Abstract
Minor metal-free sodium iron dioxide, NaFeO2, is a promising cathode material in sodium-ion batteries. Computational simulations based on the classical potentials were used to study the defects, sodium diffusion paths and cation doping behaviour in the α- and β-NaFeO2 polymorphs. The present simulations show good reproduction of both α- and β-NaFeO2. The most thermodynamically favourable defect is Na Frenkel, whereas the second most favourable defect is the cation antisite, in which Na and Fe exchange their positions. The migration energies suggest that there is a very small difference in intrinsic Na mobility between the two polymorphs but their migration paths are completely different. A variety of aliovalent and isovalent dopants were examined. Subvalent doping by Co and Zn on the Fe site is calculated to be energetically favourable in α- and β-NaFeO2, respectively, suggesting the interstitial Na concentration can be increased by using this defect engineering strategy. Conversely, doping by Ge on Fe in α-NaFeO2 and Si (or Ge) on Fe in β-NaFeO2 is energetically favourable to introduce a high concentration of Na vacancies that act as vehicles for the vacancy-assisted Na diffusion in NaFeO2. Electronic structure calculations by using density functional theory (DFT) reveal that favourable dopants lead to a reduction in the band gap.
1. Introduction
There is a demand for high-capacity rechargeable batteries to be used in large scale energy storage devices such as electric vehicles and grid-scale energy storage systems. Lithium ion batteries were of intense interest to achieve this and significant effort has been devoted to explore novel materials to produce high capacity Li-ion batteries [1,2,3,4,5]. However, there is a significant challenge to manufacture Li-ion batteries at large scale because of the low abundance and inhomogeneous distribution of lithium in the world. Furthermore, many lithium rich ores are found in remote areas leaving extraction and transportation difficult. After the commercial success of Li-ion batteries in portable applications, a considerable effort is currently devoted to Li-based supercapacitors, as they exhibit higher energy and power density compared to that of Li-ion batteries and can be used in large scale applications [6,7,8,9,10].
Sodium-ion batteries (NIBs) have recently attracted considerable interest because of the high elemental abundance with broader global distribution and the low cost of sodium [11,12,13]. A variety of sodium-based cathode materials, including NaFePO4 [14,15,16], Na2FePO4F [17,18], Na3V2(PO4)3 [19,20], Na3V(PO4)2 [21,22], Na4Co3(PO4)2P2O7 [23] and Na2CoSiO4 [24], have been synthesized and their electrochemical properties studied. There is a continuous active research on synthesizing novel cathode materials for NIBs to improve their capacity and its applicability in electrical vehicles.
Layered sodium transition metal dioxides—NaMO2 (M = Ti, V, Cr, Mn, Fe, Co and Ni) [25,26,27,28,29,30,31,32]—have been proposed as promising electrode materials for rechargeable NIBs due to their high volumetric and gravimetric densities. In addition, NaMO2 materials exhibit more transition metal redox compared to their Li analogue, owing to the larger radius of Na ions and multiple staking sequences [33].
NaFeO2 is an important cathode material for large-scale NIBs, owing to its low cost and environmentally benign nature [34,35]. There are two main polymorphs available for NaFeO2: α-NaFeO2 (hexagonal) [36] is a layered structure containing sheets of edge sharing FeO6 and NaO6 octahedrons. Electrode performance of α-NaFeO2 was first reported experimentally by Zhao et al. [37]. The electrochemical study by Yabuuchi et al. [38] showed that a reversible capacity of 80–100 mg−1 where the flat voltage of 3.3 V vs. Na metal can be delivered. Furthermore, its cycling performance was shown to be a reversible retention of 75% after 30 cycles [38]. Topotactic reaction studies (Fe3+/Fe4+ redox without the destruction of crystal) reveal that theoretical capacity of 241.8mAhg−1 can be achieved in α-NaFeO2 [39]. Another experimental study by Okada et al. [40] shows that operating voltage of more than 3.4 V versus Na metal is possible with the Fe3+/Fe4+ redox. β-NaFeO2 (orthorhombic) [41] has not been reported yet as an electrochemically active material for NIBs though there are other studies [42,43,44] on this material.
Atomistic scale simulation simulations based on the classical interatomic potentials can give useful information to the experimentalist on defect chemistry and the Na-ion transport mechanism, together with the activation energies and favourable cation doping of both polymorphs of NaFeO2. In previous work [45,46,47,48,49,50,51,52,53,54,55,56,57,58,59,60,61,62], we applied this methodology to promising cathode materials for lithium and sodium-ion batteries. Here, we examine both hexagonal and orthorhombic polymorphs of NaFeO2 and calculate the intrinsic defect formation energies, solution energies for a variety of dopants and possible diffusion pathways for sodium-ion conduction. Further, DFT calculations were performed to examine the electronic properties of doped and undoped α-NaFeO2.
2. Computational Methods
Classical pair potential calculations based on the Born model for ionic crystals were performed using the generalized utility lattice program (GULP) code [63]. The interionic interactions consist of long-range attraction (Coulombic) and short-range electron–electron repulsion. We used the well-established Buckingham potentials (refer to supplementary information) to model short-range interactions. The atomic positions and the simulation boxes were optimised using the Broyden–Fletcher–Goldfarb–Shanno (BFGS) algorithm [64]. Lattice relaxation around point defects and migrating ions were modelled using the Mott–Littleton method [65]. Vacancy-assisted Na ion diffusion was calculated considering seven interstitial Na ions between local Na hops. Activation energy reported in this study is the local maximum energy along the diffusion path. The present calculation is based on the full ionic charge model within the dilute limit. Therefore, the defect energies will be overestimated, however, the relative energies, and the trends will be consistent [66,67,68].
DFT calculations were applied for the electronic properties of NaFeO2 by means of the CASTEP plane wave code [69,70]. The generalized gradient approximation (GGA) was applied with the gradient correction added by Perdew, Burke and Ernzerhof (PBE) to the exchange–correlation energy functional [71]. The kinetic energy cut-off of the plane wave basis functions was set at 500 eV and the k-point grid at 3 × 3 × 3 for the geometry optimization calculations. After energy relaxation of the cell, the lattice constants obtained for α-NaFeO2 (a = b = 2.96 Å, c = 15.82 Å) are in good agreement with experiment (refer to Table 1). For the calculations of the density of states (DOS), a denser grid (5 × 5 × 5) was applied with a Gaussian smearing of 0.1 eV, taking into account spin polarization. The addition of the Hubbard model is necessary to derive a band gap closer to the experimental one, as the GGA method is expected to underestimate the band gap of insulators [72,73]. The Hubbard+U model was included to account for the Coulombic (repulsive) interaction of the on-site d electrons. It is well known that the GGA description cannot accurately predict the band gap due to electron delocalization overestimation. Therefore, the Hubbard correction term (+U) used for the 3d electrons of Fe and Co in this study is an established empirical method, to account for the strong on-site Coulomb interactions for the calculation of electronic properties. The U parameter was set at 4 eV for the Fe 3d states and at 3.4 eV for the 3d states of the dopant Co, according to literature [72]. This correction brings the energy band gap value closer to the experimental one.

Table 1.
Experimental and calculated structural parameters for hexagonal (α) and orthorhombic (β) NaFeO2.
3. Results and Discussion
3.1. NaFeO2 Crystal Structures
NaFeO2 has two different crystallographic structures: α (hexagonal, space group Rm) [32] and β (orthorhombic, space group Pn21a). Hexagonal phase consists of alternate layers of edge-sharing NaO6 and FeO6 octahedral units along the ab plane, as reported by Takeda et al. [32] (see Figure 1a). The crystal structure of orthorhombic NaFeO2 [41] forms corner-sharing tetrahedral units (both NaO4 and FeO4) in the ac plane as shown in Figure 1b. Using classical pair potentials selected from previous work (refer to Table S1), we first reproduced the experimental structures of both polymorphs. The experimental and calculated structural parameters are listed in Table 1. There is a good agreement between experimental and calculated lattice constants for both structures. Overestimation or underestimation of lattice constants is only within the error margin of ~2% suggesting that defect, diffusion and dopant calculation results would be enough accurate to compare with available experimental data. Furthermore, our calculation suggests that hexagonal phase is 0.31 eV lower in energy than orthorhombic phase.
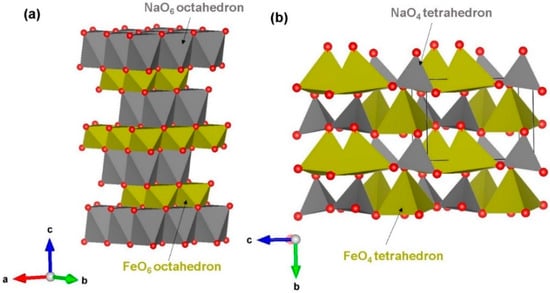
Figure 1.
Crystal structures of NaFeO2 polymorphs: (a) hexagonal (space group (m) and (b) orthorhombic (space group Pn21a).
3.2. Intrinsic Defect Processes
Possible defect processes in both α- and β-NaFeO2 were calculated. Point defect energies including vacancy and interstitial formation energies were first calculated and then they were combined to calculate the Frenkel- and Schottky-type defect formation energies. These intrinsic defect energies are useful in predicting the electrochemical behaviour of NaFeO2. Here we use the Kröger–Vink notation [74] to write equations for the Frenkel, Schottky and antisite defect formation:
Reaction energies for these intrinsic defect processes (refer to Table S2) are reported in Figure 2. The Na Frenkel is calculated to be the most energetically favourable intrinsic defect in both forms of NaFeO2. The second lowest defect energy process is found to be the Na–Fe anti-site, suggesting that a small percentage of Na on Fe sites () and Fe on Na sites ( will be observed at high temperatures. A small distortion is observed in the cation-oxygen bond distances and bond angles in the relaxed structure, but the lattice structure was not altered significantly. There are experimental and theoretical studies showing the presence of anti-site defects in many Li-ion cathode battery materials and in some as-prepared Na ion cathode materials [45,46,48,49,50,51,52,75,76,77,78,79]. There is, however, no experimental report on cation mixing of NaFeO2 yet. Nevertheless, in the future experimental preparations of as-prepared structure using different synthetic conditions or during cycling of this material, this defect may be observed. The Frenkel and Schottky defect energies were found to be highly endoergic suggesting that they are unlikely to form at low temperatures. The enthalpy to form Na2O Schottky (relation 5) is calculated as 2.76 eV/defect and 2.14 eV/defect for α- and β-NaFeO2, respectively. This process can introduce further and in the lattice at elevated temperatures. Conversely, lower defect energetics are observed for β-NaFeO2, but the overall trend is retained in both polymorphs. The difference in energetics is mainly due to the different crystal structures and difference in the coordination number of Na and Fe.
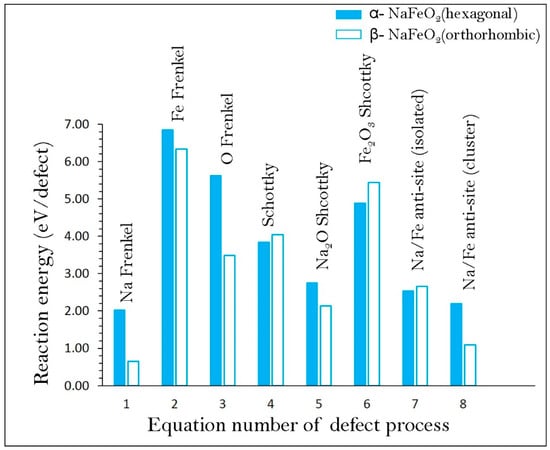
Figure 2.
Energetics of intrinsic defect process in α- and β-NaFeO2.
3.3. Sodium-Ion Diffusion
In this section intrinsic sodium-ion diffusion of NaFeO2 is discussed. Sodium-ion migration with low activation energy is one of the key requirements for a promising high-rate cathode material. The present computational technique allows us to calculate the Na vacancy migration paths together with activation energies, which are difficult to examine by experimental work alone.
For the Na vacancy migration in α-NaFeO2, we identified Na local hops (P) with the jump distance of 3.07 Å, and the migration energy was calculated to be 0.64 eV (refer to Table 2). Long range diffusion paths were then constructed. Sodium-ions migrate in the ab plane forming curved paths with overall activation energy of 0.64 eV (refer to Figure 3a). We considered Na hops between the layers but Na–Na migration distances were found to be >5 Å. Figure 3b reports the energy profile diagram for the Na local hop with the activation energy.
Table 2.
Calculated Na–Na separation and activation energy for the sodium-ion migration between two adjacent Na sites in α-NaFeO2 (refer to Figure 3a).
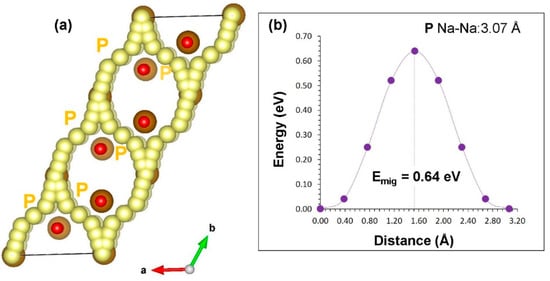
Figure 3.
Na ion migration in α-NaFeO2: (a) long migration paths and (b) energy profile of Na vacancy hopping between two adjacent Na sites.
Two different local hops, namely, A and B, were identified in β-NaFeO2 (refer to Figure 4). The energy profile diagrams for these two hops are shown in Figure 5. The migration path for hop A is in the bc plane with the jump distance of 3.51 Å and Na ion moves via a curved trajectory. The activation energy for the hop A is 0.65 eV. In the hop B, Na ions migrate in the ac plane with a curved trajectory, but with a jump distance of 3.26 Å and migration energy of 0.67 eV. Three two dimensional long range paths [(A→A→A→A), (B→B→B→B) and (A→B→A→B)] joining local Na hops were identified (see Figure 4). The lowest activation energy (0.65 eV) long range path (A→A→A→A) forms a zig-zag pattern in the bc plane. The other two paths have an overall activation energy of 0.67 eV, owing to the presence of local hop B which has an activation energy of 0.67 eV. Here, ions were treated as fully charged. Point defects in a highly ionic material might be expected to be in their fully ionic charge states. The activation energy of migration is defined as the position of the highest potential energy along the migration.
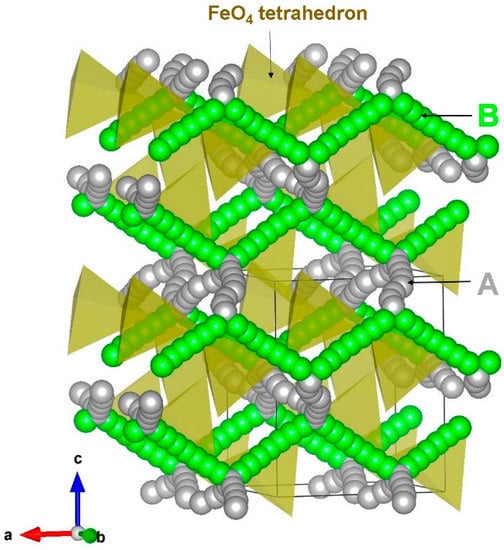
Figure 4.
Possible long range sodium vacancy migration paths considered in β-NaFeO2. Local Na migration paths are shown in green and grey atoms.
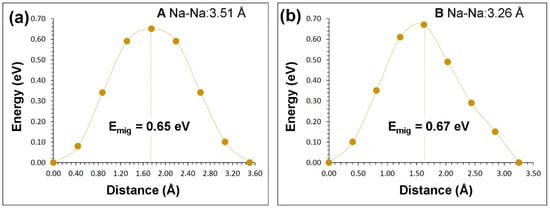
Figure 5.
Energy profile diagrams (A and B as shown in Figure 4) for local Na vacancy hopping with Na-Na separation of (a) 3.51 Å and (b) 3.26 Å β-NaFeO2.
3.4. Dopant Substitution
A variety of aliovalent and isovalent dopants were considered on the Fe site. Aliovalent dopant substitutions were charge-compensated by introducing necessary vacancies and interstitials. In all cases, appropriate lattice energies were calculated using the same Buckingham potentials used in this study and used in the solution energy calculations (refer to Table S3).
First, divalent dopants (M = Ca2+, Sr2+, Ba2+, Mn2+, Co2+, Ni2+, Cu2+ and Zn2+) were considered. The following reaction equations were used to calculate solution energies by compensating Na interstitials and O vacancies, respectively.
In the first charge compensation scheme, Na interstitials ions are introduced in the lattice. This can be an efficient way to increase the probability of Na+ ion intercalation/de-intercalation processes in the as-prepared NaFeO2. Figure 6a reports the solution energies of M2+ dopants on the Fe site. Lower solution energies are observed for β-NaFeO2. This can be due to the different crystal structures containing different coordination numbers of Fe. The most favourable dopant solution energy (1.22 eV/dopant) is calculated for Co2+ in α-NaFeO2, suggesting that a possible synthesis–doping strategy to introduce additional sodium into NaFeO2 can be achieved by doping Co on Fe sites at elevated temperatures, although the exact amount of Na incorporation cannot be determined. In the case of β-NaFeO2, Zn is the energetically favourable dopant with exothermic solution energy (‒0.09 eV/dopant). Other promising dopants are Co2+ (0.17 eV/dopant) and Ni2+ (0.19 eV/dopant). The possible composition of Co-doped NaFeO2 would be Na1+xFe1−xCoxO2 (x = 0.0–1.0). The high solution enthalpy for BaO suggests that Ba2+ is an unfavourable dopant to increase Na+ ions in both NaFeO2 polymorphs.
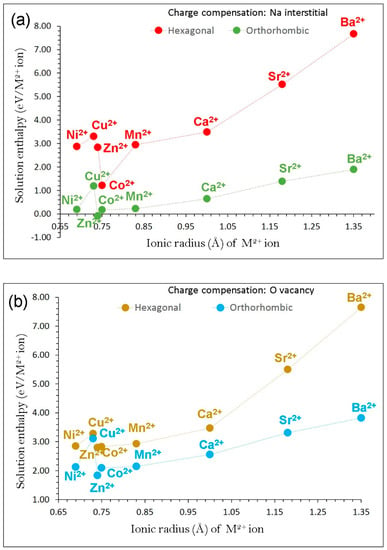
Figure 6.
Calculated enthalpy of solution of MO (M = Ni, Cu, Zn, Co, Mn, Ca, Sr and Ba) with respect to the M2+ ionic radius in NaFeO2 using (a) Na interstitial and (b) O vacancy as charge compensation.
In the second charge compensation scheme, the formation of oxygen vacancies is favoured by Zn incorporation in both α- and β-NaFeO2 (see Figure 6b). Again, lower solution energies are observed for β-NaFeO2 though the values are endoergic.
Next, we considered trivalent dopants (M = Al3+, Co3+, Ga3+, Mn3+, Sc3+, In3+, Yb3+, Y3+ and Gd3+). Equation 11 was used to calculate the solution enthalpy:
Favourable solution energies (0.00–0.20 eV) were noted for Ga, Co and Mn (see Figure 7) in α-NaFeO2. Interestingly, exothermic solution energies are observed for all dopants except for Y and Gd in β-NaFeO2. The most energetically favourable solution energy (‒0.99 eV/dopant) is observed for Co.
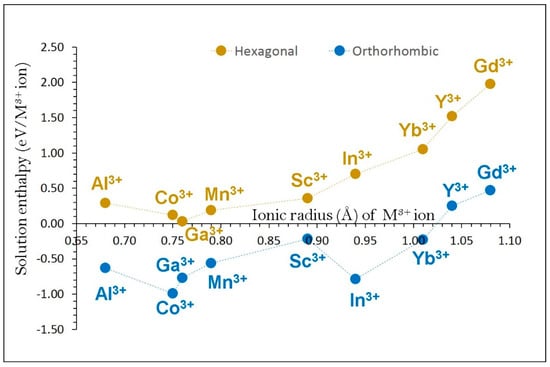
Figure 7.
Enthalpy of solution of M2O3 (M = Al, Co, Ga, Mn, Sc, In, Yb, Y and Gd) with respect to the M3+ ionic radius in NaFeO2.
We considered M4+ dopants on the Fe site to increase the concentration ofin NaFeO2. This strategy can facilitate Na self-diffusion via vacancy mechanism. Here, we calculate the solution of MO2 via the following equation,
Figure 8 reports the solution energies of MO2. It is observed that Ge exhibits the lowest solution energy (0.78 eV/dopant) in α-NaFeO2. Exothermic solution energies are calculated for SiO2 (−1.47 eV/dopant) and GeO2 (−0.87 eV/dopant) in β-NaFeO2 suggesting that these two dopants should be considered for experimental investigation.
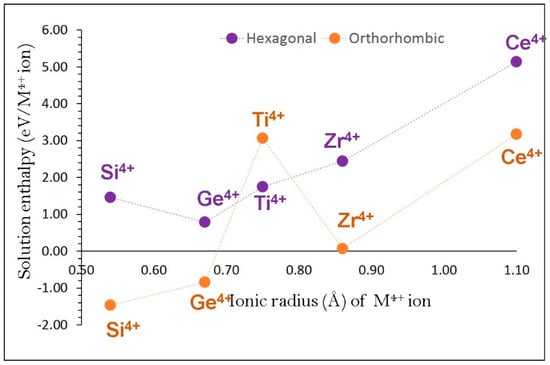
Figure 8.
Enthalpy of solution of MO2 (M = Si, Ge, Ti, Zr and Ce) with respect to the M4+ ionic radius in NaFeO2.
As redox couples Fe2+/Fe3+ and Fe3+/Fe4+ are important for the Na+ de-intercalation process and high operating voltage, respectively, a disproportionation reaction was considered according to the following equation,
Defect energy for this defect process is −3.47 eV/defect for α-phase and −4.19 eV/defect for β-phase respectively indicating that this process is likely to take place.
3.5. Density of States
The electronic structures of doped and undoped α-NaFeO2 were calculated by using first principles calculations, as described in methodology section. A supercell of 2 × 2 × 1 cells was used and the dopants of Co, Ge, Si and Zn were substitutional in the Fe position. This translates to a doping concentration of 8.3%. The density of states (DOS) plots are shown in Figure 9 for α-NaFeO2. The incorporation of defects in a substitutional Fe position leads to a band gap reduction and the appearance of defect states near the valence band. The band gap of the perfect structure is calculated to be 1.53 eV. Doping with Co decreases significantly the calculated band gap at 0.8 eV, due to a band tail that is formed near the valence band with a peak at 0.35 eV. Doping with Ge or Si further decreases the band gap at 0.7 eV and 0.6 eV, respectively, due to an appearance of states with peaks at 0.48 eV for Ge and 0.14 eV and 0.56 eV for Si. Doping with Zn also decreases in the same way as the band gap at 0.70 eV, due to states that appear 0.34 eV and 0.60 eV higher that the valence band of the perfect structure.
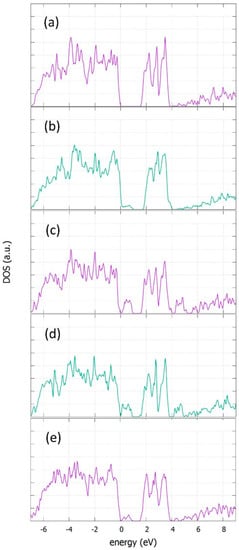
Figure 9.
Density of states of (a) perfect, (b) CoFe- (c) GeFe- (d) SiFe- and (e) ZnFe-doped α-NaFeO2.
4. Conclusions
In conclusion, using atomistic simulation techniques, we carried out a systematic survey of both α- and β-NaFeO2 to investigate intrinsic defects, sodium-ion diffusion paths and favourable aliovalent and isovalent dopants on the Fe site. The present simulations reasonably reproduce the observed polymorphs of NaFeO2. The most favourable intrinsic defect type is Na Frenkel. The second most favourable energy defect process is Na–Fe antisite, suggesting that there will be a small population of Na on Fe site and vice versa. The lowest migration energies for long-range Na ion migration in hexagonal (α-) and orthorhombic (β-) NaFeO2 are 0.65 eV and 0.67 eV, respectively, suggesting that both polymorphs exhibit favourable electrode kinetics. The present calculations further suggest that favourable dopants for creating additional Na in the α- and β-NaFeO2 are Co2+ and Zn2+ on the Fe site, respectively. A high concentration of Na vacancies can be introduced by doping Ge on Fe in α-NaFeO2 and Si (and Ge) on Fe in β-NaFeO2 to facilitate the vacancy-assisted Na diffusion in NaFeO2. Electronic structure calculations predict that in all cases substitutional doping leads to a reduction in the band gap.
Supplementary Materials
The following are available online at https://www.mdpi.com/1996-1944/12/19/3243/s1, Table S1: Interatomic potential parameters used in the atomistic simulations of NaFeO2; Table S2: Energetics of intrinsic defect process in NaFeO2; Table S3: Solution enthalpy for dopant substation at Fe site in NaFeO2.
Author Contributions
Computation, N.K. (Navaratnarajah Kuganathan) and N.K. (Nikolaos Kelaidis); Writing, N.K. (Navaratnarajah Kuganathan); Analysis and Editing, N.K. (Navaratnarajah Kuganathan) and A.C.
Funding
This research was financially supported by European Union’s H2020 Programme under Grant Agreement no 824072– HARVESTORE
Acknowledgments
Computational facilities and support were provided by the High Performance Computing Centre at Imperial College London and Coventry University.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Armand, M.; Tarascon, J.M. Building better batteries. Nature 2008, 451, 652. [Google Scholar] [CrossRef] [PubMed]
- Padhi, A.K.; Nanjundaswamy, K.S.; Goodenough, J.B. Phospho-olivines as Positive-Electrode Materials for Rechargeable Lithium Batteries. J. Electrochem. Soc. 1997, 144, 1188–1194. [Google Scholar] [CrossRef]
- Nytén, A.; Abouimrane, A.; Armand, M.; Gustafsson, T.; Thomas, J.O. Electrochemical performance of Li2FeSiO4 as a new Li-battery cathode material. Electrochem. Commun. 2005, 7, 156–160. [Google Scholar] [CrossRef]
- Masquelier, C.; Croguennec, L. Polyanionic (Phosphates, Silicates, Sulfates) Frameworks as Electrode Materials for Rechargeable Li (or Na) Batteries. Chem. Rev. 2013, 113, 6552–6591. [Google Scholar] [CrossRef] [PubMed]
- Afyon, S.; Wörle, M.; Nesper, R. A Lithium-Rich Compound Li7Mn(BO3)3 Containing Mn2+ in Tetrahedral Coordination: A Cathode Candidate for Lithium-Ion Batteries. Angew. Chem. Int. Ed. 2013, 52, 12541–12544. [Google Scholar] [CrossRef]
- Simon, P.; Gogotsi, Y.; Dunn, B. Where Do Batteries End and Supercapacitors Begin? Science 2014, 343, 1210–1211. [Google Scholar] [CrossRef] [PubMed]
- Jeżowski, P.; Fic, K.; Crosnier, O.; Brousse, T.; Béguin, F. Lithium rhenium(vii) oxide as a novel material for graphite pre-lithiation in high performance lithium-ion capacitors. J. Mater. Chem. A 2016, 4, 12609–12615. [Google Scholar] [CrossRef]
- Jeżowski, P.; Fic, K.; Crosnier, O.; Brousse, T.; Béguin, F. Use of sacrificial lithium nickel oxide for loading graphitic anode in Li-ion capacitors. Electrochim. Acta 2016, 206, 440–445. [Google Scholar] [CrossRef]
- Jeżowski, P.; Crosnier, O.; Deunf, E.; Poizot, P.; Béguin, F.; Brousse, T. Safe and recyclable lithium-ion capacitors using sacrificial organic lithium salt. Nat. Mater. 2017, 17, 167. [Google Scholar] [CrossRef]
- Jagadale, A.; Zhou, X.; Xiong, R.; Dubal, D.P.; Xu, J.; Yang, S. Lithium ion capacitors (LICs): Development of the materials. Energy Storage Mater. 2019, 19, 314–329. [Google Scholar] [CrossRef]
- Ellis, B.L.; Nazar, L.F. Sodium and sodium-ion energy storage batteries. Curr. Opin. Solid State Mater. Sci. 2012, 16, 168–177. [Google Scholar] [CrossRef]
- Yabuuchi, N.; Kubota, K.; Dahbi, M.; Komaba, S. Research Development on Sodium-Ion Batteries. Chem. Rev. 2014, 114, 11636–11682. [Google Scholar] [CrossRef] [PubMed]
- Palomares, V.; Casas-Cabanas, M.; Castillo-Martinez, E.; Han, M.H.; Rojo, T. Update on Na-based battery materials. A growing research path. Energy Environ. Sci. 2013, 6, 2312–2337. [Google Scholar] [CrossRef]
- Oh, S.-M.; Myung, S.-T.; Hassoun, J.; Scrosati, B.; Sun, Y.-K. Reversible NaFePO4 electrode for sodium secondary batteries. Electrochem. Commun. 2012, 22, 149–152. [Google Scholar] [CrossRef]
- Fang, Y.; Liu, Q.; Xiao, L.; Ai, X.; Yang, H.; Cao, Y. High-Performance Olivine NaFePO4 Microsphere Cathode Synthesized by Aqueous Electrochemical Displacement Method for Sodium Ion Batteries. ACS Appl. Mater. Interfaces 2015, 7, 17977–17984. [Google Scholar] [CrossRef] [PubMed]
- Tang, W.; Song, X.; Du, Y.; Peng, C.; Lin, M.; Xi, S.; Tian, B.; Zheng, J.; Wu, Y.; Pan, F.; et al. High-performance NaFePO4 formed by aqueous ion-exchange and its mechanism for advanced sodium ion batteries. J. Mater. Chem. A 2016, 4, 4882–4892. [Google Scholar] [CrossRef]
- Kawabe, Y.; Yabuuchi, N.; Kajiyama, M.; Fukuhara, N.; Inamasu, T.; Okuyama, R.; Nakai, I.; Komaba, S. Synthesis and electrode performance of carbon coated Na2FePO4F for rechargeable Na batteries. Electrochem. Commun. 2011, 13, 1225–1228. [Google Scholar] [CrossRef]
- Kosova, N.V.; Podugolnikov, V.R.; Devyatkina, E.T.; Slobodyuk, A.B. Structure and electrochemistry of NaFePO4 and Na2FePO4F cathode materials prepared via mechanochemical route. Mater. Res. Bull. 2014, 60, 849–857. [Google Scholar] [CrossRef]
- Jian, Z.; Zhao, L.; Pan, H.; Hu, Y.-S.; Li, H.; Chen, W.; Chen, L. Carbon coated Na3V2(PO4)3 as novel electrode material for sodium ion batteries. Electrochem. Commun. 2012, 14, 86–89. [Google Scholar] [CrossRef]
- Jiang, X.; Yang, L.; Ding, B.; Qu, B.; Ji, G.; Lee, J.Y. Extending the cycle life of Na3V2(PO4)3 cathodes in sodium-ion batteries through interdigitated carbon scaffolding. J. Mater. Chem. 2016, 4, 14669–14674. [Google Scholar] [CrossRef]
- Kovrugin, V.M.; David, R.; Chotard, J.-N.; Recham, N.; Masquelier, C. A High Voltage Cathode Material for Sodium Batteries: Na3V(PO4)2. Inorg. Chem. 2018, 57, 8760–8768. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Yoon, G.; Kim, H.; Park, Y.-U.; Kang, K. Na3V(PO4)2: A New Layered-Type Cathode Material with High Water Stability and Power Capability for Na-Ion Batteries. Chem. Mater. 2018, 30, 3683–3689. [Google Scholar] [CrossRef]
- Nose, M.; Nakayama, H.; Nobuhara, K.; Yamaguchi, H.; Nakanishi, S.; Iba, H. Na4Co3(PO4)2P2O7: A novel storage material for sodium-ion batteries. J. Power Sources 2013, 234, 175–179. [Google Scholar] [CrossRef]
- Treacher, J.C.; Wood, S.M.; Islam, M.S.; Kendrick, E. Na2CoSiO4 as a cathode material for sodium-ion batteries: Structure, electrochemistry and diffusion pathways. Phys. Chem. Chem. Phys. 2016, 18, 32744–32752. [Google Scholar] [CrossRef] [PubMed]
- Vassilaras, P.; Ma, X.; Li, X.; Ceder, G. Electrochemical Properties of Monoclinic NaNiO2. J. Electrochem. Soc. 2013, 160, A207–A211. [Google Scholar] [CrossRef]
- Kikkawa, S.; Miyazaki, S.; Koizumi, M. Deintercalated NaCoO2 and LiCoO2. J. Solid State Chem. 1986, 62, 35–39. [Google Scholar] [CrossRef]
- Ma, X.; Chen, H.; Ceder, G. Electrochemical Properties of Monoclinic NaMnO2. J. Electrochem. Soc. 2011, 158, A1307–A1312. [Google Scholar] [CrossRef]
- Komaba, S.; Takei, C.; Nakayama, T.; Ogata, A.; Yabuuchi, N. Electrochemical intercalation activity of layered NaCrO2 vs. LiCrO2. Electrochem. Commun. 2010, 12, 355–358. [Google Scholar] [CrossRef]
- Didier, C.; Guignard, M.; Denage, C.; Szajwaj, O.; Ito, S.; Saadoune, I.; Darriet, J.; Delmas, C. Electrochemical Na-Deintercalation from NaVO2. Electrochem. Solid-State Lett. 2011, 14, A75–A78. [Google Scholar] [CrossRef]
- Wu, D.; Li, X.; Xu, B.; Twu, N.; Liu, L.; Ceder, G. NaTiO2: A layered anode material for sodium-ion batteries. Energy Environ. Sci. 2015, 8, 195–202. [Google Scholar] [CrossRef]
- Maazaz, A.; Delmas, C.; Hagenmuller, P. A study of the NaxTiO2 system by electrochemical deintercalation. J. Inclusion Phenom. 1983, 1, 45–51. [Google Scholar] [CrossRef]
- Takeda, Y.; Nakahara, K.; Nishijima, M.; Imanishi, N.; Yamamoto, O.; Takano, M.; Kanno, R. Sodium deintercalation from sodium iron oxide. Mater. Res. Bull. 1994, 29, 659–666. [Google Scholar] [CrossRef]
- Guo, S.; Yi, J.; Sun, Y.; Zhou, H. Recent advances in titanium-based electrode materials for stationary sodium-ion batteries. Energy Environ. Sci. 2016, 9, 2978–3006. [Google Scholar] [CrossRef]
- Pan, H.; Hu, Y.-S.; Chen, L. Room-temperature stationary sodium-ion batteries for large-scale electric energy storage. Energy Environ. Sci. 2013, 6, 2338–2360. [Google Scholar] [CrossRef]
- Fang, Y.; Chen, Z.; Xiao, L.; Ai, X.; Cao, Y.; Yang, H. Recent Progress in Iron-Based Electrode Materials for Grid-Scale Sodium-Ion Batteries. Small 2018, 14, 1703116. [Google Scholar] [CrossRef]
- Kikkawa, S.; Miyazaki, S.; Koizumi, M. Sodium deintercalation from α-NaFeO2. Mater. Res. Bull. 1985, 20, 373–377. [Google Scholar] [CrossRef]
- Zhao, J.; Zhao, L.; Dimov, N.; Okada, S.; Nishida, T. Electrochemical and Thermal Properties of α-NaFeO2 Cathode for Na-Ion Batteries. J. Electrochem. Soc. 2013, 160, A3077–A3081. [Google Scholar] [CrossRef]
- Yabuuchi, N.; Yoshida, H.; Komaba, S. Crystal Structures and Electrode Performance of Alpha-NaFeO2 for Rechargeable Sodium Batteries. Electrochemistry 2012, 80, 716–719. [Google Scholar] [CrossRef]
- Ohzuku, T.; Ueda, A. Why transition metal (di) oxides are the most attractive materials for batteries. Solid State Ionics 1994, 69, 201–211. [Google Scholar] [CrossRef]
- Okada, S.; Yamaki, J. Iron-Based Rare-Metal-Free Cathodes. In Lithium Ion Rechargeable Battries; Ozawa, K., Ed.; Wiley-VCH: Weinheim, Germany, 2009; p. 57. [Google Scholar]
- Grey, I.E.; Hill, R.J.; Hewat, A.W. A neutron powder diffraction study of the β to γ phase transformation in NaFeO2. Z. Kristallog. 1990, 193, 51–69. [Google Scholar] [CrossRef]
- Singh, S.; Tovstolytkin, A.; Lotey, G.S. Magnetic properties of superparamagnetic β-NaFeO2 nanoparticles. J. Magn. Magn. Mater. 2018, 458, 62–65. [Google Scholar] [CrossRef]
- Viret, M.; Rubi, D.; Colson, D.; Lebeugle, D.; Forget, A.; Bonville, P.; Dhalenne, G.; Saint-Martin, R.; André, G.; Ott, F. β-NaFeO2, a new room-temperature multiferroic material. Mater. Res. Bull. 2012, 47, 2294–2298. [Google Scholar] [CrossRef]
- Watanabe, H.; Fukase, M. Weak Ferromagnetism in β-NaFeO2. J. Phys. Soc. Japan 1961, 16, 1181–1184. [Google Scholar] [CrossRef]
- Kuganathan, N.; Islam, M.S. Li2MnSiO4 Lithium Battery Material: Atomic-Scale Study of Defects, Lithium Mobility, and Trivalent Dopants. Chem. Mater. 2009, 21, 5196–5202. [Google Scholar] [CrossRef]
- Armstrong, A.R.; Kuganathan, N.; Islam, M.S.; Bruce, P.G. Structure and Lithium Transport Pathways in Li2FeSiO4 Cathodes for Lithium Batteries. JACS 2011, 133, 13031–13035. [Google Scholar] [CrossRef] [PubMed]
- Fisher, C.A.J.; Kuganathan, N.; Islam, M.S. Defect chemistry and lithium-ion migration in polymorphs of the cathode material Li2MnSiO4. J. Mater. Chem. A 2013, 1, 4207–4214. [Google Scholar] [CrossRef]
- Kuganathan, N.; Iyngaran, P.; Chroneos, A. Lithium diffusion in Li5FeO4. Sci. Rep. 2018, 8, 5832. [Google Scholar] [CrossRef]
- Kordatos, A.; Kuganathan, N.; Kelaidis, N.; Iyngaran, P.; Chroneos, A. Defects and lithium migration in Li2CuO2. Sci. Rep. 2018, 8, 6754. [Google Scholar] [CrossRef]
- Kuganathan, N.; Ganeshalingam, S.; Chroneos, A. Defects, Dopants and Lithium Mobility in Li9V3(P2O7)3 (PO4)2. Sci. Rep. 2018, 8, 8140. [Google Scholar] [CrossRef]
- Kuganathan, N.; Kordatos, A.; Chroneos, A. Li2SnO3 as a Cathode Material for Lithium-ion Batteries: Defects, Lithium Ion Diffusion and Dopants. Sci. Rep. 2018, 8, 12621. [Google Scholar] [CrossRef]
- Kuganathan, N.; Chroneos, A. Defects, Dopants and Sodium Mobility in Na2MnSiO4. Sci. Rep. 2018, 8, 14669. [Google Scholar] [CrossRef] [PubMed]
- Kuganathan, N.; Chroneos, A. Defects and dopant properties of Li3V2(PO4)3. Sci. Rep. 2019, 9, 333. [Google Scholar] [CrossRef] [PubMed]
- Kuganathan, N.; Kordatos, A.; Chroneos, A. Defect Chemistry and Li-ion Diffusion in Li2RuO3. Sci. Rep. 2019, 9, 550. [Google Scholar] [CrossRef]
- Kuganathan, N.; Kordatos, A.; Fitzpatrick, M.E.; Vovk, R.V.; Chroneos, A. Defect process and lithium diffusion in Li2TiO3. Solid State Ionics 2018, 327, 93–98. [Google Scholar] [CrossRef]
- Kuganathan, N.; Kordatos, A.; Anurakavan, S.; Iyngaran, P.; Chroneos, A. Li3SbO4 lithium-ion battery material: Defects, lithium ion diffusion and tetravalent dopants. Mater. Chem. Phys. 2019, 225, 34–41. [Google Scholar] [CrossRef]
- Kuganathan, N.; Iyngaran, P.; Vovk, R.; Chroneos, A. Defects, dopants and Mg diffusion in MgTiO3. Sci. Rep. 2019, 9, 4394. [Google Scholar] [CrossRef]
- Kuganathan, N.; Kordatos, A.; Kelaidis, N.; Chroneos, A. Defects, Lithium Mobility and Tetravalent Dopants in the Li3NbO4 Cathode Material. Sci. Rep. 2019, 9, 2192. [Google Scholar] [CrossRef]
- Kuganathan, N.; Tsoukalas, L.H.; Chroneos, A. Defects, dopants and Li-ion diffusion in Li2SiO3. Solid State Ionics 2019, 335, 61–66. [Google Scholar] [CrossRef]
- Kuganathan, N.; Sgourou, E.N.; Panayiotatos, Y.; Chroneos, A. Defect Process, Dopant Behaviour and Li Ion Mobility in the Li2MnO3 Cathode Material. Energies 2019, 12, 1329. [Google Scholar] [CrossRef]
- Kuganathan, N.; Chroneos, A. Defect Chemistry and Na-Ion Diffusion in Na3Fe2(PO4)3 Cathode Material. Materials 2019, 12, 1348. [Google Scholar] [CrossRef]
- Kuganathan, N.; Chroneos, A. Na3V(PO4)2 cathode material for Na ion batteries: Defects, dopants and Na diffusion. Solid State Ionics 2019, 336, 75–79. [Google Scholar] [CrossRef]
- Gale, J.D.; Rohl, A.L. The General Utility Lattice Program (GULP). Mol. Simulat. 2003, 29, 291–341. [Google Scholar] [CrossRef]
- Gale, J.D. GULP: A computer program for the symmetry-adapted simulation of solids. J. Chem. Soc. Faraday Trans. 1997, 93, 629–637. [Google Scholar] [CrossRef]
- Mott, N.F.; Littleton, M.J. Conduction in polar crystals. I. Electrolytic conduction in solid salts. Trans. Faraday Soc. 1938, 34, 485–499. [Google Scholar] [CrossRef]
- Grimes, R.W.; Busker, G.; McCoy, M.A.; Chroneos, A.; Kilner, J.A.; Chen, S.-P. The Effect of Ion Size on Solution Mechanism and Defect Cluster Geometry. Ber. Bunsengest. Phys. Chem. 1997, 101, 1204–1210. [Google Scholar] [CrossRef]
- Seymour, I.D.; Chroneos, A.; Kilner, J.A.; Grimes, R.W. Defect processes in orthorhombic LnBaCo2O5.5 double perovskites. Phys. Chem. Chem. Phys. 2011, 13, 15305–15310. [Google Scholar] [CrossRef]
- Jay, E.E.; Rushton, M.J.D.; Chroneos, A.; Grimes, R.W.; Kilner, J.A. Genetics of superionic conductivity in lithium lanthanum titanates. Phys. Chem. Chem. Phys. 2015, 17, 178–183. [Google Scholar] [CrossRef]
- Segall, M.D.; Philip, J.D.L.; Probert, M.J.; Pickard, C.J.; Hasnip, P.J.; Clark, S.J.; Payne, M.C. First-principles simulation: Ideas, illustrations and the CASTEP code. J. Phys. Condens. Matter 2002, 14, 2717. [Google Scholar] [CrossRef]
- Clark Stewart, J.; Segall Matthew, D.; Pickard Chris, J.; Hasnip Phil, J.; Probert Matt, I.J.; Refson, K.; Payne Mike, C. First principles methods using CASTEP. In Zeitschrift für Kristallographie Crystalline Materials; The University of York: York, UK, 2005; Volume 220, p. 567. [Google Scholar]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef]
- Kubota, K.; Asari, T.; Yoshida, H.; Yaabuuchi, N.; Shiiba, H.; Nakayama, M.; Komaba, S. Understanding the Structural Evolution and Redox Mechanism of a NaFeO2–NaCoO2 Solid Solution for Sodium-Ion Batteries. Adv. Funct. Mater. 2016, 26, 6047–6059. [Google Scholar] [CrossRef]
- Perdew, J.P. Density functional theory and the band gap problem. Int. J. Quantum Chem. 1985, 28, 497–523. [Google Scholar] [CrossRef]
- Kröger, F.A.; Vink, H.J. Relations between the Concentrations of Imperfections in Crystalline Solids. In Solid State Physics; Seitz, F., Turnbull, D., Eds.; Academic Press: Eindhoven, The Netherlands, 1956; Volume 3, pp. 307–435. [Google Scholar]
- Nytén, A.; Kamali, S.; Häggström, L.; Gustafsson, T.; Thomas, J.O. The lithium extraction/insertion mechanism in Li2FeSiO4. J. Mater. Chem. 2006, 16, 2266–2272. [Google Scholar] [CrossRef]
- Liu, H.; Choe, M.-J.; Enrique, R.A.; Orvañanos, B.; Zhou, L.; Liu, T.; Thornton, K.; Grey, C.P. Effects of Antisite Defects on Li Diffusion in LiFePO4 Revealed by Li Isotope Exchange. J. Phys. Chem. C 2017, 121, 12025–12036. [Google Scholar] [CrossRef]
- Kempaiah Devaraju, M.; Duc Truong, Q.; Hyodo, H.; Sasaki, Y.; Honma, I. Synthesis, characterization and observation of antisite defects in LiNiPO4 nanomaterials. Sci. Rep. 2015, 5, 11041. [Google Scholar] [CrossRef] [PubMed]
- Politaev, V.V.; Petrenko, A.A.; Nalbandyan, V.B.; Medvedev, B.S.; Shvetsova, E.S. Crystal structure, phase relations and electrochemical properties of monoclinic Li2MnSiO4. J. Solid State Chem. 2007, 180, 1045–1050. [Google Scholar] [CrossRef]
- Guo, S.; Sun, Y.; Liu, P.; Yi, J.; He, P.; Zhang, X.; Zhu, Y.; Senga, R.; Suenaga, K.; Chen, M.; et al. Cation-mixing stabilized layered oxide cathodes for sodium-ion batteries. Sci. Bull. 2018, 63, 376–384. [Google Scholar] [CrossRef]
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).