Characterization of Avian Influenza Virus H10–H12 Subtypes Isolated from Wild Birds in Shanghai, China from 2016 to 2019

 , ,
, ,
Abstract
1. Introduction
2. Materials and Methods
2.1. Sample Collection
2.2. Virus Identification and Genome Sequencing
2.3. Sequence Analysis
3. Results
3.1. Prevalence of H10–H12 AIVs in Wild Birds
3.2. Molecular Characterization
3.3. Sequence and Phylogenetic Analysis of H10 Isolates
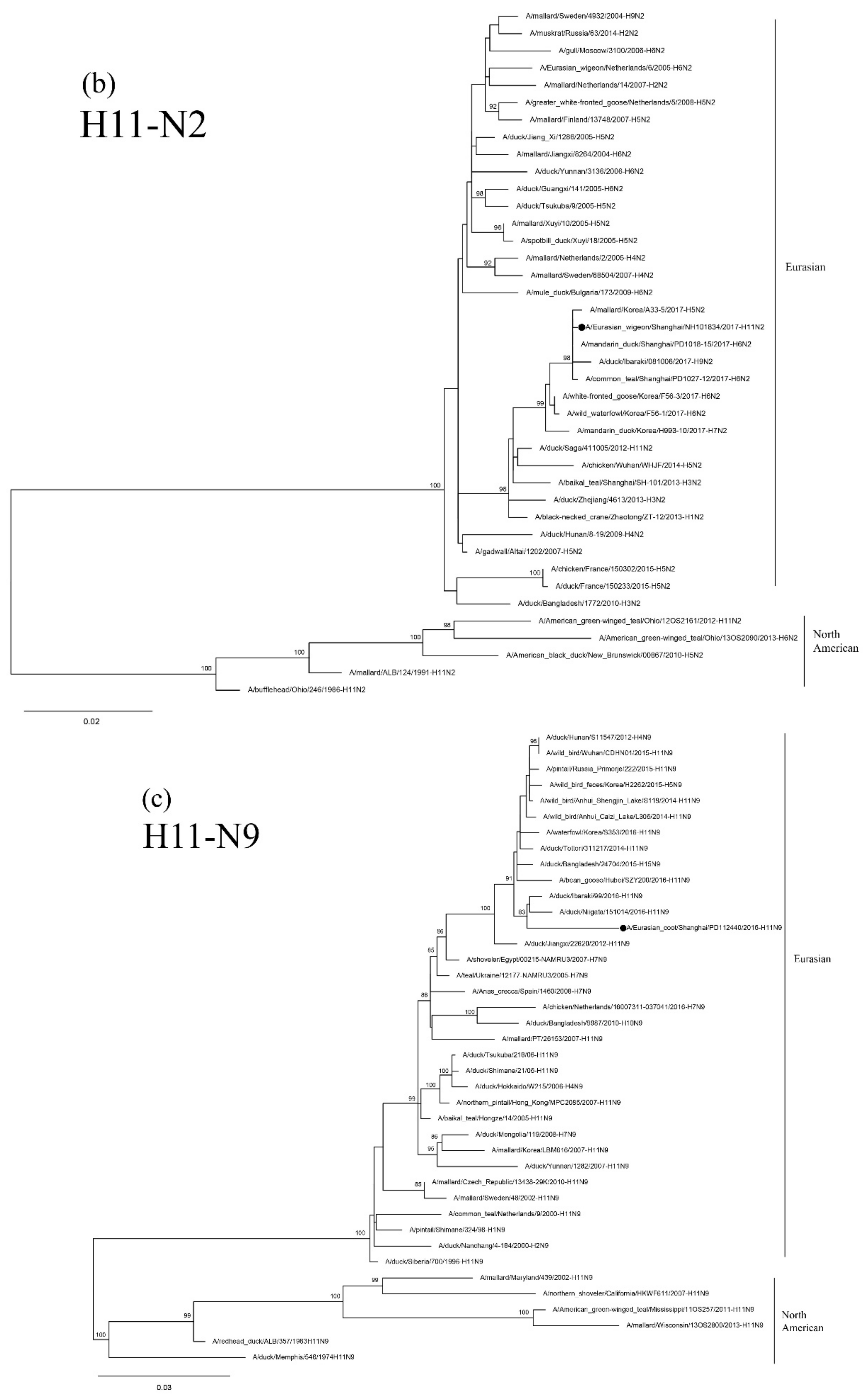
3.4. Sequence and Phylogenetic Analysis of H11 Isolates
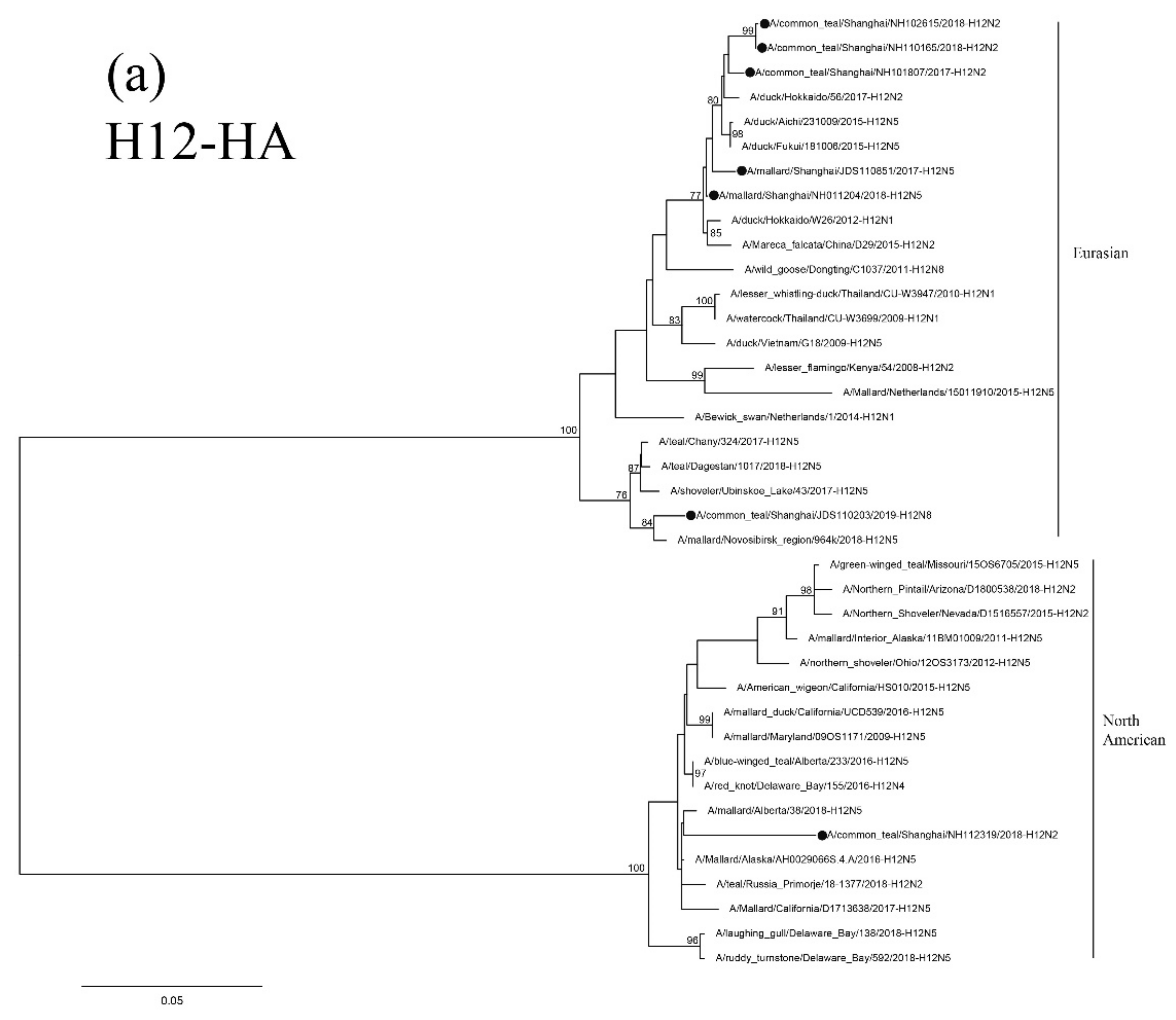
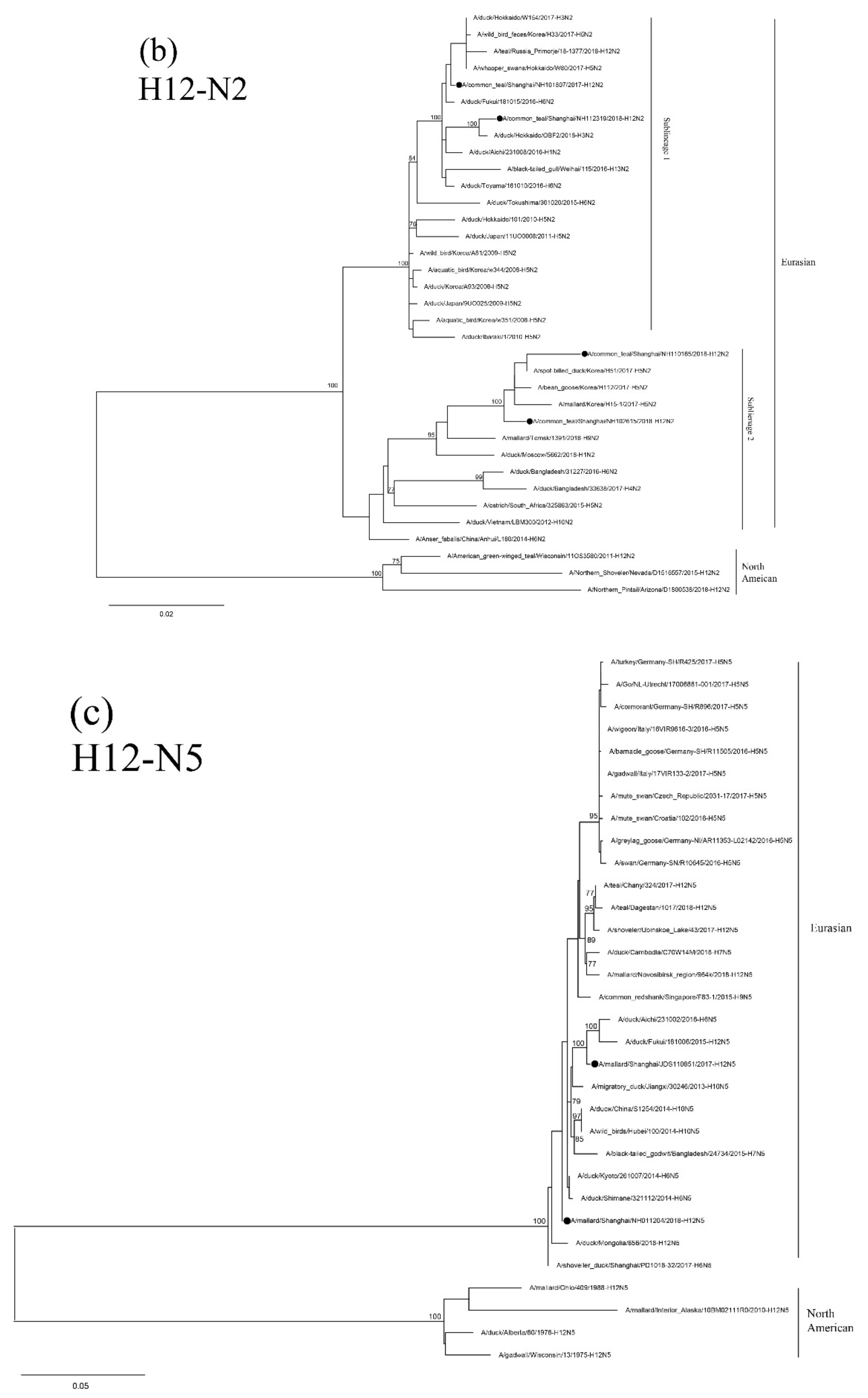
3.5. Sequence and Phylogenetic Analysis of H12 Isolates
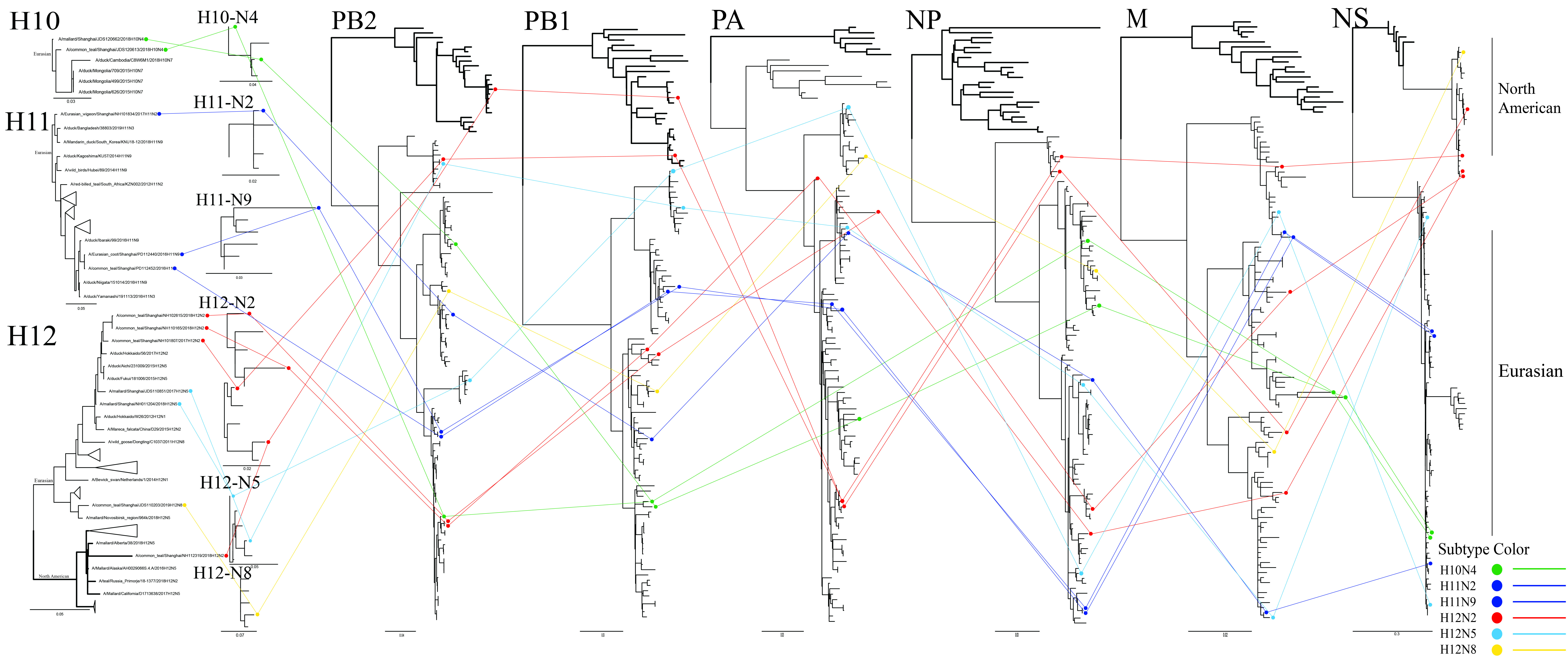
3.6. Reassortment Relationships of H10–H12 Isolates
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Data Availability
References
- Webster, R.G.; Bean, W.J.; Gorman, O.T.; Chambers, T.M.; Kawaoka, Y. Evolution and ecology of influenza A viruses. Microbiol. Immunol. 1992, 56, 359–375. [Google Scholar] [CrossRef]
- Kawaoka, Y.; Chambers, T.M.; Sladen, W.L.; Gwebster, R. Is the gene pool of influenza viruses in shorebirds and gulls different from that in wild ducks. Virology 1988, 163, 247–250. [Google Scholar] [CrossRef]
- Guan, M.; Hall, J.S.; Zhang, X.; Dusek, R.J.; Olivier, A.K.; Liu, L.; Li, L.; Krauss, S.; Danner, A.; Li, T.; et al. Aerosol transmission of Gull-Origin Iceland subtype H10N7 influenza A virus in ferrets. J. Virol. 2019, 93. [Google Scholar] [CrossRef] [PubMed]
- Brown, J.D.; Swayne, D.E.; Cooper, R.J.; Burns, R.E.; Stallknecht, D.E. Persistence of H5 and H7 avian influenza viruses in water. Avian Dis. 2007, 51, 285–289. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.J.; Kwon, J.S.; Lee, D.H.; Lee, Y.N.; Youn, H.N.; Lee, Y.J.; Kim, M.C.; Jeong, O.M.; Kang, H.M.; Kwon, J.H.; et al. Continuing evolution and interspecies transmission of influenza viruses in live bird markets in Korea. Avian Dis. 2010, 54 (Suppl. 1), 738–748. [Google Scholar] [CrossRef]
- Krauss, S.; Walker, D.; Pryor, S.P.; Niles, L.; Chenghong, L.; Hinshaw, V.S.; Webster, R.G. Influenza A viruses of migrating wild aquatic birds in North America. Vector Borne Zoonot 2004, 4, 177–189. [Google Scholar] [CrossRef]
- Latorre-Margalef, N.; Tolf, C.; Grosbois, V.; Avril, A.; Bengtsson, D.; Wille, M.; Osterhaus, A.D.; Fouchier, R.A.; Olsen, B.; Waldenstrom, J. Long-term variation in influenza A virus prevalence and subtype diversity in migratory mallards in northern Europe. Proc. Biol. 2014, 281, 20140098. [Google Scholar] [CrossRef]
- Munster, V.J.; Baas, C.; Lexmond, P.; Waldenstrom, J.; Wallensten, A.; Fransson, T.; Rimmelzwaan, G.F.; Beyer, W.E.; Schutten, M.; Olsen, B.; et al. Spatial, temporal, and species variation in prevalence of influenza A viruses in wild migratory birds. PLoS Pathog. 2007, 3, e61. [Google Scholar] [CrossRef]
- Wille, M.; Latorre-Margalef, N.; Tolf, C.; Halpin, R.; Wentworth, D.; Fouchier, R.A.M.; Raghwani, J.; Pybus, O.G.; Olsen, B.; Waldenstrom, J. Where do all the subtypes go? Temporal dynamics of H8-H12 influenza A viruses in waterfowl. Virus Evol. 2018, 4, vey025. [Google Scholar] [CrossRef]
- Vachieri, S.G.; Xiong, X.; Collins, P.J.; Walker, P.A.; Martin, S.R.; Haire, L.F.; Zhang, Y.; McCauley, J.W.; Gamblin, S.J.; Skehel, J.J. Receptor binding by H10 influenza viruses. Nature 2014, 511, 475–477. [Google Scholar] [CrossRef]
- Zohari, S.; Neimanis, A.; Harkonen, T.; Moreaus, C.; Valaracher, J.F. Avian influenza A(H10N7) virus involvement in mass mortality of harbour seals (Phoca vitulina) in Sweden, March through October 2014. Euro. Survll. 2014, 19, 20967–20973. [Google Scholar] [CrossRef] [PubMed]
- Klingeborn, B.; Englund, L.; Rott, R.; Juntti, N.; Rockborn, G. An avian influenza A virus killing a mammalian species-the mink. Arch. Virol. 1985, 86, 347–351. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.Y.; Yuan, H.; Gao, R.B.; Zhang, J.X.; Wang, D.Y.; Xiong, Y.; Fan, G.Y.; Yang, F.; Li, X.D.; Zhou, J.F.; et al. Clinical and epidemiological characteristics of a fatal case of avian influenza A H10N8 virus infection: A descriptive study. Lancet 2014, 383, 714–721. [Google Scholar] [CrossRef]
- Lam, T.T.; Wang, J.; Shen, Y.; Zhou, B.; Duan, L.; Cheung, C.L.; Ma, C.; Lycett, S.J.; Leung, C.Y.; Chen, X.; et al. The genesis and source of the H7N9 influenza viruses causing human infections in China. Nature 2013, 502, 241–244. [Google Scholar] [CrossRef]
- Velarde, R.; Calvin, S.E.; Ojkic, D.; Barker, I.K.; Nagy, E. Avian influenza virus H13 circulating in ring-billed gulls (Larus delawarensis) in southern Ontario, Canada. Avian Dis. Digest. 2010, 54, 411–419. [Google Scholar] [CrossRef]
- Bui, V.N.; Ogawa, H.; Hussein, I.T.; Hill, N.J.; Trinh, D.Q.; AboElkhair, M.; Sultan, S.; Ma, E.; Saito, K.; Watanabe, Y.; et al. Genetic characterization of a rare H12N3 avian influenza virus isolated from a green-winged teal in Japan. Virus Genes 2015, 50, 316–320. [Google Scholar] [CrossRef]
- Wilcox, B.R.; Knutsen, G.A.; Berdeen, J.; Goekjian, V.; Poulson, R.; Goyal, S.; Sreevatsan, S.; Cardona, C.; Berghaus, R.D.; Swayne, D.E.; et al. Influenza-A viruses in ducks in northwestern Minnesota: Fine scale spatial and temporal variation in prevalence and subtype diversity. PLoS ONE 2011, 6, e24010. [Google Scholar] [CrossRef]
- Sharp, G.B.; Kawaoka, Y.; Jones, D.J.; Bean, W.J.; Pryor, S.P.; Hinshaw, V.; Webster, R.G. Coinfection of wild ducks by influenza A viruses distribution patterns and biological significance. J. Virol. 1997, 71, 6128–6135. [Google Scholar] [CrossRef]
- Huang, Y.; Khan, M.I.; Mandoiu, I. Neuraminidase subtyping of avian influenza viruses with primer hunter-designed primers and quadruplicate primer pools. PLoS ONE 2013, 8, e81842. [Google Scholar] [CrossRef]
- Shi, B.; Zhan, X.M.; Zheng, J.X.; Qiu, H.; Liang, D.; Ye, Y.M.; Yang, G.J.; Liu, Y.; Liu, J. Identifying key bird species and geographical hotspots of avian influenza A (H7N9) virus in China. Infect. Dis. Poverty 2018, 7, 97. [Google Scholar] [CrossRef]
- He, G.; Zhou, L.; Zhu, C.; Shi, H.; Li, X.; Wu, D.; Liu, J.; Lv, J.; Hu, C.; Li, Z.; et al. Identification of two novel avian influenza a (H5N6) viruses in wild birds, Shanghai, in 2016. Vet. Microbiol. 2017, 208, 53–57. [Google Scholar] [CrossRef] [PubMed]
- Kim, G.S.; Kim, T.S.; Son, J.S.; Lai, V.D.; Park, J.E.; Wang, S.J.; Jheong, W.H.; Mo, I.P. The difference of detection rate of avian influenza virus in the wild bird surveillance using various methods. J. Vet. Sci. 2019, 20, e56. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, E.; Stech, J.; Guan, Y.; Webster, R.G.; Perez, D.R. Universal primer set for the full-length amplification of all influenza A viruses. Arch. Virol. 2001, 146, 2275–2289. [Google Scholar] [CrossRef] [PubMed]
- Matrosovich, M.; Tuzikov, A.; Bovin, N.; Gambaryan, A.; Klimov, A.; Castrucci, M.R.; Donatelli, I.; Kawaoka, Y. Early alterations of the receptor-binding properties of H1, H2, and H3 avian influenza virus hemagglutinins after their introduction into mammals. J. Virol. 2000, 74, 8502–8512. [Google Scholar] [CrossRef] [PubMed]
- Hay, A.J.; Wolstenholme, A.J.; Skehel, J.J.; Smith, M.H. The molecular basis of the specific anti-influenza action of amantadine. EMBO J. 1985, 4, 3021–3024. [Google Scholar] [CrossRef]
- Czudai-Matwich, V.; Otte, A.; Matrosovich, M.; Gabriel, G.; Klenk, H.D. PB2 mutations D701N and S714R promote adaptation of an influenza H5N1 virus to a mammalian host. J. Virol. 2014, 88, 8735–8742. [Google Scholar] [CrossRef]
- Schat, K.A.; Bingham, J.; Butler, J.M.; Chen, L.M.; Lowther, S.; Crowley, T.M.; Moore, R.J.; Donis, R.O.; Lowenthal, J.W. Role of position 627 of PB2 and the multibasic cleavage site of the hemagglutinin in the virulence of H5N1 avian influenza virus in chickens and ducks. PLoS ONE 2012, 7, e30960. [Google Scholar] [CrossRef]
- Globig, A.; Baumer, A.; Revilla-Fernandez, S.; Beer, M.; Wodak, E.; Fink, M.; Greber, N.; Harder, T.C.; Wilking, H.; Brunhart, I.; et al. Ducks as sentinels for avian influenza in wild birds. Emerg. Infect. Dis. 2009, 15, 1633–1636. [Google Scholar] [CrossRef]
- Schneider, E.K.; Li, J.; Velkov, T. A Portrait of the Sialyl Glycan Receptor Specificity of the H10 Influenza Virus Hemagglutinin-A Picture of an Avian Virus on the Verge of Becoming a Pandemic? Vaccines 2017, 5, 51–66. [Google Scholar] [CrossRef]
- Veits, J.; Weber, S.; Stech, O.; Breithaupt, A.; Stech, J. Avian influenza virus hemagglutinins H2, H4, H8, and H14 support a highly pathogenic phenotype. Proc. Natl. Acad. Sci. USA 2012, 109, 2579–2584. [Google Scholar] [CrossRef]
- Bodewes, R.; Zohari, S.; Krog, J.S.; Hall, M.D.; Harder, T.C.; Bestebroer, T.M.; van de Bildt, M.W.G.; Spronken, M.I.; Larsen, L.E.; Siebert, U.; et al. Spatiotemporal analysis of the genetic diversity of seal influenza A(H10N7) virus, Northwestern Europe. J. Virol. 2016, 90, 4269–4277. [Google Scholar] [CrossRef] [PubMed]
- Gill, J.S.; Webby, R.; Gilchrist, M.J.R.; Gray, G.C. Avian influenza among waterfowl hunters and wildlife professionals. Emerg. Infect. Dis. 2006, 12, 1284–1286. [Google Scholar] [CrossRef] [PubMed]
- Adams, S.C.; Xing, Z.; Li, J.L.; Cardona, C.J. Immune-related gene expression in response to H11N9 low pathogenic avian influenza virus infection in chicken and Pekin duck peripheral blood mononuclear cells. Mol. Immunol. 2009, 46, 1744–1749. [Google Scholar] [CrossRef] [PubMed]
- Shi, J.; Deng, G.; Liu, P.; Zhou, J.; Guan, L.; Li, W.; Li, X.; Guo, J.; Wang, G.; Fan, J.; et al. Isolation and characterization of H7N9 viruses from live poultry markets—Implication of the source of current H7N9 infection in humans. Chin. Sci. Bull. 2013, 58, 1857–1863. [Google Scholar] [CrossRef]
- Lu, L.; Lycett, S.J.; Brown, A.J.L. Reassortment patterns of avian influenza virus internal segments among different subtypes. BMC Evol. Biol. 2014, 14, 16–31. [Google Scholar] [CrossRef]
- Koehler, A.V.; Pearce, J.M.; Flint, P.L.; Franson, J.C.; Ip, H.S. Genetic evidence of intercontinental movement of avian influenza in a migratory bird: The northern pintail (Anas acuta). Mol. Ecol. 2008, 17, 4754–4762. [Google Scholar] [CrossRef]
- Ramey, A.M.; Reeves, A.B.; Donnelly, T.; Poulson, R.L.; Stallknecht, D.E. Introduction of Eurasian-Origin influenza A(H8N4) virus into North America by migratory birds. Emerg. Infect. Dis. 2018, 24, 1950–1953. [Google Scholar] [CrossRef]
- Ramey, A.M.; Reeves, A.B.; Sonsthagen, S.A.; TeSlaa, J.L.; Nashold, S.; Donnelly, T.; Casler, B.; Hallb, J.S. Dispersal of H9N2 influenza A viruses between East Asia and North America by wild birds. Virology 2015, 482, 79–83. [Google Scholar] [CrossRef]
- Krauss, S.; Obert, C.A.; Franks, J.; Walker, D.; Jones, K.; Seiler, P.; Niles, L.; Pryor, S.P.; Obenauer, J.C.; Naeve, C.W.; et al. Influenza in migratory birds and evidence of limited intercontinental virus exchange. PLoS Pathog. 2007, 3, e167. [Google Scholar] [CrossRef]
- Winker, K.; McCracken, K.G.; Gibson, D.D.; Pruett, C.L.; Meier, R.; Huettmann, K.; Wege, M.; Kulikova, I.V.; Zhuravlev, Y.N.; Perdue, M.L.; et al. Movements of birds and avian influenza from Asia into Alaska. Emerg. Infect. Dis. 2007, 13, 547–552. [Google Scholar] [CrossRef]
- Lee, D.H.; Torchetti, M.K.; Winker, K.; Hon, S.I.; Song, C.S.; Swayne, D.E. Intercontinental spread of asian-origin H5N8 to North America through Beringia by migratory birds. J. Virol. 2015, 89, 6521–6524. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Isolates | Abbreviation | Subtype | Accession Numbers in GenBank |
|---|---|---|---|
| A/Common Teal/Shanghai/JDS120613/2018(H10N4) | JDS120613-H10N4 | H10N4 | MN049531–MN049537 |
| A/Mallard/Shanghai/JDS120662/2018(H10N4) | JDS120662-H10N4 | H10N4 | MN049523–MN049530 |
| A/Eurasian Coot/Shanghai/PD112440/2016(H11N9) | PD112440-H11N9 | H11N9 | MN049550–MN059557 |
| A/Common Teal/Shanghai/PD112452/2016(H11Nx) | PD112452-H11Nx | H11Nx | MN044998–MN045004 |
| A/Eurasian Wigeon/Shanghai/NH101834/2017(H11N2) | NH101834-H11N2 | H11N2 | MN044910–MN044917 |
| A/Common Teal/Shanghai/NH101807/2017(H12N2) | NH101807-H12N2 | H12N2 | MN049563–MN049569 |
| A/Mallard/Shanghai/JDS110851/2017(H12N5) | JDS110851-H12N5 | H12N5 | MN049575–MN049581 |
| A/Common Teal/Shanghai/NH102615/2018(H12N2) | NH102615-H12N2 | H12N2 | MN121558–MN121565 |
| A/Common Teal/Shanghai/NH110165/2018(H12N2) | NH110165-H12N2 | H12N2 | MN122300–MN122307 |
| A/Common Teal/Shanghai/NH112319/2018(H12N2) | NH112319-H12N2 | H12N2 | MN049593–MN049600 |
| A/Mallard/Shanghai/NH011204/2018(H12N5) | NH011204-H12N5 | H12N5 | MN049584–MN049591 |
| A/Common Teal/Shanghai/JDS110203/2019(H12N8) | JDS110203-H12N8 | H12N8 | MN795764–MN795771 |
| Virus | Amino Acid Sequence at HA Cleavage Site | HA Receptor-Binding Site | M2 Key Site | PB2 | ||
|---|---|---|---|---|---|---|
| 226 | 228 | 31 | 627 | 701 | ||
| JDS120613-H10N4 | ELTQGR↓GLF | Q | G | S | E | D |
| JDS120662-H10N4 | ELMQGR↓GLF | Q | G | S | E | D |
| PD112440-H11N9 | PAIASR↓GLF | Q | G | S | E | D |
| PD112452-H11Nx | PAIASR↓GLF | Q | G | S | E | D |
| NH101834-H11N2 | PAIASR↓GLF | Q | G | S | E | D |
| NH101807-H12N2 | PQAQDR↓GLF | Q | G | S | E | D |
| JDS110851-H12N5 | PQAQDR↓GLF | Q | G | S | E | D |
| NH102615-H12N2 | PQAQDR↓GLF | Q | G | S | E | D |
| NH110165-H12N2 | PQAQDR↓GLF | Q | G | S | E | D |
| NH112319-H12N2 | PQAQGR↓GLF | Q | G | S | E | D |
| NH011204-H12N5 | PQAQDR↓GLF | Q | G | S | E | D |
| JDS110203-H12N8 | PQVQNR↓GLF | Q | G | S | E | D |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tang, L.; Tang, W.; Ming, L.; Gu, J.; Qian, K.; Li, X.; Wang, T.; He, G. Characterization of Avian Influenza Virus H10–H12 Subtypes Isolated from Wild Birds in Shanghai, China from 2016 to 2019. Viruses 2020, 12, 1085. https://doi.org/10.3390/v12101085
Tang L, Tang W, Ming L, Gu J, Qian K, Li X, Wang T, He G. Characterization of Avian Influenza Virus H10–H12 Subtypes Isolated from Wild Birds in Shanghai, China from 2016 to 2019. Viruses. 2020; 12(10):1085. https://doi.org/10.3390/v12101085
Chicago/Turabian StyleTang, Ling, Wangjun Tang, Le Ming, Jianming Gu, Kai Qian, Xiaofang Li, Tianhou Wang, and Guimei He. 2020. "Characterization of Avian Influenza Virus H10–H12 Subtypes Isolated from Wild Birds in Shanghai, China from 2016 to 2019" Viruses 12, no. 10: 1085. https://doi.org/10.3390/v12101085
APA StyleTang, L., Tang, W., Ming, L., Gu, J., Qian, K., Li, X., Wang, T., & He, G. (2020). Characterization of Avian Influenza Virus H10–H12 Subtypes Isolated from Wild Birds in Shanghai, China from 2016 to 2019. Viruses, 12(10), 1085. https://doi.org/10.3390/v12101085