B cell-mediated transfer infection. PLoS Pathog. 2011, 7, e1001338. [Google Scholar] [CrossRef]
- Parkin, D.M. The global health burden of infection-associated cancers in the year 2002. Int. J. Cancer 2006, 118, 3030–3044. [Google Scholar] [CrossRef]
- Bouvard, V.; Baan, R.; Straif, K.; Grosse, Y.; Secretan, B.; El Ghissassi, F.; Benbrahim-Tallaa, L.; Guha, N.; Freeman, C.; Galichet, L.; et al. A review of human carcinogens-part b: Biological agents. Lancet Oncol. 2009, 10, 321–322. [Google Scholar] [CrossRef]
- Charostad, J.; Nakhaie, M.; Dehghani, A.; Faghihloo, E. The interplay between EBV and KSHV viral products and NF-kappaB pathway in oncogenesis. Infect. Agents Cancer 2020, 15, 62. [Google Scholar] [CrossRef]
- Shannon-Lowe, C.; Rickinson, A. The global landscape of EBV-associated tumors. Front. Oncol. 2019, 9, 713. [Google Scholar] [CrossRef] [PubMed]
- Mizushima, N.; Levine, B. Autophagy in Human Diseases. N. Engl. J. Med. 2020, 383, 1564–1576. [Google Scholar] [CrossRef] [PubMed]
- Mizushima, N.; Yoshimori, T.; Ohsumi, Y. The role of ATG proteins in autophagosome formation. Annu. Rev. Cell Dev. Biol. 2011, 27, 107–132. [Google Scholar] [CrossRef] [PubMed]
- Hu, Z.; Raucci, S.; Jaquenoud, M.; Hatakeyama, R.; Stumpe, M.; Rohr, R.; Reggiori, F.; De Virgilio, C.; Dengjel, J. Multilayered control of protein turnover by TORC1 and ATG1. Cell Rep. 2019, 28, 3486–3496.e3486. [Google Scholar] [CrossRef]
- Birgisdottir, A.B.; Johansen, T. Autophagy and endocytosis—Interconnections and interdependencies. J. Cell Sci. 2020, 133, jcs228114. [Google Scholar] [CrossRef] [PubMed]
- Itakura, E.; Kishi-Itakura, C.; Mizushima, N. The hairpin-type tail-anchored snare syntaxin 17 targets to autophagosomes for fusion with endosomes/lysosomes. Cell 2012, 151, 1256–1269. [Google Scholar] [CrossRef]
- Matsui, T.; Jiang, P.; Nakano, S.; Sakamaki, Y.; Yamamoto, H.; Mizushima, N. Autophagosomal YKT6 is required for fusion with lysosomes independently of syntaxin 17. J. Cell Biol. 2018, 217, 2633–2645. [Google Scholar] [CrossRef]
- Orvedahl, A.; Sumpter, R., Jr.; Xiao, G.; Ng, A.; Zou, Z.; Tang, Y.; Narimatsu, M.; Gilpin, C.; Sun, Q.; Roth, M.; et al. Image-based genome-wide siRNA screen identifies selective autophagy factors. Nature 2011, 480, 113–117. [Google Scholar] [CrossRef]
- Lee, D.Y.; Sugden, B. The latent membrane protein 1 oncogene modifies B-cell physiology by regulating autophagy. Oncogene 2008, 27, 2833–2842. [Google Scholar] [CrossRef]
- Bhattacharjee, S.; Bose, P.; Patel, K.; Roy, S.G.; Gain, C.; Gowda, H.; Robertson, E.S.; Saha, A. Transcriptional and epigenetic modulation of autophagy promotes ebv oncoprotein EBNA3C induced B-cell survival. Cell Death Dis 2018, 9, 605. [Google Scholar] [CrossRef]
- De Leo, A.; Colavita, F.; Ciccosanti, F.; Fimia, G.M.; Lieberman, P.M.; Mattia, E. Inhibition of autophagy in EBV-positive Burkitt’s lymphoma cells enhances EBV lytic genes expression and replication. Cell Death Dis. 2015, 6, e1876. [Google Scholar] [CrossRef] [PubMed]
- Nowag, H.; Guhl, B.; Thriene, K.; Romao, S.; Ziegler, U.; Dengjel, J.; Münz, C. Macroautopphagy proteins assist Epstein Barr virus production and get incorporated into the virus particles. EBioMedicine 2014, 1, 116–125. [Google Scholar] [CrossRef] [PubMed]
- Granato, M.; Santarelli, R.; Farina, A.; Gonnella, R.; Lotti, L.V.; Faggioni, A.; Cirone, M. EBV blocks the autophagic flux and appropriates the autophagic machinery to enhance viral replication. J. Virol. 2014, 88, 12715–12726. [Google Scholar] [CrossRef] [PubMed]
- Vilmen, G.; Glon, D.; Siracusano, G.; Lussignol, M.; Shao, Z.; Hernandez, E.; Perdiz, D.; Quignon, F.; Mouna, L.; Pous, C.; et al. BHRF1, a Bcl2 viral homolog, disturbs mitochondrial dynamics and stimulates mitophagy to dampen type i ifn induction. Autophagy 2020, in press. [Google Scholar] [CrossRef] [PubMed]
- Yla-Anttila, P.; Gupta, S.; Masucci, M.G. The Epstein-Barr virus deubiquitinase BPLF1 targets SQSTM1/p62 to inhibit selective autophagy. Autophagy 2021, in press. [Google Scholar] [CrossRef] [PubMed]
- Wen, H.J.; Yang, Z.; Zhou, Y.; Wood, C. Enhancement of autophagy during lytic replication by the Kaposi’s sarcoma-associated herpesvirus replication and transcription activator. J. Virol. 2010, 84, 7448–7458. [Google Scholar] [CrossRef]
- Pattingre, S.; Tassa, A.; Qu, X.; Garuti, R.; Liang, X.H.; Mizushima, N.; Packer, M.; Schneider, M.D.; Levine, B. Bcl-2 antiapoptotic proteins inhibit Beclin 1-dependent autophagy. Cell 2005, 122, 927–939. [Google Scholar] [CrossRef]
- Dong, X.; Cheng, A.; Zou, Z.; Yang, Y.S.; Sumpter, R.M., Jr.; Huang, C.L.; Bhagat, G.; Virgin, H.W.; Lira, S.A.; Levine, B. Endolysosomal trafficking of viral G protein-coupled receptor functions in innate immunity and control of viral oncogenesis. Proc. Natl. Acad. Sci. USA 2016, 113, 2994–2999. [Google Scholar] [CrossRef] [PubMed]
- Leidal, A.M.; Cyr, D.P.; Hill, R.J.; Lee, P.W.; McCormick, C. Subversion of autophagy by Kaposi’s sarcoma-associated herpesvirus impairs oncogene-induced senescence. Cell Host Microbe 2012, 11, 167–180. [Google Scholar] [CrossRef]
- Lee, J.S.; Li, Q.; Lee, J.Y.; Lee, S.H.; Jeong, J.H.; Lee, H.R.; Chang, H.; Zhou, F.C.; Gao, S.J.; Liang, C.; et al. FLIP-mediated autophagy regulation in cell death control. Nat. Cell Biol. 2009, 11, 1355–1362. [Google Scholar] [CrossRef] [PubMed]
- Liang, Q.; Chang, B.; Brulois, K.F.; Castro, K.; Min, C.K.; Rodgers, M.A.; Shi, M.; Ge, J.; Feng, P.; Oh, B.H.; et al. Kaposi’s sarcoma-associated herpesvirus K7 modulates Rubicon-mediated inhibition of autophagosome maturation. J. Virol. 2013, 87, 12499–12503. [Google Scholar] [CrossRef]
- Romeo, M.A.; Santarelli, R.; Gilardini Montani, M.S.; Gonnella, R.; Benedetti, R.; Faggioni, A.; Cirone, M. Viral infection and autophagy dysregulation: The case of HHV-6, EBV and KSHV. Cells 2020, 9, 2624. [Google Scholar] [CrossRef]
- Kulwichit, W.; Edwards, R.H.; Davenport, E.M.; Baskar, J.F.; Godfrey, V.; Raab-Traub, N. Expression of the Epstein-Barr virus latent membrane protein 1 induces B cell lymphoma in transgenic mice. Proc. Natl. Acad. Sci. USA 1998, 95, 11963–11968. [Google Scholar] [CrossRef] [PubMed]
- Wilson, J.B.; Bell, J.L.; Levine, A.J. Expression of Epstein-Barr virus nuclear antigen-1 induces B cell neoplasia in transgenic mice. EMBO J. 1996, 15, 3117–3126. [Google Scholar] [CrossRef]
- AlQarni, S.; Al-Sheikh, Y.; Campbell, D.; Drotar, M.; Hannigan, A.; Boyle, S.; Herzyk, P.; Kossenkov, A.; Armfield, K.; Jamieson, L.; et al. Lymphomas driven by Epstein-Barr virus nuclear antigen-1 (EBNA1) are dependant upon MDM2. Oncogene 2018, 37, 3998–4012. [Google Scholar] [CrossRef] [PubMed]
- Sin, S.H.; Kim, Y.; Eason, A.; Dittmer, D.P. KSHV latency locus cooperates with myc to drive lymphoma in mice. PLoS Pathog. 2015, 11, e1005135. [Google Scholar] [CrossRef]
- Altmann, M.; Hammerschmidt, W. Epstein-Barr virus provides a new paradigm: A requirement for the immediate inhibition of apoptosis. PLoS Biol. 2005, 3, e404. [Google Scholar] [CrossRef]
- Mrozek-Gorska, P.; Buschle, A.; Pich, D.; Schwarzmayr, T.; Fechtner, R.; Scialdone, A.; Hammerschmidt, W. Epstein-Barr virus reprograms human B lymphocytes immediately in the prelatent phase of infection. Proc. Natl. Acad. Sci. USA 2019, 116, 16046–16055. [Google Scholar] [CrossRef]
- Pich, D.; Mrozek-Gorska, P.; Bouvet, M.; Sugimoto, A.; Akidil, E.; Grundhoff, A.; Hamperl, S.; Ling, P.D.; Hammerschmidt, W. First days in the life of naive human B lymphocytes infected with Epstein-Barr virus. mBio 2019, 10, e01723-19. [Google Scholar] [CrossRef] [PubMed]
- Price, A.M.; Luftig, M.A. To be or not IIb: A multi-step process for Epstein-Barr virus latency establishment and consequences for B cell tumorigenesis. PLoS Pathog. 2015, 11, e1004656. [Google Scholar] [CrossRef] [PubMed]
- Price, A.M.; Tourigny, J.P.; Forte, E.; Salinas, R.E.; Dave, S.S.; Luftig, M.A. Analysis of Epstein-Barr virus-regulated host gene expression changes through primary B-cell outgrowth reveals delayed kinetics of latent membrane protein 1-mediated NF-kappaB activation. J. Virol. 2012, 86, 11096–11106. [Google Scholar] [CrossRef] [PubMed]
- Allday, M.J.; Bazot, Q.; White, R.E. The ebna3 family: Two oncoproteins and a tumour suppressor that are central to the biology of EBV in B cells. Curr. Top. Microbiol. Immunol. 2015, 391, 61–117. [Google Scholar]
- Kempkes, B.; Ling, P.D. EBNA2 and its coactivator EBNA-LP. Curr. Top. Microbiol. Immunol. 2015, 391, 35–59. [Google Scholar] [PubMed]
- Nikitin, P.A.; Yan, C.M.; Forte, E.; Bocedi, A.; Tourigny, J.P.; White, R.E.; Allday, M.J.; Patel, A.; Dave, S.S.; Kim, W.; et al. An ATM/chk2-mediated DNA damage-responsive signaling pathway suppresses Epstein-Barr virus transformation of primary human B cells. Cell Host Microbe 2010, 8, 510–522. [Google Scholar] [CrossRef]
- Anderton, E.; Yee, J.; Smith, P.; Crook, T.; White, R.E.; Allday, M.J. Two Epstein-Barr virus (EBV) oncoproteins cooperate to repress expression of the proapoptotic tumour-suppressor Bim: Clues to the pathogenesis of Burkitt’s lymphoma. Oncogene 2008, 27, 421–433. [Google Scholar] [CrossRef] [PubMed]
- Skalska, L.; White, R.E.; Parker, G.A.; Turro, E.; Sinclair, A.J.; Paschos, K.; Allday, M.J. Induction of p16(ink4a) is the major barrier to proliferation when Epstein-Barr virus (EBV) transforms primary B cells into lymphoblastoid cell lines. PLoS Pathog. 2013, 9, e1003187. [Google Scholar] [CrossRef]
- Paschos, K.; Parker, G.A.; Watanatanasup, E.; White, R.E.; Allday, M.J. Bim promoter directly targeted by EBNA3C in polycomb-mediated repression by EBV. Nucleic. Acids Res. 2012, 40, 7233–7246. [Google Scholar] [CrossRef]
- Kieser, A.; Sterz, K.R. The latent membrane protein 1 (LMP1). Curr. Top. Microbiol. Immunol. 2015, 391, 119–149. [Google Scholar]
- Cen, O.; Longnecker, R. Latent membrane protein 2 (LMP2). Curr. Top. Microbiol. Immunol. 2015, 391, 151–180. [Google Scholar]
- Thorley-Lawson, D.A. Epstein-Barr virus: Exploiting the immune system. Nat. Rev. Immunol. 2001, 1, 75–82. [Google Scholar] [CrossRef]
- Heuts, F.; Rottenberg, M.E.; Salamon, D.; Rasul, E.; Adori, M.; Klein, G.; Klein, E.; Nagy, N. T cells modulate Epstein-Barr virus latency phenotypes during infection of humanized mice. J. Virol. 2014, 88, 3235–3245. [Google Scholar] [CrossRef]
- Nagy, N.; Adori, M.; Rasul, A.; Heuts, F.; Salamon, D.; Ujvari, D.; Madapura, H.S.; Leveau, B.; Klein, G.; Klein, E. Soluble factors produced by activated CD4+ T cells modulate EBV latency. Proc. Natl. Acad. Sci. USA 2012, 109, 1512–1517. [Google Scholar] [CrossRef]
- Murer, A.; McHugh, D.; Caduff, N.; Kalchschmidt, J.S.; Barros, M.H.; Zbinden, A.; Capaul, R.; Niedobitek, G.; Allday, M.J.; Chijioke, O.; et al. EBV persistence without its EBNA3A and 3C oncogenes in vivo. PLoS Pathog. 2018, 14, e1007039. [Google Scholar] [CrossRef] [PubMed]
- Sin, S.H.; Dittmer, D.P. Viral latency locus augments B-cell response in vivo to induce chronic marginal zone enlargement, plasma cell hyperplasia, and lymphoma. Blood 2013, 121, 2952–2963. [Google Scholar] [CrossRef] [PubMed]
- Klein, U.; Gloghini, A.; Gaidano, G.; Chadburn, A.; Cesarman, E.; Dalla-Favera, R.; Carbone, A. Gene expression profile analysis of AIDS-related primary effusion lymphoma (PEL) suggests a plasmablastic derivation and identifies PEL-specific transcripts. Blood 2003, 101, 4115–4121. [Google Scholar] [CrossRef]
- McDonald, C.; Karstegl, C.E.; Kellam, P.; Farrell, P.J. Regulation of the Epstein-Barr virus Zp promoter in B lymphocytes during reactivation from latency. J. Gen. Virol. 2010, 91, 622–629. [Google Scholar] [CrossRef]
- Reusch, J.A.; Nawandar, D.M.; Wright, K.L.; Kenney, S.C.; Mertz, J.E. Cellular differentiation regulator BLIMP1 induces Epstein-Barr virus lytic reactivation in epithelial and B cells by activating transcription from both the R and Z promoters. J. Virol. 2015, 89, 1731–1743. [Google Scholar] [CrossRef] [PubMed]
- Ersing, I.; Nobre, L.; Wang, L.W.; Soday, L.; Ma, Y.; Paulo, J.A.; Narita, Y.; Ashbaugh, C.W.; Jiang, C.; Grayson, N.E.; et al. A temporal proteomic map of Epstein-Barr virus lytic replication in B cells. Cell Rep. 2017, 19, 1479–1493. [Google Scholar] [CrossRef]
- Ma, S.D.; Hegde, S.; Young, K.H.; Sullivan, R.; Rajesh, D.; Zhou, Y.; Jankowska-Gan, E.; Burlingham, W.J.; Sun, X.; Gulley, M.L.; et al. A new model of Epstein-Barr virus infection reveals an important role for early lytic viral protein expression in the development of lymphomas. J. Virol. 2011, 85, 165–177. [Google Scholar] [CrossRef]
- Ma, S.D.; Yu, X.; Mertz, J.E.; Gumperz, J.E.; Reinheim, E.; Zhou, Y.; Tang, W.; Burlingham, W.J.; Gulley, M.L.; Kenney, S.C. An Epstein-Barr virus (EBV) mutant with enhanced BZLF1 expression causes lymphomas with abortive lytic EBV infection in a humanized mouse model. J. Virol. 2012, 86, 7976–7987. [Google Scholar] [CrossRef]
- Bristol, J.A.; Djavadian, R.; Albright, E.R.; Coleman, C.B.; Ohashi, M.; Hayes, M.; Romero-Masters, J.C.; Barlow, E.A.; Farrell, P.J.; Rochford, R.; et al. A cancer-associated Epstein-Barr virus BZLF1 promoter variant enhances lytic infection. PLoS Pathog. 2018, 14, e1007179. [Google Scholar] [CrossRef]
- Okuno, Y.; Murata, T.; Sato, Y.; Muramatsu, H.; Ito, Y.; Watanabe, T.; Okuno, T.; Murakami, N.; Yoshida, K.; Sawada, A.; et al. Defective Epstein-Barr virus in chronic active infection and haematological malignancy. Nat. Microbiol. 2019, 4, 404–413. [Google Scholar] [CrossRef]
- Antsiferova, O.; Müller, A.; Rämer, P.; Chijioke, O.; Chatterjee, B.; Raykova, A.; Planas, R.; Sospedra, M.; Shumilov, A.; Tsai, M.H.; et al. Adoptive transfer of EBV specific CD8+ T cell clones can transiently control EBV infection in humanized mice. PLoS Pathog. 2014, 10, e1004333. [Google Scholar] [CrossRef] [PubMed]
- Tsai, M.H.; Lin, X.; Shumilov, A.; Bernhardt, K.; Feederle, R.; Poirey, R.; Kopp-Schneider, A.; Pereira, B.; Almeida, R.; Delecluse, H.J. The biological properties of different Epstein-Barr virus strains explain their association with various types of cancers. Oncotarget 2017, 8, 10238–10254. [Google Scholar] [CrossRef] [PubMed]
- Tsai, M.H.; Raykova, A.; Klinke, O.; Bernhardt, K.; Gartner, K.; Leung, C.S.; Geletneky, K.; Sertel, S.; Münz, C.; Feederle, R.; et al. Spontaneous lytic replication and epitheliotropism define an Epstein-Barr virus strain found in carcinomas. Cell Rep. 2013, 5, 458–470. [Google Scholar] [CrossRef]
- Schuren, A.B.; Costa, A.I.; Wiertz, E.J. Recent advances in viral evasion of the MHC class I processing pathway. Curr. Opin. Immunol. 2016, 40, 43–50. [Google Scholar] [CrossRef] [PubMed]
- Ressing, M.E.; Keating, S.E.; van Leeuwen, D.; Koppers-Lalic, D.; Pappworth, I.Y.; Wiertz, E.J.; Rowe, M. Impaired transporter associated with antigen processing-dependent peptide transport during productive EBV infection. J. Immunol. 2005, 174, 6829–6838. [Google Scholar] [CrossRef]
- Pudney, V.A.; Leese, A.M.; Rickinson, A.B.; Hislop, A.D. CD8+ immunodominance among Epstein-Barr virus lytic cycle antigens directly reflects the efficiency of antigen presentation in lytically infected cells. J. Exp. Med. 2005, 201, 349–360. [Google Scholar] [CrossRef] [PubMed]
- Hislop, A.D.; Ressing, M.E.; van Leeuwen, D.; Pudney, V.A.; Horst, D.; Koppers-Lalic, D.; Croft, N.P.; Neefjes, J.J.; Rickinson, A.B.; Wiertz, E.J. A CD8+ T cell immune evasion protein specific to Epstein-Barr virus and its close relatives in old world primates. J. Exp. Med. 2007, 204, 1863–1873. [Google Scholar] [CrossRef]
- Croft, N.P.; Shannon-Lowe, C.; Bell, A.I.; Horst, D.; Kremmer, E.; Ressing, M.E.; Wiertz, E.J.; Middeldorp, J.M.; Rowe, M.; Rickinson, A.B.; et al. Stage-specific inhibition of MHC class I presentation by the Epstein-Barr virus BNLF2a protein during virus lytic cycle. PLoS Pathog. 2009, 5, e1000490. [Google Scholar] [CrossRef]
- Horst, D.; van Leeuwen, D.; Croft, N.P.; Garstka, M.A.; Hislop, A.D.; Kremmer, E.; Rickinson, A.B.; Wiertz, E.J.; Ressing, M.E. Specific targeting of the EBV lytic phase protein BNLF2a to the transporter associated with antigen processing results in impairment of HLA class I-restricted antigen presentation. J. Immunol. 2009, 182, 2313–2324. [Google Scholar] [CrossRef]
- Jochum, S.; Moosmann, A.; Lang, S.; Hammerschmidt, W.; Zeidler, R. The EBV immunoevasins vIL-10 and BNLF2a protect newly infected B cells from immune recognition and elimination. PLoS Pathog. 2012, 8, e1002704. [Google Scholar] [CrossRef] [PubMed]
- Zuo, J.; Currin, A.; Griffin, B.D.; Shannon-Lowe, C.; Thomas, W.A.; Ressing, M.E.; Wiertz, E.J.; Rowe, M. The Epstein-Barr virus G-protein-coupled receptor contributes to immune evasion by targeting MHC class I molecules for degradation. PLoS Pathog. 2009, 5, e1000255. [Google Scholar] [CrossRef]
- Rowe, M.; Glaunsinger, B.; van Leeuwen, D.; Zuo, J.; Sweetman, D.; Ganem, D.; Middeldorp, J.; Wiertz, E.J.; Ressing, M.E. Host shutoff during productive Epstein-Barr virus infection is mediated by BGLF5 and may contribute to immune evasion. Proc. Natl. Acad. Sci. USA 2007, 104, 3366–3371. [Google Scholar] [CrossRef]
- Zuo, J.; Thomas, W.; van Leeuwen, D.; Middeldorp, J.M.; Wiertz, E.J.; Ressing, M.E.; Rowe, M. The DNAse of gammaherpesviruses impairs recognition by virus-specific CD8+ T cells through an additional host shutoff function. J. Virol. 2008, 82, 2385–2393. [Google Scholar] [CrossRef]
- Quinn, L.L.; Zuo, J.; Abbott, R.J.; Shannon-Lowe, C.; Tierney, R.J.; Hislop, A.D.; Rowe, M. Cooperation between Epstein-Barr virus immune evasion proteins spreads protection from CD8+ T cell recognition across all three phases of the lytic cycle. PLoS Pathog. 2014, 10, e1004322. [Google Scholar] [CrossRef] [PubMed]
- Albanese, M.; Tagawa, T.; Bouvet, M.; Maliqi, L.; Lutter, D.; Hoser, J.; Hastreiter, M.; Hayes, M.; Sugden, B.; Martin, L.; et al. Epstein-Barr virus microRNAs reduce immune surveillance by virus-specific CD8+ T cells. Proc. Natl. Acad. Sci. USA 2016, 113, E6467–E6475. [Google Scholar] [CrossRef]
- Murer, A.; Ruhl, J.; Zbinden, A.; Capaul, R.; Hammerschmidt, W.; Chijioke, O.; Münz, C. MicroRNAs of Epstein-Barr virus attenuate T-cell-mediated immune control in vivo. mBio 2019, 10, e01941-01918. [Google Scholar] [CrossRef]
- Levitskaya, J.; Coram, M.; Levitsky, V.; Imreh, S.; Steigerwald-Mullen, P.M.; Klein, G.; Kurilla, M.G.; Masucci, M.G. Inhibition of antigen processing by the internal repeat region of the Epstein-Barr virus nuclear antigen-1. Nature 1995, 375, 685–688. [Google Scholar] [CrossRef] [PubMed]
- Levitskaya, J.; Sharipo, A.; Leonchiks, A.; Ciechanover, A.; Masucci, M.G. Inhibition of ubiquitin/proteasome-dependent protein degradation by the Gly-Ala repeat domain of the Epstein-Barr virus nuclear antigen 1. Proc. Natl. Acad. Sci. USA 1997, 94, 12616–12621. [Google Scholar] [CrossRef] [PubMed]
- Yin, Y.; Manoury, B.; Fahraeus, R. Self-inhibition of synthesis and antigen presentation by Epstein-Barr virus-encoded EBNA1. Science 2003, 301, 1371–1374. [Google Scholar] [CrossRef] [PubMed]
- Tomescu, C.; Law, W.K.; Kedes, D.H. Surface downregulation of major histocompatibility complex class I, PE-CAM, and ICAM-1 following de novo infection of endothelial cells with Kaposi’s sarcoma-associated herpesvirus. J. Virol. 2003, 77, 9669–9684. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Coscoy, L.; Ganem, D. Kaposi’s sarcoma-associated herpesvirus encodes two proteins that block cell surface display of MHC class I chains by enhancing their endocytosis. Proc. Natl. Acad. Sci. USA 2000, 97, 8051–8056. [Google Scholar] [CrossRef]
- Ishido, S.; Wang, C.; Lee, B.S.; Cohen, G.B.; Jung, J.U. Downregulation of major histocompatibility complex class I molecules by Kaposi’s sarcoma-associated herpesvirus K3 and K5 proteins. J. Virol. 2000, 74, 5300–5309. [Google Scholar] [CrossRef] [PubMed]
- Duncan, L.M.; Nathan, J.A.; Lehner, P.J. Stabilization of an E3 ligase-E2-ubiquitin complex increases cell surface MHC class I expression. J. Immunol. 2010, 184, 6978–6985. [Google Scholar] [CrossRef] [PubMed]
- Hewitt, E.W.; Duncan, L.; Mufti, D.; Baker, J.; Stevenson, P.G.; Lehner, P.J. Ubiquitylation of MHC class I by the K3 viral protein signals internalization and tsg101-dependent degradation. EMBO J. 2002, 21, 2418–2429. [Google Scholar] [CrossRef]
- Damania, B.; Münz, C. Immunodeficiencies that predispose to pathologies by human oncogenic gamma-herpesviruses. FEMS Microbiol. Rev. 2019, 43, 181–192. [Google Scholar] [CrossRef] [PubMed]
- Strowig, T.; Gurer, C.; Ploss, A.; Liu, Y.F.; Arrey, F.; Sashihara, J.; Koo, G.; Rice, C.M.; Young, J.W.; Chadburn, A.; et al. Priming of protective T cell responses against virus-induced tumors in mice with human immune system components. J. Exp. Med. 2009, 206, 1423–1434. [Google Scholar] [CrossRef]
- Chijioke, O.; Marcenaro, E.; Moretta, A.; Capaul, R.; Münz, C. The SAP-dependent 2B4 receptor mediates CD8+ T cell dependent immune control of Epstein Barr virus infection in mice with reconstituted human immune system components. J. Infect. Dis. 2015, 212, 803–807. [Google Scholar] [CrossRef] [PubMed]
- McHugh, D.; Myburgh, R.; Caduff, N.; Spohn, M.; Kok, Y.L.; Keller, C.W.; Murer, A.; Chatterjee, B.; Rühl, J.; Engelmann, C.; et al. EBV renders B cells susceptible to HIV-1 in humanized mice. Life Sci. Alliance 2020, 3, e202000640. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Spriggs, M.K.; Kovats, S.; Turk, S.M.; Comeau, M.R.; Nepom, B.; Hutt-Fletcher, L.M. Epstein-Barr virus uses HLA class II as a cofactor for infection of B lymphocytes. J. Virol. 1997, 71, 4657–4662. [Google Scholar] [CrossRef]
- Ressing, M.E.; van Leeuwen, D.; Verreck, F.A.; Gomez, R.; Heemskerk, B.; Toebes, M.; Mullen, M.M.; Jardetzky, T.S.; Longnecker, R.; Schilham, M.W.; et al. Interference with T cell receptor-HLA-DR interactions by Epstein-Barr virus gp42 results in reduced T helper cell recognition. Proc. Natl. Acad. Sci. USA 2003, 100, 11583–11588. [Google Scholar] [CrossRef] [PubMed]
- Ressing, M.E.; van Leeuwen, D.; Verreck, F.A.; Keating, S.; Gomez, R.; Franken, K.L.; Ottenhoff, T.H.; Spriggs, M.; Schumacher, T.N.; Hutt-Fletcher, L.M.; et al. Epstein-Barr virus gp42 is posttranslationally modified to produce soluble gp42 that mediates HLA class II immune evasion. J. Virol. 2005, 79, 841–852. [Google Scholar] [CrossRef] [PubMed]
- Tagawa, T.; Albanese, M.; Bouvet, M.; Moosmann, A.; Mautner, J.; Heissmeyer, V.; Zielinski, C.; Lutter, D.; Hoser, J.; Hastreiter, M.; et al. Epstein-Barr viral miRNAs inhibit antiviral CD4+ T cell responses targeting IL-12 and peptide processing. J. Exp. Med. 2016, 213, 2065–2080. [Google Scholar] [CrossRef] [PubMed]
- Nathan, J.A.; Lehner, P.J. The trafficking and regulation of membrane receptors by the RING-CH ubiquitin E3 ligases. Exp. Cell Res. 2009, 315, 1593–1600. [Google Scholar] [CrossRef]
- Zuo, J.; Hislop, A.D.; Leung, C.S.; Sabbah, S.; Rowe, M. Kaposi’s sarcoma-associated herpesvirus-encoded viral IRF3 modulates major histocompatibility complex class II (MHC-II) antigen presentation through MHC-II transactivator-dependent and -independent mechanisms: Implications for oncogenesis. J. Virol. 2013, 87, 5340–5350. [Google Scholar] [CrossRef] [PubMed]
- El Hawary, R.E.; Mauracher, A.A.; Meshaal, S.S.; Eldash, A.; Abd Elaziz, D.S.; Alkady, R.; Lotfy, S.; Opitz, L.; Galal, N.M.; Boutros, J.A.; et al. MHC-II deficiency among Egyptians: Novel mutations and unique phenotypes. J. Allergy Clin. Immunol. Pract. 2019, 7, 856–863. [Google Scholar] [CrossRef]
- Chijioke, O.; Muller, A.; Feederle, R.; Barros, M.H.; Krieg, C.; Emmel, V.; Marcenaro, E.; Leung, C.S.; Antsiferova, O.; Landtwing, V.; et al. Human natural killer cells prevent infectious mononucleosis features by targeting lytic Epstein-Barr virus infection. Cell Rep. 2013, 5, 1489–1498. [Google Scholar] [CrossRef]
- Azzi, T.; Lunemann, A.; Murer, A.; Ueda, S.; Beziat, V.; Malmberg, K.J.; Staubli, G.; Gysin, C.; Berger, C.; Münz, C.; et al. Role for early-differentiated natural killer cells in infectious mononucleosis. Blood 2014, 124, 2533–2543. [Google Scholar] [CrossRef]
- Landtwing, V.; Raykova, A.; Pezzino, G.; Beziat, V.; Marcenaro, E.; Graf, C.; Moretta, A.; Capaul, R.; Zbinden, A.; Ferlazzo, G.; et al. Cognate HLA absence in trans diminishes human NK cell education. J. Clin. Investig. 2016, 126, 3772–3782. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Lam, J.K.P.; Azzi, T.; Hui, K.F.; Wong, A.M.G.; McHugh, D.; Caduff, N.; Chan, K.H.; Münz, C.; Chiang, A.K.S. Co-infection of cytomegalovirus and Epstein-Barr virus diminishes the frequency of CD56dimNKG2A+KIR− NK cells and contributes to suboptimal control of EBV in immunosuppressed children with post-transplant lymphoproliferative disorder. Front. Immunol. 2020, 11, 1231. [Google Scholar] [CrossRef] [PubMed]
- Biron, C.A.; Byron, K.S.; Sullivan, J.L. Severe herpesvirus infections in an adolescent without natural killer cells. N. Engl. J. Med. 1989, 320, 1731–1735. [Google Scholar] [CrossRef]
- Eidenschenk, C.; Dunne, J.; Jouanguy, E.; Fourlinnie, C.; Gineau, L.; Bacq, D.; McMahon, C.; Smith, O.; Casanova, J.L.; Abel, L.; et al. A novel primary immunodeficiency with specific natural-killer cell deficiency maps to the centromeric region of chromosome 8. Am. J. Hum. Genet. 2006, 78, 721–727. [Google Scholar] [CrossRef] [PubMed]
- Gineau, L.; Cognet, C.; Kara, N.; Lach, F.P.; Dunne, J.; Veturi, U.; Picard, C.; Trouillet, C.; Eidenschenk, C.; Aoufouchi, S.; et al. Partial MCM4 deficiency in patients with growth retardation, adrenal insufficiency, and natural killer cell deficiency. J. Clin. Investig. 2012, 122, 821–832. [Google Scholar] [CrossRef]
- Pappworth, I.Y.; Wang, E.C.; Rowe, M. The switch from latent to productive infection in Epstein-Barr virus-infected B cells is associated with sensitization to NK cell killing. J. Virol. 2007, 81, 474–482. [Google Scholar] [CrossRef] [PubMed]
- Nachmani, D.; Stern-Ginossar, N.; Sarid, R.; Mandelboim, O. Diverse herpesvirus microRNAs target the stress-induced immune ligand MICB to escape recognition by natural killer cells. Cell Host Microbe 2009, 5, 376–385. [Google Scholar] [CrossRef] [PubMed]
- Thomas, M.; Boname, J.M.; Field, S.; Nejentsev, S.; Salio, M.; Cerundolo, V.; Wills, M.; Lehner, P.J. Down-regulation of NKG2D and NKp80 ligands by Kaposi’s sarcoma-associated herpesvirus K5 protects against NK cell cytotoxicity. Proc. Natl. Acad. Sci. USA 2008, 105, 1656–1661. [Google Scholar] [CrossRef]
- Caduff, N.; McHugh, D.; Rieble, L.; Forconi, C.S.; Ong’echa, J.M.; Oluoch, P.O.; Raykova, A.; Murer, A.; Böni, M.; Zuppiger, L.; et al. KSHV infection drives poorly cytotoxic CD56 negative natural killer cell differentiation in vivo upon KSHV/EBV dual infection. Cell Rep. 2021, 35, 109056. [Google Scholar] [CrossRef]
- Liu, Y.; de Waal Malefyt, R.; Briere, F.; Parham, C.; Bridon, J.M.; Banchereau, J.; Moore, K.W.; Xu, J. The EBV IL-10 homologue is a selective agonist with impaired binding to the IL-10 receptor. J. Immunol. 1997, 158, 604–613. [Google Scholar]
- Bejarano, M.T.; Masucci, M.G. Interleukin-10 abrogates the inhibition of Epstein-Barr virus-induced B-cell transformation by memory T-cell responses. Blood 1998, 92, 4256–4262. [Google Scholar] [CrossRef] [PubMed]
- Salek-Ardakani, S.; Arrand, J.R.; Mackett, M. Epstein-Barr virus encoded interleukin-10 inhibits HLA-class I, ICAM-1, and B7 expression on human monocytes: Implications for immune evasion by EBV. Virology 2002, 304, 342–351. [Google Scholar] [CrossRef] [PubMed]
- Zeidler, R.; Eissner, G.; Meissner, P.; Uebel, S.; Tampe, R.; Lazis, S.; Hammerschmidt, W. Downregulation of TAP1 in B lymphocytes by cellular and Epstein-Barr virus-encoded interleukin-10. Blood 1997, 90, 2390–2397. [Google Scholar] [CrossRef] [PubMed]
- Nicholas, J.; Ruvolo, V.R.; Burns, W.H.; Sandford, G.; Wan, X.; Ciufo, D.; Hendrickson, S.B.; Guo, H.G.; Hayward, G.S.; Reitz, M.S. Kaposi’s sarcoma-associated human herpesvirus-8 encodes homologues of macrophage inflammatory protein-1 and interleukin-6. Nat. Med. 1997, 3, 287–292. [Google Scholar] [CrossRef]
- Moore, P.S.; Boshoff, C.; Weiss, R.A.; Chang, Y. Molecular mimicry of human cytokine and cytokine response pathway genes by KSHV. Science 1996, 274, 1739–1744. [Google Scholar] [CrossRef]
- Yamin, R.; Kaynan, N.S.; Glasner, A.; Vitenshtein, A.; Tsukerman, P.; Bauman, Y.; Ophir, Y.; Elias, S.; Bar-On, Y.; Gur, C.; et al. The viral KSHV chemokine vMIP-II inhibits the migration of naive and activated human NK cells by antagonizing two distinct chemokine receptors. PLoS Pathog. 2013, 9, e1003568. [Google Scholar] [CrossRef] [PubMed]
- Xia, T.; O’Hara, A.; Araujo, I.; Barreto, J.; Carvalho, E.; Sapucaia, J.B.; Ramos, J.C.; Luz, E.; Pedroso, C.; Manrique, M.; et al. EBV microRNAs in primary lymphomas and targeting of CXCL-11 by EBV-miR-BHRF1-3. Cancer Res. 2008, 68, 1436–1442. [Google Scholar] [CrossRef] [PubMed]
- White, R.E.; Ramer, P.C.; Naresh, K.N.; Meixlsperger, S.; Pinaud, L.; Rooney, C.; Savoldo, B.; Coutinho, R.; Bodor, C.; Gribben, J.; et al. EBNA3B-deficient EBV promotes B cell lymphomagenesis in humanized mice and is found in human tumors. J. Clin. Investig. 2012, 122, 1487–1502. [Google Scholar] [CrossRef] [PubMed]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Münz, C. Regulation of the Macroautophagic Machinery, Cellular Differentiation, and Immune Responses by Human Oncogenic γ-Herpesviruses. Viruses 2021, 13, 859. https://doi.org/10.3390/v13050859
Münz C. Regulation of the Macroautophagic Machinery, Cellular Differentiation, and Immune Responses by Human Oncogenic γ-Herpesviruses. Viruses. 2021; 13(5):859. https://doi.org/10.3390/v13050859
Chicago/Turabian StyleMünz, Christian. 2021. "Regulation of the Macroautophagic Machinery, Cellular Differentiation, and Immune Responses by Human Oncogenic γ-Herpesviruses" Viruses 13, no. 5: 859. https://doi.org/10.3390/v13050859
APA StyleMünz, C. (2021). Regulation of the Macroautophagic Machinery, Cellular Differentiation, and Immune Responses by Human Oncogenic γ-Herpesviruses. Viruses, 13(5), 859. https://doi.org/10.3390/v13050859