Congenital fusion of the gums is an extremely rare disorder. This pathology is hardly ever seen in isolation [
1], usually occurring together with syndromes such as Van der Woude, cleft palate alveolar synechiae, and oromandibular limb hypogenesis syndrome. Only 27 cases of syngnathia have been reported since the first case reported in 1936 by Burket [
2]. Laster et al. [
3] counted 24 cases in the literature and reported another case. Daniels [
4] added a case of bony fusion of the mandible to the maxilla and to the zygomatic complex in a neonate; more recently, Parkins and Boamah [
5] reported a case of fusion of the jaws, synechiae of the buccal mucosa and the gingiva, with cleft of the hard palate.
The etiology of this congenital malformation is unclear. A review of the literature identifies eight cases with similar malformations after in utero exposure to immunosuppressive therapy as mother’s treatment for renal transplantation (cleft lip and palate, bilateral microtia and external auditory canal atresia, chorioretinal coloboma, hypertelorism, and micrognathia).
Early treatment to mobilize the jaw is essential not only for the restriction of mouth opening that affects infant growth and development due to problems in feeding, swallowing, and respiration, but also because in the early stages this condition can be treated more easily.
This report is the first case documented in the literature that includes not only fibrous maxillomandibular fusion of the jaws at midline, synechiae, and complete cleft of the median palate, but also facial dysmorphism with duplication of craniofacial midline, including hypophyseal duplication and agenesis of corpus callosum.
Case Report
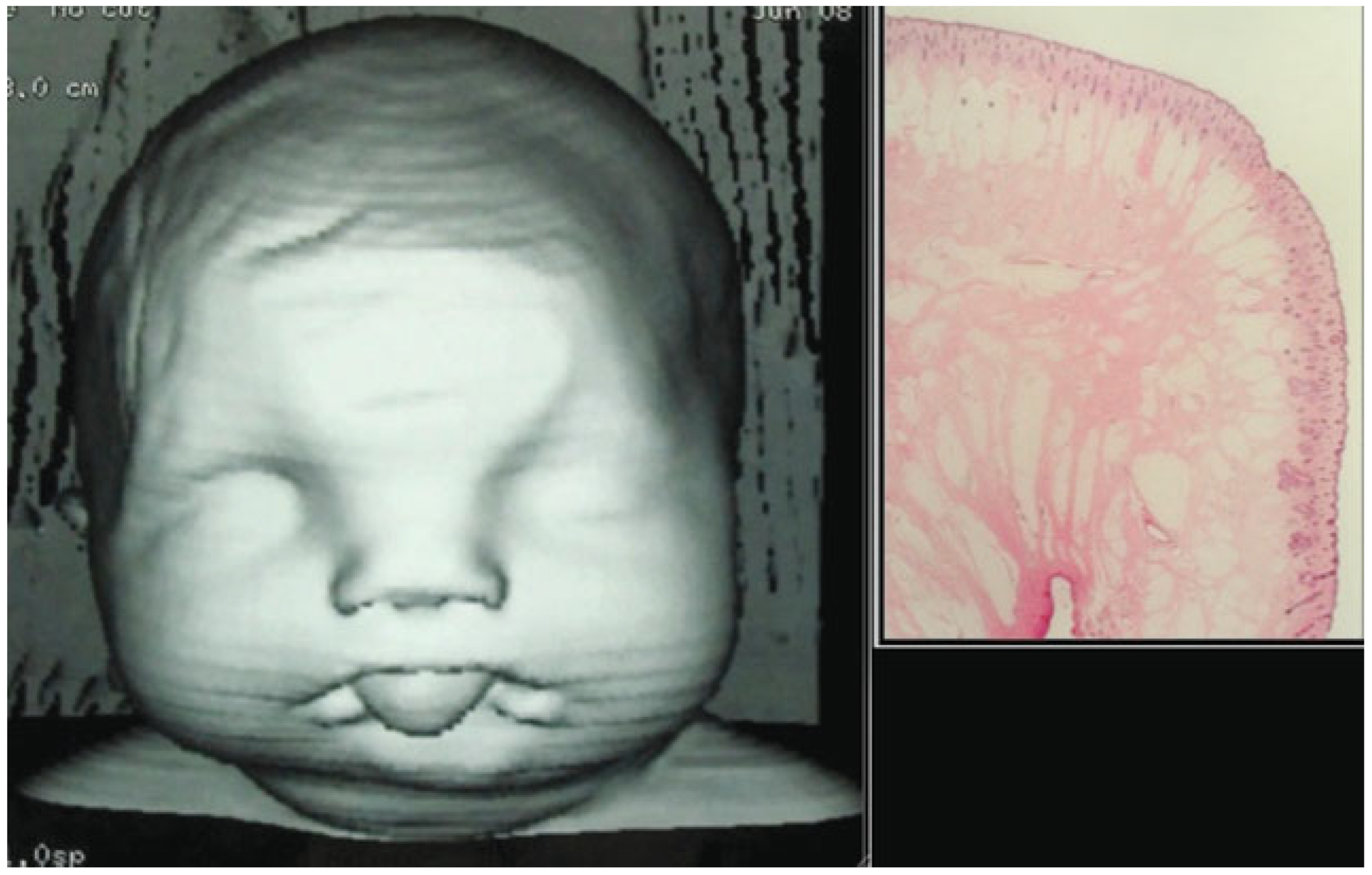
A 1-day-old female baby born at 40 weeks of amenorrhea by cesarean because of cephalopelvic disproportion was referred to Maxillofacial Department in La Paz University Hospital, when the obstetrician noticed that she could not open her mouth after birth because the upper and lower gums were fused (
Figure 1). The patient weighed 3450 g and was 49 cm in height with 37.2-cm head circumference. She was born to healthy, nonconsanguineous parents, neither of whom smoked or drank alcohol.
Neonatal examination exhibited frontonasal dysmorphism (bifid anterior cranium, hypertelorism, blepharophimosis, broad nasal bridge, and broad nasal tip), low implantation of the ears, a short neck, bifid and protrusive tongue covered with lanugo, inferior lip and buccolabial sulci malformation, macrostomia, microretrognathia, and except for a small slit in the lateral regions, through which only the end of alveolar ridge could be seen, the mandible and maxilla were fused at gum level (
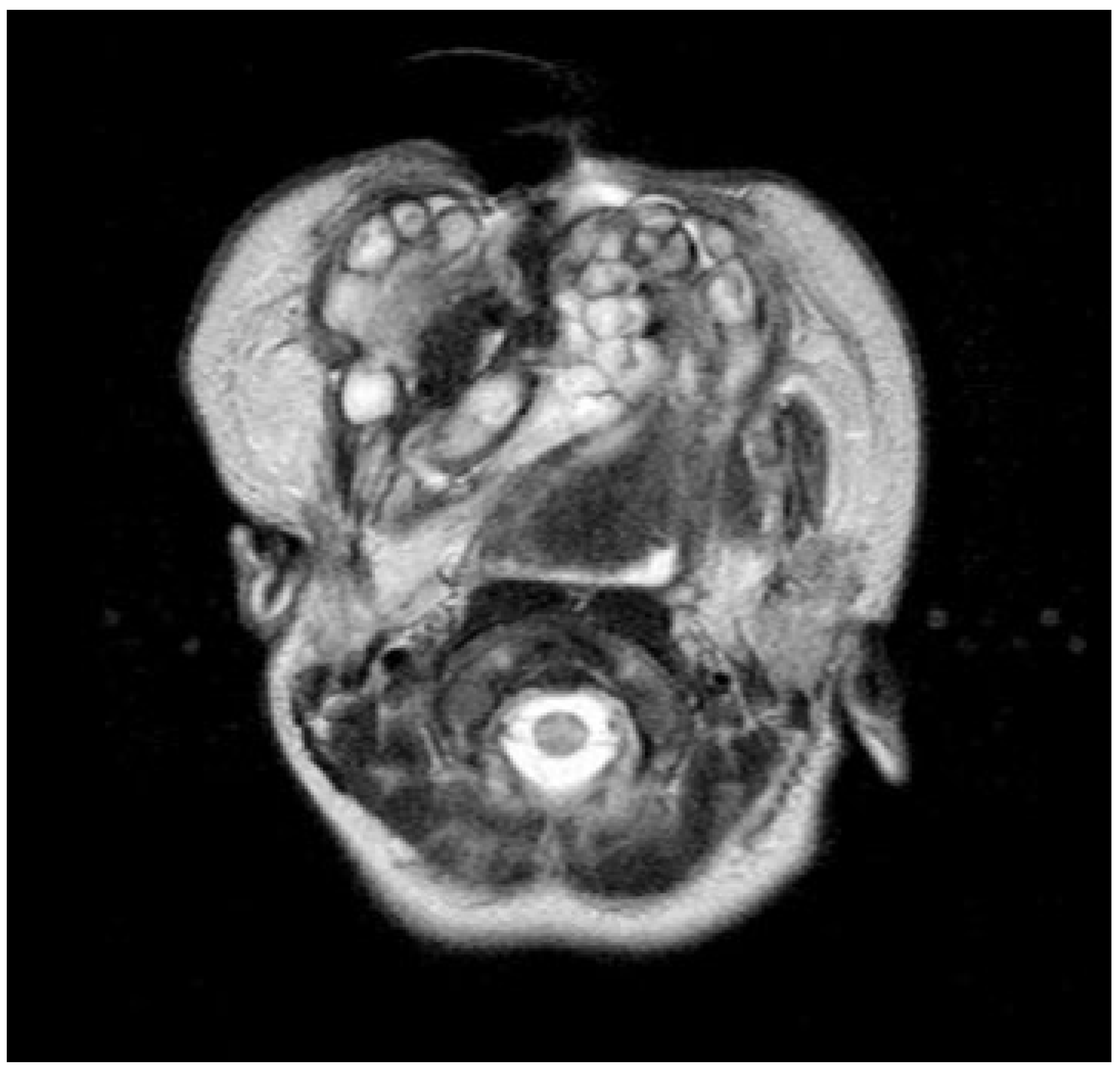
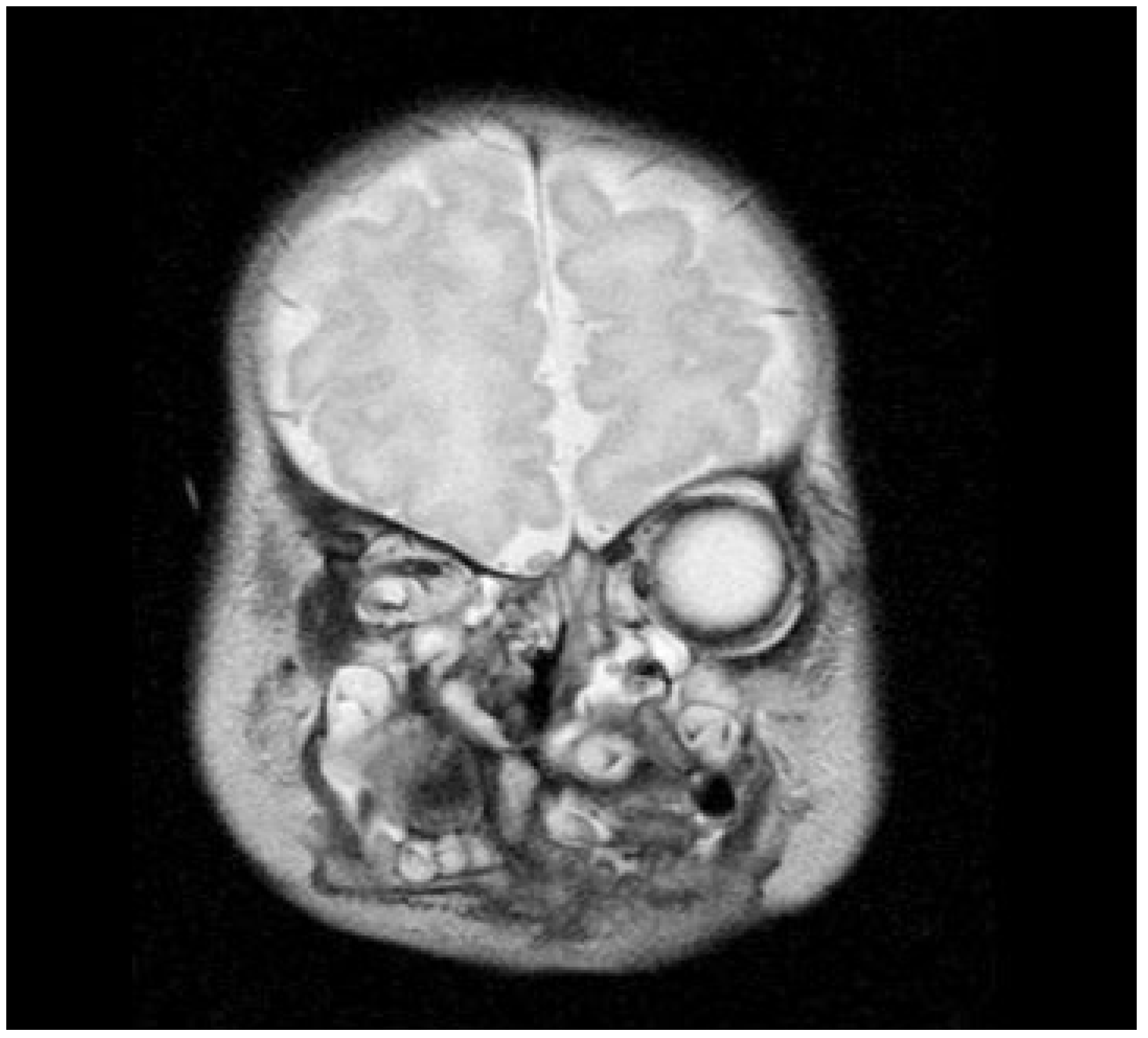
Figure 2). Nasoendoscopy showed complete cleft of the median palate. Magnetic resonance imaging (MRI) revealed orbital hypertelorism, midline cranial cleft ending just superior to the nasal dorsum, agenesis of the corpus callosum, and maxillomandibular fusion (
Figure 3 and
Figure 4).
She exhibited no respiratory distress, and nasotracheal intubation or tracheotomy was unnecessary; intervention was planned 1 week later. Airway management made it necessary to perform a tracheotomy. Neurologically, she was conscious with correct muscular tone. The laboratory analysis revealed no pathology. Thorax radiology showed a double image of vertebral bodies in the cervical level. Ultrasound revealed the agenesis of corpus callosum, as suggested by the MRI. Computed tomography showed the same pathology in addition to complete frontonasal dysplasia (
Figure 5). Ophthalmologic study described a severe hypertelorism with blepharophimosis. Pupillary and foveal reflexes were normal. Transparent corneas were normal in size with absence of cataracts. Abdominal ultrasonography discarded any other associated pathology. Surgical treatment was planned with a stereolithographic model when the child was 2 weeks old.
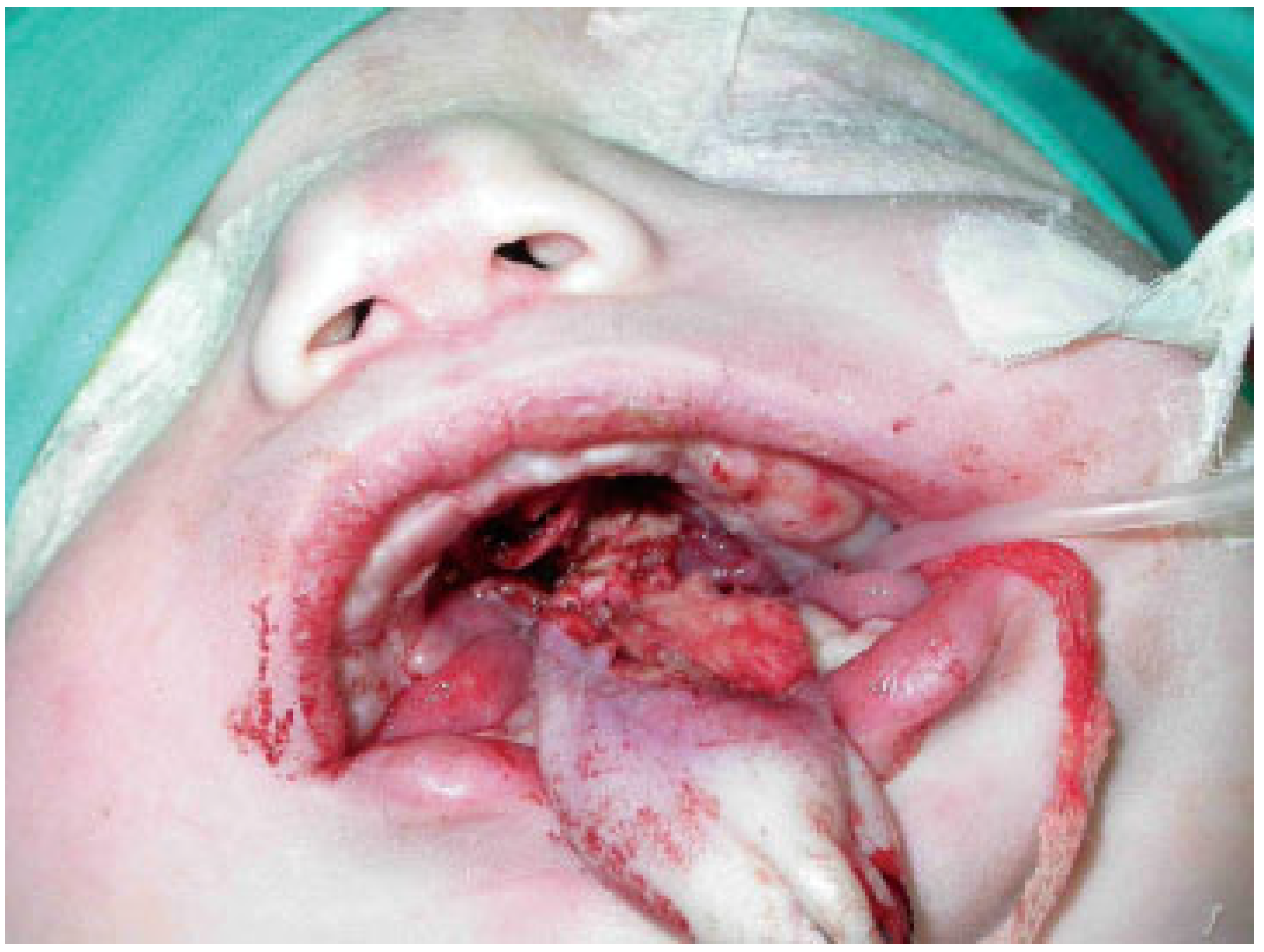
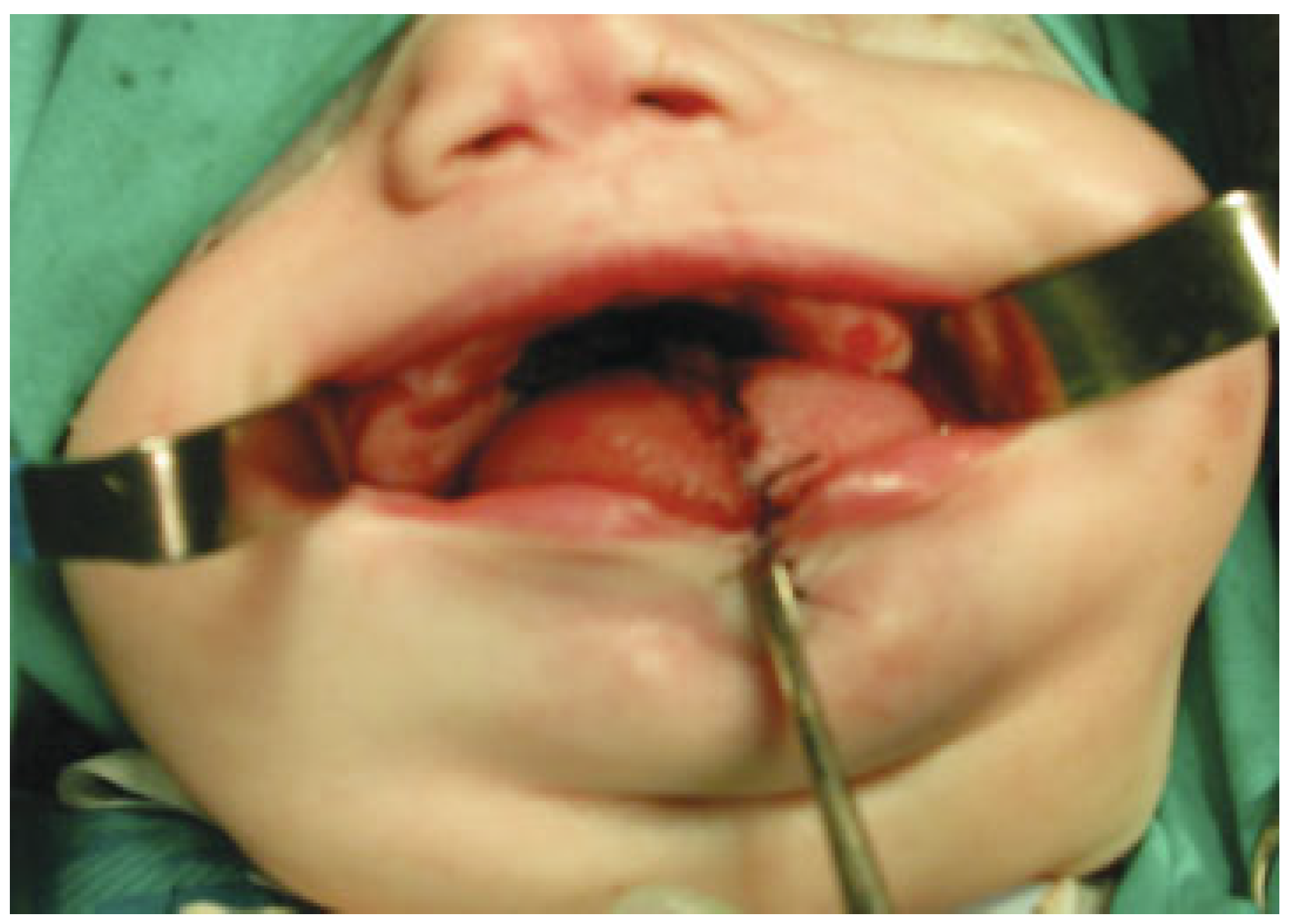
Under general anesthesia and ventilation by tracheostomy, the gums were separated through blunt dissection and a periosteal elevator to separate the fibrous union of the gingiva and also the synechiae of the buccal mucosa (
Figure 6 and
Figure 7).
The third day after the intervention, a gastroesophageal study was perform. The gastrografin contrast revealed no existence of tracheoesophageal fistulae. A minor hiatal hernia was described. With the surgical success of the maxillomandibular pathology, the newborn died of the serious associated neurological disorders.
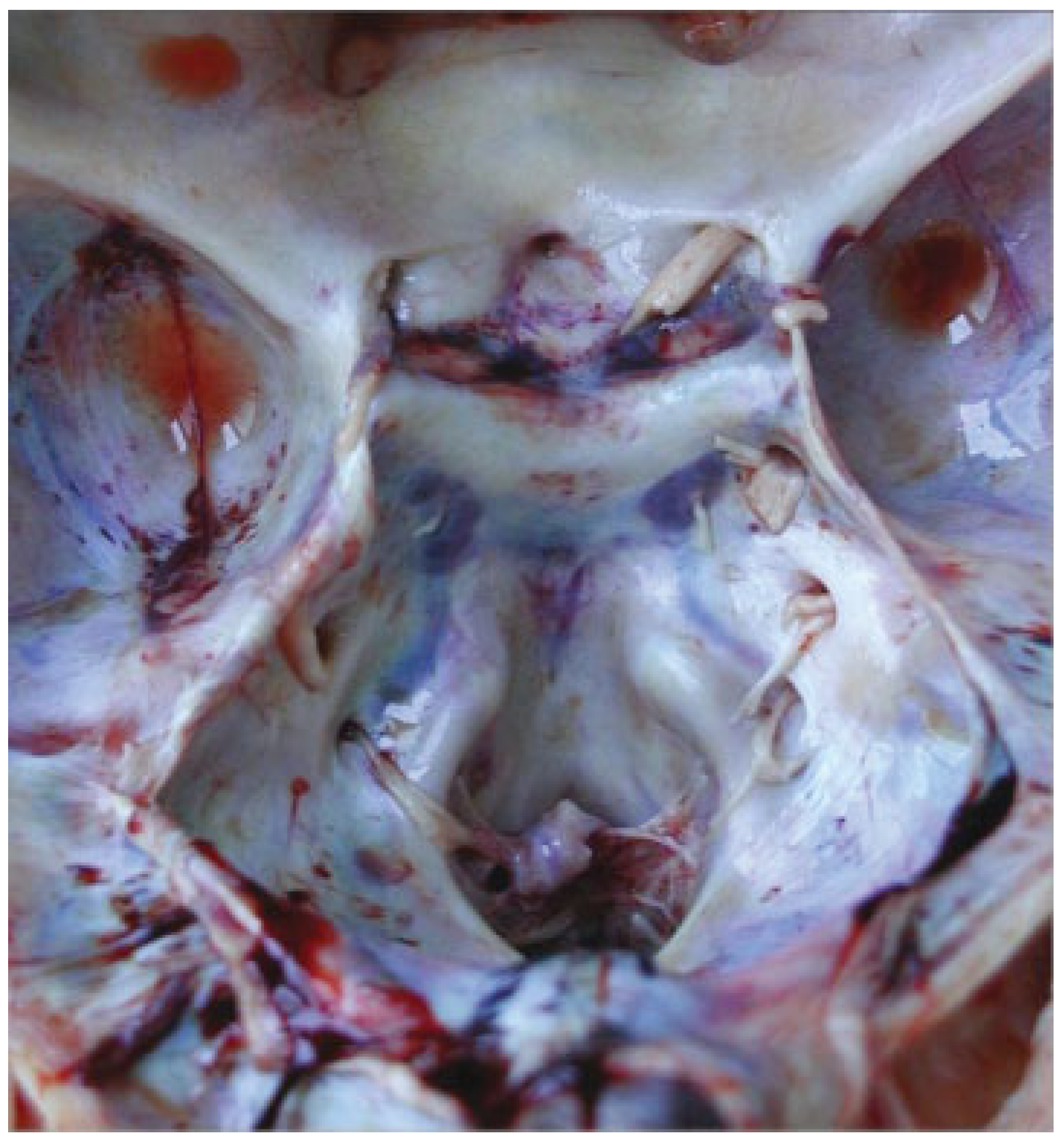
The autopsy study described a girl, 2 months and 20 days old, with karyotype (46,XX). She had frontonasal malformation (hypertelorism, broad nasal bridge, bifid nasal tip and anterior cranium), macrostomia, retrognathia, and bifid tongue. She also presented maxillomandibular bony fusion and fusion of the tongue to the palate and to the inferior lip by a hairy polyp (
Figure 8), both corrected surgically. In the cranium, the sella turcica was broadened (
Figure 9), with little prominence of the clinoid apophyses and with two cavities that contained two complete hypophyses with two infundibulums (
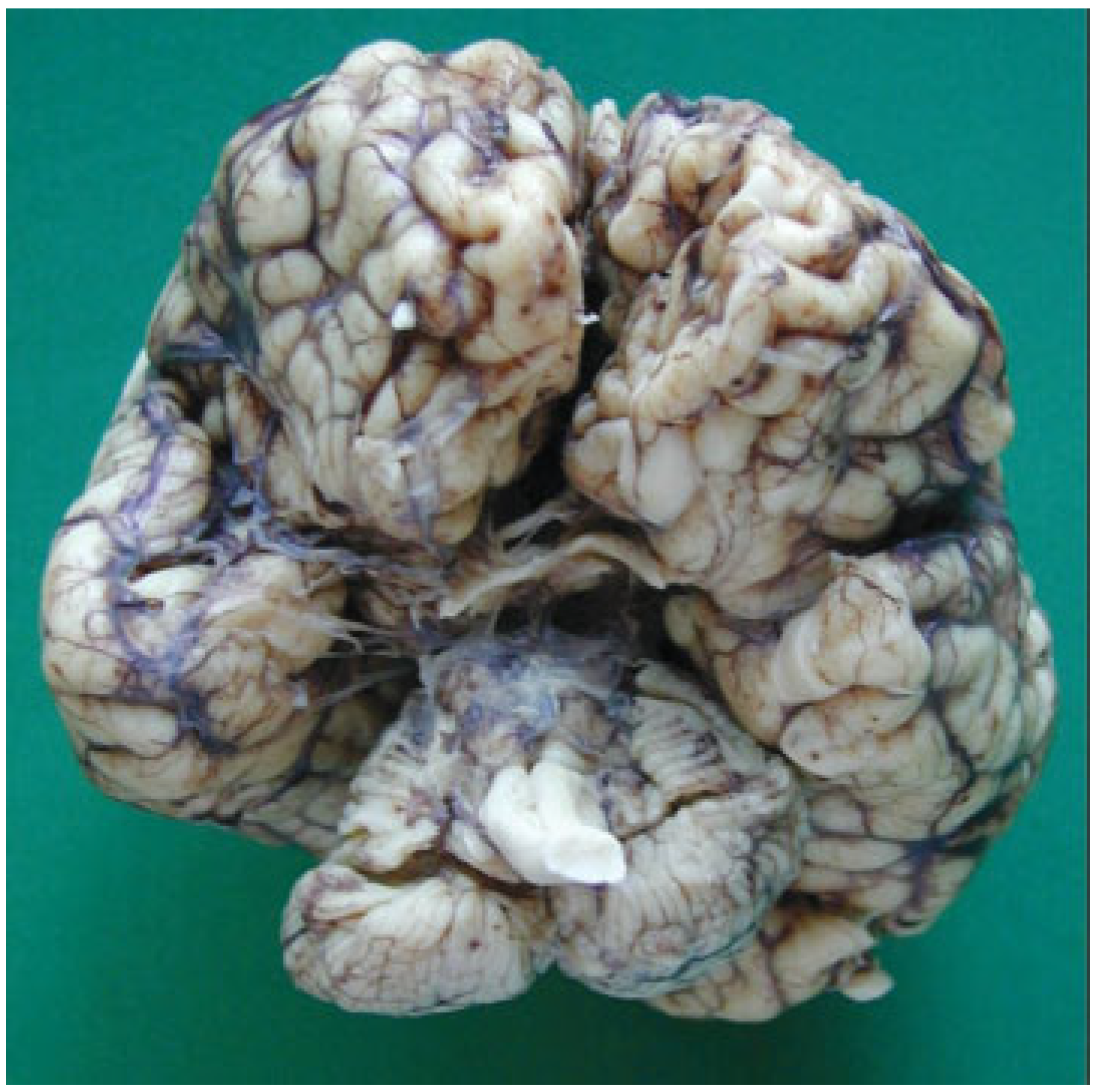
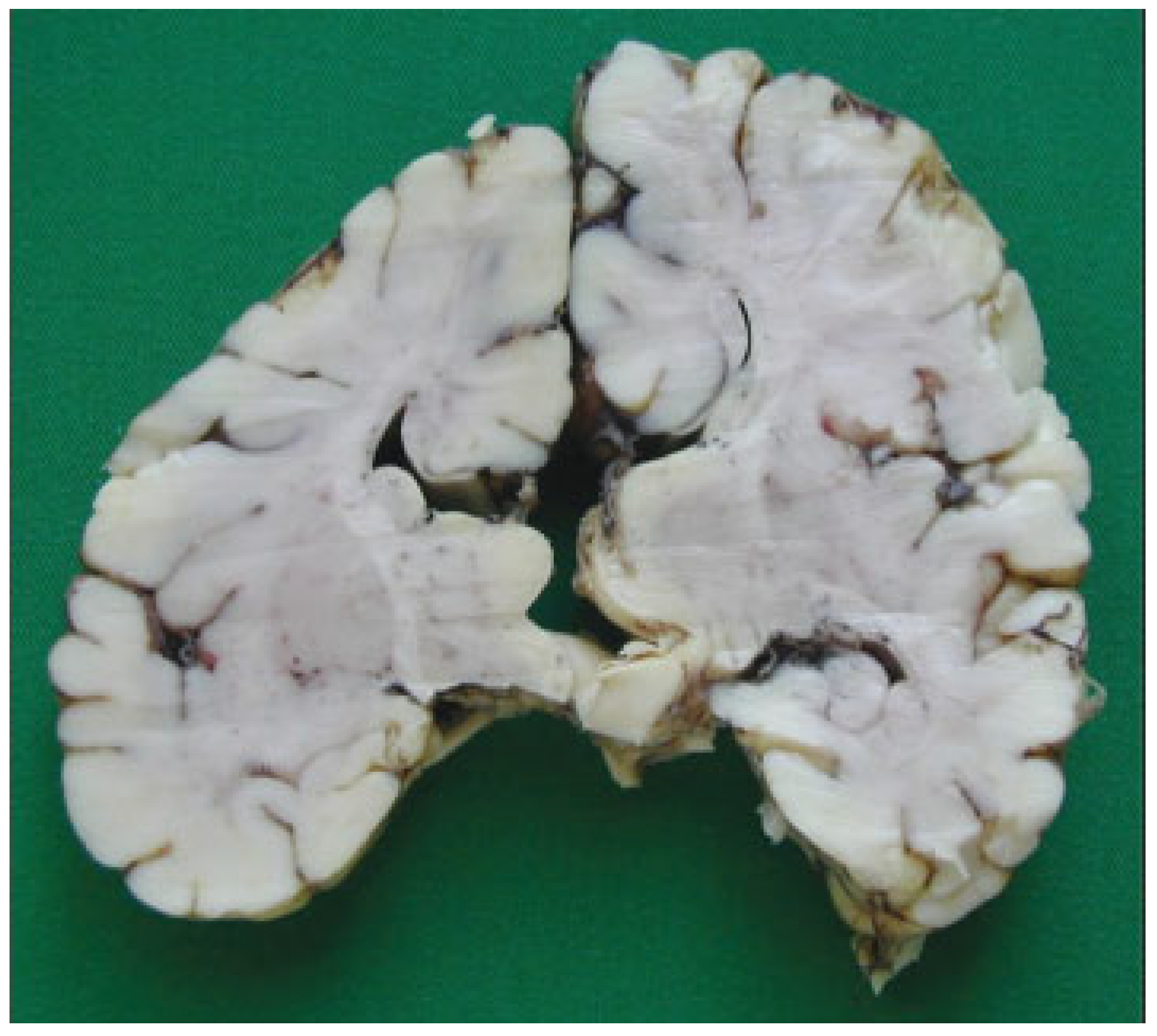
Figure 10). The three cranial fossae were narrowed anteroposteriorly. The central nervous system (CNS) had both olfactory bulbs and corpus callosum agenesis (
Figure 11 and
Figure 12), anomalous morphology of the brain stem and spinal cord (
Figure 13), and neuronal heterotopia in occipital white matter and leptomeningeal heterotopia.
Discussion
The median craniofacial dysraphia, one of the most frequent atypical facial clefts, represents ~40% of all atypical clefts. Clinical exam of the patient may reveal orbital hypertelorism, bifid nose, V-shaped frontal hairline, duplication of nasal septum and anterior nasal spine, and median cleft of the upper lip (premaxilla and palate) [
6].
DeMyer [
7] proposed that if any two or more of these components are present, the median cleft face syndrome diagnosis is justified. The components of this syndrome include bifid cranium, hypertelorism, median cleft nose, median cleft lip and premaxilla. Hypophyseal duplication is not included in it.
Review of the embryology of these parts of the face leads to the presumption that the primary defect is the fusion of the alveolar ridges. By the fifth week of fetal life, the mandibular arch is quite distinct, developing from the first branchial arch. Between the weeks 8 and 12, there is a marked acceleration of the mandibular growth. At the same time, the two maxillary processes laterally and the nasal process medially form the palate and maxillary alveolar ridge. The lateral segments grow toward the midline by differential proliferations. The nasal septum proliferates downward and unites in the midline with the lateral palatal segments. Fusion progresses from anterior to posterior [
8]. It seems, therefore, that the probable sequence of events in this baby was: (1) fusion of the gums; (2) posterior cleft palate due to failure of the tongue mass to drop fully because of the micrognathia and the fused gums.
Maxillomandibular fusion is usually discovered immediately after birth because the baby is not able to open the mouth or feed normally, unless the patient had partial fusion of the gums and was able to feed by sucking through the small opening on the sides between the teeth [
9]. Duplication of the pituitary gland is a very rare malformation [
10].
There are seven previous autopsy cases of hypophyseal duplication associated with the following frontonasal malformations, in order of frequency (
Table 1): hypertelorism (six cases), an intraoral mass (epignathus) [
4], bifid tongue [
3], duplication of the anterior groove of the spinal cord [
1], and ‘‘V’’-shaped hairline [
1]. The associated anomalies of the CNS reported were broadened sella turcica and hypothalamic anomalies [
7], corpus callosum agenesis [
5] and olfactory bulbs agenesis [
2], and glial and/or neuronal heterotopias [
1].
The present case has several common features with the one described by Hori in 1983 [
11], which, in addition, had cleft palate, abnormal circle of Willis, and duplication of the spinal cord groove extended to the thoracic level but had no olfactory bulbs agenesis. There are several hypotheses about the origin of the hypophyseal duplication and the associated malformations, yet none of them is definitive.
This case includes features not reported in the other case reports: the agenesis of corpus callosum with duplication of craniofacial facial midline. The corpus callosum, which comprises the largest fiber tract in the brain, connects the left and right cerebral hemispheres. The flow of information facilitated by this tract plays an integral role in many critical functions, including behavior, emotion, and higherorder cognition. Indeed, defective development of this tract in humans is correlated with a large number of syndromes, such as Mowat-Wilson syndrome, acrocallosal syndrome, and Aicardi syndrome, as well as disorders including mycophenolate-associated embryopathy, autism, and schizophrenia [
12]. Formation of the corpus callosum requires a series of dynamic events to be coordinated both spatially and temporally during both embryogenesis and the postnatal period. These include correct patterning of the midline, differentiation and specification of callosal neurons within the nascent cortical plate, development of distinct midline glial populations, targeting of axons to the contralateral hemisphere, and elimination of those supernumerary axons overproduced during development [
12]. These conditional factors for the development of the corpus callosum could explain its association with pathologies of the craniofacial midline such as duplication of the pituitary gland [
10] and median cleft face syndromes [
6].
Burket in 1936 [
2] was the first to report a patient who also had temporomandibular joint ankylosis, fusion of gums, and associated facial hemiatrophy as well as Horner’s syndrome. Laster et al. [
3] counted 24 cases in the literature and reported another case. The last case was reported by Parkins and Boamah [
5]; thus there are a total of 27 in the literature. This report is a case of fibrous maxillomandibular fusion of the jaws, synechiae of the buccal mucosa and the gingiva with cleft of the hard palate, with the unusual feature of agenesis of corpus callosum, hypertelorism, and facial dysmorphism due to midline cranial duplication.
The classification for syngnathia regarding other anomalies was proposed by Dawson et al. [
13] as follows: type 1: simple syngnathia, with no other anomalies in the head and neck; type 2: complex syngnathia, with subgroups, type 2a: syngnathia coexistent with aglossia and type 2b: syngnathia coexistent with agenesis or hypoplasia of proximal mandible.
However, Laster et al. [
3] proposed a modified classification for bony syngnathia after their experience with a case of fusion of the mandible to the zygomatic complex and maxillary tuberosity: type 1a: simple anterior syngnathia, characterized by bony fusion of the alveolar ridge only and without other congenital deformity in the head and neck; type 1b: complex anterior syngnathia, characterized by bony fusion of the alveolar ridges only and associated with other congenital deformity in the head and neck; type 2a: simple zygomaticomandibular syngnathia characterized by bony fusion of the mandible to the zygomatic complex causing only mandibular micrognathia; type 2b: complex zygomaticomandibular syngnathia, characterized by bony fusion of the mandible to the zygomatic complex and associated with clefts or temporomandibular joint ankylosis.
The severity of this disorder has no fixed pattern. The range of jaw fusion extends from complete fusion [
8], unilateral fusion [
14], or bilateral fusion with a small anterior opening [
1,
4].
One of the most relevant surgical aspects is the airway maintenance during the anesthesia, including tracheotomy [
15,
16], and the fiber-optic intubation [
17], which was the most appropriate method for the proper management of the airway in this case.






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}