Heterogeneous Coordination Environments in Lithium-Neutralized Ionomers Identified Using 1H and 7Li MAS NMR


Abstract
:
1. Introduction
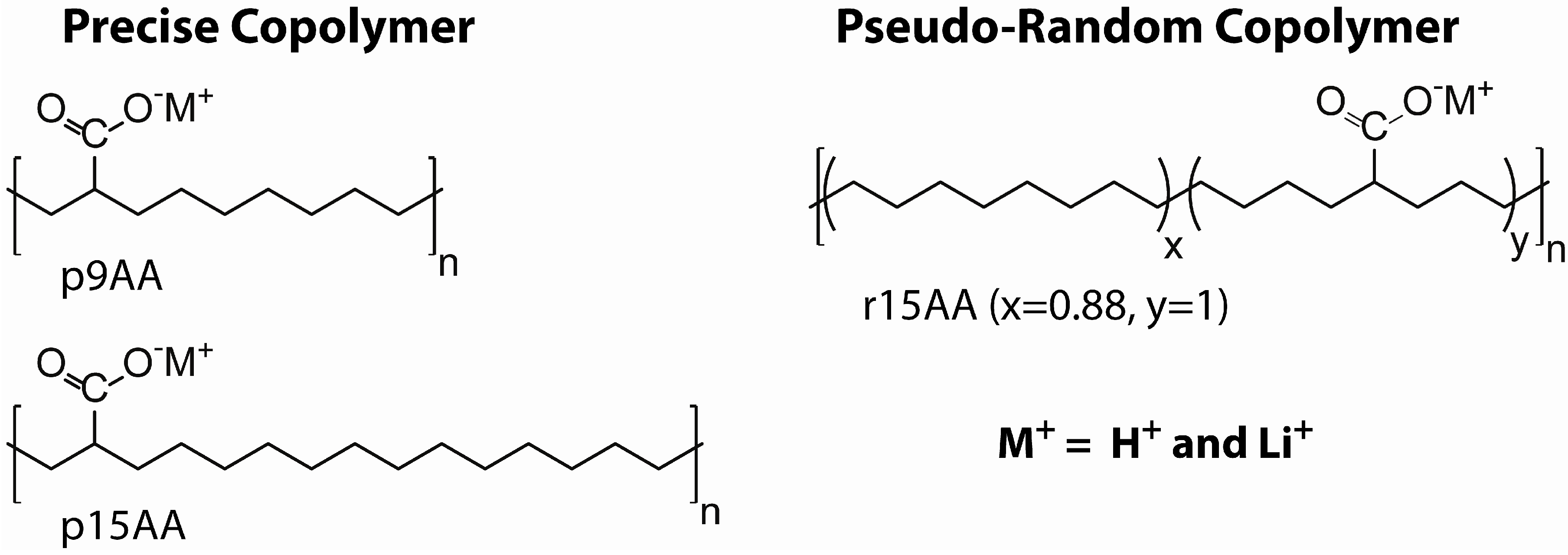
2. Results and Discussion
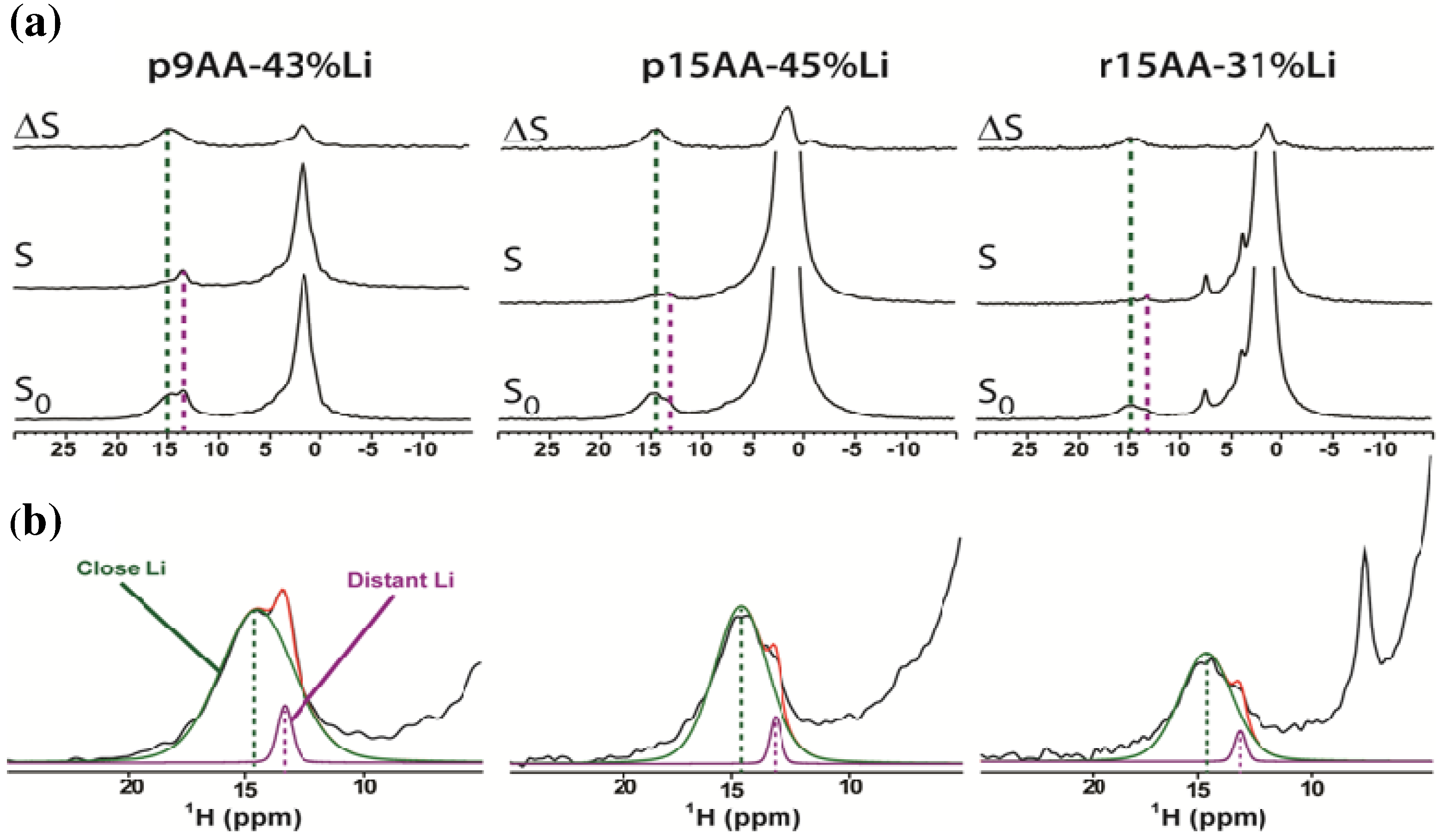
2.1. Solid-State 1H MAS NMR

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Tg (K) | δ(1H) (ppm) a | FWHM (1H) (Hz) a | Fraction (%) a | δ(7Li) (ppm) b | FWHM (7Li) (Hz) b |
|---|---|---|---|---|---|---|
| p9AA-43%Li | 346 | +14.7
+13.4 | 2050
520 | 89%
11% | 0.20 | 280 |
| p15AA-45%Li | 347 | +14.8
+13.3 | 1680
360 | 94
6 | 0.21 | 290 |
| r15AA-31%Li | 313 (amorphous)
344 (crystalline) | +14.9
+13.3 | 1790
360 | 94
6 | 0.35 | 265 |
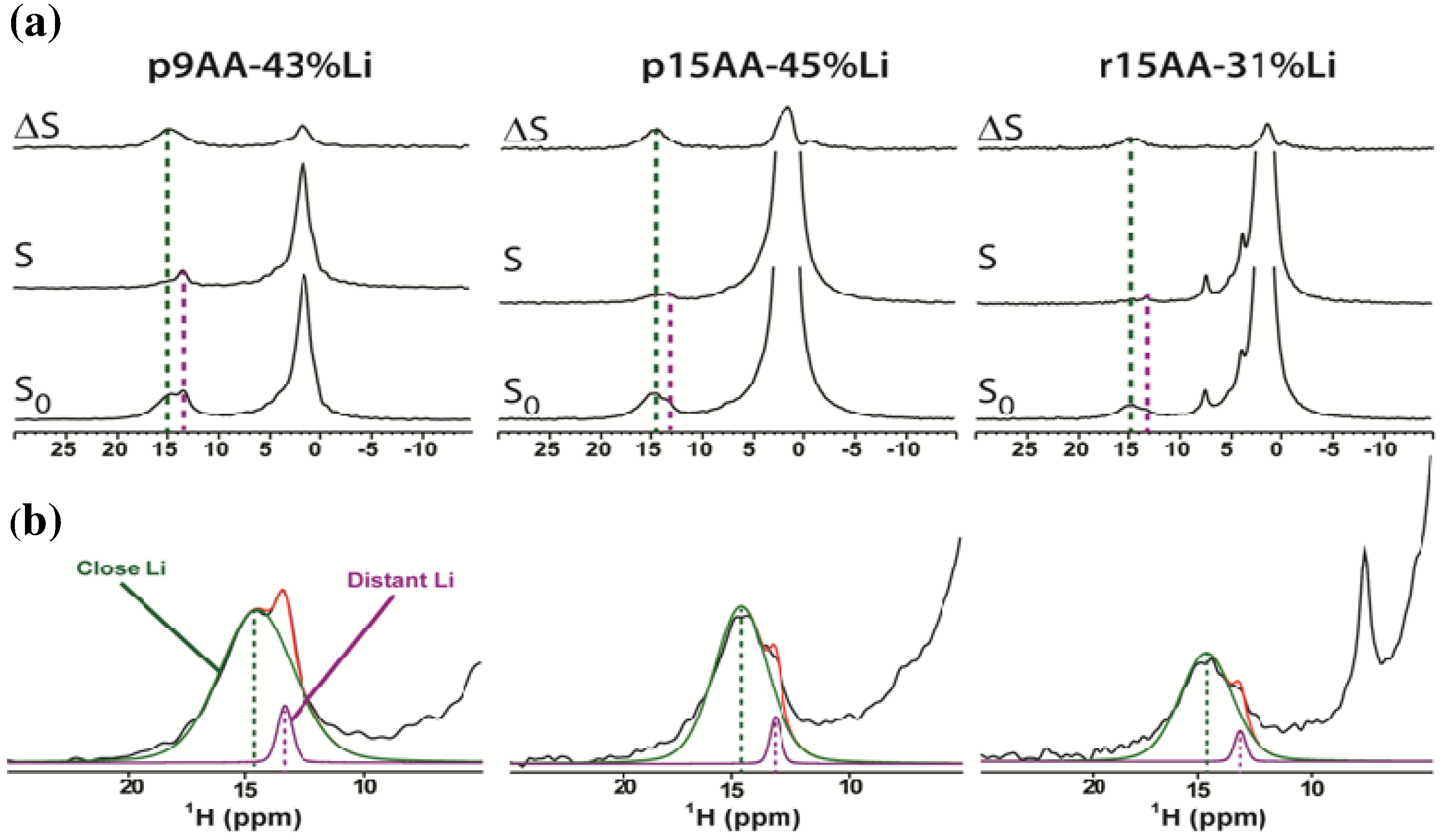
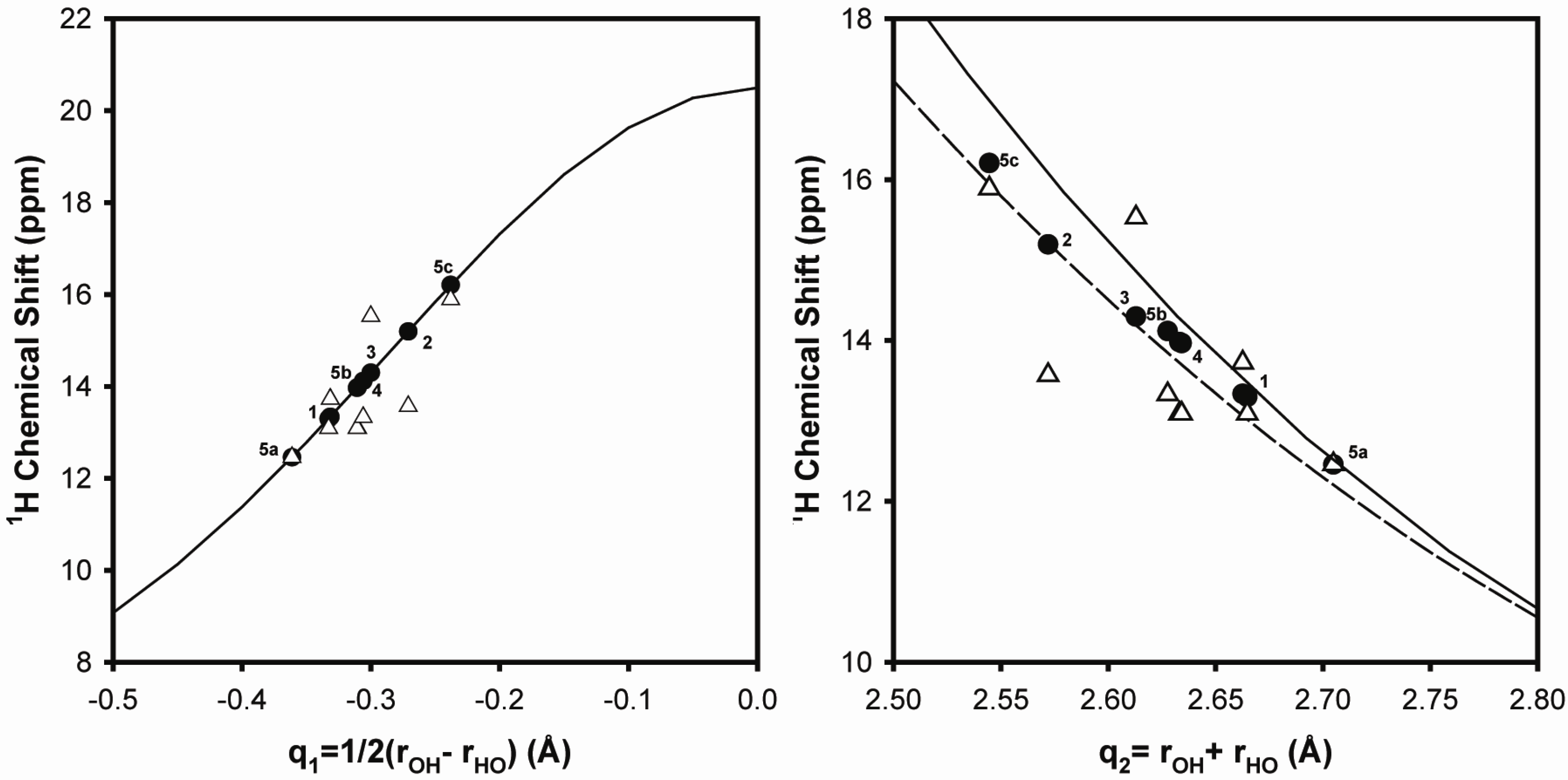
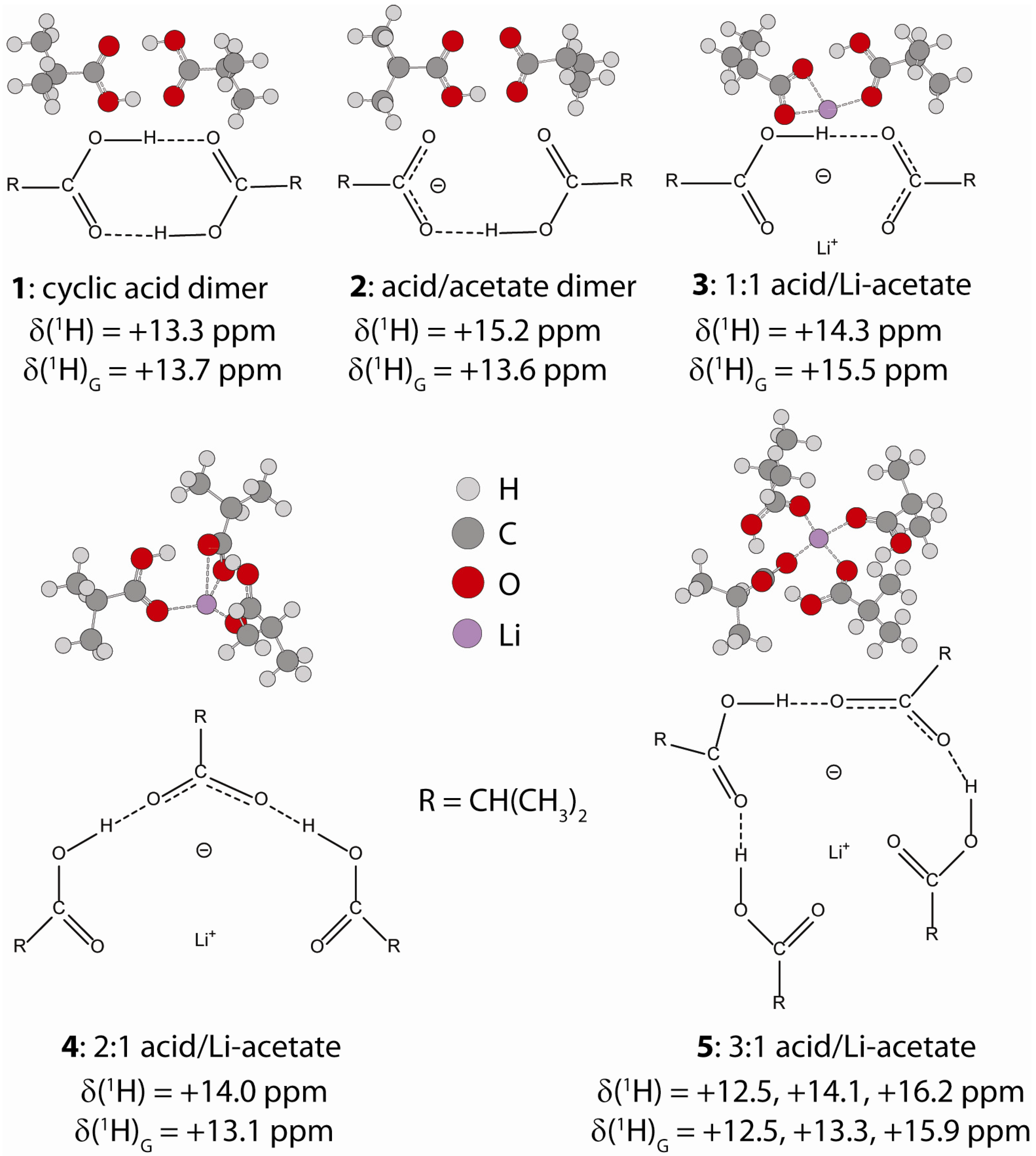
2.1.1. Hydrogen Bond Correlations

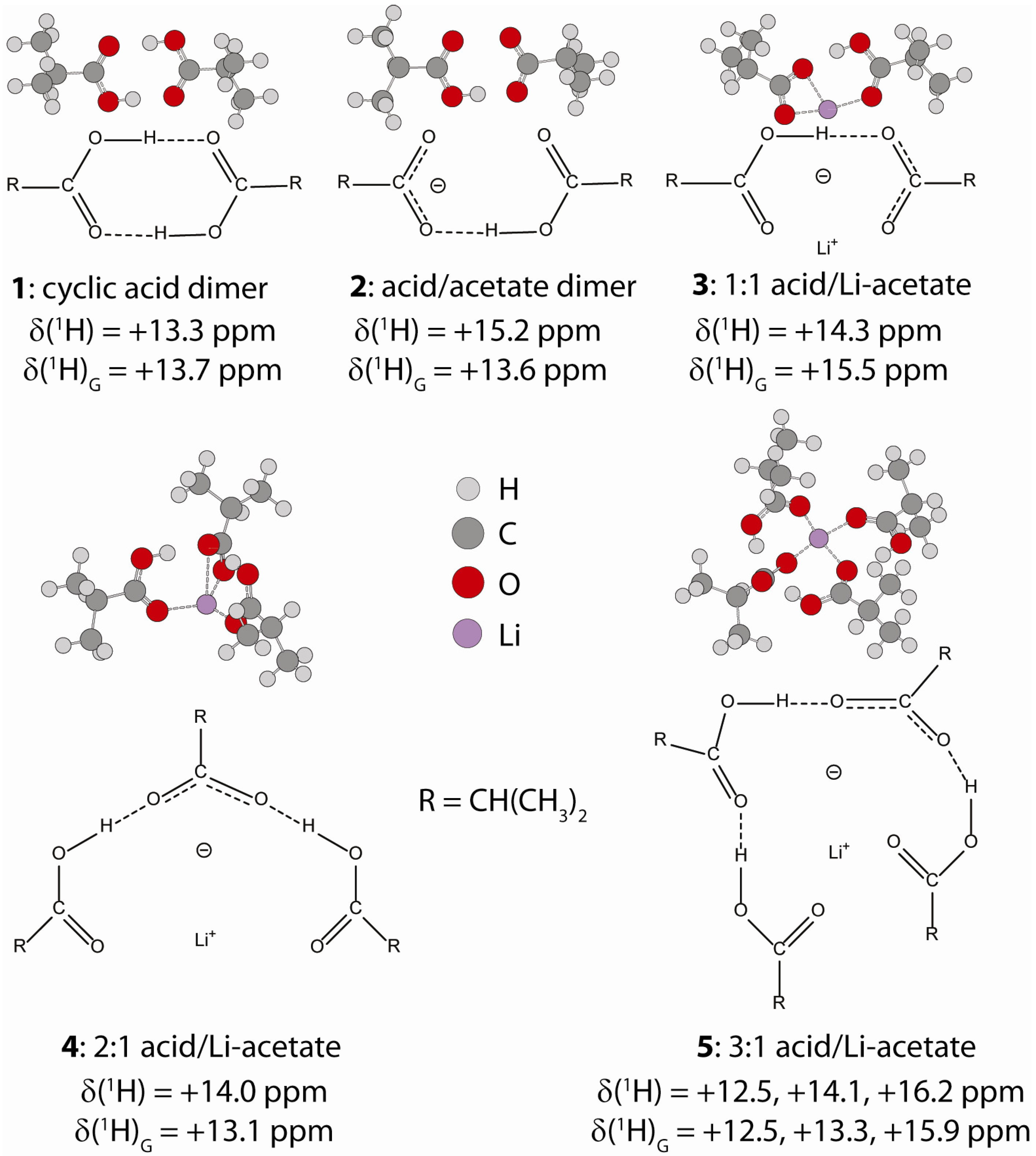
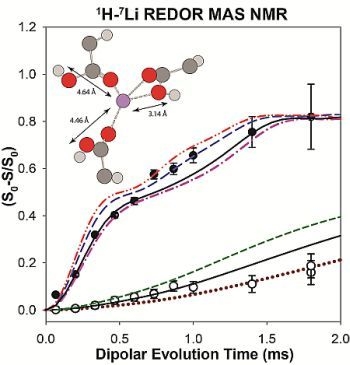
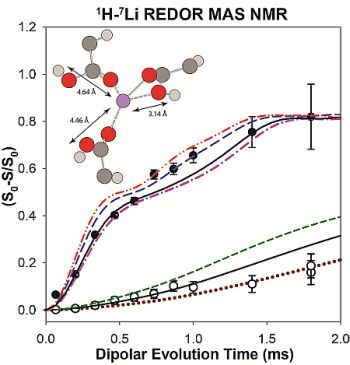
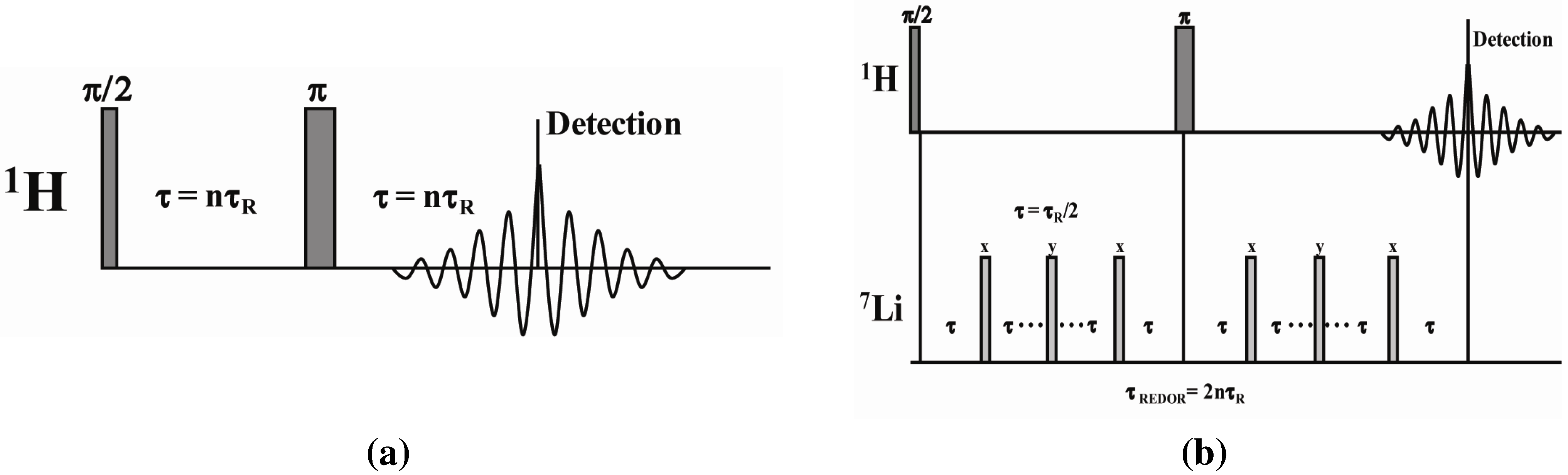
2.2. 1H-7Li REDOR MAS NMR
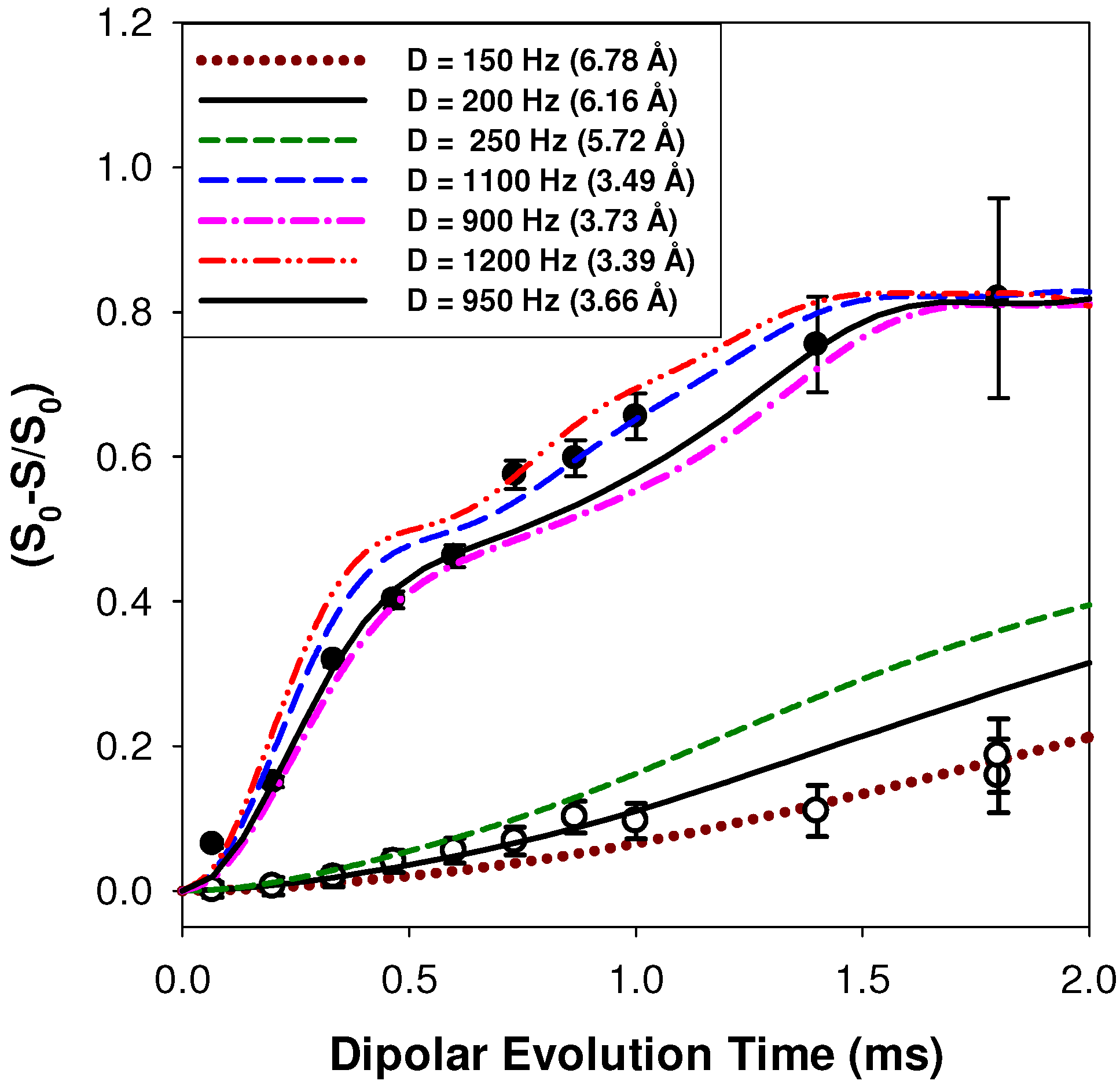
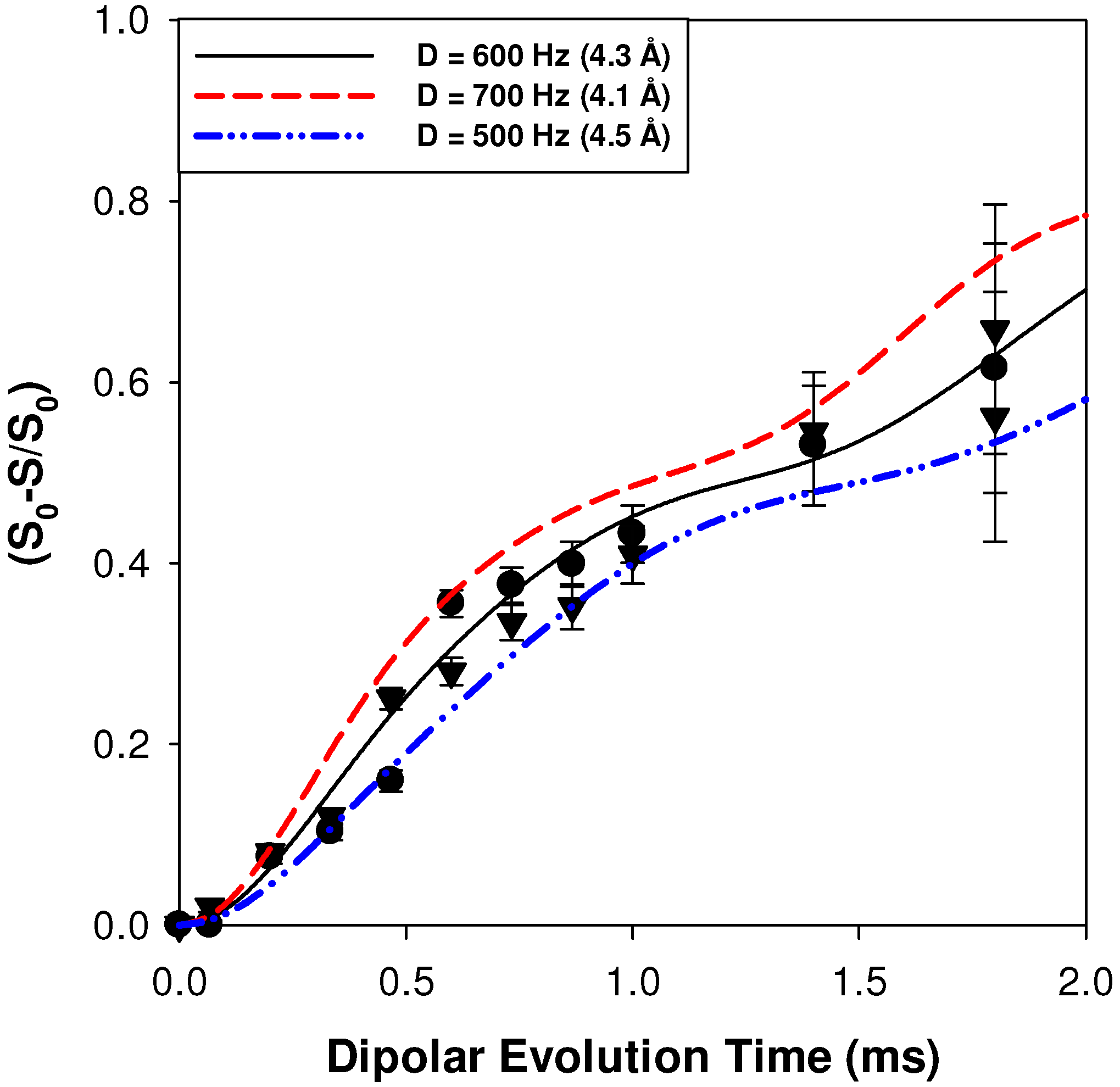
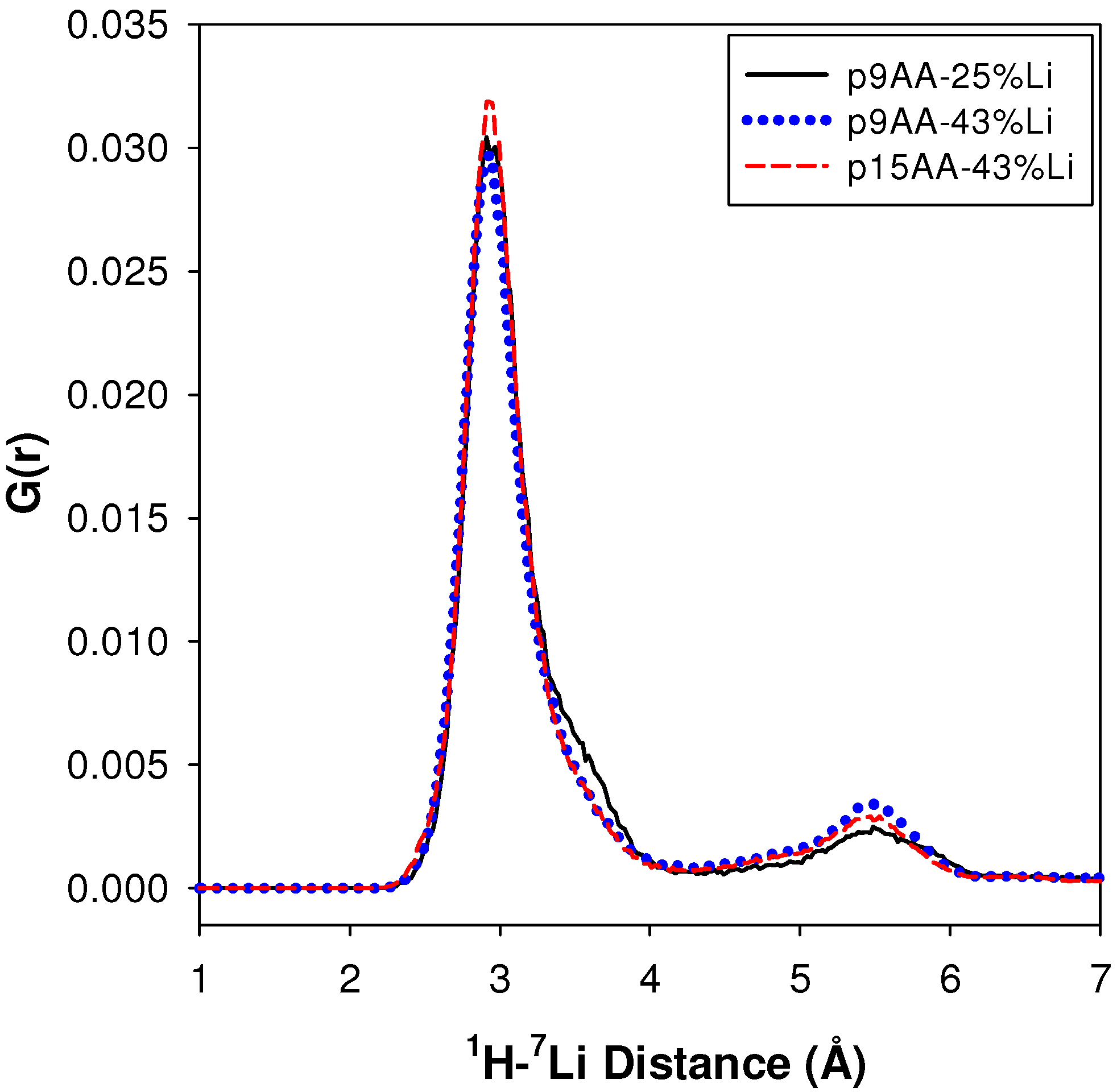
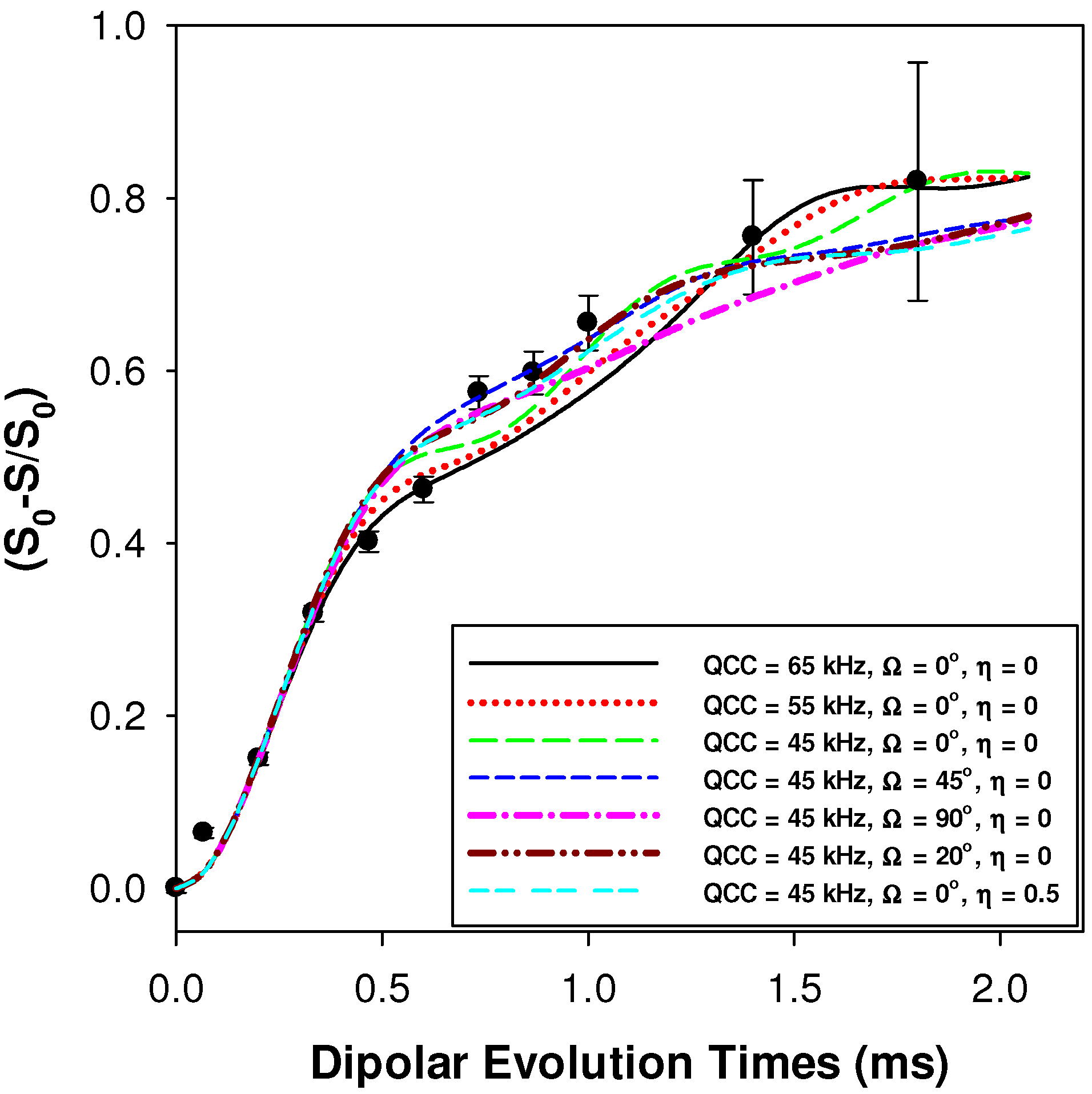
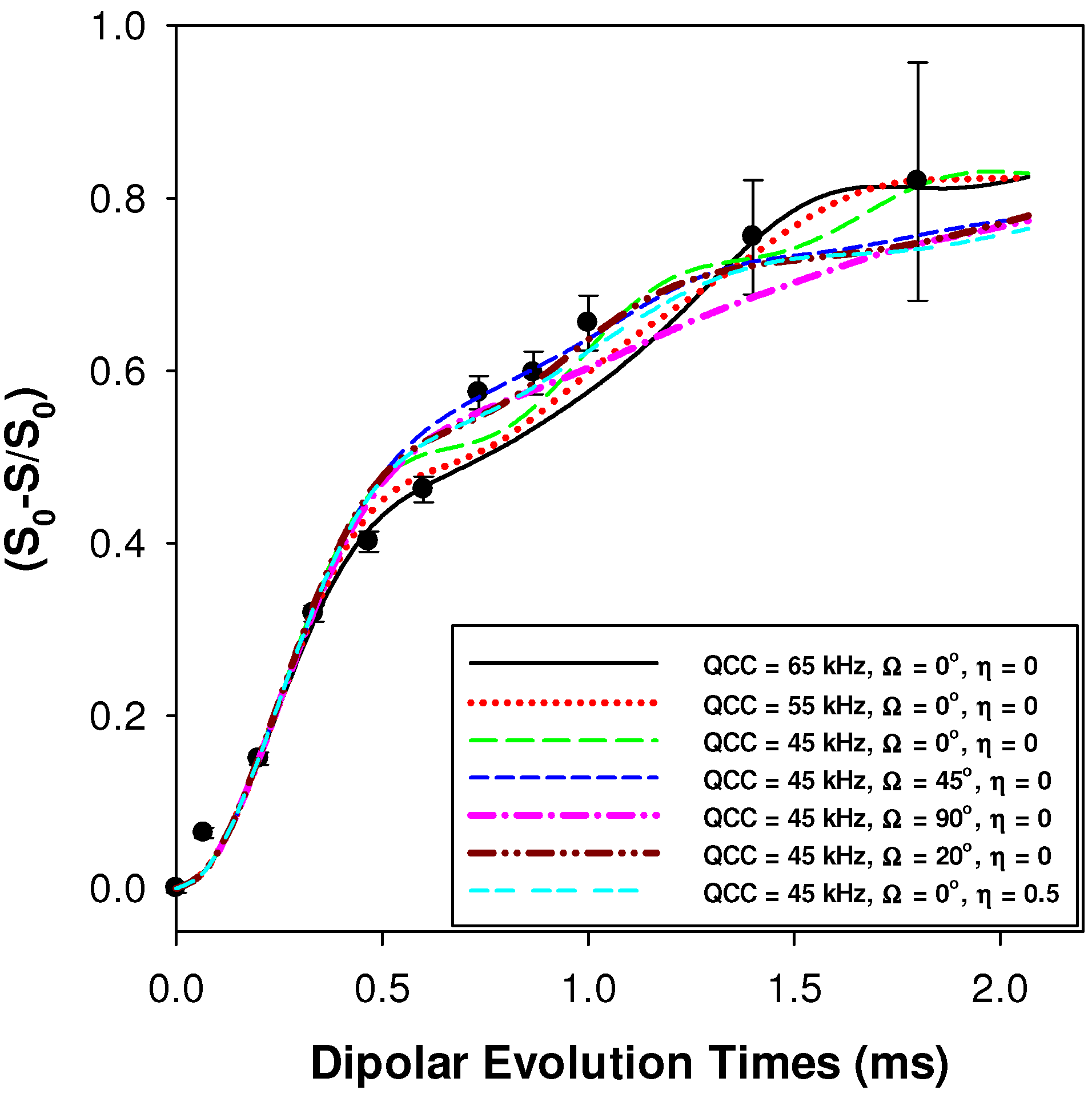
2.2.1. Measuring the Average 1H-7Li Distance
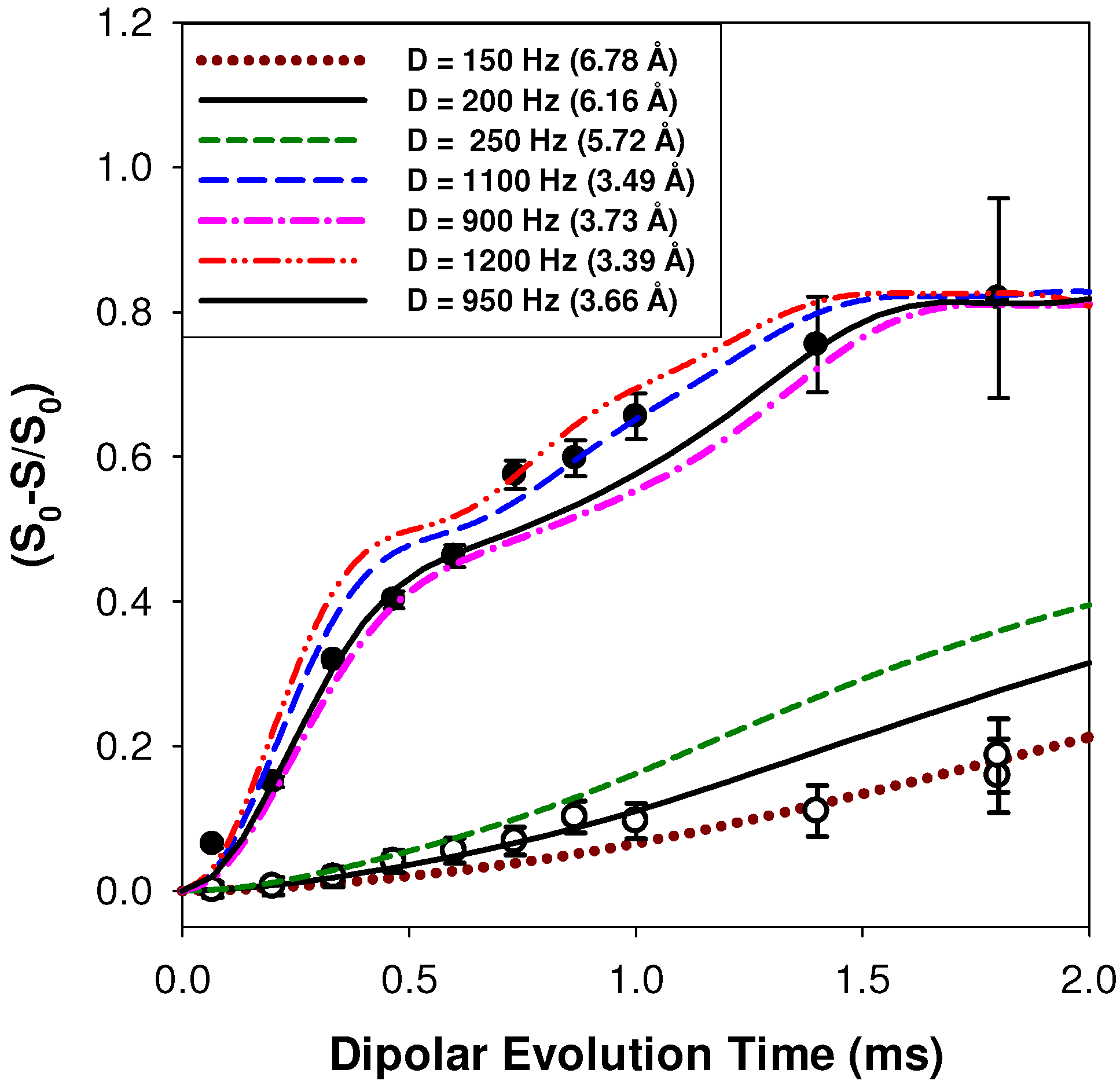

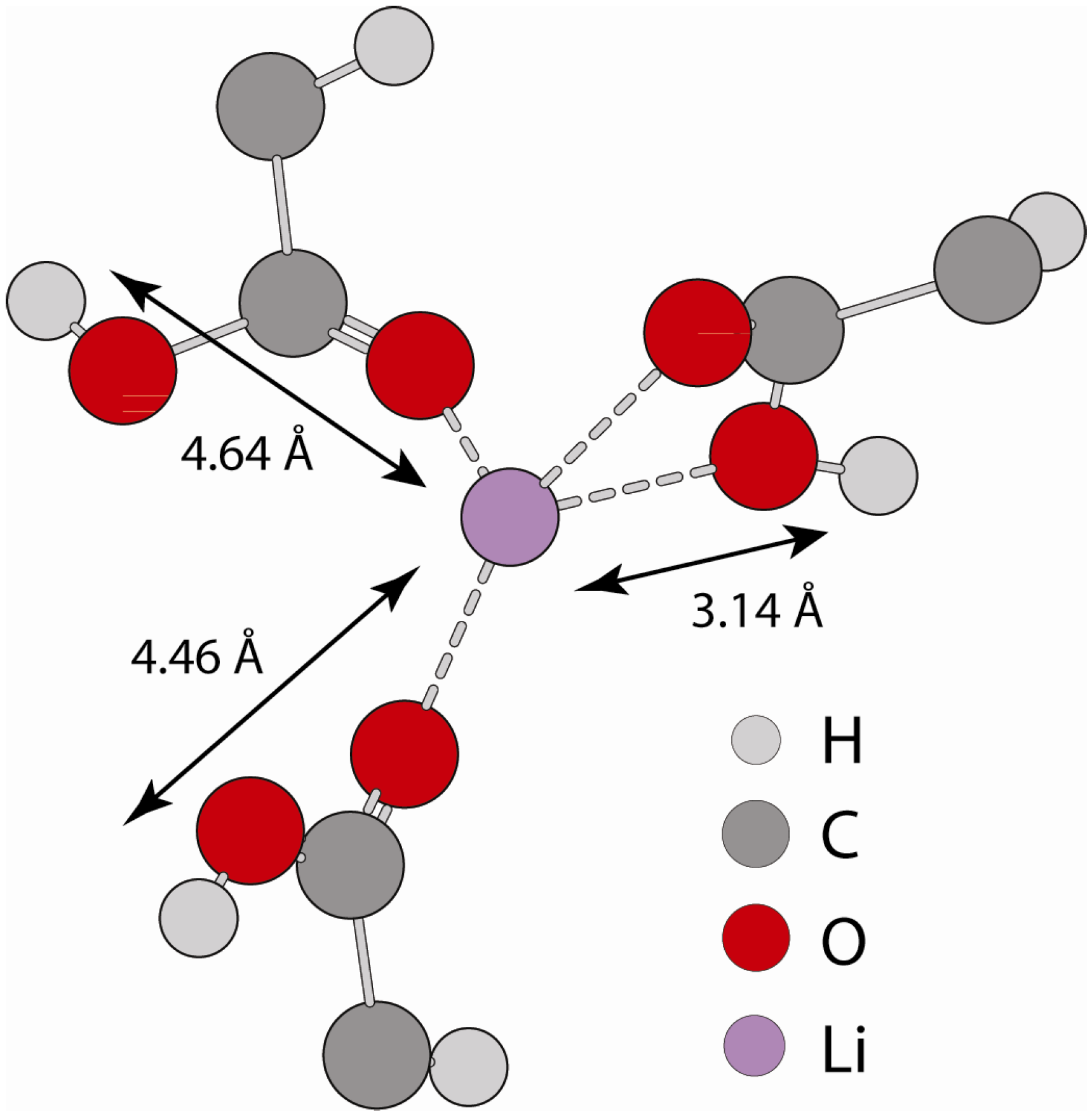
2.2.2. MD Structural Motifs
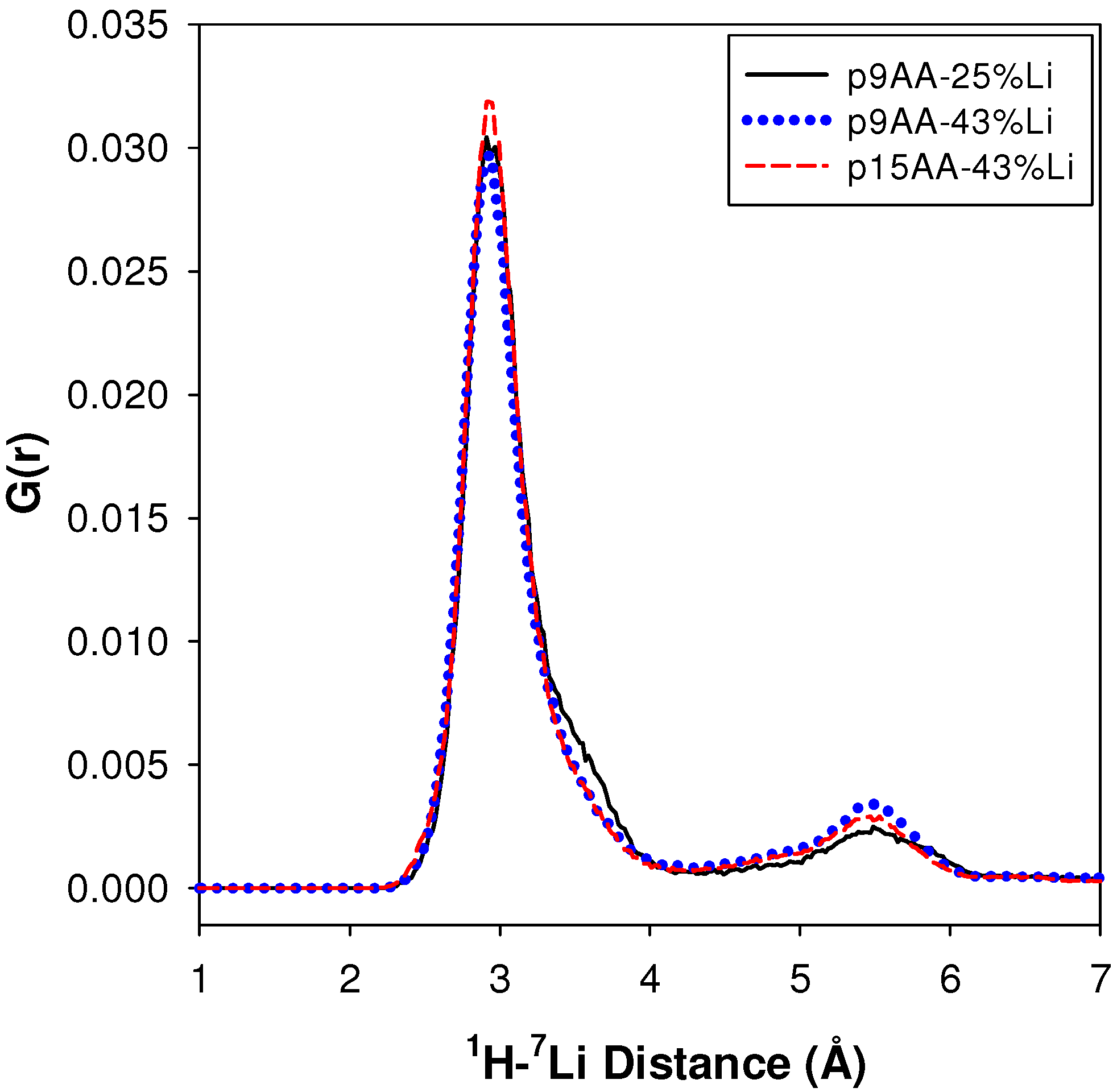
| Sample | 1H-7Li (Å) | Major Fraction(%) a | 1H-7Li (Å) | Minor Fraction (%) a | <1H-7Li> (Å) |
|---|---|---|---|---|---|
| p9AA-25% Li | 2.91 | 84 | 5.49 | 16 | 3.43 |
| P9AA-43% Li | 2.93 | 80 | 5.43 | 20 | 3.49 |
| p15AA-43% Li | 2.95 | 83 | 5.53 | 17 | 3.42 |
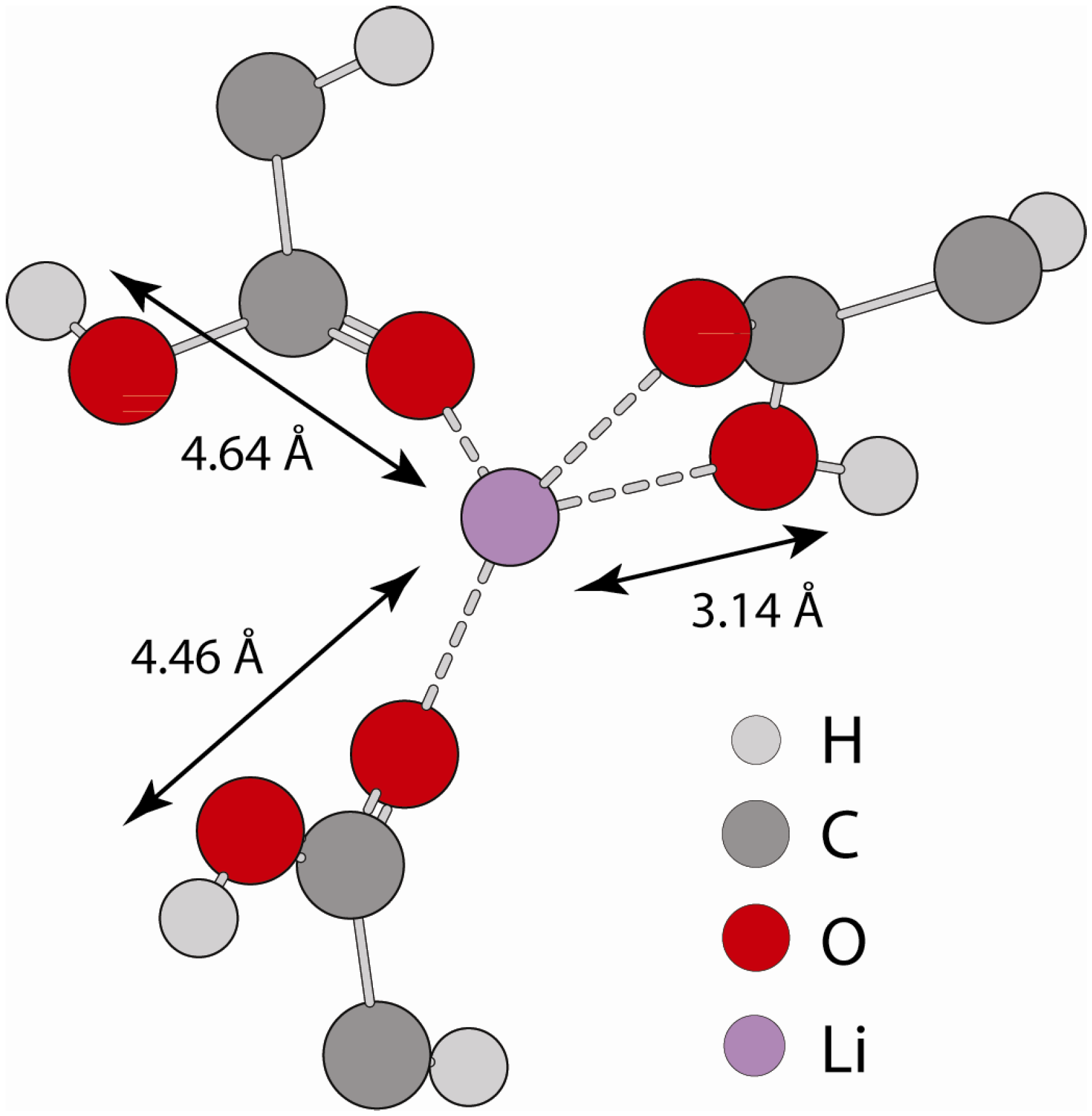
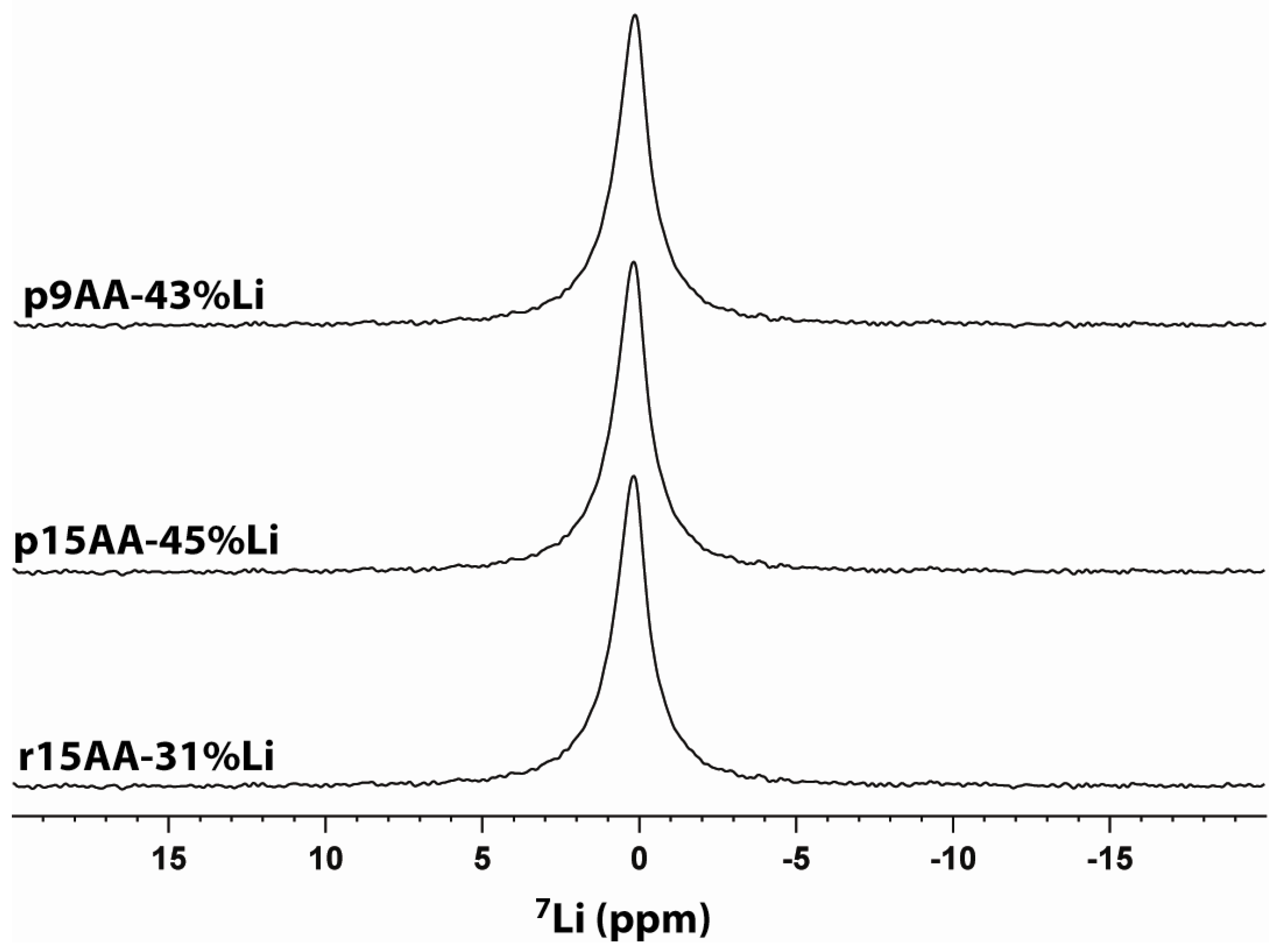
2.3. 7Li MAS NMR
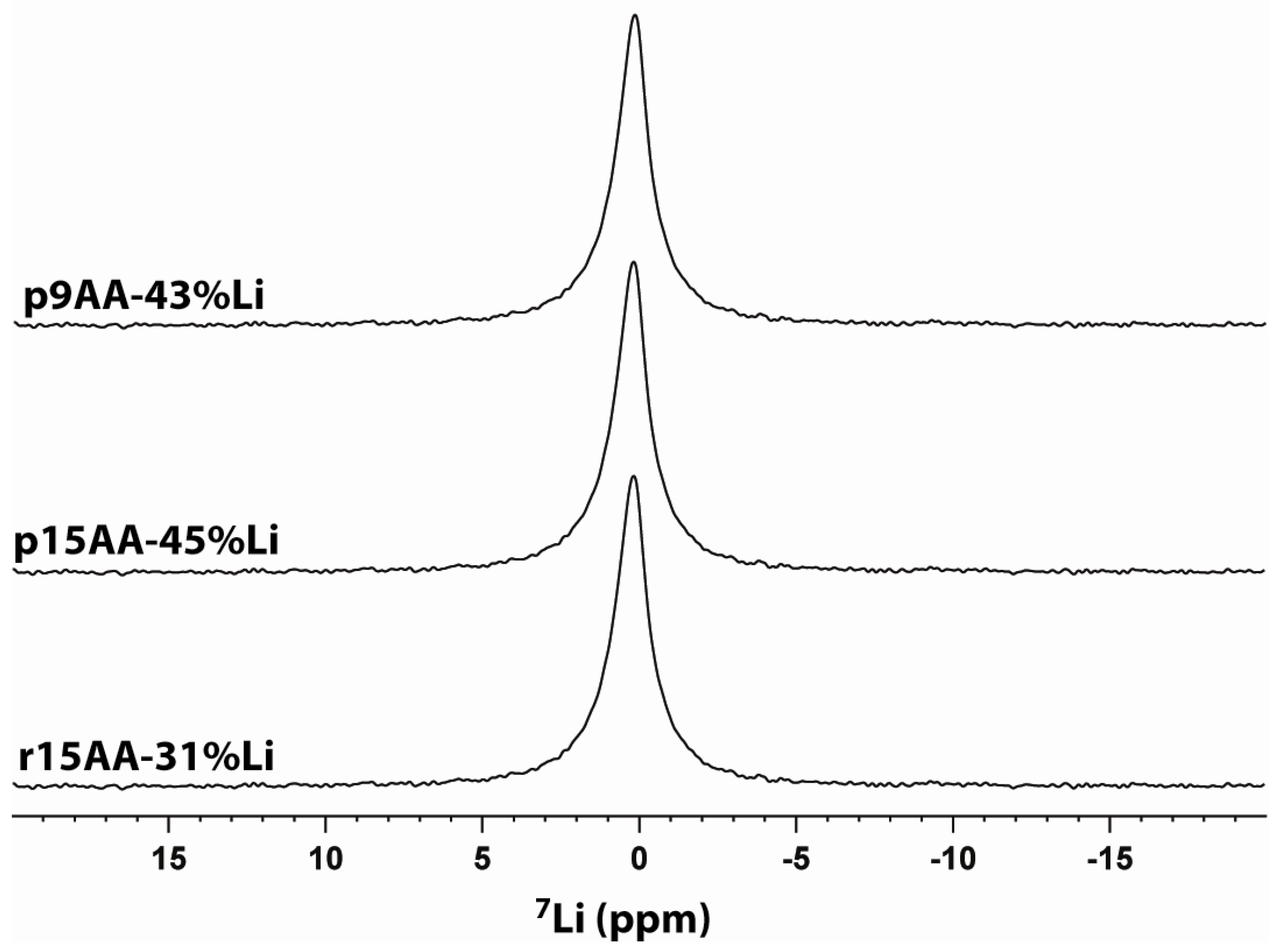
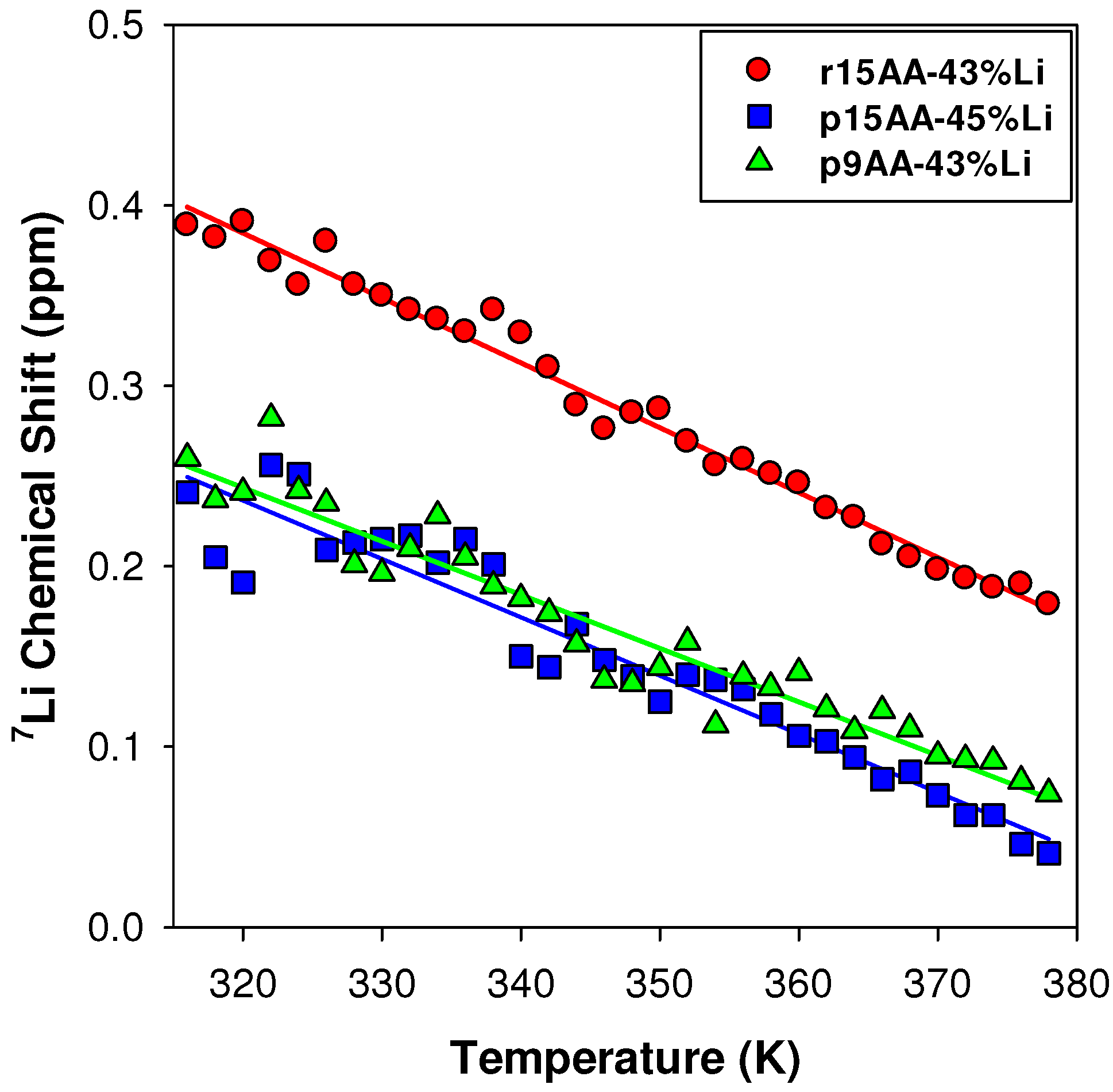
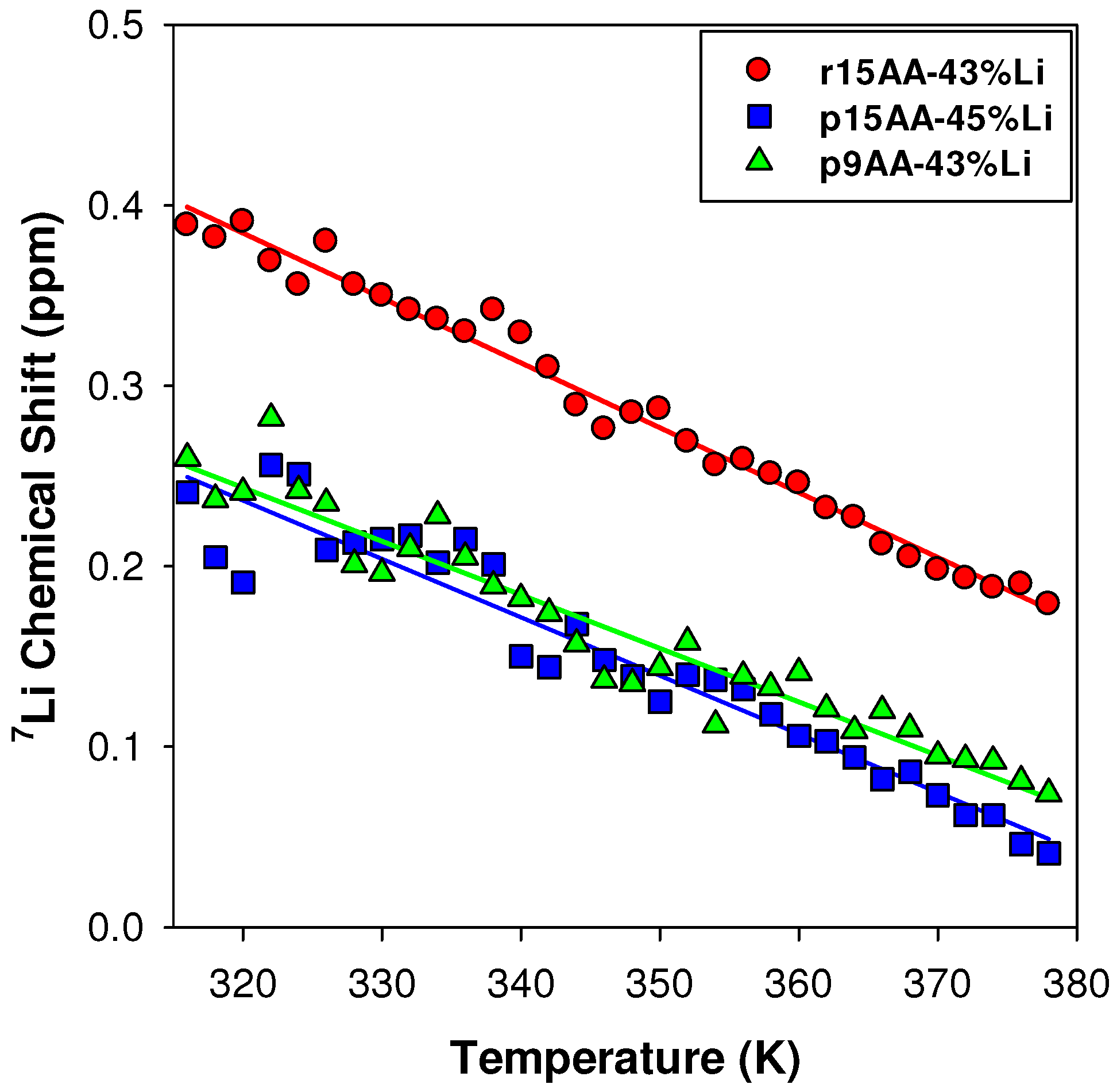
2.3.1. Temperature Variation
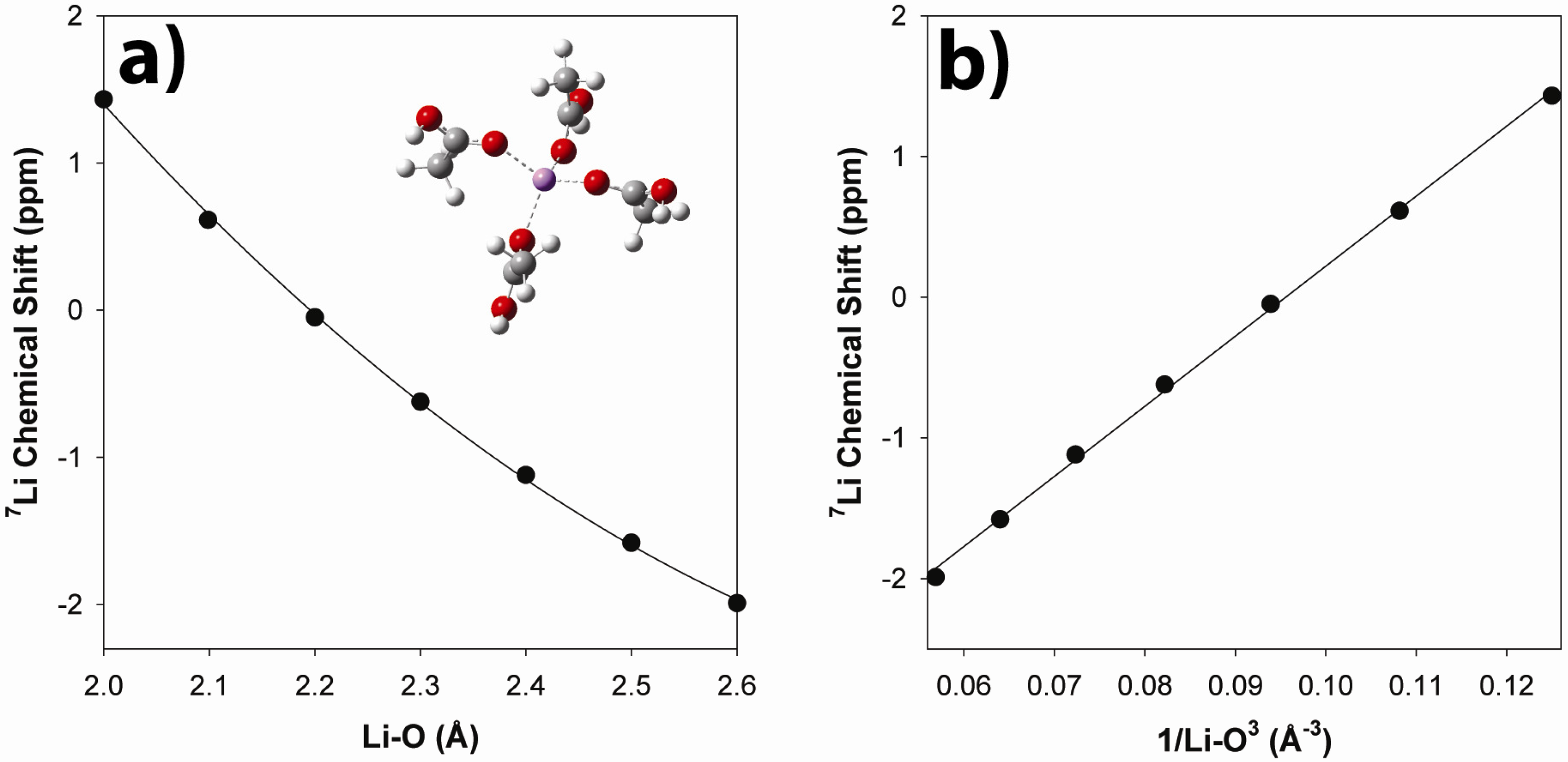
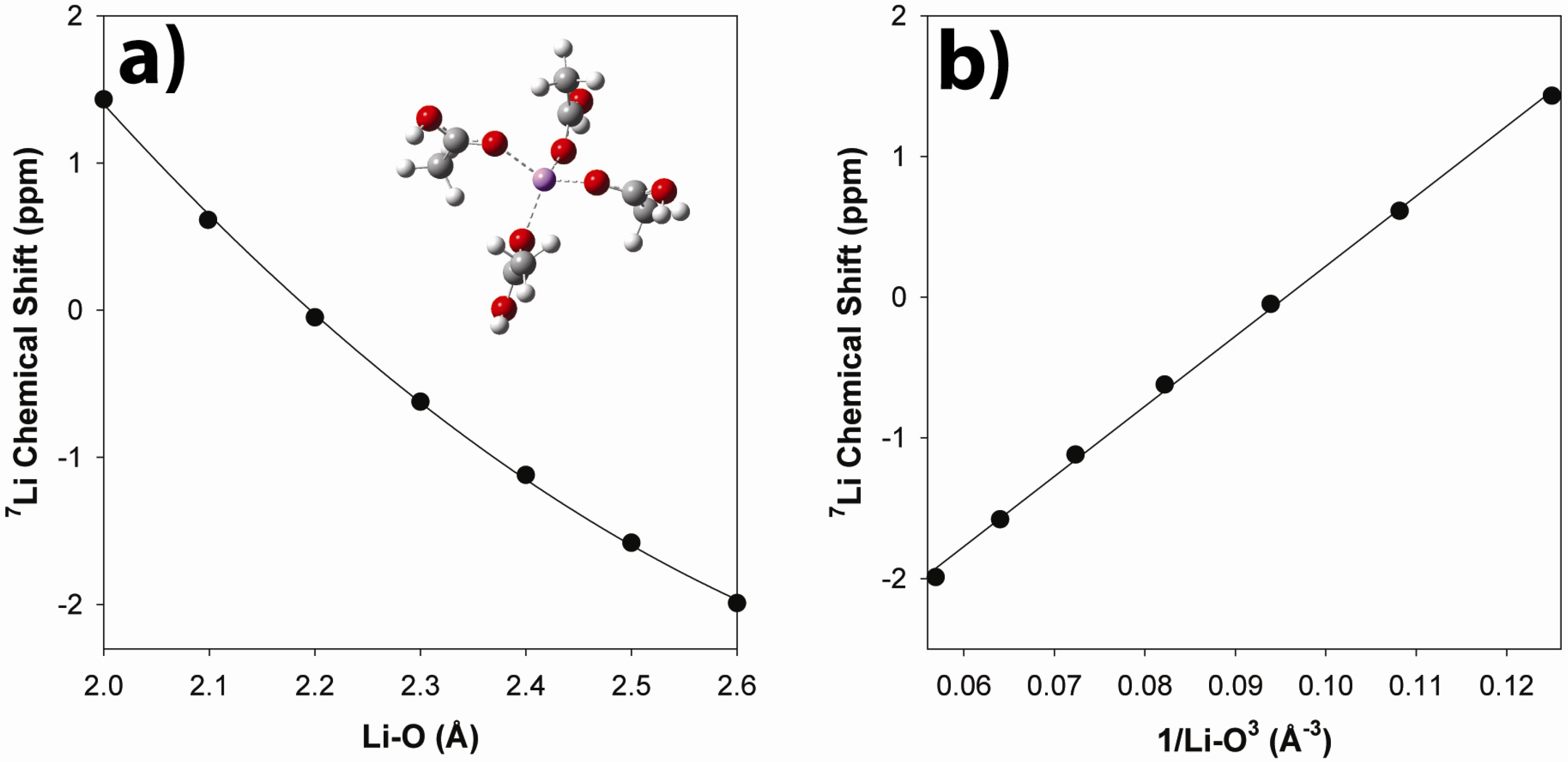
2.3.2. Ab Initio Prediction of 7Li NMR Chemical Shifts
2.4 Summary of Local Structure and Impact on Tg
3. Experimental Section
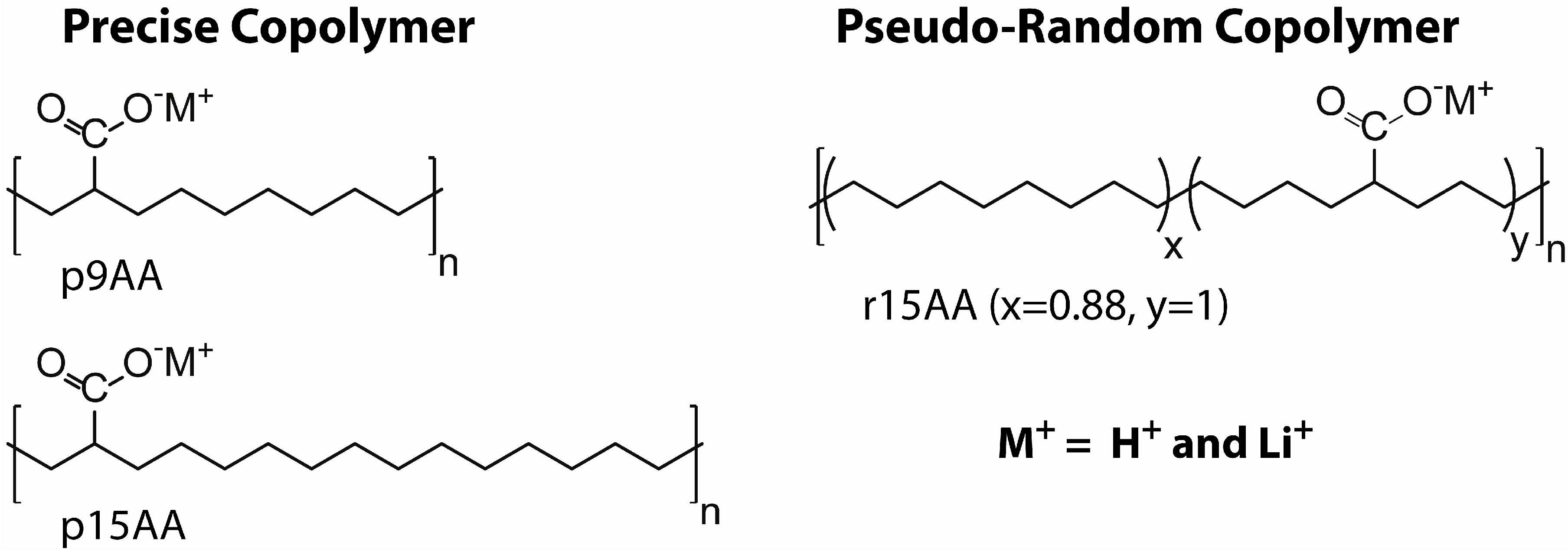
3.1. Ionomer Material Preparation
3.2. Solid-State 1H NMR Spectroscopy
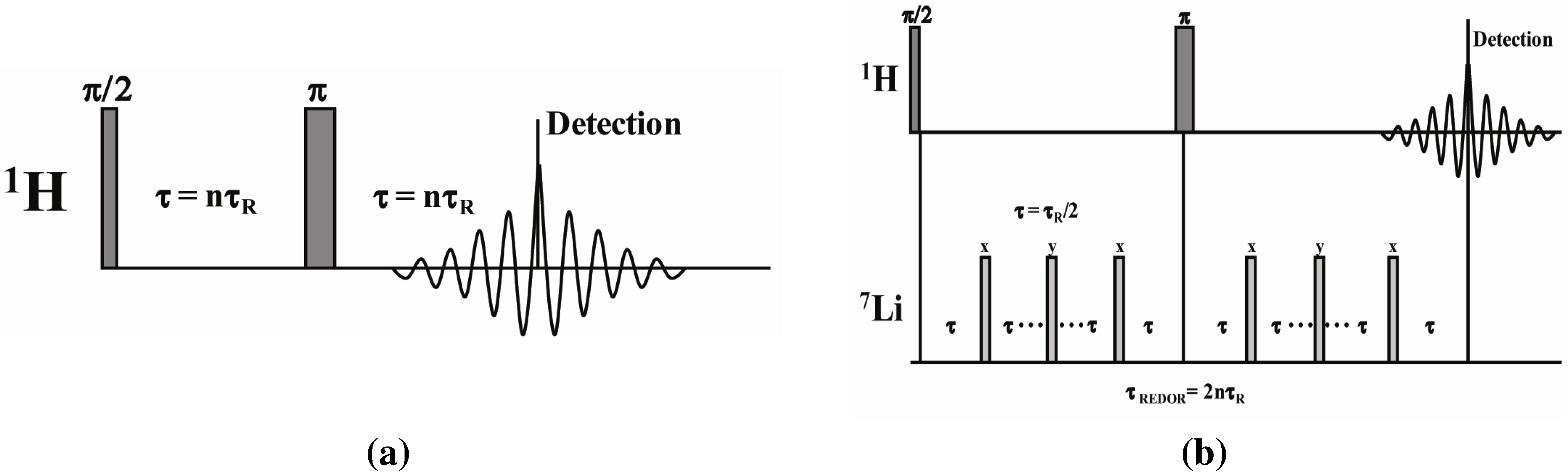
3.3. MD Simulations
3.4. Ab Initio Chemical Shift Calculations
4. Conclusions
Acknowledgments
References
- Zhang, H.; Shen, P.K. Recent developments of polymer electrolyte membranes for fuel cells. Chem. Rev. 2012, 112, 2780–2832. [Google Scholar] [CrossRef] [PubMed]
- Alloin, F.; Iojoiu, C. Composite Polymer Electrolytes for Electrochemical Devices. In Polymer Electrolytes: Fundamentals and Applications; Sequeira, C.A.C., Santos, D.M.F., Eds.; Woodhead Publishing Limited: Cambridge, UK, 2010; p. 632. [Google Scholar]
- Jaudouin, O.; Robin, J.J.; Lopez-Cuesta, J.M.; Perrin, D.; Imbert, C. Ionomer-based polyurethanes: A comparative study of properties and applications. Polym. Int. 2011, 61, 495–510. [Google Scholar] [CrossRef]
- Iojoiu, C.; Sood, R. Polysulfone-based ionomers. In High Performance Polymers and Engineering Plastics; Mittal, V., Ed.; Scrivener Publishing LLC: Salem, MA, USA, 2011; pp. 81–110. [Google Scholar]
- Li, X.F.; Paoloni, F.P.V.; Weiber, E.A.; Jiang, Z.H.; Jannasch, P. Fully aromatic ionomers with precisely sequenced sulfonated moieties for enhanced proton conductivity. Macromolecules 2012, 45, 1447–1459. [Google Scholar] [CrossRef]
- Baughman, T.W.; Chan, C.D.; Winey, K.I.; Wagener, K.B. Synthesis and morphology of well-defined poly(ethylene-co-acrylic acid) copolymers. Macromolecules 2007, 40, 6564–6571. [Google Scholar] [CrossRef]
- Alamo, R.G.; Jeon, K.; Smith, R.L.; Boz, E.; Wagener, K.B.; Bockstaller, M.R. Crystallization of polyethylenes containing chlorines: Precise vs random placement. Macromolecules 2008, 41, 7141–7151. [Google Scholar] [CrossRef]
- Opper, K.L.; Fassbender, B.; Brunklaus, G.; Spiess, H.W.; Wagener, K.B. Polyethylene functionalized with precisely spaced phosphonic acid groups. Macromolecules 2009, 42, 4407–4409. [Google Scholar] [CrossRef]
- Wei, Y.; Graf, R.; Sworen, J.C.; Cheng, C.Y.; Bowers, C.R.; Wagener, K.B.; Spiess, H.W. Local and collective motions in precise polyolefins with alkyl branches: A combination of 2H and 13C solid-state NMR spectroscopy. Angew. Chem. Int. Ed. 2009, 48, 4617–4620. [Google Scholar] [CrossRef]
- Aitken, B.S.; Buitrago, C.F.; Heffley, J.D.; Lee, M.; Gibson, H.W.; Winey, K.I.; Wagener, K.B. Precision ionomers: Synthesis and thermal/mechanical characterization. Macromolecules 2012, 45, 681–687. [Google Scholar] [CrossRef]
- Leonard, J.K.; Wei, Y.; Wagener, K.B. Synthesis and thermal characterization of precision poly(ethylene-co-vinyl amine) copolymers. Macromolecules 2012, 45, 671–680. [Google Scholar] [CrossRef]
- Seitz, M.E.; Chan, C.D.; Opper, K.L.; Baughman, T.W.; Wagener, K.B.; Winey, K.I. Nanoscale morphology in precisely sequenced poly(ethylene-coacrylic acid) zinc ionomers. J. Am. Chem. Soc. 2010, 132, 8165–8174. [Google Scholar] [CrossRef] [PubMed]
- Boz, E.; Wagener, K.B.; Ghosal, A.; Fu, R.; Alamo, R.G. Synthesis and crystallization of precision ADMET polyolefins containing halogens. Macromolecules 2006, 39, 4437–4447. [Google Scholar] [CrossRef]
- Jenkins, J.E.; Seitz, M.E.; Buitrago, C.F.; Winey, K.I.; Opper, K.L.; Baughman, T.W.; Wagener, K.B.; Alam, T.M. Solid-state 13C NMR structure and dynamics characterization of zinc neutralized polyethylene acrylic acid ionomers. Polymer 2012, 53, 3917–3927. [Google Scholar] [CrossRef]
- Alam, T.M.; Jenkins, J.; Seitz, M.E.; Buitrago, C.F.; Winey, K.I.; Opper, K.L.; Baughman, T.W.; Wagener, K.B. 1H MAS NMR Spectroscopy of Polyethylene Acrylic Acid Copolymers and Ionomers. In NMR Spectroscopy of Polymers: Innovative Stratagies for Complex Macromolecules; Cheng, H., Ed.; American Chemical Society: Washington, DC, USA, 2011; pp. 115–131. [Google Scholar]
- Fortier-McGill, B.; Toader, V.; Reven, L. 1H solid state NMR study of poly(methacrylic acid) hydrogen-bonded complexes. Macromolecules 2012, 45, 6015–6026. [Google Scholar] [CrossRef]
- Tolstoy, P.M.; Schah-Mohammed, P.; Smirnov, S.N.; Golubev, N.; Denisov, G.S.; Limbach, H.H. Characterization of fluxional hydrogen-bonded complexes of acetic acid and acetate by NMR: Geometries and isotope and solvent effects. J. Am. Chem. Soc. 2004, 126, 5621–5634. [Google Scholar] [CrossRef] [PubMed]
- Jeffrey, G.A.; Yeon, Y. The correlation between hydrogen-bond length and proton chemical shifts in crystals. Acta Cryst. 1986, B42, 410–413. [Google Scholar] [CrossRef]
- Harris, T.K.; Zhao, Q.; Mildvan, A.S. NMR studies of strong hydrogen bonds in enzymes and in a model compound. J. Mol. Struct. 2000, 552, 97–109. [Google Scholar] [CrossRef]
- Sternberg, U.; Brunner, E. The influence of short-range geometry on the chemical shift of protons in hydrogen bonds. J. Magn. Reson. 1994, 142–150. [Google Scholar] [CrossRef]
- Limbach, H.-H.; Tolstoy, P.M.; Perez-Hernandez, N.; Guo, J.; Shenderovich, I.G.; Denisov, G.S. OHO hydrogen bond geometries and NMR chemical shifts: From equilibrium structures to geometric H/D isotope effects, with application for water, protonated water and compressed ice. Israel J. Chem. 2009, 49, 199–216. [Google Scholar] [CrossRef]
- Benedict, H.; Limbach, H.H.; Wehlan, M.; Fehlhammer, W.P.; Golubev, N.; Janoschek, R. Solid state 15N NMR and theoretical studies of primary and secondary H/D isotope effects on the low-barrier NHN-hydrogen bonds. J. Am. Chem. Soc. 1998, 120, 2939–2950. [Google Scholar] [CrossRef]
- Steiner, T. Lengthening of the covalent X-H bond in heteronuclear hydrogen bonds quantified from organic and organometallic neutron crystal structures. J. Phys. Chem. A 1998, 102, 7041–7052. [Google Scholar] [CrossRef]
- Hall, L.M.; Seitz, M.E.; Winey, K.I.; Opper, K.L.; Wagener, K.B.; Stevens, M.J.; Frischknecht, A.L. Ionic aggregate structure in ionomer melts: Effect of molecular architecture on aggregates and the ionomer peak. J. Am. Chem. Soc. 2012, 134, 574–587. [Google Scholar] [CrossRef] [PubMed]
- Hall, L.M.; Stevens, M.J.; Frischknecht, A.L. Effect of polymer architecture and ionic aggregation on the scattering peak in model ionomers. Phys. Rev. Lett. 2011, 106. [Google Scholar] [CrossRef]
- Alam, T.M.; Hart, D.; Rempe, S.L.B. Computing the 7Li NMR chemical shielding of hydrated Li+ using cluster calculations and time-averaged configurations from Ab Initio molecular dynamics simulations. Phys. Chem. Chem. Phys. 2011, 13, 13629–13637. [Google Scholar] [CrossRef] [PubMed]
- Bolinitineanu, D.S.; Stevens, M.J.; Frischknecht, A.L. Sandia National Laboratories, Albuquerque, NM, USA. Atomistic Simulations Predict a Surprising Variety of Morphologies in Percise Ionomers. Unpublished work. 2012. [Google Scholar]
- Bielecki, A.; Burum, D.P. Temperature dependence of 207Pb MAS spectra of solid lead nitrate: An accurate, sensitive thermometer for variable-temperature MAS. J. Magn. Reson. Ser. A 1995, 116, 215–220. [Google Scholar] [CrossRef]
- Takahashi, T.; Kawashima, H.; Sugisawa, H.; Baba, T. 207Pb chemical shift thermometer at high temperature for magic angle spinning experiments. Solid State Nucl. Magn. Reson. 1999, 15, 119–123. [Google Scholar] [CrossRef] [PubMed]
- Gullion, T.; Schaefer, J. Rotational-echo double-resonance NMR. J. Magn. Reson. 1989, 81, 196–200. [Google Scholar]
- Bak, M.; Rasmussen, J.T.; Nielsen, N.C. SIMPSON: A general simulation program for solid-state NMR spectroscopy. J. Magn. Reson. 2000, 147, 296–380. [Google Scholar] [CrossRef] [PubMed]
- Plimpton, S.J. Fast parallel algorithms for short-range molecular dynamics. J. Comp. Phys. 1995, 117, 1–19. [Google Scholar] [CrossRef]
- Jorgensen, W.L.; Maxwell, D.S.; Triado-Rives, J. Development and testing of the OPLS ALL-atom force field on conformational energetics and properties of organic liquids. J. Am. Chem. Soc. 1996, 118, 11225–11236. [Google Scholar] [CrossRef]
- Frisch, M.J. Gaussian 09, Revision A.02, Gaussian Inc.: Wallingford, CT, USA, 2009.
- McLean, A.D.; Chandler, G.S. Contracted gaussian basis sets for molecular calculations. I. Second row atoms, Z = 11–18. J. Chem. Phys. 1980, 72, 5639–5648. [Google Scholar]
- Krishnan, R.; Binkley, J.S.; Pople, J.A. Self-consistent molecular orbital methods. XX. A basis set for correlated wave functions. J. Chem. Phys. 1980, 72, 650–654. [Google Scholar]
- Becke, A.D. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1992, 98, 5648–5652. [Google Scholar]
- Lee, C.; Yang, W.; Parr, R.G. Development of the colle-salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B 1988, 37, 785–789. [Google Scholar] [CrossRef]
- Cheeseman, J.R.; Trucks, G.W.; Keith, T.A.; Frisch, M.J. A comparison of models for calculating nuclear magnetic resonance shielding tensors. J. Chem. Phys. 1996, 104, 5497–5509. [Google Scholar] [CrossRef]
- Antušek, A.; Kedziera, D.; Kaczmarek-Kedziera, A.; Jaszunski, M. Coupled Cluster study of NMR shielding of alkali metal ions in water complexes and magnetic moments of alkali metal nuclei. Chem. Phys. Lett. 2012, 532, 1–8. [Google Scholar] [CrossRef]
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Alam, T.M.; Jenkins, J.E.; Bolintineanu, D.S.; Stevens, M.J.; Frischknecht, A.L.; Buitrago, C.F.; Winey, K.I.; Opper, K.L.; Wagener, K.B. Heterogeneous Coordination Environments in Lithium-Neutralized Ionomers Identified Using 1H and 7Li MAS NMR. Materials 2012, 5, 1508-1527. https://doi.org/10.3390/ma5081508
Alam TM, Jenkins JE, Bolintineanu DS, Stevens MJ, Frischknecht AL, Buitrago CF, Winey KI, Opper KL, Wagener KB. Heterogeneous Coordination Environments in Lithium-Neutralized Ionomers Identified Using 1H and 7Li MAS NMR. Materials. 2012; 5(8):1508-1527. https://doi.org/10.3390/ma5081508
Chicago/Turabian StyleAlam, Todd M., Janelle E. Jenkins, Dan S. Bolintineanu, Mark J. Stevens, Amalie L. Frischknecht, C. Francisco Buitrago, Karen I. Winey, Kathleen L. Opper, and Kenneth B. Wagener. 2012. "Heterogeneous Coordination Environments in Lithium-Neutralized Ionomers Identified Using 1H and 7Li MAS NMR" Materials 5, no. 8: 1508-1527. https://doi.org/10.3390/ma5081508
APA StyleAlam, T. M., Jenkins, J. E., Bolintineanu, D. S., Stevens, M. J., Frischknecht, A. L., Buitrago, C. F., Winey, K. I., Opper, K. L., & Wagener, K. B. (2012). Heterogeneous Coordination Environments in Lithium-Neutralized Ionomers Identified Using 1H and 7Li MAS NMR. Materials, 5(8), 1508-1527. https://doi.org/10.3390/ma5081508