Massively Parallel Profiling of HIV-1 Resistance to the Fusion Inhibitor Enfuvirtide


{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Generation of Env Mutant Virus Libraries
2.2. Resistance Profiling
2.3. Analysis of Deep Sequencing Data
2.4. Data Availability and Source Code
2.5. TZM-BL Inhibition Assays
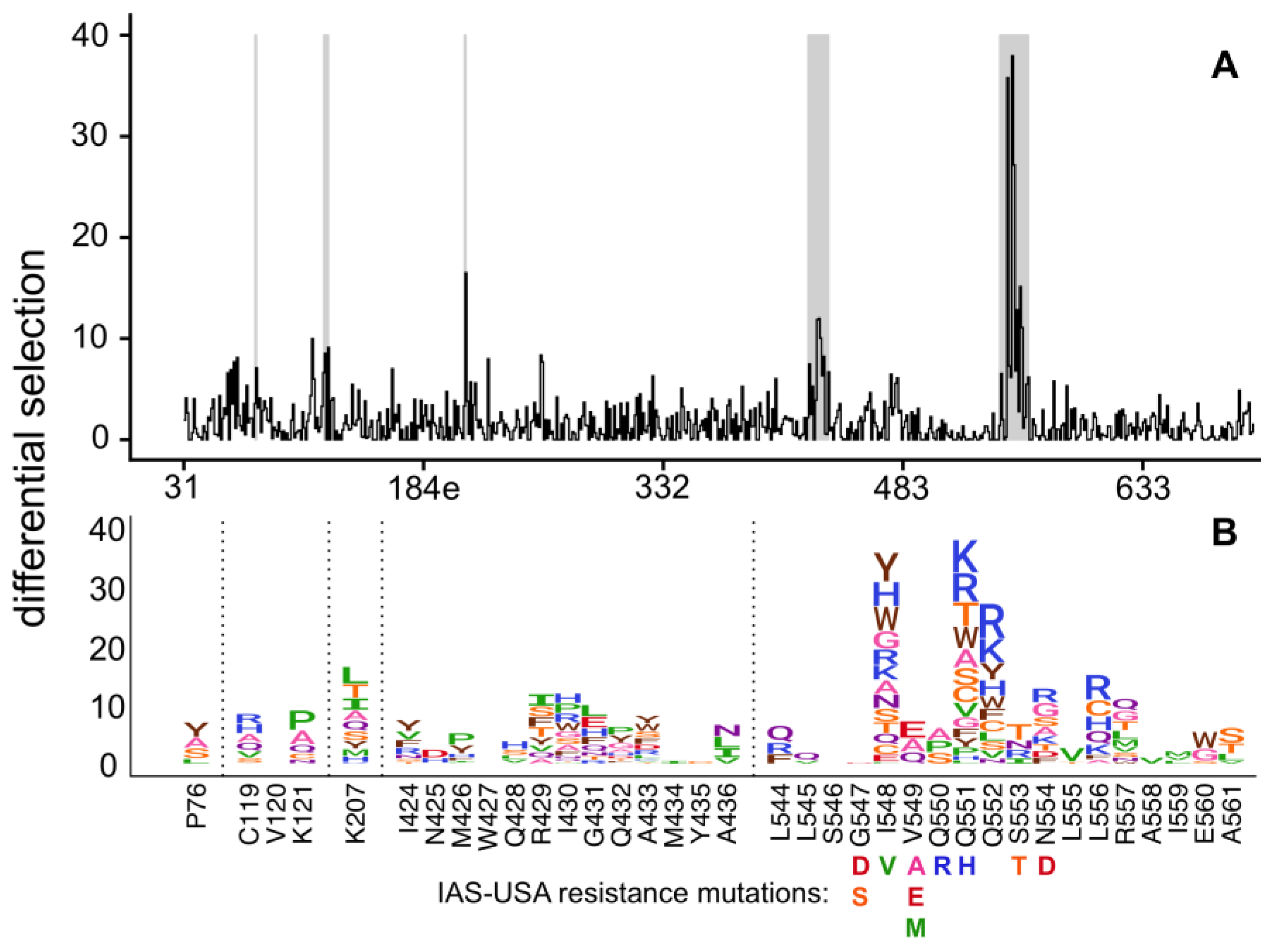
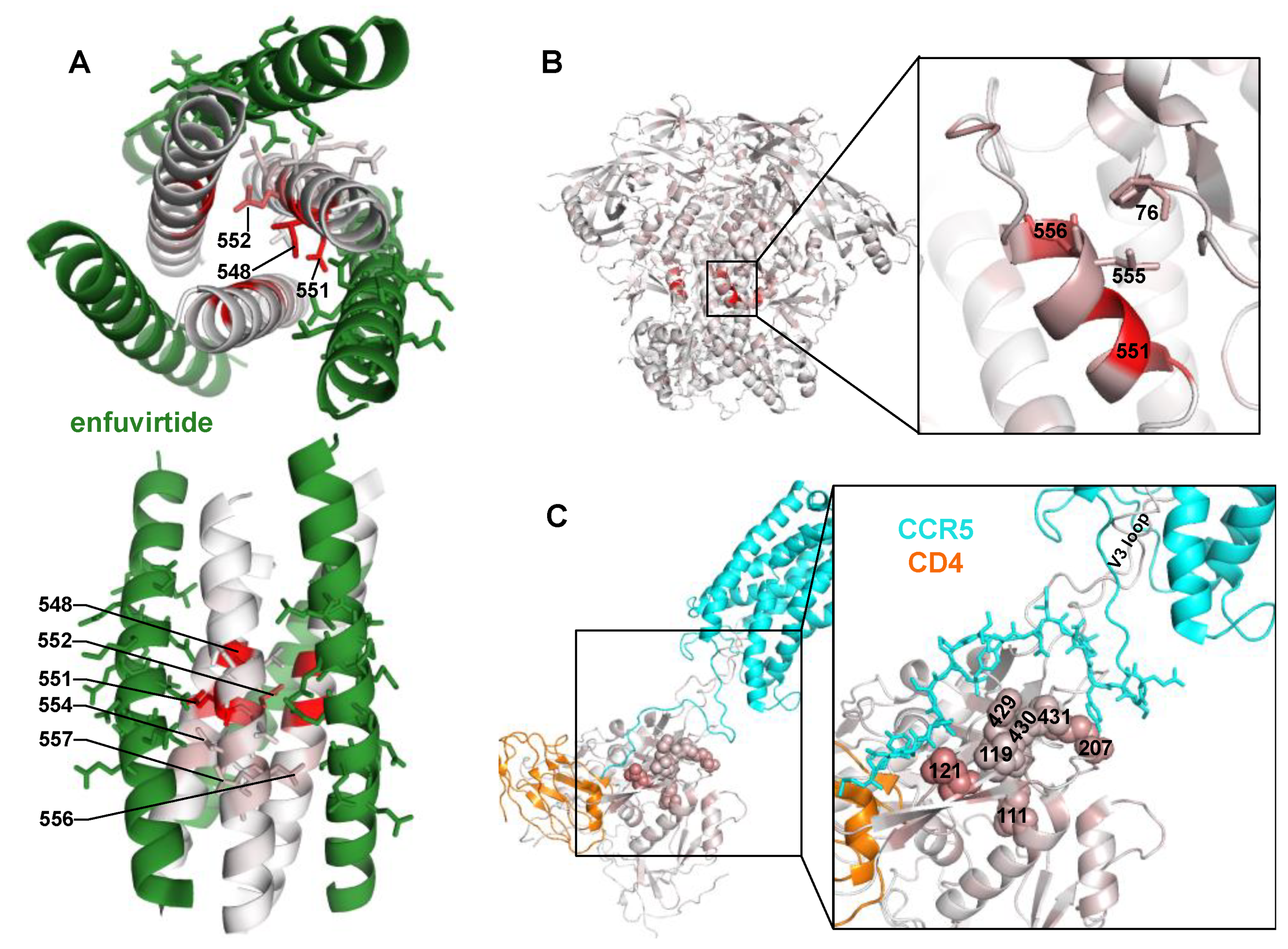
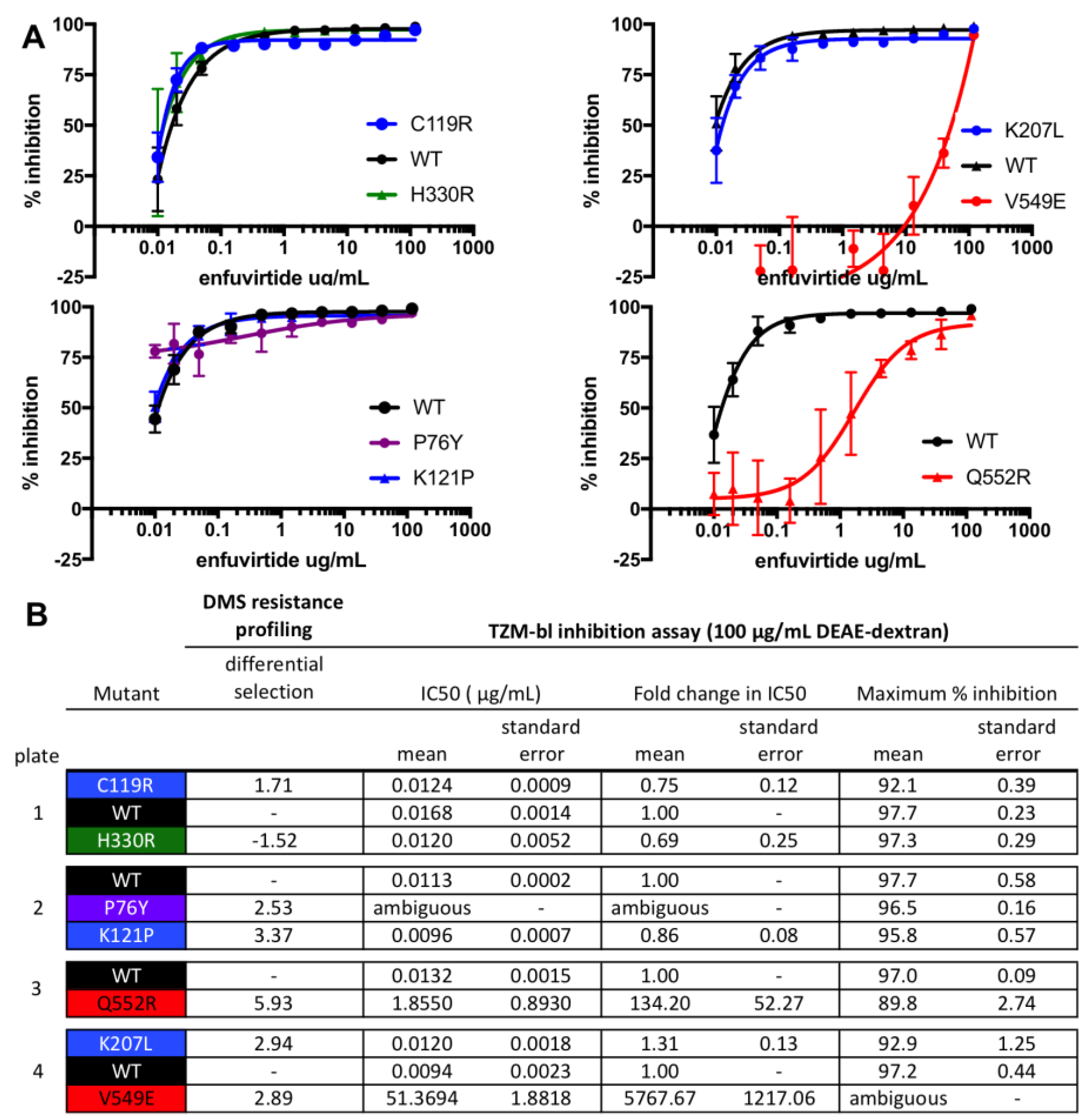
3. Results
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Wild, C.T.; Shugars, D.C.; Greenwell, T.K.; McDanal, C.B.; Matthews, T.J. Peptides corresponding to a predictive alpha-helical domain of human immunodeficiency virus type 1 gp41 are potent inhibitors of virus infection. Proc. Natl. Acad. Sci. USA 1994, 91, 9770–9774. [Google Scholar] [CrossRef]
- Jiang, S.; Lin, K.; Strick, N.; Neurath, A.R. HIV-1 inhibition by a peptide. Nature 1993, 365, 113. [Google Scholar] [CrossRef] [PubMed]
- Chan, D.C.; Kim, P.S. HIV entry and its inhibition. Cell 1998, 93, 681–684. [Google Scholar] [CrossRef]
- Zhang, X.; Ding, X.; Zhu, Y.; Chong, H.; Cui, S.; He, J.; Wang, X.; He, Y. Structural and functional characterization of HIV-1 cell fusion inhibitor T20. AIDS 2019, 33, 1–11. [Google Scholar] [CrossRef] [PubMed]
- De Feo, C.J.; Weiss, C.D. Escape from human immunodeficiency virus type 1 (HIV-1) entry inhibitors. Viruses 2012, 4, 3859–3911. [Google Scholar] [CrossRef]
- Keller, P.W.; Morrison, O.; Vassell, R.; Weiss, C.D. HIV-1 gp41 Residues Modulate CD4-Induced Conformational Changes in the Envelope Glycoprotein and Evolution of a Relaxed Conformation of gp120. J. Virol. 2018, 92, e00583-18. [Google Scholar] [CrossRef] [PubMed]
- Reeves, J.D.; Gallo, S.A.; Ahmad, N.; Miamidian, J.L.; Harvey, P.E.; Sharron, M.; Pohlmann, S.; Sfakianos, J.N.; Derdeyn, C.A.; Blumenthal, R.; et al. Sensitivity of HIV-1 to entry inhibitors correlates with envelope/coreceptor affinity, receptor density, and fusion kinetics. Proc. Natl. Acad. Sci. USA 2002, 99, 16249–16254. [Google Scholar] [CrossRef]
- Khasnis, M.D.; Halkidis, K.; Bhardwaj, A.; Root, M.J. Receptor Activation of HIV-1 Env Leads to Asymmetric Exposure of the gp41 Trimer. PLoS Pathog. 2016, 12, e1006098. [Google Scholar] [CrossRef]
- Zhu, X.; Zhu, Y.; Ye, S.; Wang, Q.; Xu, W.; Su, S.; Sun, Z.; Yu, F.; Liu, Q.; Wang, C.; et al. Improved Pharmacological and Structural Properties of HIV Fusion Inhibitor AP3 over Enfuvirtide: Highlighting Advantages of Artificial Peptide Strategy. Sci. Rep. 2015, 5, 13028. [Google Scholar] [CrossRef]
- He, Y.; Xiao, Y.; Song, H.; Liang, Q.; Ju, D.; Chen, X.; Lu, H.; Jing, W.; Jiang, S.; Zhang, L. Design and Evaluation of Sifuvirtide, a Novel HIV-1 Fusion Inhibitor. J. Biol. Chem. 2008, 283, 11126–11134. [Google Scholar] [CrossRef]
- He, Y.; Cheng, J.; Li, J.; Qi, Z.; Lu, H.; Dong, M.; Jiang, S.; Dai, Q. Identification of a Critical Motif for the Human Immunodeficiency Virus Type 1 (HIV-1) gp41 Core Structure: Implications for Designing Novel Anti-HIV Fusion Inhibitors. J. Virol. 2008, 82, 6349–6358. [Google Scholar] [CrossRef]
- Dwyer, J.J.; Wilson, K.L.; Davison, D.K.; Freel, S.A.; Seedorff, J.E.; Wring, S.A.; Tvermoes, N.A.; Matthews, T.J.; Greenberg, M.L.; Delmedico, M.K. Design of helical, oligomeric HIV-1 fusion inhibitor peptides with potent activity against enfuvirtide-resistant virus. Proc. Natl. Acad. Sci. USA 2007, 104, 12772–12777. [Google Scholar] [CrossRef]
- Xu, W.; Pu, J.; Su, S.; Hua, C.; Su, X.; Wang, Q.; Jiang, S.; Lu, L. Revisiting the mechanism of enfuvirtide and designing an analog with improved fusion inhibitory activity by targeting triple sites in gp41. AIDS 2019, 1. [Google Scholar] [CrossRef]
- Clotet, B.; Cooper, D. Clinical management of treatment-experienced, HIV- infected patients with the fusion inhibitor enfuvirtide: Consensus recommendations. AIDS 2004. [Google Scholar] [CrossRef]
- Miller, M.D.; Hazuda, D.J. HIV resistance to the fusion inhibitor enfuvirtide: Mechanisms and clinical implications. Drug Resist. Updat. 2004, 7, 89–95. [Google Scholar] [CrossRef]
- Wei, X.; Decker, J.M.; Liu, H.; Zhang, Z.; Arani, R.B.; Kilby, J.M.; Saag, M.S.; Wu, X.; Shaw, G.M.; Kappes, J.C. Emergence of resistant human immunodeficiency virus type 1 in patients receiving fusion inhibitor (T-20) monotherapy. Antimicrob. Agents Chemother. 2002, 46, 1896–1905. [Google Scholar] [CrossRef]
- Melby, T.; Sista, P.; DeMasi, R.; Kirkland, T.; Roberts, N.; Salgo, M.; Heilek-Snyder, G.; Cammack, N.; Matthews, T.J.; Greenberg, M.L. Characterization of envelope glycoprotein gp41 genotype and phenotypic susceptibility to enfuvirtide at baseline and on treatment in the phase III clinical trials TORO-1 and TORO-2. AIDS Res. Hum. Retrovir. 2006, 22, 375–385. [Google Scholar] [CrossRef]
- Su, C.; Melby, T.; DeMasi, R.; Ravindran, P.; Heilek-Snyder, G. Genotypic changes in human immunodeficiency virus type 1 envelope glycoproteins on treatment with the fusion inhibitor enfuvirtide and their influence on changes in drug susceptibility in vitro. J. Clin. Virol. 2006, 36, 249–257. [Google Scholar] [CrossRef]
- Poveda, E.; Rodés, B.; Lebel-Binay, S.; Faudon, J.L.; Jimenez, V.; Soriano, V. Dynamics of enfuvirtide resistance in HIV-infected patients during and after long-term enfuvirtide salvage therapy. J. Clin. Virol. 2005, 34, 295–301. [Google Scholar] [CrossRef]
- Greenberg, M.L.; Cammack, N. Resistance to enfuvirtide, the first HIV fusion inhibitor. J. Antimicrob. Chemother. 2004, 54, 333–340. [Google Scholar] [CrossRef]
- Marcelin, A.G.; Reynes, J.; Yerly, S.; Ktorza, N.; Segondy, M.; Piot, J.C.; Delfraissy, J.F.; Kaiser, L.; Perrin, L.; Katlama, C.; et al. Characterization of genotypic determinants in HR-1 and HR-2 gp41 domains in individuals with persistent HIV viraemia under T-20. AIDS 2004. [Google Scholar] [CrossRef]
- Sista, P.R.; Melby, T.; Davison, D.; Jin, L.; Mosier, S.; Mink, M.; Nelson, E.L.; DeMasi, R.; Cammack, N.; Salgo, M.P.; et al. Characterization of determinants of genotypic and phenotypic resistance to enfuvirtide in baseline and on-treatment HIV-1 isolates. AIDS 2004, 18, 1787–1794. [Google Scholar] [CrossRef] [PubMed]
- Mink, M.; Mosier, S.M.; Janumpalli, S.; Davison, D.; Jin, L.; Melby, T.; Sista, P.; Erickson, J.; Lambert, D.; Stanfield-Oakley, S.A.; et al. Impact of Human Immunodeficiency Virus Type 1 gp41 Amino Acid Substitutions Selected during Enfuvirtide Treatment on gp41 Binding and Antiviral Potency of Enfuvirtide In Vitro. J. Virol. 2005, 79, 12447–12454. [Google Scholar] [CrossRef] [PubMed]
- Rimsky, L.T.; Shugars, D.C.; Matthews, T.J. Determinants of Human Immunodeficiency Virus Type 1 Resistance to gp41-Derived Inhibitory Peptides. J. Virol. 1998, 72, 986–993. [Google Scholar]
- Xu, L.; Ratcliffe, D.; Workman, J.; Joall, A.; Myers, R.; Smit, E.; Cane, P.A.; Greenberg, M.L.; Pillay, D.; Pozniak, A.; et al. Emergence and Evolution of Enfuvirtide Resistance following Long-Term Therapy Involves Heptad Repeat 2 Mutations within Emergence and Evolution of Enfuvirtide Resistance following Long-Term Therapy Involves Heptad Repeat 2 Mutations within gp41. Antimicrob. Agents Chemother. 2005, 49, 1113–1119. [Google Scholar] [CrossRef] [PubMed]
- Derdeyn, C.A.; Decker, J.M.; Sfakianos, J.N.; Wu, X.; O’Brien, W.A.; Ratner, L.; Kappes, J.C.; Shaw, G.M.; Hunter, E. Sensitivity of Human Immunodeficiency Virus Type 1 to the Fusion Inhibitor T-20 Is Modulated by Coreceptor Specificity Defined by the V3 Loop of gp120. J. Virol. 2000, 74, 8358–8367. [Google Scholar] [CrossRef]
- Derdeyn, C.A.; Decker, J.M.; Sfakianos, J.N.; Zhang, Z.; O’Brien, W.A.; Ratner, L.; Shaw, G.M.; Hunter, E. Sensitivity of human immunodeficiency virus type 1 to fusion inhibitors targeted to the gp41 first heptad repeat involves distinct regions of gp41 and is consistently modulated by gp120 interactions with the coreceptor. J. Virol. 2001, 75, 8605–8614. [Google Scholar] [CrossRef]
- Reeves, J.D.; Miamidian, J.L.; Biscone, M.J.; Lee, F.; Ahmad, N.; Pierson, T.C.; Doms, R.W. Impact of Mutations in the Coreceptor Binding Site on Human Immunodeficiency Virus Type 1 Fusion, Infection, and Entry Inhibitor Sensitivity. J. Virol. 2004, 78, 5476–5485. [Google Scholar] [CrossRef] [PubMed]
- Roman, F.; Gonzalez, D.; Lambert, C.; Deroo, S.; Fischer, A.; Baurith, T.; Staub, T.; Boulmé, R.; Arendt, V.; Schneider, F.; et al. Uncommon mutations at residue positions critical for enfuvirtide (T-20) resistance in enfuvirtide-naive patients infected with subtype B and non-B HIV-1 strains. J. Acquir. Immune Defic. Syndr. 2003, 33, 134–139. [Google Scholar] [CrossRef]
- Yu, X.; Yuan, L.; Huang, Y.; Xu, W.; Fang, Z.; Liu, S.; Shao, Y.; Jiang, S.; Ma, L. Susceptibility of HIV-1 subtypes B′, CRF07_BC and CRF01_AE that are predominantly circulating in China to HIV-1 entry inhibitors. PLoS ONE 2011, 6, 1–8. [Google Scholar] [CrossRef]
- Cilliers, T.; Patience, T.; Pillay, C.; Papathanasopoulos, M.; Morris, L. Sensitivity of HIV Type 1 Subtype C Isolates to the Entry Inhibitor T-20. AIDS Res. Hum. Retrovir. 2004. [Google Scholar] [CrossRef]
- Taylor, B.S.; Sobieszczyk, M.E.; McCutchan, F.E.; Hammer, S.M. The Challenge of HIV-1 Subtype Diversity. N. Engl. J. Med. 2008, 358, 1590–1602. [Google Scholar] [CrossRef]
- D’Arrigo, R.; Ciccozzi, M.; Gori, C.; Montieri, S.; Aquaro, S.; Bellagamba, R.; Boumis, E.; Di Perri, G.; Pizzi, D.; Antinori, A.; et al. gp41 Sequence Variability in HIV Type 1 Non-B Subtypes Infected Patients Undergoing Enfuvirtide Pressure. AIDS Res. Hum. Retrovir. 2007, 23, 1296–1302. [Google Scholar] [CrossRef]
- Haddox, H.K.; Dingens, A.S.; Hilton, S.K.; Overbaugh, J.; Bloom, J.D. Mapping mutational effects along the evolutionary landscape of HIV envelope. Elife 2018, 7, e34420. [Google Scholar] [CrossRef]
- Haddox, H.K.; Dingens, A.S.; Bloom, J.D. Experimental Estimation of the Effects of All Amino-Acid Mutations to HIV’s Envelope Protein on Viral Replication in Cell Culture. PLoS Pathog. 2016, 12, e1006114. [Google Scholar] [CrossRef]
- Dingens, A.S.; Haddox, H.K.; Overbaugh, J.; Bloom, J.D. Comprehensive Mapping of HIV-1 Escape from a Broadly Neutralizing Antibody. Cell Host Microbe 2017, 21, 777–787.e4. [Google Scholar] [CrossRef]
- Dingens, A.S.; Arenz, D.; Weight, H.; Overbaugh, J.; Bloom, J.D. An Antigenic Atlas of HIV-1 Escape from Broadly Neutralizing Antibodies Distinguishes Functional and Structural Epitopes. Immunity 2019, 50, 520–532.e3. [Google Scholar] [CrossRef]
- Dingens, A.S.; Acharya, P.; Haddox, H.K.; Rawi, R.; Xu, K.; Chuang, G.-Y.; Wei, H.; Zhang, B.; Mascola, J.R.; Carragher, B.; et al. Complete functional mapping of infection- and vaccine-elicited antibodies against the fusion peptide of HIV. PLoS Pathog. 2018, 14, e1007159. [Google Scholar] [CrossRef]
- Bloom, J.D. Software for the analysis and visualization of deep mutational scanning data. BMC Bioinform. 2015, 16, 1–13. [Google Scholar] [CrossRef]
- Doud, M.B.; Hensley, S.E.; Bloom, J.D. Complete mapping of viral escape from neutralizing antibodies. PLoS Pathog. 2017, 13, e1006271. [Google Scholar] [CrossRef]
- Wensing, A.M.; Calvez, V.; Günthard, H.F.; Johnson, V.A.; Paredes, R.; Pillay, D.; Shafer, R.W.; Richman, D.D. 2017 Update of the Drug Resistance Mutations in HIV-1. Top. Antivir. Med. 2016, 24, 132–133. [Google Scholar]
- Shaik, M.; Peng, H.; Lu, J.; Rits-volloch, S.; Xu, C.; Liao, M.; Chen, B. Structural basis of coreceptor recognition by HIV-1 envelope spike. Nature 2018, 4. [Google Scholar] [CrossRef] [PubMed]
- Sampah, M.E.S.; Shen, L.; Jilek, B.L.; Siliciano, R.F. Dose-response curve slope is a missing dimension in the analysis of HIV-1 drug resistance. Proc. Natl. Acad. Sci. USA 2011, 108, 7613–7618. [Google Scholar] [CrossRef]
- Herschhorn, A.; Gu, C.; Moraca, F.; Ma, X.; Farrell, M.; Smith, A.B.; Pancera, M.; Kwong, P.D.; Schön, A.; Freire, E.; et al. The β20–β21 of gp120 is a regulatory switch for HIV-1 Env conformational transitions. Nat. Commun. 2017, 8, 1049. [Google Scholar] [CrossRef]
- Alam, S.M.; Paleos, C.A.; Liao, H.-X.; Scearce, R.; Robinson, J.; Haynes, B.F.; Alam, S.M.; Paleos, C.A.; Liao, H.X.; Scearce, R.; et al. An inducible HIV type 1 gp41 HR-2 peptide-binding site on HIV type 1 envelope gp120. AIDS Res. Hum. Retrovir. 2004, 20, 836–845. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Lu, H.; Niu, J.; Xu, Y.; Wu, S.; Jiang, S. Different from the HIV fusion inhibitor C34, the anti-HIV drug fuzeon (T-20) inhibits HIV-1 entry by targeting multiple sites in gp41 and gp120. J. Biol. Chem. 2005, 280, 11259–11273. [Google Scholar] [CrossRef]
- McCoy, L.E.; Falkowska, E.; Doores, K.J.; Le, K.; Sok, D.; van Gils, M.J.; Euler, Z.; Burger, J.A.; Seaman, M.S.; Sanders, R.W.; et al. Incomplete Neutralization and Deviation from Sigmoidal Neutralization Curves for HIV Broadly Neutralizing Monoclonal Antibodies. PLoS Pathog. 2015, 11, 1–19. [Google Scholar] [CrossRef]
- Melby, T.; DeSpirito, M.; DeMasi, R.; Heilek-Snyder, G.; Greenberg, M.L.; Graham, N. HIV-1 Coreceptor Use in Triple-Class Treatment–Experienced Patients: Baseline Prevalence, Correlates, and Relationship to Enfuvirtide Response. J. Infect. Dis. 2006, 194, 238–246. [Google Scholar] [CrossRef]



© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dingens, A.S.; Arenz, D.; Overbaugh, J.; Bloom, J.D. Massively Parallel Profiling of HIV-1 Resistance to the Fusion Inhibitor Enfuvirtide. Viruses 2019, 11, 439. https://doi.org/10.3390/v11050439
Dingens AS, Arenz D, Overbaugh J, Bloom JD. Massively Parallel Profiling of HIV-1 Resistance to the Fusion Inhibitor Enfuvirtide. Viruses. 2019; 11(5):439. https://doi.org/10.3390/v11050439
Chicago/Turabian StyleDingens, Adam S., Dana Arenz, Julie Overbaugh, and Jesse D. Bloom. 2019. "Massively Parallel Profiling of HIV-1 Resistance to the Fusion Inhibitor Enfuvirtide" Viruses 11, no. 5: 439. https://doi.org/10.3390/v11050439
APA StyleDingens, A. S., Arenz, D., Overbaugh, J., & Bloom, J. D. (2019). Massively Parallel Profiling of HIV-1 Resistance to the Fusion Inhibitor Enfuvirtide. Viruses, 11(5), 439. https://doi.org/10.3390/v11050439