Manipulation of JAK/STAT Signalling by High-Risk HPVs: Potential Therapeutic Targets for HPV-Associated Malignancies



Abstract
1. Introduction
2. Human Papillomaviruses
2.1. HPV Life Cycle
2.2. HPV in Cancer Development
3. Janus Kinase/Signal Transducer and Activator of Transcription (JAK/STAT) Signalling Pathways
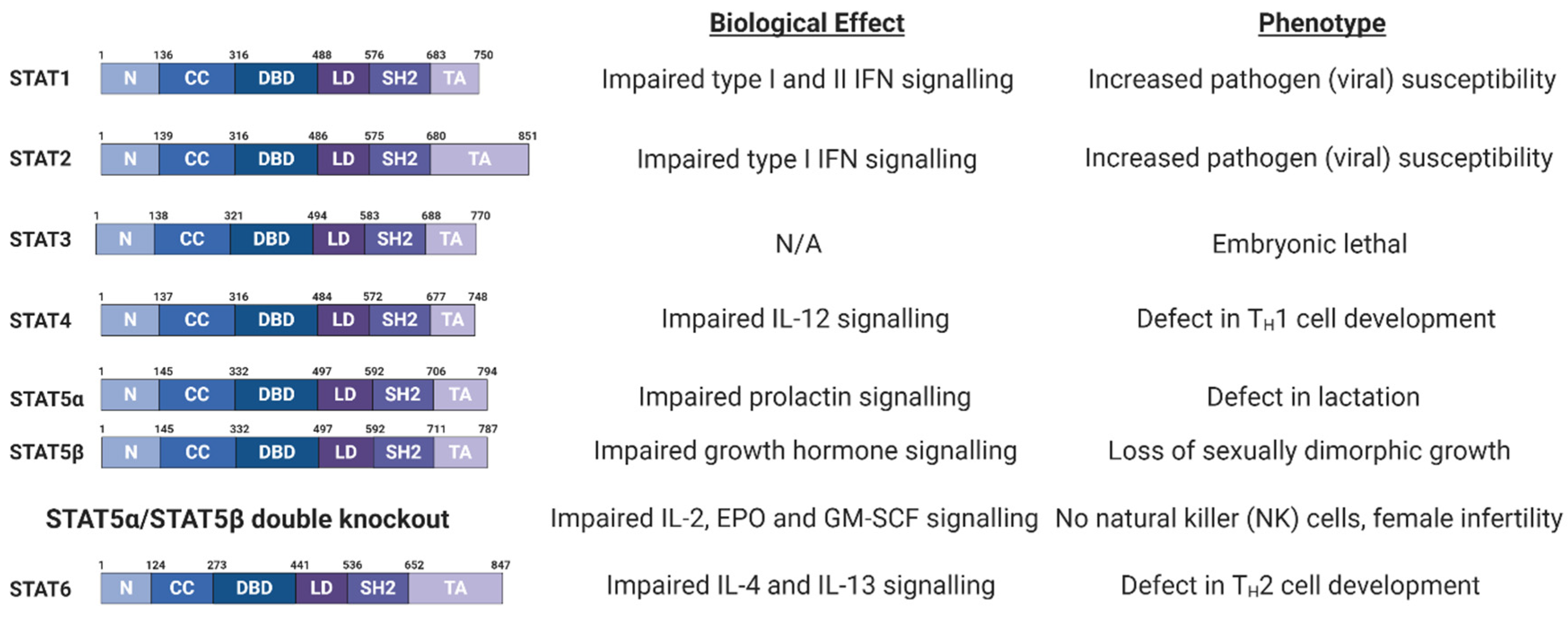
3.1. Signal Transducer and Activator of Transcription (STAT) Family
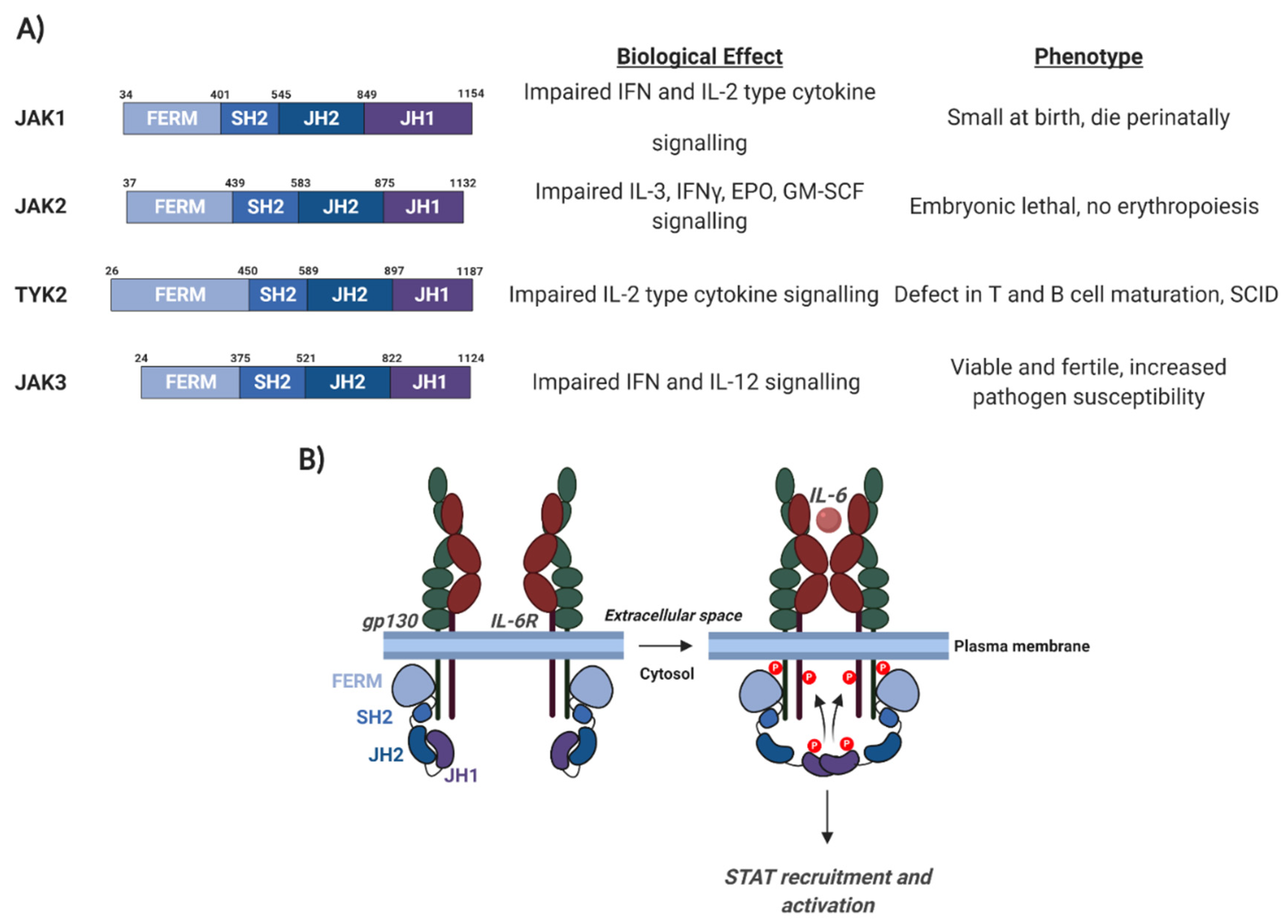
3.2. Janus Kinases (JAKs)
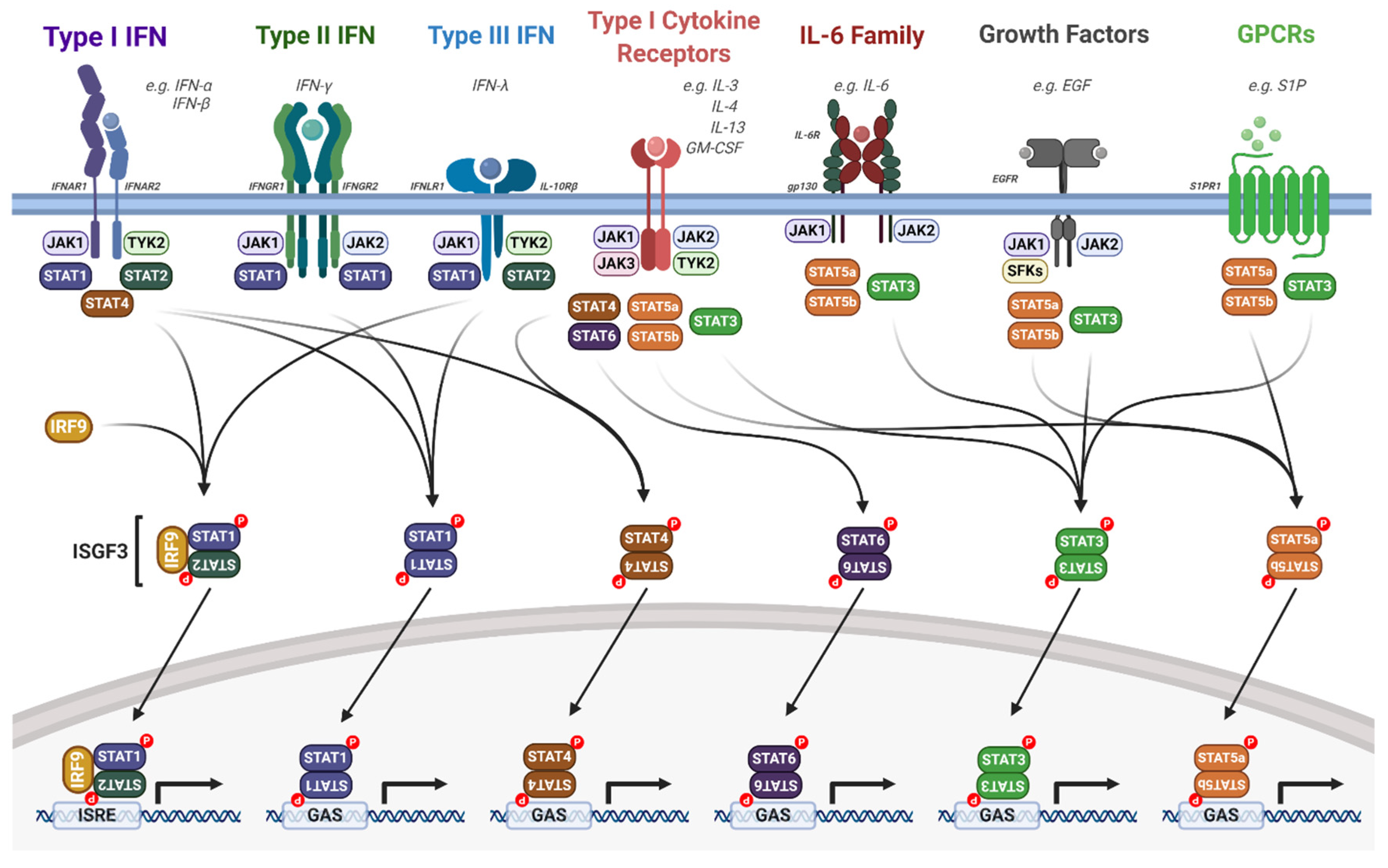
3.3. Activation of JAK/STAT Signalling
4. JAK/STAT Signalling in the Immune Response
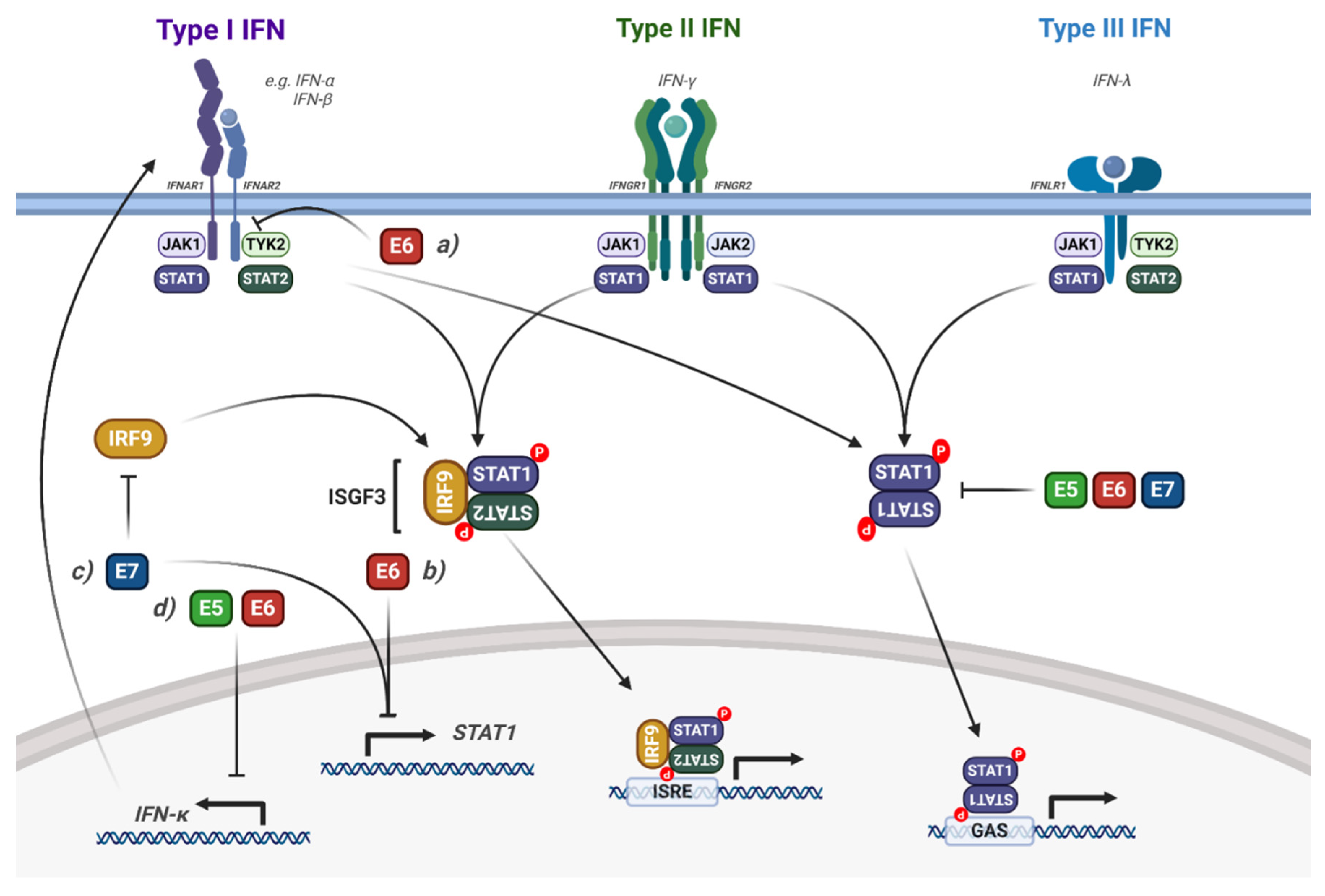
Interaction of HPV with STAT1/2 Signalling
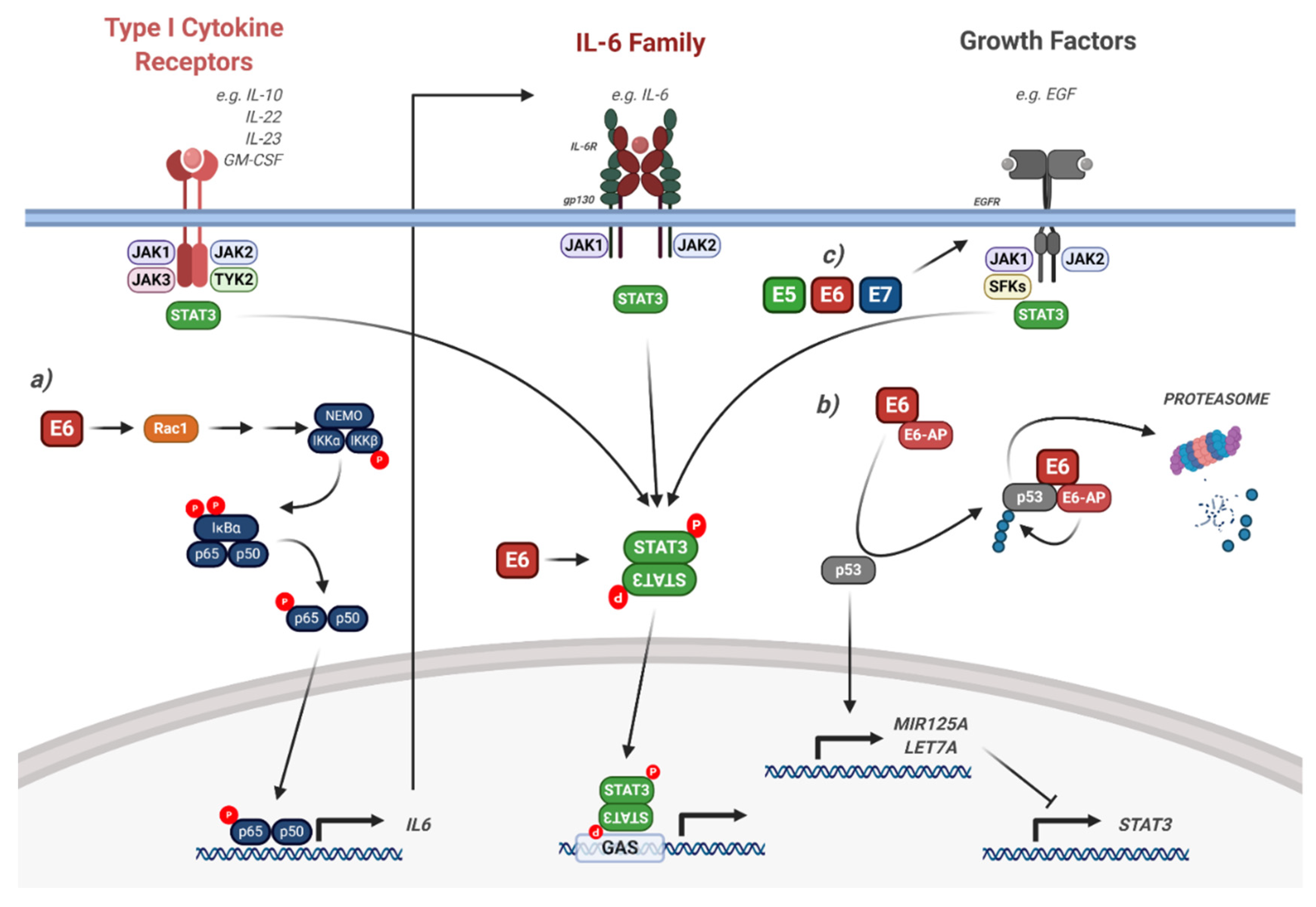
5. STAT3 and STAT5 are Critical Drivers of HPV-Induced Malignancy
5.1. Interaction of HPV with STAT3 Signalling
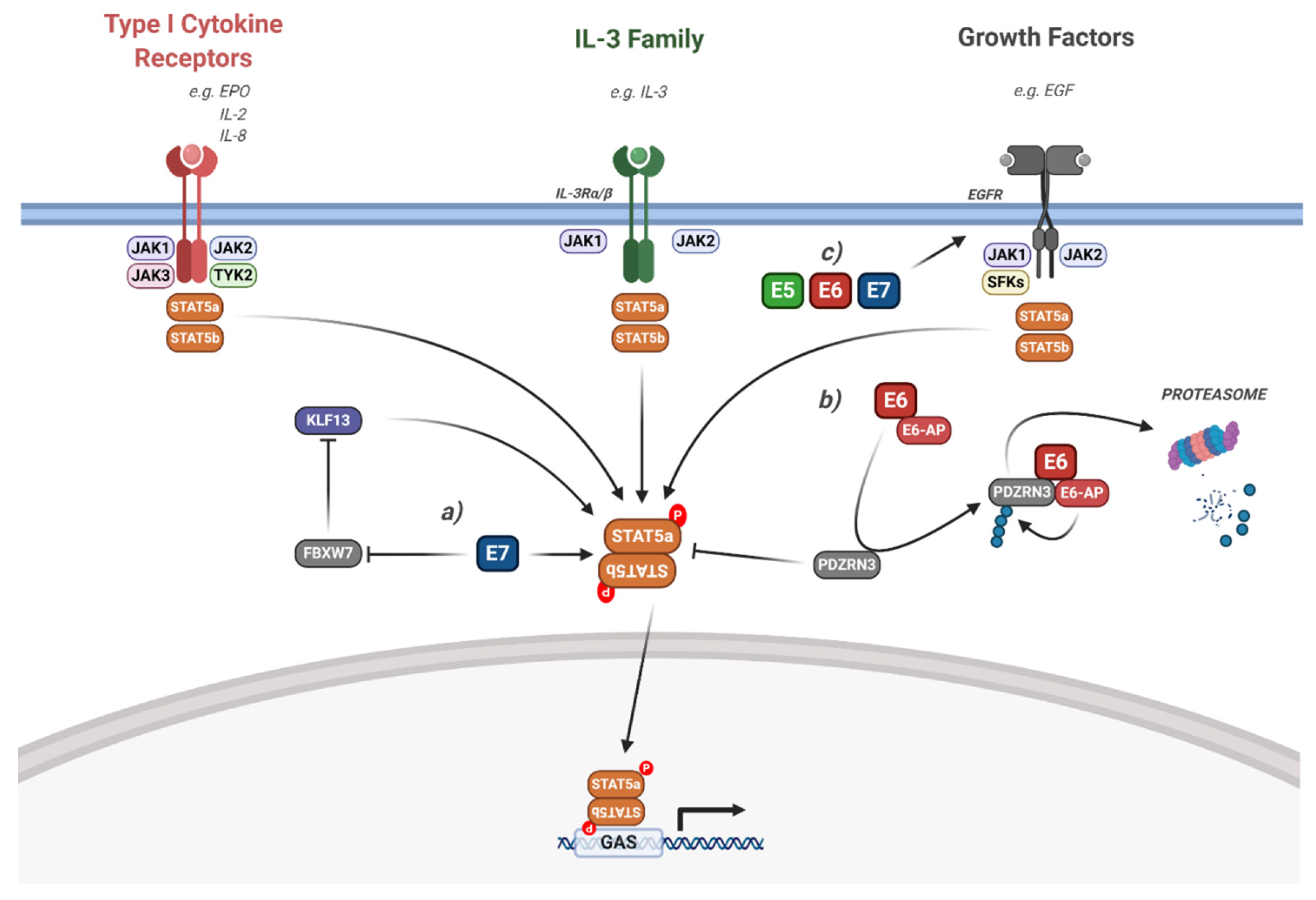
5.2. Interaction of HPV with STAT5 Signalling
6. Targeting the JAK/STAT Pathway in HPV-Associated Cancers
6.1. Direct Targeting
6.1.1. Small Molecule Inhibitors
6.1.2. Nucleotide Therapeutics Targeting STAT3/STAT5
6.2. Indirect Targeting
6.2.1. Targeting IL-6 Signalling
6.2.2. Targeting Janus Kinases
7. Conclusions
Funding
Conflicts of Interest
References
- Chow, L.Q.M. Head and Neck Cancer. N. Engl. J. Med. 2020, 382, 60–72. [Google Scholar] [CrossRef] [PubMed]
- Lowy, D.R.; Schiller, J.T. Reducing HPV-associated cancer globally. Cancer Prev. Res. 2012, 5, 18–23. [Google Scholar] [CrossRef] [PubMed]
- Gupta, S.; Kumar, P.; Das, B.C. HPV: Molecular pathways and targets. Curr. Probl. Cancer 2018, 42, 161–174. [Google Scholar] [CrossRef] [PubMed]
- Cramer, J.D.; Burtness, B.; Le, Q.T.; Ferris, R.L. The changing therapeutic landscape of head and neck cancer. Nat. Rev. Clin. Oncol. 2019, 16, 669–683. [Google Scholar] [CrossRef] [PubMed]
- Zhu, H.; Luo, H.; Zhang, W.; Shen, Z.; Hu, X.; Zhu, X. Molecular mechanisms of cisplatin resistance in cervical cancer. Drug Des. Dev. Ther. 2016, 10, 1885–1895. [Google Scholar] [CrossRef]
- Doorbar, J. The papillomavirus life cycle. J. Clin. Virol. 2005, 32, 7–15. [Google Scholar] [CrossRef]
- Ronco, L.V.; Karpova, A.Y.; Vidal, M.; Howley, P.M. Human papillomavirus 16 E6 oncoprotein binds to interferon regulatory factor-3 and inhibits its transcriptional activity. Genes Dev. 1998, 12, 2061–2072. [Google Scholar] [CrossRef]
- Lau, L.; Gray, E.E.; Brunette, R.L.; Stetson, D.B. DNA tumor virus oncogenes antagonize the cGAS-STING DNA-sensing pathway. Science 2015, 350, 568–571. [Google Scholar] [CrossRef]
- Moody, C.A.; Laimins, L.A. Human papillomavirus oncoproteins: Pathways to transformation. Nat. Rev. Cancer 2010, 10, 550–560. [Google Scholar] [CrossRef]
- Pim, D.; Massimi, P.; Dilworth, S.M.; Banks, L. Activation of the protein kinase B pathway by the HPV-16 E7 oncoprotein occurs through a mechanism involving interaction with PP2A. Oncogene 2005, 24, 7830–7838. [Google Scholar] [CrossRef]
- Brand, T.M.; Hartmann, S.; Bhola, N.E.; Peyser, N.D.; Li, H.; Zeng, Y.; Wechsler, E.I.; Ranall, M.V.; Bandyopadhyay, S.; Duvvuri, U.; et al. Human Papillomavirus Regulates HER3 Expression in Head and Neck Cancer: Implications for Targeted HER3 Therapy in HPV+ Patients. Clin. Cancer Res. 2017, 23, 3072–3083. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Srirangam, A.; A Potter, D.; Roman, A. HPV16 E5 protein disrupts the c-Cbl–EGFR interaction and EGFR ubiquitination in human foreskin keratinocytes. Oncogene 2005, 24, 2585–2588. [Google Scholar] [CrossRef] [PubMed]
- Spangle, J.M.; Munger, K. The HPV16 E6 Oncoprotein Causes Prolonged Receptor Protein Tyrosine Kinase Signaling and Enhances Internalization of Phosphorylated Receptor Species. PLoS Pathog. 2013, 9, e1003237. [Google Scholar] [CrossRef] [PubMed]
- Wasson, C.; Morgan, E.L.; Müller, M.; Ross, R.L.; Hartley, M.; Roberts, S.; Macdonald, A. Human papillomavirus type 18 E5 oncogene supports cell cycle progression and impairs epithelial differentiation by modulating growth factor receptor signalling during the virus life cycle. Oncotarget 2017, 8, 103581–103600. [Google Scholar] [CrossRef] [PubMed]
- Muñoz-Bello, J.O.; Olmedo-Nieva, L.; Contreras-Paredes, A.; Gonzalez, A.M.F.; Rocha-Zavaleta, L.; Lizano, M. Regulation of the Wnt/β-Catenin Signaling Pathway by Human Papillomavirus E6 and E7 Oncoproteins. Viruses 2015, 7, 4734–4755. [Google Scholar] [CrossRef] [PubMed]
- He, C.; Mao, D.; Hua, G.; Lv, X.; Chen, X.; Angeletti, P.C.; Dong, J.; Remmenga, S.W.; Rodabaugh, K.J.; Zhou, J.; et al. The Hippo/ YAP pathway interacts with EGFR signaling and HPV oncoproteins to regulate cervical cancer progression. EMBO Mol. Med. 2015, 7, 1426–1449. [Google Scholar] [CrossRef]
- Morgan, E.L.; Patterson, M.R.; Ryder, E.L.; Lee, S.Y.; Wasson, C.W.; Harper, K.L.; Li, Y.; Griffin, S.; Blair, E.; Whitehouse, A.; et al. MicroRNA-18a targeting of the STK4/MST1 tumour suppressor is necessary for transformation in HPV positive cervical cancer. PLoS Pathog. 2020, 16, e1008624. [Google Scholar] [CrossRef]
- Yu, H.; Jove, R. The STATs of cancer—New molecular targets come of age. Nat. Rev. Cancer 2004, 4, 97–105. [Google Scholar] [CrossRef]
- Reich, N.C. STATs get their move on. JAK-STAT 2013, 2, e27080. [Google Scholar] [CrossRef]
- Yu, H.; Pardoll, E.; Jove, R. STATs in cancer inflammation and immunity: A leading role for STAT3. Nat. Rev. Cancer 2009, 9, 798–809. [Google Scholar] [CrossRef]
- Van Doorslaer, K.; Chen, Z.; Bernard, H.-U.; Chan, P.K.S.; DeSalle, R.; Dillner, J.; Forslund, O.; Haga, T.; McBride, A.A.; Villa, L.L.; et al. ICTV Virus Taxonomy Profile: Papillomaviridae. J. Gen. Virol. 2018, 99, 989–990. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Z.-M.; Baker, C.C. Papillomavirus genome structure, expression, and post-transcriptional regulation. Front. Biosci. 2006, 11, 2286. [Google Scholar] [CrossRef] [PubMed]
- Cripe, T.P.; Haugen, T.H.; Turk, J.P.; Tabatabai, F.; Schmid, P.G.; Dürst, M.; Gissmann, L.; Roman, A.; Turek, L.P. Transcriptional regulation of the human papillomavirus-16 E6-E7 promoter by a keratinocyte-dependent enhancer, and by viral E2 trans-activator and repressor gene products: Implications for cervical carcinogenesis. EMBO J. 1987, 6, 3745–3753. [Google Scholar] [CrossRef] [PubMed]
- Cheng, S.; Schmidt-Grimminger, D.C.; Murant, T.; Broker, T.R.; Chow, L.T. Differentiation-dependent up-regulation of the human papillomavirus E7 gene reactivates cellular DNA replication in suprabasal differentiated keratinocytes. Genes Dev. 1995, 9, 2335–2349. [Google Scholar] [CrossRef] [PubMed]
- Klumpp, D.J.; Laimins, L.A. Differentiation-Induced Changes in Promoter Usage for Transcripts Encoding the Human Papillomavirus Type 31 Replication Protein E1. Virology 1999, 257, 239–246. [Google Scholar] [CrossRef]
- Ruesch, M.N.; Stubenrauch, F.; Laimins, L.A. Activation of Papillomavirus Late Gene Transcription and Genome Amplification upon Differentiation in Semisolid Medium Is Coincident with Expression of Involucrin and Transglutaminase but Not Keratin-10. J. Virol. 1998, 72, 5016–5024. [Google Scholar] [CrossRef] [PubMed]
- Aydin, I.; Villalonga-Planells, R.; Greune, L.; Bronnimann, M.P.; Calton, C.M.; Becker, M.; Lai, K.-Y.; Campos, S.; Schmidt, M.A.; Schelhaas, M. A central region in the minor capsid protein of papillomaviruses facilitates viral genome tethering and membrane penetration for mitotic nuclear entry. PLoS Pathog. 2017, 13, e1006308. [Google Scholar] [CrossRef]
- Calton, C.M.; Bronnimann, M.P.; Manson, A.R.; Li, S.; Chapman, J.A.; Suarez-Berumen, M.; Williamson, T.R.; Molugu, S.K.; Bernal, R.A.; Campos, S.K. Translocation of the papillomavirus L2/vDNA complex across the limiting membrane requires the onset of mitosis. PLoS Pathog. 2017, 13, e1006200. [Google Scholar] [CrossRef]
- Evander, M.; Frazer, I.H.; Payne, E.; Qi, Y.M.; Hengst, K.; A McMillan, N. Identification of the alpha6 integrin as a candidate receptor for papillomaviruses. J. Virol. 1997, 71, 2449–2456. [Google Scholar] [CrossRef]
- Drobni, P.; Mistry, N.; McMillan, N.A.; Evander, M. Carboxy-fluorescein diacetate, succinimidyl ester labeled papillomavirus virus-like particles fluoresce after internalization and interact with heparan sulfate for binding and entry. Virology 2003, 310, 163–172. [Google Scholar] [CrossRef]
- Young, J.M.; El Abidine, A.Z.; Gómez-Martinez, R.A.; Ozbun, M.A. The Known and Potential Intersections of Rab-GTPases in Human Papillomavirus Infections. Front. Cell Dev. Boil. 2019, 7, 139. [Google Scholar] [CrossRef] [PubMed]
- Richards, R.M.; Lowy, U.R.; Schiller, J.T.; Day, P.M. Cleavage of the papillomavirus minor capsid protein, L2, at a furin consensus site is necessary for infection. Proc. Natl. Acad. Sci. USA 2006, 103, 1522–1527. [Google Scholar] [CrossRef] [PubMed]
- Day, P.M.; Lowy, D.R.; Schiller, J.T. Heparan Sulfate-Independent Cell Binding and Infection with Furin-Precleaved Papillomavirus Capsids. J. Virol. 2008, 82, 12565–12568. [Google Scholar] [CrossRef] [PubMed]
- Bronnimann, M.P.; Calton, C.M.; Chiquette, S.F.; Li, S.; Lu, M.; Chapman, J.A.; Bratton, K.N.; Schlegel, A.M.; Campos, S. Furin Cleavage of L2 during Papillomavirus Infection: Minimal Dependence on Cyclophilins. J. Virol. 2016, 90, 6224–6234. [Google Scholar] [CrossRef]
- Surviladze, Z.; Dziduszko, A.; Ozbun, M.A. Essential Roles for Soluble Virion-Associated Heparan Sulfonated Proteoglycans and Growth Factors in Human Papillomavirus Infections. PLoS Pathog. 2012, 8, e1002519. [Google Scholar] [CrossRef] [PubMed]
- Surviladze, Z.; Sterkand, R.T.; Ozbun, M.A. Interaction of human papillomavirus type 16 particles with heparan sulfate and syndecan-1 molecules in the keratinocyte extracellular matrix plays an active role in infection. J. Gen. Virol. 2015, 96, 2232–2241. [Google Scholar] [CrossRef] [PubMed]
- Schelhaas, M.; Shah, B.; Holzer, M.; Blattmann, P.; Kühling, L.; Day, P.M.; Schiller, J.T.; Helenius, A. Entry of Human Papillomavirus Type 16 by Actin-Dependent, Clathrin- and Lipid Raft-Independent Endocytosis. PLoS Pathog. 2012, 8, e1002657. [Google Scholar] [CrossRef] [PubMed]
- Scheffer, K.D.; Gawlitza, A.; Spoden, G.A.; Zhang, X.A.; Lambert, C.; Berditchevski, F.; Florin, L. Tetraspanin CD151 Mediates Papillomavirus Type 16 Endocytosis. J. Virol. 2013, 87, 3435–3446. [Google Scholar] [CrossRef]
- Smith, J.L.; Lidke, D.S.; Ozbun, M.A. Virus activated filopodia promote human papillomavirus type 31 uptake from the extracellular matrix. Virology 2008, 381, 16–21. [Google Scholar] [CrossRef]
- Schelhaas, M.; Ewers, H.; Rajamäki, M.-L.; Day, P.M.; Schiller, J.T.; Helenius, A. Human Papillomavirus Type 16 Entry: Retrograde Cell Surface Transport along Actin-Rich Protrusions. PLoS Pathog. 2008, 4, e1000148. [Google Scholar] [CrossRef]
- Abban, C.Y.; Meneses, P.I. Usage of heparan sulfate, integrins, and FAK in HPV16 infection. Virology 2010, 403, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Surviladze, Z.; Sterk, R.T.; Deharo, S.A.; Ozbun, M.A. Cellular Entry of Human Papillomavirus Type 16 Involves Activation of the Phosphatidylinositol 3-Kinase/Akt/mTOR Pathway and Inhibition of Autophagy. J. Virol. 2013, 87, 2508–2517. [Google Scholar] [CrossRef] [PubMed]
- Mikuličić, S.; Finke, J.; Boukhallouk, F.; Wüstenhagen, E.; Sons, D.; Homsi, Y.; Reiss, K.; Lang, T.; Florin, L. ADAM17-dependent signaling is required for oncogenic human papillomavirus entry platform assembly. eLife 2019, 8, e44345. [Google Scholar] [CrossRef] [PubMed]
- Lipovsky, A.; Popa, A.; Pimienta, G.; Wyler, M.; Bhan, A.; Kuruvilla, L.; Guie, M.-A.; Poffenberger, A.C.; Nelson, C.D.S.; Atwood, W.J.; et al. Genome-wide siRNA screen identifies the retromer as a cellular entry factor for human papillomavirus. Proc. Natl. Acad. Sci. USA 2013, 110, 7452–7457. [Google Scholar] [CrossRef] [PubMed]
- Popa, A.; Zhang, W.; Harrison, M.S.; Goodner, K.; Kazakov, T.; Goodwin, E.C.; Lipovsky, A.; Burd, C.; DiMaio, D. Direct Binding of Retromer to Human Papillomavirus Type 16 Minor Capsid Protein L2 Mediates Endosome Exit during Viral Infection. PLoS Pathog. 2015, 11, e1004699. [Google Scholar] [CrossRef] [PubMed]
- Bodily, J.M.; Meyers, C. Genetic Analysis of the Human Papillomavirus Type 31 Differentiation-Dependent Late Promoter. J. Virol. 2005, 79, 3309–3321. [Google Scholar] [CrossRef]
- Spink, K.M.; Laimins, L.A. Induction of the Human Papillomavirus Type 31 Late Promoter Requires Differentiation but Not DNA Amplification. J. Virol. 2005, 79, 4918–4926. [Google Scholar] [CrossRef]
- Stoler, M.H.; Whitbeck, A.; Wolinsky, S.M.; Broker, T.R.; Chow, L.T.; Howett, M.K.; Kreider, J.W. Infectious cycle of human papillomavirus type 11 in human foreskin xenografts in nude mice. J. Virol. 1990, 64, 3310–3318. [Google Scholar] [CrossRef]
- Egawa, N.; Wang, Q.; Griffin, H.M.; Murakami, I.; Jackson, D.; Mahmood, R.; Doorbar, J. HPV16 and 18 genome amplification show different E4-dependence, with 16E4 enhancing E1 nuclear accumulation and replicative efficiency via its cell cycle arrest and kinase activation functions. PLoS Pathog. 2017, 13, e1006282. [Google Scholar] [CrossRef]
- Münger, K.; Werness, B.; Dyson, N.; Phelps, W.; Harlow, E.; Howley, P. Complex formation of human papillomavirus E7 proteins with the retinoblastoma tumor suppressor gene product. EMBO J. 1989, 8, 4099–4105. [Google Scholar] [CrossRef]
- Boyer, S.N.; E Wazer, D.; Band, V. E7 protein of human papilloma virus-16 induces degradation of retinoblastoma protein through the ubiquitin-proteasome pathway. Cancer Res. 1996, 56, 4620–4624. [Google Scholar] [PubMed]
- Morgan, E.L.; Wasson, C.; Hanson, L.; Kealy, D.; Pentland, I.; McGuire, V.; Scarpini, C.; Coleman, N.; Arthur, J.S.C.; Parish, J.L.; et al. STAT3 activation by E6 is essential for the differentiation-dependent HPV18 life cycle. PLoS Pathog. 2018, 14, e1006975. [Google Scholar] [CrossRef] [PubMed]
- Doorbar, J. The E4 protein; structure, function and patterns of expression. Virology 2013, 445, 80–98. [Google Scholar] [CrossRef] [PubMed]
- Buck, C.B.; Cheng, N.; Thompson, C.D.; Lowy, D.R.; Steven, A.C.; Schiller, J.T.; Trus, B.L. Arrangement of L2 within the Papillomavirus Capsid. J. Virol. 2008, 82, 5190–5197. [Google Scholar] [CrossRef] [PubMed]
- Venuti, A.; Paolini, F.; Nasir, L.; Corteggio, A.; Roperto, S.; Campo, M.S.; Borzacchiello, G. Papillomavirus E5: The smallest oncoprotein with many functions. Mol. Cancer 2011, 10, 140. [Google Scholar] [CrossRef] [PubMed]
- Genther, S.M.; Sterling, S.; Duensing, S.; Munger, K.; Sattler, C.; Lambert, P.F. Quantitative Role of the Human Papillomavirus Type 16 E5 Gene during the Productive Stage of the Viral Life Cycle. J. Virol. 2003, 77, 2832–2842. [Google Scholar] [CrossRef] [PubMed]
- Fehrmann, F.; Klumpp, D.J.; Laimins, L.A. Human Papillomavirus Type 31 E5 Protein Supports Cell Cycle Progression and Activates Late Viral Functions upon Epithelial Differentiation. J. Virol. 2003, 77, 2819–2831. [Google Scholar] [CrossRef]
- Ashrafi, G.H.; Haghshenas, M.R.; Marchetti, B.; O’Brien, P.M.; Campo, M.S. E5 protein of human papillomavirus type 16 selectively downregulates surface HLA class I. Int. J. Cancer 2005, 113, 276–283. [Google Scholar] [CrossRef]
- Zhang, B.; Li, P.; Wang, E.; Brahmi, Z.; Dunn, K.; Blum, J.S.; Roman, A. The E5 protein of human papillomavirus type 16 perturbs MHC class II antigen maturation in human foreskin keratinocytes treated with interferon-gamma. Virology 2003, 310, 100–108. [Google Scholar] [CrossRef]
- Miura, S.; Kawana, K.; Schust, D.J.; Fujii, T.; Yokoyama, T.; Iwasawa, Y.; Nagamatsu, T.; Adachi, K.; Tomio, A.; Tomio, K.; et al. CD1d, a Sentinel Molecule Bridging Innate and Adaptive Immunity, Is Downregulated by the Human Papillomavirus (HPV) E5 Protein: A Possible Mechanism for Immune Evasion by HPV. J. Virol. 2010, 84, 11614–11623. [Google Scholar] [CrossRef]
- Maufort, J.P.; Shai, A.; Pitot, H.C.; Lambert, P.F. A role for HPV16 E5 in cervical carcinogenesis. Cancer Res. 2010, 70, 2924–2931. [Google Scholar] [CrossRef] [PubMed]
- Straight, S.W.; Hinkle, P.M.; Jewers, R.J.; McCance, D.J. The E5 oncoprotein of human papillomavirus type 16 transforms fibroblasts and effects the downregulation of the epidermal growth factor receptor in keratinocytes. J. Virol. 1993, 67, 4521–4532. [Google Scholar] [CrossRef] [PubMed]
- Crusius, K.; Auvinen, E.; Alonso, A. Enhancement of EGF- and PMA-mediated MAP kinase activation in cells expressing the human papillomavirus type 16 E5 protein. Oncogene 1997, 15, 1437–1444. [Google Scholar] [CrossRef] [PubMed]
- Valle, G.F.; Banks, L. The human papillomavirus (HPV)-6 and HPV-16 E5 proteins co-operate with HPV-16 E7 in the transformation of primary rodent cells. J. Gen. Virol. 1995, 76, 1239–1245. [Google Scholar] [CrossRef] [PubMed]
- Pim, D.; Collins, M.; Banks, L. Human papillomavirus type 16 E5 gene stimulates the transforming activity of the epidermal growth factor receptor. Oncogene 1992, 7, 27–32. [Google Scholar]
- I Rodríguez, M.; E Finbow, M.; Alonso, A. Binding of human papillomavirus 16 E5 to the 16 kDa subunit c (proteolipid) of the vacuolar H+-ATPase can be dissociated from the E5-mediated epidermal growth factor receptor overactivation. Oncogene 2000, 19, 3727–3732. [Google Scholar] [CrossRef]
- Thomas, J.T.; Hubert, W.G.; Ruesch, M.N.; Laimins, L.A. Human papillomavirus type 31 oncoproteins E6 and E7 are required for the maintenance of episomes during the viral life cycle in normal human keratinocytes. Proc. Natl. Acad. Sci. USA 1999, 96, 8449–8454. [Google Scholar] [CrossRef]
- Flores, E.R.; Allen-Hoffmann, B.L.; Lee, D.; Lambert, P.F. The Human Papillomavirus Type 16 E7 Oncogene Is Required for the Productive Stage of the Viral Life Cycle. J. Virol. 2000, 74, 6622–6631. [Google Scholar] [CrossRef]
- Park, R.B.; Androphy, E.J. Genetic Analysis of High-Risk E6 in Episomal Maintenance of Human Papillomavirus Genomes in Primary Human Keratinocytes. J. Virol. 2002, 76, 11359–11364. [Google Scholar] [CrossRef]
- Niebler, M.; Qian, X.; Höfler, D.; Kogosov, V.; Kaewprag, J.; Kaufmann, A.M.; Ly, R.; Böhmer, G.; Zawatzky, R.; Rösl, F.; et al. Post-Translational Control of IL-1β via the Human Papillomavirus Type 16 E6 Oncoprotein: A Novel Mechanism of Innate Immune Escape Mediated by the E3-Ubiquitin Ligase E6-AP and p53. PLoS Pathog. 2013, 9, e1003536. [Google Scholar] [CrossRef]
- Cicchini, L.; Westrich, J.A.; Xu, T.; Vermeer, D.W.; Berger, J.N.; Clambey, E.T.; Lee, D.; Song, J.I.; Lambert, P.F.; Greer, R.O.; et al. Suppression of Antitumor Immune Responses by Human Papillomavirus through Epigenetic Downregulation of CXCL14. mBio 2016, 7, e00270-16. [Google Scholar] [CrossRef] [PubMed]
- Westrich, J.A.; Vermeer, D.W.; Silva, A.; Bonney, S.; Berger, J.N.; Cicchini, L.; Greer, R.O.; Song, J.I.; Raben, D.; Slansky, J.E.; et al. CXCL14 suppresses human papillomavirus-associated head and neck cancer through antigen-specific CD8+ T-cell responses by upregulating MHC-I expression. Oncogene 2019, 38, 7166–7180. [Google Scholar] [CrossRef] [PubMed]
- Lambert, P.F.; Pan, H.; Pitot, H.C.; Liem, A.; Jackson, M.; Griep, A.E. Epidermal cancer associated with expression of human papillomavirus type 16 E6 and E7 oncogenes in the skin of transgenic mice. Proc. Natl. Acad. Sci. USA 1993, 90, 5583–5587. [Google Scholar] [CrossRef] [PubMed]
- Arbeit, J.M.; Munger, K.; Howley, P.M.; Hanahan, D. Progressive squamous epithelial neoplasia in K14-human papillomavirus type 16 transgenic mice. J. Virol. 1994, 68, 4358–4368. [Google Scholar] [CrossRef] [PubMed]
- Jeon, S.; Lambert, P.F. Integration of human papillomavirus type 16 DNA into the human genome leads to increased stability of E6 and E7 mRNAs: Implications for cervical carcinogenesis. Proc. Natl. Acad. Sci. USA 1995, 92, 1654–1658. [Google Scholar] [CrossRef] [PubMed]
- Herber, R.; Liem, A.; Pitot, H.; Lambert, P.F. Squamous epithelial hyperplasia and carcinoma in mice transgenic for the human papillomavirus type 16 E7 oncogene. J. Virol. 1996, 70, 1873–1881. [Google Scholar] [CrossRef]
- Song, S.; Pitot, H.C.; Lambert, P.F. The Human Papillomavirus Type 16 E6 Gene Alone Is Sufficient To Induce Carcinomas in Transgenic Animals. J. Virol. 1999, 73, 5887–5893. [Google Scholar] [CrossRef]
- Song, S.; Liem, A.; Miller, J.A.; Lambert, P.F. Human Papillomavirus Types 16 E6 and E7 Contribute Differently to Carcinogenesis. Virology 2000, 267, 141–150. [Google Scholar] [CrossRef]
- Duensing, S.; Munger, K. The human papillomavirus type 16 E6 and E7 oncoproteins independently induce numerical and structural chromosome instability. Cancer Res. 2002, 62, 7075–7082. [Google Scholar]
- Riley, R.R.; Duensing, S.; Brake, T.; Munger, K.; Lambert, P.F.; Arbeit, J.M. Dissection of human papillomavirus E6 and E7 function in transgenic mouse models of cervical carcinogenesis. Cancer Res. 2003, 63, 4862–4871. [Google Scholar]
- Hoppe-Seyler, K.; Bossler, F.; Braun, J.A.; Herrmann, A.L.; Hoppe-Seyler, F. The HPV E6/E7 Oncogenes: Key Factors for Viral Carcinogenesis and Therapeutic Targets. Trends Microbiol. 2018, 26, 158–168. [Google Scholar] [CrossRef] [PubMed]
- Kho, E.-Y.; Wang, H.-K.; Banerjee, N.S.; Broker, T.R.; Chow, L.T. HPV-18 E6 mutants reveal p53 modulation of viral DNA amplification in organotypic cultures. Proc. Natl. Acad. Sci. USA 2013, 110, 7542–7549. [Google Scholar] [CrossRef] [PubMed]
- Thomas, M.; Narayan, N.; Pim, D.; Tomaić, V.; Massimi, P.; Nagasaka, K.; Kranjec, C.; Gammoh, N.; Banks, L.; Tomai, V. Human papillomaviruses, cervical cancer and cell polarity. Oncogene 2008, 27, 7018–7030. [Google Scholar] [CrossRef] [PubMed]
- Simonson, S.J.; Difilippantonio, M.J.; Lambert, P.F. Two Distinct Activities Contribute to Human Papillomavirus 16 E6’s Oncogenic Potential. Cancer Res. 2005, 65, 8266–8273. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, M.L.; Nguyen, M.M.; Lee, D.; Griep, A.E.; Lambert, P.F. The PDZ Ligand Domain of the Human Papillomavirus Type 16 E6 Protein Is Required for E6’s Induction of Epithelial Hyperplasia In Vivo. J. Virol. 2003, 77, 6957–6964. [Google Scholar] [CrossRef] [PubMed]
- Shin, M.-K.; Sage, J.; Lambert, P.F. Inactivating All Three Rb Family Pocket Proteins Is Insufficient to Initiate Cervical Cancer. Cancer Res. 2012, 72, 5418–5427. [Google Scholar] [CrossRef] [PubMed][Green Version]
- McLaughlin-Drubin, M.E.; Munger, K. The human papillomavirus E7 oncoprotein. Virology 2009, 384, 335–344. [Google Scholar] [CrossRef]
- Balsitis, S.J.; Sage, J.; Duensing, S.; Munger, K.; Jacks, T.; Lambert, P.F. Recapitulation of the Effects of the Human Papillomavirus Type 16 E7 Oncogene on Mouse Epithelium by Somatic Rb Deletion and Detection of pRb-Independent Effects of E7 In Vivo. Mol. Cell. Biol. 2003, 23, 9094–9103. [Google Scholar] [CrossRef]
- Moody, C.A.; Laimins, L.A. Human Papillomaviruses Activate the ATM DNA Damage Pathway for Viral Genome Amplification upon Differentiation. PLoS Pathog. 2009, 5, e1000605. [Google Scholar] [CrossRef]
- Takeda, K.; Noguchi, K.; Shi, W.; Tanaka, T.; Matsumoto, M.; Yoshida, N.; Kishimoto, T.; Akira, S. Targeted disruption of the mouse Stat3 gene leads to early embryonic lethality. Proc. Natl. Acad. Sci. USA 1997, 94, 3801–3804. [Google Scholar] [CrossRef]
- Meraz, M.A.; White, J.; Sheehan, K.C.; Bach, E.A.; Rodig, S.J.; Dighe, A.S.; Kaplan, D.H.; Riley, J.K.; Greenlund, A.C.; Campbell, D.; et al. Targeted Disruption of the Stat1 Gene in Mice Reveals Unexpected Physiologic Specificity in the JAK–STAT Signaling Pathway. Cell 1996, 84, 431–442. [Google Scholar] [CrossRef]
- Park, C.; Li, S.; Cha, E.; Schindler, C. Immune Response in Stat2 Knockout Mice. Immunity 2000, 13, 795–804. [Google Scholar] [CrossRef]
- Wang, Y.; Feng, D.; Wang, H.; Xu, M.-J.; Park, O.; Li, Y.; Gao, B. STAT4 knockout mice are more susceptible to concanavalin A-induced T-cell hepatitis. Am. J. Pathol. 2014, 184, 1785–1794. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Robinson, G.W.; Wagner, K.-U.; Garrett, L.; Wynshaw-Boris, A.; Hennighausen, L. Stat5a is mandatory for adult mammary gland development and lactogenesis. Genes Dev. 1997, 11, 179–186. [Google Scholar] [CrossRef]
- Udy, G.B.; Towers, R.P.; Snell, R.; Wilkins, R.J.; Park, S.-H.; Ram, P.A.; Waxman, D.J.; Davey, H.W. Requirement of STAT5b for sexual dimorphism of body growth rates and liver gene expression. Proc. Natl. Acad. Sci. USA 1997, 94, 7239–7244. [Google Scholar] [CrossRef]
- Teglund, S.; McKay, C.; Schuetz, E.; Van Deursen, J.M.; Stravopodis, D.J.; Wang, D.; Brown, M.P.; Bodner, S.; Grosveld, G.; Ihle, J.N. Stat5a and Stat5b Proteins Have Essential and Nonessential, or Redundant, Roles in Cytokine Responses. Cell 1998, 93, 841–850. [Google Scholar] [CrossRef]
- Zhu, J.; Guo, L.; Watson, C.J.; Hu-Li, J.; Paul, W.E. Stat6 is necessary and sufficient for IL-4’s role in Th2 differentiation and cell expansion. J. Immunol. 2001, 166, 7276–7281. [Google Scholar] [CrossRef]
- Rodig, S.J.; A Meraz, M.; White, J.; A Lampe, P.; Riley, J.K.; Arthur, C.D.; King, K.L.; Sheehan, K.C.; Yin, L.; Pennica, D.; et al. Disruption of the Jak1 Gene Demonstrates Obligatory and Nonredundant Roles of the Jaks in Cytokine-Induced Biologic Responses. Cell 1998, 93, 373–383. [Google Scholar] [CrossRef]
- Parganas, E.; Wang, D.; Stravopodis, D.J.; Topham, D.J.; Marine, J.-C.; Teglund, S.; Vanin, E.F.; Bodner, S.; Colamonici, O.R.; Van Deursen, J.M.; et al. Jak2 Is Essential for Signaling through a Variety of Cytokine Receptors. Cell 1998, 93, 385–395. [Google Scholar] [CrossRef]
- Park, S.Y.; Saijo, K.; Takahashi, T.; Osawa, M.; Arase, H.; Hirayama, N.; Miyake, K.; Nakauchi, H.; Shirasawa, T.; Saito, T. Developmental defects of lymphoid cells in Jak3 kinase-deficient mice. Immunity 1995, 3, 771–782. [Google Scholar] [CrossRef]
- Karaghiosoff, M.; Neubauer, H.; Lassnig, C.; Kovarik, P.; Schindler, H.; Pircher, H.; McCoy, B.; Bogdan, C.; Decker, T.; Brem, G.; et al. Partial Impairment of Cytokine Responses in Tyk2-Deficient Mice. Immunity 2000, 13, 549–560. [Google Scholar] [CrossRef]
- Darnell, J.; Kerr, I.; Stark, G. Jak-STAT pathways and transcriptional activation in response to IFNs and other extracellular signaling proteins. Science 1994, 264, 1415–1421. [Google Scholar] [CrossRef] [PubMed]
- Akira, S. Functional Roles of STAT Family Proteins: Lessons from Knockout Mice. Stem Cells 1999, 17, 138–146. [Google Scholar] [CrossRef] [PubMed]
- Meissl, K.; Macho-Maschler, S.; Müller, M.; Strobl, B. The good and the bad faces of STAT1 in solid tumours. Cytokine 2017, 89, 12–20. [Google Scholar] [CrossRef] [PubMed]
- Bousoik, E.; Aliabadi, H.M. “Do We Know Jack” About JAK? A Closer Look at JAK/STAT Signaling Pathway. Front. Oncol. 2018, 8, 287. [Google Scholar] [CrossRef] [PubMed]
- Lupardus, P.J.; Ultsch, M.; Wallweber, H.; Kohli, P.B.; Johnson, A.R.; Eigenbrot, C. Structure of the pseudokinase-kinase domains from protein kinase TYK2 reveals a mechanism for Janus kinase (JAK) autoinhibition. Proc. Natl. Acad. Sci. USA 2014, 111, 8025–8030. [Google Scholar] [CrossRef]
- Ferrao, R.D.; Wallweber, H.J.; Lupardus, P.J. Receptor-mediated dimerization of JAK2 FERM domains is required for JAK2 activation. eLife 2018, 7, e38089. [Google Scholar] [CrossRef]
- Decker, T.; Kovarik, P. Serine phosphorylation of STATs. Oncogene 2000, 19, 2628–2637. [Google Scholar] [CrossRef]
- Johnson, D.E.; O’Keefe, R.A.; Grandis, J.R. Targeting the IL-6/JAK/STAT3 signalling axis in cancer. Nat. Rev. Clin. Oncol. 2018, 15, 234–248. [Google Scholar] [CrossRef]
- Reich, N.C.; Liu, L. Tracking STAT nuclear traffic. Nat. Rev. Immunol. 2006, 6, 602–612. [Google Scholar] [CrossRef]
- Sekimoto, T.; Imamoto, N.; Nakajima, K.; Hirano, T.; Yoneda, Y. Extracellular signal-dependent nuclear import of Stat1 is mediated by nuclear pore-targeting complex formation with NPI-1, but not Rch1. EMBO J. 1997, 16, 7067–7077. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Moczygemba, M.; Gutch, M.J.; French, D.L.; Reich, N.C. Distinct STAT Structure Promotes Interaction of STAT2 with the p48 Subunit of the Interferon-α-stimulated Transcription Factor ISGF3. J. Biol. Chem. 1997, 272, 20070–20076. [Google Scholar] [CrossRef] [PubMed]
- Lau, J.F.; Parisien, J.-P.; Horvath, C.M. Interferon regulatory factor subcellular localization is determined by a bipartite nuclear localization signal in the DNA-binding domain and interaction with cytoplasmic retention factors. Proc. Natl. Acad. Sci. USA 2000, 97, 7278–7283. [Google Scholar] [CrossRef] [PubMed]
- Banninger, G.; Reich, N.C. STAT2 Nuclear Trafficking. J. Biol. Chem. 2004, 279, 39199–39206. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; McBride, K.M.; Reich, N.C. STAT3 nuclear import is independent of tyrosine phosphorylation and mediated by importin-alpha3. Proc. Natl. Acad. Sci. USA 2005, 102, 8150–8155. [Google Scholar] [CrossRef] [PubMed]
- Zeng, R.; Aoki, Y.; Yoshida, M.; Arai, K.-I.; Watanabe, S. Stat5B shuttles between cytoplasm and nucleus in a cytokine-dependent and -independent manner. J. Immunol. 2002, 168, 4567–4575. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Chatterjee-Kishore, M.; Staugaitis, S.M.; Nguyen, H.; Schlessinger, K.; Levy, D.E.; Stark, G.R. Novel roles of unphosphorylated STAT3 in oncogenesis and transcriptional regulation. Cancer Res. 2005, 65, 939–947. [Google Scholar]
- Yang, J.; Liao, X.; Agarwal, M.K.; Barnes, L.; Auron, P.E.; Stark, G.R. Unphosphorylated STAT3 accumulates in response to IL-6 and activates transcription by binding to NFkappaB. Genes Dev. 2007, 21, 1396–1408. [Google Scholar] [CrossRef]
- Safran, M.; Chalifa-Caspi, M.V.; Shmueli, O.; Olender, T.; Lapidot, M.; Rosen, N.; Shmoish, M.; Peter, Y.; Glusman, G.; Feldmesser, E.; et al. Human Gene-Centric Databases at the Weizmann Institute of Science: GeneCards, UDB, CroW 21 and HORDE. Nucleic Acids Res. 2003, 31, 142–146. [Google Scholar] [CrossRef]
- Platanias, L.C. Mechanisms of type-I- and type-II-interferon-mediated signalling. Nat. Rev. Immunol. 2005, 5, 375–386. [Google Scholar] [CrossRef]
- Borden, E.C.; Sen, G.C.; Uzé, G.; Silverman, R.H.; Ransohoff, R.M.; Foster, G.R.; Stark, G.R. Interferons at age 50: Past, current and future impact on biomedicine. Nat. Rev. Drug Discov. 2007, 6, 975–990. [Google Scholar] [CrossRef] [PubMed]
- Assil, S.; Webster, B.; Dreux, M. Regulation of the Host Antiviral State by Intercellular Communications. Viruses 2015, 7, 4707–4733. [Google Scholar] [CrossRef] [PubMed]
- Bordignon, V.; Di Domenico, E.G.; Trento, E.; D’Agosto, G.; Cavallo, I.; Pontone, M.; Pimipinelli, F.; Mariani, L.; Ensoli, F. How Human Papillomavirus Replication and Immune Evasion Strategies Take Advantage of the Host DNA Damage Repair Machinery. Viruses 2017, 9, 390. [Google Scholar] [CrossRef] [PubMed]
- Nees, M.; Geoghegan, J.M.; Hyman, T.; Frank, S.; Miller, L.; Woodworth, C.D. Papillomavirus type 16 oncogenes downregulate expression of interferon-responsive genes and upregulate proliferation-associated and NF-kappaB-responsive genes in cervical keratinocytes. J. Virol. 2001, 75, 4283–4296. [Google Scholar] [CrossRef] [PubMed]
- Barnard, P.; McMillan, N.A. The Human Papillomavirus E7 Oncoprotein Abrogates Signaling Mediated by Interferon-α. Virology 1999, 259, 305–313. [Google Scholar] [CrossRef] [PubMed]
- Hong, S.; Mehta, K.P.; Laimins, L.A. Suppression of STAT-1 Expression by Human Papillomaviruses Is Necessary for Differentiation-Dependent Genome Amplification and Plasmid Maintenance. J. Virol. 2011, 85, 9486–9494. [Google Scholar] [CrossRef]
- Evans, M.R.; James, C.D.; Bristol, M.L.; Nulton, T.J.; Wang, X.; Kaur, N.; White, E.A.; Windle, B.; Morgan, I.M. Human Papillomavirus 16 E2 Regulates Keratinocyte Gene Expression Relevant to Cancer and the Viral Life Cycle. J. Virol. 2019, 93, e01067-19. [Google Scholar] [CrossRef]
- James, C.D.; Fontan, C.T.; Otoa, R.; Das, D.; Prabhakar, A.T.; Wang, X.; Bristol, M.L.; Morgan, I.M. Human Papillomavirus 16 E6 and E7 Synergistically Repress Innate Immune Gene Transcription. mSphere 2020, 5, 660. [Google Scholar] [CrossRef]
- LaFleur, D.W.; Nardelli, B.; Tsareva, T.; Mather, D.; Feng, P.; Semenuk, M.; Taylor, K.; Buergin, M.; Chinchilla, D.; Roshke, V.; et al. Interferon-κ, a Novel Type I Interferon Expressed in Human Keratinocytes. J. Biol. Chem. 2001, 276, 39765–39771. [Google Scholar] [CrossRef]
- Rincon-Orozco, B.; Halec, G.; Rosenberger, S.; Muschik, D.; Nindl, I.; Bachmann, A.; Ritter, T.M.; Dondog, B.; Ly, R.; Bosch, F.X.; et al. Epigenetic silencing of interferon-kappa in human papillomavirus type 16-positive cells. Cancer Res. 2009, 69, 8718–8725. [Google Scholar] [CrossRef]
- DeCarlo, C.A.; Severini, A.; Edler, L.; Escott, N.G.; Lambert, P.F.; Ulanova, M.; Zehbe, I. IFN-κ, a novel type I IFN, is undetectable in HPV-positive human cervical keratinocytes. Lab. Investig. 2010, 90, 1482–1491. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Reiser, J.; Hurst, J.; Voges, M.; Krauss, P.; Münch, P.; Iftner, T.; Stubenrauch, F. High-Risk Human Papillomaviruses Repress Constitutive Kappa Interferon Transcription via E6 To Prevent Pathogen Recognition Receptor and Antiviral-Gene Expression. J. Virol. 2011, 85, 11372–11380. [Google Scholar] [CrossRef] [PubMed]
- Sunthamala, N.; Thierry, F.; Teissier, S.; Pientong, C.; Kongyingyoes, B.; Tangsiriwatthana, T.; Sangkomkamhang, U.; Ekalaksananan, T. E2 Proteins of High Risk Human Papillomaviruses Down-Modulate STING and IFN-κ Transcription in Keratinocytes. PLoS ONE 2014, 9, e91473. [Google Scholar] [CrossRef] [PubMed]
- Habiger, C.; Jäger, G.; Walter, M.; Iftner, T.; Stubenrauch, F. Interferon Kappa Inhibits Human Papillomavirus 31 Transcription by Inducing Sp100 Proteins. J. Virol. 2016, 90, 694–704. [Google Scholar] [CrossRef] [PubMed]
- Raikhy, G.; Woodby, B.L.; Scott, M.L.; Shin, G.; Myers, J.E.; Scott, R.S.; Bodily, J.M. Suppression of Stromal Interferon Signaling by Human Papillomavirus 16. J. Virol. 2019, 93, e00458-19. [Google Scholar] [CrossRef] [PubMed]
- Scott, M.L.; Woodby, B.L.; Ulicny, J.; Raikhy, G.; Orr, A.W.; Songock, W.K.; Bodily, J.M. Human Papillomavirus 16 E5 Inhibits Interferon Signaling and Supports Episomal Viral Maintenance. J. Virol. 2020, 94. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Labrecque, S.; Gauzzi, M.C.; Cuddihy, A.R.; Wong, A.H.; Pellegrini, S.; Matlashewski, G.J.; E Koromilas, A. The human papilloma virus (HPV)-18 E6 oncoprotein physically associates with Tyk2 and impairs Jak-STAT activation by interferon-α. Oncogene 1999, 18, 5727–5737. [Google Scholar] [CrossRef] [PubMed]
- Igelmann, S.; Neubauer, H.A.; Ferbeyre, G. STAT3 and STAT5 Activation in Solid Cancers. Cancers 2019, 11, 1428. [Google Scholar] [CrossRef] [PubMed]
- Sano, S.; Itami, S.; Takeda, K.; Tarutani, M.; Yamaguchi, Y.; Miura, H.; Yoshikawa, K.; Akira, S.; Takeda, J. Keratinocyte-specific ablation of Stat3 exhibits impaired skin remodeling, but does not affect skin morphogenesis. EMBO J. 1999, 18, 4657–4668. [Google Scholar] [CrossRef]
- Miyoshi, K.; Takaishi, M.; Nakajima, K.; Ikeda, M.; Kanda, T.; Tarutani, M.; Iiyama, T.; Asao, N.; DiGiovanni, J.; Sano, S. Stat3 as a Therapeutic Target for the Treatment of Psoriasis: A Clinical Feasibility Study with STA-21, a Stat3 Inhibitor. J. Investig. Dermatol. 2011, 131, 108–117. [Google Scholar] [CrossRef]
- Orecchia, V.; Regis, G.; Tassone, B.; Valenti, C.; Avalle, L.; Saoncella, S.; Calautti, E.; Poli, V. Constitutive STAT3 activation in epidermal keratinocytes enhances cell clonogenicity and favours spontaneous immortalization by opposing differentiation and senescence checkpoints. Exp. Dermatol. 2015, 24, 29–34. [Google Scholar] [CrossRef] [PubMed]
- Carpenter, R.L.; Lo, H.-W. STAT3 Target Genes Relevant to Human Cancers. Cancers 2014, 6, 897–925. [Google Scholar] [CrossRef] [PubMed]
- Silver, J.S.; Hunter, C.A. gp130 at the nexus of inflammation, autoimmunity, and cancer. J. Leukoc. Biol. 2010, 88, 1145–1156. [Google Scholar] [CrossRef] [PubMed]
- Arany, I.; Grattendick, K.G.; Tyring, S.K. Interleukin-10 induces transcription of the early promoter of human papillomavirus type 16 (HPV16) through the 5′-segment of the upstream regulatory region (URR). Antivir. Res. 2002, 55, 331–339. [Google Scholar] [CrossRef]
- Tao, Y.; Shen, H.; Liu, Y.; Li, G.; Huang, Z.; Liu, Y. IL-23R in laryngeal cancer: A cancer immunoediting process that facilitates tumor cell proliferation and results in cisplatin resistance. Carcinogenesis 2020, 69. [Google Scholar] [CrossRef]
- Wang, Y.Z.; Wharton, W.; Garcia, R.; Kraker, A.; Jove, R.; Pledger, W.J. Activation of Stat3 preassembled with platelet-derived growth factor beta receptors requires Src kinase activity. Oncogene 2000, 19, 2075–2085. [Google Scholar] [CrossRef] [PubMed]
- Chan, K.S.; Carbajal, S.; Kiguchi, K.; Clifford, J.; Sano, S.; DiGiovanni, J. Epidermal growth factor receptor-mediated activation of Stat3 during multistage skin carcinogenesis. Cancer Res. 2004, 64, 2382–2389. [Google Scholar] [CrossRef]
- Lee, H.; Deng, J.; Kujawski, M.; Yang, C.; Liu, Y.; Herrmann, A.; Kortylewski, M.; Horne, D.; Somlo, G.; Forman, S.; et al. STAT3-induced S1PR1 expression is crucial for persistent STAT3 activation in tumors. Nat. Med. 2010, 16, 1421–1428. [Google Scholar] [CrossRef]
- Kira, M.; Sano, S.; Takagi, S.; Yoshikawa, K.; Takeda, J.; Itami, S. STAT3 Deficiency in Keratinocytes Leads to Compromised Cell Migration through Hyperphosphorylation of p130cas. J. Biol. Chem. 2002, 277, 12931–12936. [Google Scholar] [CrossRef]
- Wu, R.; Sun, S.; Steinberg, B.M. Requirement of STAT3 Activation for Differentiation of Mucosal Stratified Squamous Epithelium. Mol. Med. 2003, 9, 77–84. [Google Scholar] [CrossRef]
- Gartsbein, M.; Alt, A.; Hashimoto, K.; Nakajima, K.; Kuroki, T.; Tennenbaum, T. The role of protein kinase C activation and STAT3 Ser727 phosphorylation in insulin-induced keratinocyte proliferation. J. Cell Sci. 2006, 119, 470–481. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Shukla, S.; Jadli, M.; Thakur, K.; Shishodia, G.; Mahata, S.; Basir, S.F.; Das, B.C.; Bharti, A.C. Level of phospho-STAT3 (Tyr705) correlates with copy number and physical state of human papillomavirus 16 genome in cervical precancer and cancer lesions. PLoS ONE 2019, 14, e0222089. [Google Scholar] [CrossRef] [PubMed]
- Shukla, S.; Shishodia, G.; Mahata, S.; Hedau, S.; Pandey, A.; Bhambhani, S.; Batra, S.; Basir, S.F.; Das, B.C.; Bharti, A.C. Aberrant expression and constitutive activation of STAT3 in cervical carcinogenesis: Implications in high-risk human papillomavirus infection. Mol. Cancer 2010, 9, 282. [Google Scholar] [CrossRef] [PubMed]
- Ren, C.; Cheng, X.; Lu, B.; Yang, G. Activation of interleukin-6/signal transducer and activator of transcription 3 by human papillomavirus early proteins 6 induces fibroblast senescence to promote cervical tumourigenesis through autocrine and paracrine pathways in tumour microenvironment. Eur. J. Cancer 2013, 49, 3889–3899. [Google Scholar] [CrossRef] [PubMed]
- Shishodia, G.; Verma, G.; Srivastava, Y.; Mehrotra, R.; Das, B.C.; Bharti, A.C. Deregulation of microRNAs Let-7a and miR-21 mediate aberrant STAT3 signaling during human papillomavirus-induced cervical carcinogenesis: Role of E6 oncoprotein. BMC Cancer 2014, 14, 996. [Google Scholar] [CrossRef]
- Shishodia, G.; Shukla, S.; Srivastava, Y.; Masaldan, S.; Mehta, S.; Bhambhani, S.; Sharma, S.; Mehrotra, R.; Das, B.C.; Bharti, A.C. Alterations in microRNAs miR-21 and let-7a correlate with aberrant STAT3 signaling and downstream effects during cervical carcinogenesis. Mol. Cancer 2015, 14, 1–13. [Google Scholar] [CrossRef]
- Chuerduangphui, J.; Pientong, C.; Chaiyarit, P.; Patarapadungkit, N.; Chotiyano, A.; Kongyingyoes, B.; Promthet, S.; Swangphon, P.; Wongjampa, W.; Ekalaksananan, T. Effect of human papillomavirus 16 oncoproteins on oncostatin M upregulation in oral squamous cell carcinoma. Med Oncol. 2016, 33, 83. [Google Scholar] [CrossRef]
- Yadav, A.; Kumar, B.; Datta, J.; Teknos, T.N.; Kumar, P. IL-6 promotes head and neck tumor metastasis by inducing epithelial-mesenchymal transition via the JAK-STAT3-SNAIL signaling pathway. Mol. Cancer Res. 2011, 9, 1658–1667. [Google Scholar] [CrossRef]
- Winder, D.M.; Chattopadhyay, A.; Muralidhar, B.; Bauer, J.; English, W.R.; Zhang, X.; Karagavriilidou, K.; Roberts, I.; Pett, M.R.; Murphy, G.; et al. Overexpression of the oncostatin M receptor in cervical squamous cell carcinoma cells is associated with a pro-angiogenic phenotype and increased cell motility and invasiveness. J. Pathol. 2011, 225, 448–462. [Google Scholar] [CrossRef]
- Argiris, A.; Duffy, A.G.; Kummar, S.; Simone, N.L.; Arai, Y.; Kim, S.W.; Rudy, S.F.; Kannabiran, V.R.; Yang, X.; Jang, M.; et al. Early tumor progression associated with enhanced EGFR signaling with bortezomib, cetuximab, and radiotherapy for head and neck cancer. Clin. Cancer Res. 2011, 17, 5755–5764. [Google Scholar] [CrossRef]
- Shukla, S.; Mahata, S.; Shishodia, G.; Pandey, A.; Tyagi, A.; Vishnoi, K.; Basir, S.F.; Das, B.C.; Bharti, A.C. Functional Regulatory Role of STAT3 in HPV16-Mediated Cervical Carcinogenesis. PLoS ONE 2013, 8, e67849. [Google Scholar] [CrossRef] [PubMed]
- Morgan, E.L.; Macdonald, A. Autocrine STAT3 activation in HPV positive cervical cancer through a virus-driven Rac1—NFκB—IL-6 signalling axis. PLoS Pathog. 2019, 15, e1007835. [Google Scholar] [CrossRef] [PubMed]
- Morgan, E.L.; Macdonald, A. JAK2 Inhibition Impairs Proliferation and Sensitises Cervical Cancer Cells to Cisplatin-Induced Cell Death. Cancers 2019, 11, 1934. [Google Scholar] [CrossRef] [PubMed]
- Gaykalova, D.A.; Manola, J.B.; Ozawa, H.; Zizkova, V.; Morton, K.; Bishop, J.A.; Sharma, R.; Zhang, C.; Michailidi, C.; Considine, M.; et al. NF-κB and stat3 transcription factor signatures differentiate HPV-positive and HPV-negative head and neck squamous cell carcinoma. Int. J. Cancer 2015, 137, 1879–1889. [Google Scholar] [CrossRef] [PubMed]
- Fan, Z.; Cui, H.; Xu, X.; Lin, Z.; Zhang, X.; Kang, L.; Han, B.; Meng, J.; Yan, Z.; Yan, X.; et al. MiR-125a suppresses tumor growth, invasion and metastasis in cervical cancer by targeting STAT3. Oncotarget 2015, 6, 25266–25280. [Google Scholar] [CrossRef] [PubMed]
- Fan, Z.; Cui, H.; Yu, H.; Ji, Q.; Kang, L.; Han, B.; Wang, J.; Dong, Q.; Li, Y.; Yan, Z.; et al. MiR-125a promotes paclitaxel sensitivity in cervical cancer through altering STAT3 expression. Oncogenesis 2016, 5, e197. [Google Scholar] [CrossRef]
- Tian, W.-J.; Huang, M.-L.; Qin, Q.-F.; Chen, Q.; Fang, K.; Wang, P.-L. Prognostic Impact of Epidermal Growth Factor Receptor Overexpression in Patients with Cervical Cancer: A Meta-Analysis. PLoS ONE 2016, 11, e0158787. [Google Scholar] [CrossRef]
- Byeon, H.K.; Ku, M.; Yang, J. Beyond EGFR inhibition: Multilateral combat strategies to stop the progression of head and neck cancer. Exp. Mol. Med. 2019, 51, 1–14. [Google Scholar] [CrossRef]
- Sriuranpong, V.; Park, J.I.; Amornphimoltham, P.; Patel, V.; Nelkin, B.D.; Gutkind, J.S. Epidermal growth factor receptor-independent constitutive activation of STAT3 in head and neck squamous cell carcinoma is mediated by the autocrine/paracrine stimulation of the interleukin 6/gp130 cytokine system. Cancer Res. 2003, 63, 2948–2956. [Google Scholar]
- Akerman, G.S.; Tolleson, W.H.; Brown, K.L.; Zyzak, L.L.; Mourateva, E.; Engin, T.S.; Basaraba, A.; Coker, A.L.; E Creek, K.; Pirisi, L. Human papillomavirus type 16 E6 and E7 cooperate to increase epidermal growth factor receptor (EGFR) mRNA levels, overcoming mechanisms by which excessive EGFR signaling shortens the life span of normal human keratinocytes. Cancer Res. 2001, 61, 3837–3843. [Google Scholar]
- Rani, A.; Murphy, J.J. STAT5 in Cancer and Immunity. J. Interf. Cytokine Res. 2016, 36, 226–237. [Google Scholar] [CrossRef] [PubMed]
- Zhu, J.; Cote-Sierra, J.; Guo, L.; E Paul, W. Stat5 Activation Plays a Critical Role in Th2 Differentiation. Immunity 2003, 19, 739–748. [Google Scholar] [CrossRef]
- Leong, P.L.; Xi, S.; Drenning, S.D.; Dyer, K.F.; Wentzel, A.L.; Lerner, E.C.; E Smithgall, T.; Grandis, J.R. Differential function of STAT5 isoforms in head and neck cancer growth control. Oncogene 2002, 21, 2846–2853. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Xi, S.; Zhang, Q.; E Gooding, W.; E Smithgall, T.; Grandis, J.R. Constitutive activation of Stat5b contributes to carcinogenesis In Vivo. Cancer Res. 2003, 63, 6763–6771. [Google Scholar] [PubMed]
- Dai, X.; Sayama, K.; Shirakata, Y.; Hanakawa, Y.; Yamasaki, K.; Tokumaru, S.; Yang, L.; Wang, X.; Hirakawa, S.; Tohyama, M.; et al. STAT5a/PPARgamma pathway regulates involucrin expression in keratinocyte differentiation. J. Investig. Dermatol. 2007, 127, 1728–1735. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Hong, S.; Laimins, L.A. The JAK-STAT Transcriptional Regulator, STAT-5, Activates the ATM DNA Damage Pathway to Induce HPV 31 Genome Amplification upon Epithelial Differentiation. PLoS Pathog. 2013, 9, e1003295. [Google Scholar] [CrossRef] [PubMed]
- Hong, S.; Cheng, S.; Iovane, A.; Laimins, L.A. STAT-5 Regulates Transcription of the Topoisomerase IIβ-Binding Protein 1 (TopBP1) Gene to Activate the ATR Pathway and Promote Human Papillomavirus Replication. mBio 2015, 6, e02006-15. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Hong, S.; Maniar, K.P.; Cheng, S.; Jie, C.; Rademaker, A.W.; Krensky, A.M.; Clayberger, C. KLF13 regulates the differentiation-dependent human papillomavirus life cycle in keratinocytes through STAT5 and IL-8. Oncogene 2016, 35, 5565–5575. [Google Scholar] [CrossRef]
- Koppikar, P.; Lui, V.W.Y.; Man, D.; Xi, S.; Chai, R.L.; Nelson, E.; Tobey, A.B.; Grandis, J.R. Constitutive activation of signal transducer and activator of transcription 5 contributes to tumor growth, epithelial-mesenchymal transition, and resistance to epidermal growth factor receptor targeting. Clin. Cancer Res. 2008, 14, 7682–7690. [Google Scholar] [CrossRef][Green Version]
- Liu, L.-B.; Xie, F.; Chang, K.-K.; Shang, W.-Q.; Meng, Y.-H.; Yu, J.-J.; Li, H.; Sun, Q.; Yuan, M.-M.; Jin, L.-P.; et al. Chemokine CCL17 induced by hypoxia promotes the proliferation of cervical cancer cell. Am. J. Cancer Res. 2015, 5, 3072–3084. [Google Scholar]
- Valle-Mendiola, A.; Weiss-Steider, B.; Rocha-Zavaleta, L.; Soto-Cruz, I. IL-2 enhances cervical cancer cells proliferation and JAK3/STAT5 phosphorylation at low doses, while at high doses IL-2 has opposite effects. Cancer Investig. 2014, 32, 115–125. [Google Scholar] [CrossRef] [PubMed]
- Thomas, M.; Banks, L. PDZRN3/LNX3 is a novel target of human papillomavirus type 16 [HPV-16] and HPV-18 E6. J. Virol. 2015, 89, 1439–1444. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Hayakawa, F.; Sugimoto, K.; Harada, Y.; Hashimoto, N.; Ohi, N.; Kurahashi, S.; Naoe, T. A novel STAT inhibitor, OPB-31121, has a significant antitumor effect on leukemia with STAT-addictive oncokinases. Blood Cancer J. 2013, 3, e166. [Google Scholar] [CrossRef] [PubMed]
- Tolcher, A.; Flaherty, K.; Shapiro, G.I.; Berlin, J.; Witzig, T.; Habermann, T.; Bullock, A.; Rock, E.; Elekes, A.; Lin, C.; et al. A First-in-Human Phase I Study of OPB-111077, a Small-Molecule STAT3 and Oxidative Phosphorylation Inhibitor, in Patients with Advanced Cancers. Oncologist 2018, 23, 658–672. [Google Scholar] [CrossRef]
- Yoo, C.; Kang, J.; Lim, H.Y.; Kim, J.H.; Lee, M.-A.; Lee, K.-H.; Kim, T.-Y.; Ryoo, B.-Y. Phase I Dose-Finding Study of OPB-111077, a Novel STAT3 Inhibitor, in Patients with Advanced Hepatocellular Carcinoma. Cancer Res. Treat. 2019, 51, 510–518. [Google Scholar] [CrossRef]
- Liao, Z.; Gu, L.; Vergalli, J.; Mariani, S.A.; De Dominici, M.; Lokareddy, R.K.; Dagvadorj, A.; Purushottamachar, P.; McCue, P.A.; Trabulsi, E.; et al. Structure-Based Screen Identifies a Potent Small Molecule Inhibitor of Stat5a/b with Therapeutic Potential for Prostate Cancer and Chronic Myeloid Leukemia. Mol. Cancer Ther. 2015, 14, 1777–1793. [Google Scholar] [CrossRef]
- Bai, L.; Zhou, H.; Xu, R.; Zhao, Y.; Chinnaswamy, K.; McEachern, D.; Chen, J.; Yang, C.-Y.; Liu, Z.; Wang, M.; et al. A Potent and Selective Small-Molecule Degrader of STAT3 Achieves Complete Tumor Regression In Vivo. Cancer Cell 2019, 36, 498–511. [Google Scholar] [CrossRef]
- Reilley, M.J.; McCoon, P.; Cook, C.; Lyne, P.D.; Kurzrock, R.; Kim, Y.; Woessner, R.; Younes, A.; Nemunaitis, J.; Fowler, N.H.; et al. STAT3 antisense oligonucleotide AZD9150 in a subset of patients with heavily pretreated lymphoma: Results of a phase 1b trial. J. Immunother. Cancer 2018, 6, 119. [Google Scholar] [CrossRef]
- Wang, X.; Zeng, J.; Shi, M.; Zhao, S.; Bai, W.; Cao, W.; Tu, Z.; Huang, Z.; Feng, W. Targeted Blockage of Signal Transducer and Activator of Transcription 5 Signaling Pathway with Decoy Oligodeoxynucleotides Suppresses Leukemic K562 Cell Growth. DNA Cell Biol. 2011, 30, 71–78. [Google Scholar] [CrossRef]
- Hong, D.; Kurzrock, R.; Kim, Y.; Woessner, R.; Younes, A.; Nemunaitis, J.; Fowler, N.; Zhou, T.; Schmidt, J.; Jo, M.; et al. AZD9150, a next-generation antisense oligonucleotide inhibitor of STAT3 with early evidence of clinical activity in lymphoma and lung cancer. Sci. Transl. Med. 2015, 7, 314ra185. [Google Scholar] [CrossRef]
- Zhu, Q.; Jing, N. Computational study on mechanism of G-quartet oligonucleotide T40214 selectively targeting Stat3. J. Comput. Mol. Des. 2007, 21, 641–648. [Google Scholar] [CrossRef] [PubMed]
- Jing, N.; Li, Y.; Xiong, W.; Sha, W.; Jing, L.; Tweardy, D.J. G-Quartet Oligonucleotides. Cancer Res. 2004, 64, 6603–6609. [Google Scholar] [CrossRef] [PubMed]
- Rossi, J.-F.; Négrier, S.; James, N.D.; Kocak, I.; Hawkins, R.; Davis, H.; Prabhakar, U.; Qin, X.; Mulders, P.; Berns, B. A phase I/II study of siltuximab (CNTO 328), an anti-interleukin-6 monoclonal antibody, in metastatic renal cell cancer. Br. J. Cancer 2010, 103, 1154–1162. [Google Scholar] [CrossRef]
- Angevin, E.; Tabernero, J.; Elez, E.; Cohen, S.J.; Bahleda, R.; Van Laethem, J.-L.; Ottensmeier, C.H.; Lopez-Martin, J.A.; Clive, S.; Joly, F.; et al. A Phase I/II, Multiple-Dose, Dose-Escalation Study of Siltuximab, an Anti-Interleukin-6 Monoclonal Antibody, in Patients with Advanced Solid Tumors. Clin. Cancer Res. 2014, 20, 2192–2204. [Google Scholar] [CrossRef] [PubMed]
- Brighton, T.A.; Khot, A.; Harrison, S.J.; Ghez, D.; Weiss, B.M.; Kirsch, A.; Magen, H.; Gironella, M.; Oriol, A.; Streetly, M.; et al. Randomized, Double-Blind, Placebo-Controlled, Multicenter Study of Siltuximab in High-Risk Smoldering Multiple Myeloma. Clin. Cancer Res. 2019, 25, 3772–3775. [Google Scholar] [CrossRef] [PubMed]
- A Hunter, C.; Jones, S.A. IL-6 as a keystone cytokine in health and disease. Nat. Immunol. 2015, 16, 448–457. [Google Scholar] [CrossRef] [PubMed]
- Hirabayashi, Y. Tocilizumab, an anti-interleukin-6 receptor antibody, efficiently ameliorates persistent joint inflammation in rheumatoid arthritis. Mod. Rheumatol. 2020, 63, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Dijkgraaf, E.M.; Santegoets, S.J.A.M.; Reyners, A.K.L.; Goedemans, R.; Wouters, M.C.A.; Kenter, G.G.; Van Erkel, A.R.; Van Poelgeest, M.I.E.; Nijman, H.W.; Van Der Hoeven, J.J.M.; et al. A phase I trial combining carboplatin/doxorubicin with tocilizumab, an anti-IL-6R monoclonal antibody, and interferon-α2b in patients with recurrent epithelial ovarian cancer. Ann. Oncol. 2015, 26, 2141–2149. [Google Scholar] [CrossRef]
- Heo, Y.-A.; Deeks, E.D. Sarilumab: A Review in Moderate to Severe Rheumatoid Arthritis. Drugs 2018, 78, 929–940. [Google Scholar]
- Nikolaus, S.; Waetzig, G.H.; Butzin, S.; Ziolkiewicz, M.; Al-Massad, N.; Thieme, F.; Lövgren, U.; Rasmussen, B.B.; Reinheimer, T.M.; Seegert, D.; et al. Evaluation of interleukin-6 and its soluble receptor components sIL-6R and sgp130 as markers of inflammation in inflammatory bowel diseases. Int. J. Color. Dis. 2018, 33, 927–936. [Google Scholar] [CrossRef]
- Sandborn, W.J.; Ghosh, S.; Panés, J.; Vranic, I.; Su, C.; Rousell, S.; Niezychowski, W. Tofacitinib, an Oral Janus Kinase Inhibitor, in Active Ulcerative Colitis. N. Engl. J. Med. 2012, 367, 616–624. [Google Scholar] [CrossRef] [PubMed]
- Passamonti, F.; Griesshammer, M.; Palandri, F.; Egyed, M.; Benevolo, G.; Devos, T.; Callum, J.; Vannucchi, A.M.; Sivgin, S.; Bensasson, C.; et al. Ruxolitinib for the treatment of inadequately controlled polycythaemia vera without splenomegaly (RESPONSE-2): A randomised, open-label, phase 3b study. Lancet Oncol. 2017, 18, 88–99. [Google Scholar] [CrossRef]
- Giaccone, G.; Sanborn, R.E.; Waqar, S.N.; Martinez-Marti, A.; Ponce, S.; Zhen, H.; Kennealey, G.; Erickson-Viitanen, S.; Schaefer, E. A Placebo-Controlled Phase II Study of Ruxolitinib in Combination with Pemetrexed and Cisplatin for First-Line Treatment of Patients With Advanced Nonsquamous Non–Small-Cell Lung Cancer and Systemic Inflammation. Clin. Lung Cancer 2018, 19, e567–e574. [Google Scholar] [CrossRef] [PubMed]
- Mascarenhas, J.; Hoffman, R.; Talpaz, M.; Gerds, A.T.; Stein, B.; Gupta, V.; Szoke, A.; Drummond, M.; Pristupa, A.; Granston, T.; et al. Faculty Opinions recommendation of Pacritinib vs best available therapy, including ruxolitinib, in patients with myelofibrosis: A randomized clinical trial. JAMA Oncol. 2018, 4, 652–659. [Google Scholar] [CrossRef] [PubMed]
- Wong, A.L.-A.; Hirpara, J.L.; Pervaiz, S.; Eu, J.-Q.; Sethi, G.; Goh, B.C. Do STAT3 inhibitors have potential in the future for cancer therapy? Expert Opin. Investig. Drugs 2017, 26, 883–887. [Google Scholar] [CrossRef]
- Ren, X.; Duan, L.; He, Q.; Zhang, Z.; Zhou, Y.; Wu, D.; Pan, J.; Pei, D.; Ding, K. Identification of Niclosamide as a New Small-Molecule Inhibitor of the STAT3 Signaling Pathway. ACS Med. Chem. Lett. 2010, 1, 454–459. [Google Scholar] [CrossRef] [PubMed]
- Shin, D.-S.; Kim, H.-N.; Shin, K.D.; Yoon, Y.J.; Kim, S.-J.; Han, D.C.; Kwon, B.-M. Cryptotanshinone Inhibits Constitutive Signal Transducer and Activator of Transcription 3 Function through Blocking the Dimerization in DU145 Prostate Cancer Cells. Cancer Res. 2009, 69, 193–202. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Sun, Y.; Pireddu, R.; Yang, H.; Urlam, M.K.; Lawrence, H.R.; Guida, W.C.; Lawrence, N.J.; Sebti, S.M. A novel inhibitor of STAT3 homodimerization selectively suppresses STAT3 activity and malignant transformation. Cancer Res. 2013, 73, 1922–1933. [Google Scholar] [CrossRef]
- Brambilla, L.; Genini, D.; Laurini, E.; Merulla, J.; Perez, L.; Fermeglia, M.; Carbone, G.M.; Pricl, S.; Catapano, C.V. Hitting the right spot: Mechanism of action of OPB-31121, a novel and potent inhibitor of the Signal Transducer and Activator of Transcription 3 (STAT3). Mol. Oncol. 2015, 9, 1194–1206. [Google Scholar] [CrossRef]
- Kim, M.-J.; Nam, H.-J.; Kim, H.-P.; Han, S.-W.; Im, S.-A.; Kim, T.-Y.; Oh, D.; Bang, Y. OPB-31121, a novel small molecular inhibitor, disrupts the JAK2/STAT3 pathway and exhibits an antitumor activity in gastric cancer cells. Cancer Lett. 2013, 335, 145–152. [Google Scholar] [CrossRef]
- Rondanin, R.; Simoni, D.; Maccesi, M.; Romagnoli, R.; Grimaudo, S.; Pipitone, R.M.; Meli, M.; Cascio, A.; Tolomeo, M. Effects of Pimozide Derivatives on pSTAT5 in K562 Cells. ChemMedChem 2017, 12, 1183–1190. [Google Scholar] [CrossRef] [PubMed]
- Nelson, E.A.; Walker, S.R.; Weisberg, E.; Bar-Natan, M.; Barrett, R.; Gashin, L.B.; Terrell, S.; Klitgaard, J.L.; Santo, L.; Addorio, M.R.; et al. The STAT5 inhibitor pimozide decreases survival of chronic myelogenous leukemia cells resistant to kinase inhibitors. Blood 2011, 117, 3421–3429. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.; Gao, H.; Yang, Y.; He, M.; Wu, Y.; Song, Y.; Tong, Y.; Rao, Y. PROTACs: Great opportunities for academia and industry. Signal Transduct. Target. Ther. 2019, 4, 1–33. [Google Scholar] [CrossRef] [PubMed]
- Lau, Y.-T.K.; Ramaiyer, M.; Johnson, D.E.; Grandis, J.R. Targeting STAT3 in Cancer with Nucleotide Therapeutics. Cancers 2019, 11, 1681. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.; Zhang, J. STAT3 Decoy ODN Therapy for Cancer. Adv. Struct. Saf. Stud. 2015, 1317, 167–183. [Google Scholar]
- Leong, P.L.; Andrews, G.A.; Johnson, D.E.; Dyer, K.F.; Xi, S.; Mai, J.C.; Robbins, P.D.; Gadiparthi, S.; Burke, N.A.; Watkins, S.F.; et al. Targeted inhibition of Stat3 with a decoy oligonucleotide abrogates head and neck cancer cell growth. Proc. Natl. Acad. Sci. USA 2003, 100, 4138–4143. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Zhang, J.; Wang, L.; Wei, H.; Tian, Z. Therapeutic effects of STAT3 decoy oligodeoxynucleotide on human lung cancer in xenograft mice. BMC Cancer 2007, 7, 1–11. [Google Scholar] [CrossRef]
- Sen, M.; Joyce, S.; Panahandeh, M.; Li, C.; Thomas, S.M.; Maxwell, J.; Wang, L.; Gooding, W.E.; Johnson, D.E.; Grandis, J.R. Targeting Stat3 abrogates EGFR inhibitor resistance in cancer. Clin. Cancer Res. 2012, 18, 4986–4996. [Google Scholar] [CrossRef]
- Dean, N.M.; Bennett, C.F. Antisense oligonucleotide-based therapeutics for cancer. Oncogene 2003, 22, 9087–9096. [Google Scholar] [CrossRef]
- Niu, G.; Wright, K.L.; Huang, M.; Song, L.; Haura, E.; Turkson, J.; Zhang, S.; Wang, T.; Sinibaldi, D.; Coppola, D.; et al. Constitutive Stat3 activity up-regulates VEGF expression and tumor angiogenesis. Oncogene 2002, 21, 2000–2008. [Google Scholar] [CrossRef]
- Barton, B.E.; Karras, J.G.; Murphy, T.F.; Barton, A.; Huang, H.F. Signal transducer and activator of transcription 3 (STAT3) activation in prostate cancer: Direct STAT3 inhibition induces apoptosis in prostate cancer lines. Mol. Cancer Ther. 2004, 3, 11–20. [Google Scholar] [PubMed]
- Jing, N.; Zhu, Q.; Yuan, P.; Li, Y.; Mao, L.; Tweardy, D.J. Targeting signal transducer and activator of transcription 3 with G-quartet oligonucleotides: A potential novel therapy for head and neck cancer. Mol. Cancer Ther. 2006, 5, 279–286. [Google Scholar] [CrossRef] [PubMed]
- Goumas, F.A.; Holmer, R.; Egberts, J.-H.; Gontarewicz, A.; Heneweer, C.; Geisen, U.; Hauser, C.; Mende, M.-M.; Legler, K.; Röcken, C.; et al. Inhibition of IL-6 signaling significantly reduces primary tumor growth and recurrencies in orthotopic xenograft models of pancreatic cancer. Int. J. Cancer 2015, 137, 1035–1046. [Google Scholar] [CrossRef] [PubMed]
- Fisher, D.T.; Appenheimer, M.M.; Evans, S.S. The two faces of IL-6 in the tumor microenvironment. Semin. Immunol. 2014, 26, 38–47. [Google Scholar] [CrossRef]
- Greenhill, C.J.; Rose-John, S.; Ferlin, W.; O’Neill, L.; Hertzog, P.; Mansell, A.; Jenkins, B.J. IL-6 Trans-signaling modulates TLR4-dependent inflammatory responses via STAT3. J. Immunol. 2011, 186, 1199–1208. [Google Scholar] [CrossRef]
- Schumacher, N.; Rose-John, S.; Rose-John, S. ADAM17 Activity and IL-6 Trans-Signaling in Inflammation and Cancer. Cancers 2019, 11, 1736. [Google Scholar] [CrossRef]
- Gao, J.; Zhao, S.; Halstensen, T.S. Increased interleukin-6 expression is associated with poor prognosis and acquired cisplatin resistance in head and neck squamous cell carcinoma. Oncol. Rep. 2016, 35, 3265–3274. [Google Scholar] [CrossRef][Green Version]
- Squarize, C.H.; Castilho, R.M.; Sriuranpong, V.; Pinto, D.S.; Gutkind, J.S. Molecular cross-talk between the NFkappaB and STAT3 signaling pathways in head and neck squamous cell carcinoma. Neoplasia 2006, 8, 733. [Google Scholar] [CrossRef]
- Sayyah, J.; Sayeski, P.P. Jak2 inhibitors: Rationale and role as therapeutic agents in hematologic malignancies. Curr. Oncol. Rep. 2009, 11, 117–124. [Google Scholar] [CrossRef]
- Furumoto, Y.; Gadina, M. The arrival of JAK inhibitors: Advancing the treatment of immune and hematologic disorders. BioDrugs 2013, 27, 431–438. [Google Scholar] [CrossRef]
- Sen, M.; Pollock, N.I.; Black, J.; Degrave, K.A.; Wheeler, S.E.; Freilino, M.L.; Joyce, S.; Lui, V.W.Y.; Zeng, Y.; Chiosea, S.I.; et al. JAK kinase inhibition abrogates STAT3 activation and head and neck squamous cell carcinoma tumor growth. Neoplasia 2015, 17, 256–264. [Google Scholar] [CrossRef] [PubMed]
- Hoover, A.C.; Strand, G.L.; Nowicki, P.N.; E Anderson, M.; Vermeer, P.D.; Klingelhutz, A.J.; Bossler, A.D.; Pottala, J.V.; Hendriks, W.; Lee, J.H. Impaired PTPN13 phosphatase activity in spontaneous or HPV-induced squamous cell carcinomas potentiates oncogene signaling through the MAP kinase pathway. Oncogene 2009, 28, 3960–3970. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Therapeutic | Target/Mechanism | Indication | Regulatory Status | References |
|---|---|---|---|---|
| OPB-31121 | STAT3 dimerisation inhibitor | e.g., Advanced solid tumours | Phase I | [183] |
| OPB-111077 | [184,185] | |||
| IST5-002 | STAT5 dimerisation inhibitor | e.g., pancreatic cancer | Pre-clinical | [186] |
| SD-36 | STAT3 [PROTAC] | e.g., leukaemias and lymphomas | Pre-clinical | [187] |
| STAT3 decoy | STAT3 response element from FOS gene | HNSCC | Pre-clinical | [188] |
| STAT5 decoy | STAT5 decoy oligonucleotide | e.g., leukaemias | Pre-clinical | [189] |
| AZD9150 | STAT3 antisense oligonucleotide | e.g., solid tumours, metastatic HNSCC | Phase I/II | [188,190] |
| T40214 | G-quartet oligodeoxynucleotides | e.g., HNSCC, liver cancer | Pre-clinical | [191] |
| T40231 | e.g., HNSCC, prostate cancer | Pre-clinical | [192] | |
| Siltuximab | Anti-IL-6 mAb | e.g., multiple myeloma, solid tumours | Phase I/II | [193,194,195] |
| Olokizumab | e.g., RA | Phase II | [196] | |
| Tocilizumab | Anti-IL-6R mAb | e.g., RA | FDA approved | [197,198] |
| Sarilumab | [199] | |||
| Olamkicept | Soluble gp130-Fc fusion protein | e.g., RA, IBD | Phase I/II | [200] |
| Tofacitinib | JAK inhibitor | e.g., RA, psoriasis, myelofibrosis | FDA approved | [201] |
| Ruxolitinib | FDA approved | [202,203] | ||
| Pacritinib | Phase II | [204] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Morgan, E.L.; Macdonald, A. Manipulation of JAK/STAT Signalling by High-Risk HPVs: Potential Therapeutic Targets for HPV-Associated Malignancies. Viruses 2020, 12, 977. https://doi.org/10.3390/v12090977
Morgan EL, Macdonald A. Manipulation of JAK/STAT Signalling by High-Risk HPVs: Potential Therapeutic Targets for HPV-Associated Malignancies. Viruses. 2020; 12(9):977. https://doi.org/10.3390/v12090977
Chicago/Turabian StyleMorgan, Ethan L., and Andrew Macdonald. 2020. "Manipulation of JAK/STAT Signalling by High-Risk HPVs: Potential Therapeutic Targets for HPV-Associated Malignancies" Viruses 12, no. 9: 977. https://doi.org/10.3390/v12090977
APA StyleMorgan, E. L., & Macdonald, A. (2020). Manipulation of JAK/STAT Signalling by High-Risk HPVs: Potential Therapeutic Targets for HPV-Associated Malignancies. Viruses, 12(9), 977. https://doi.org/10.3390/v12090977