
Discovery of Flavonoids as Novel Inhibitors of ATP Citrate Lyase: Structure–Activity Relationship and Inhibition Profiles
 , ,
, ,
Abstract
:1. Introduction
2. Results
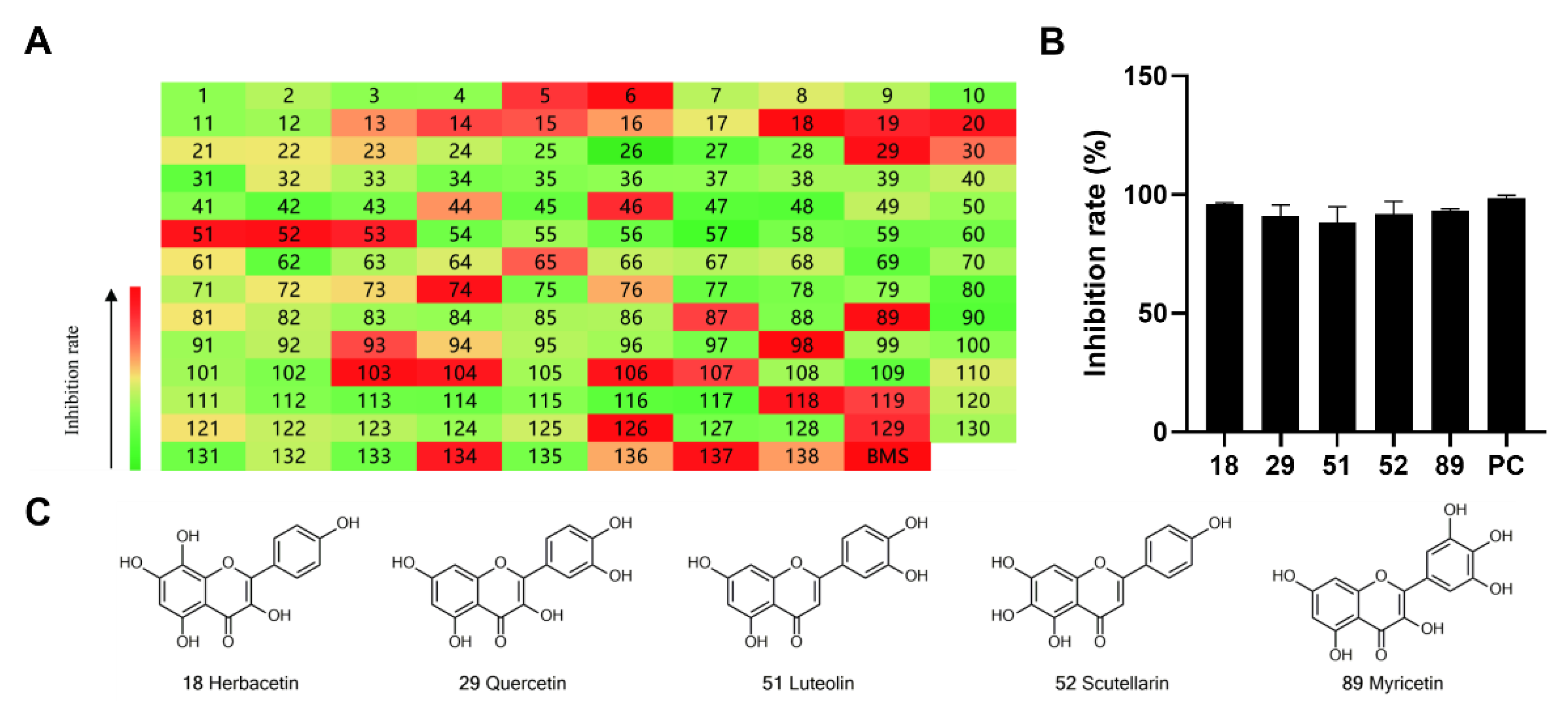
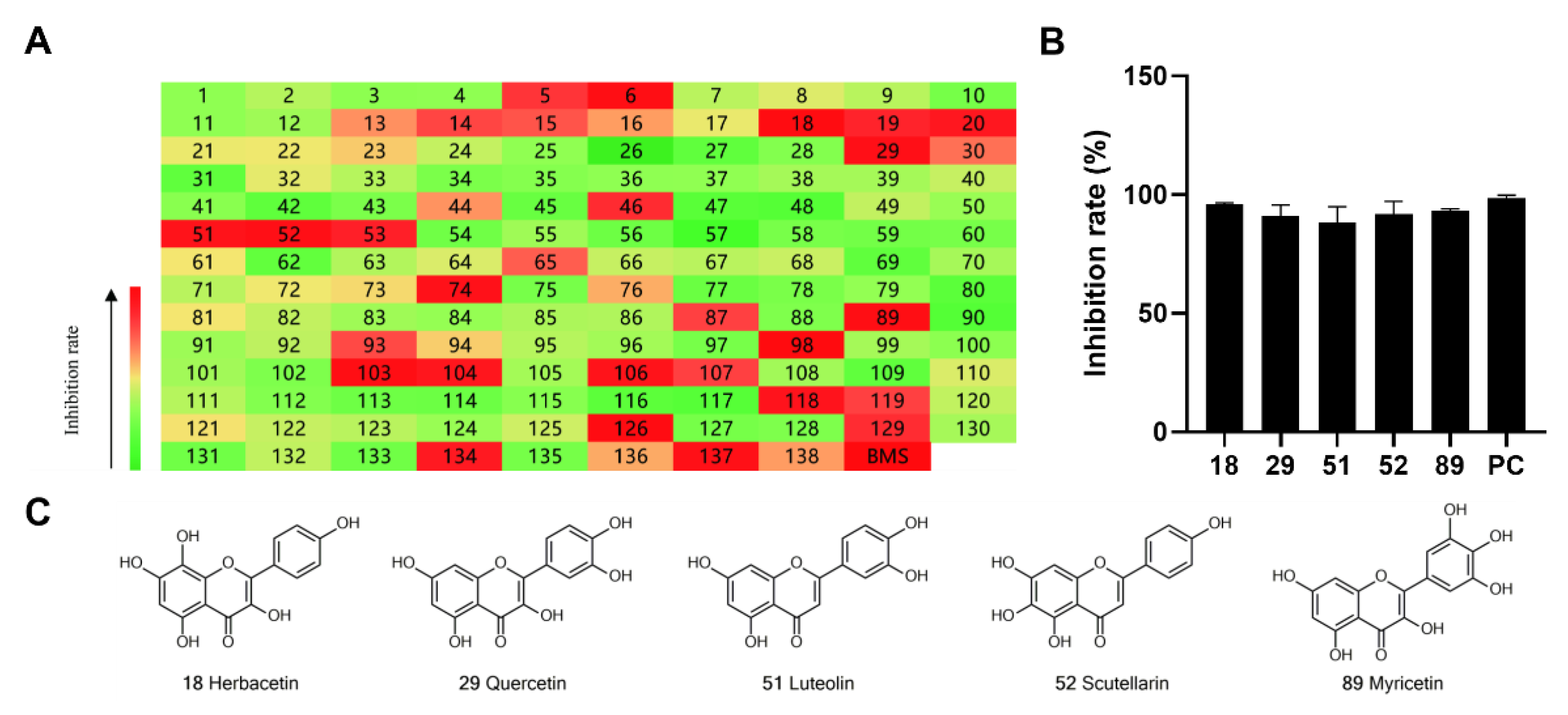
2.1. Preliminary Screening of Natural Flavonoids for ACLY Inhibitors
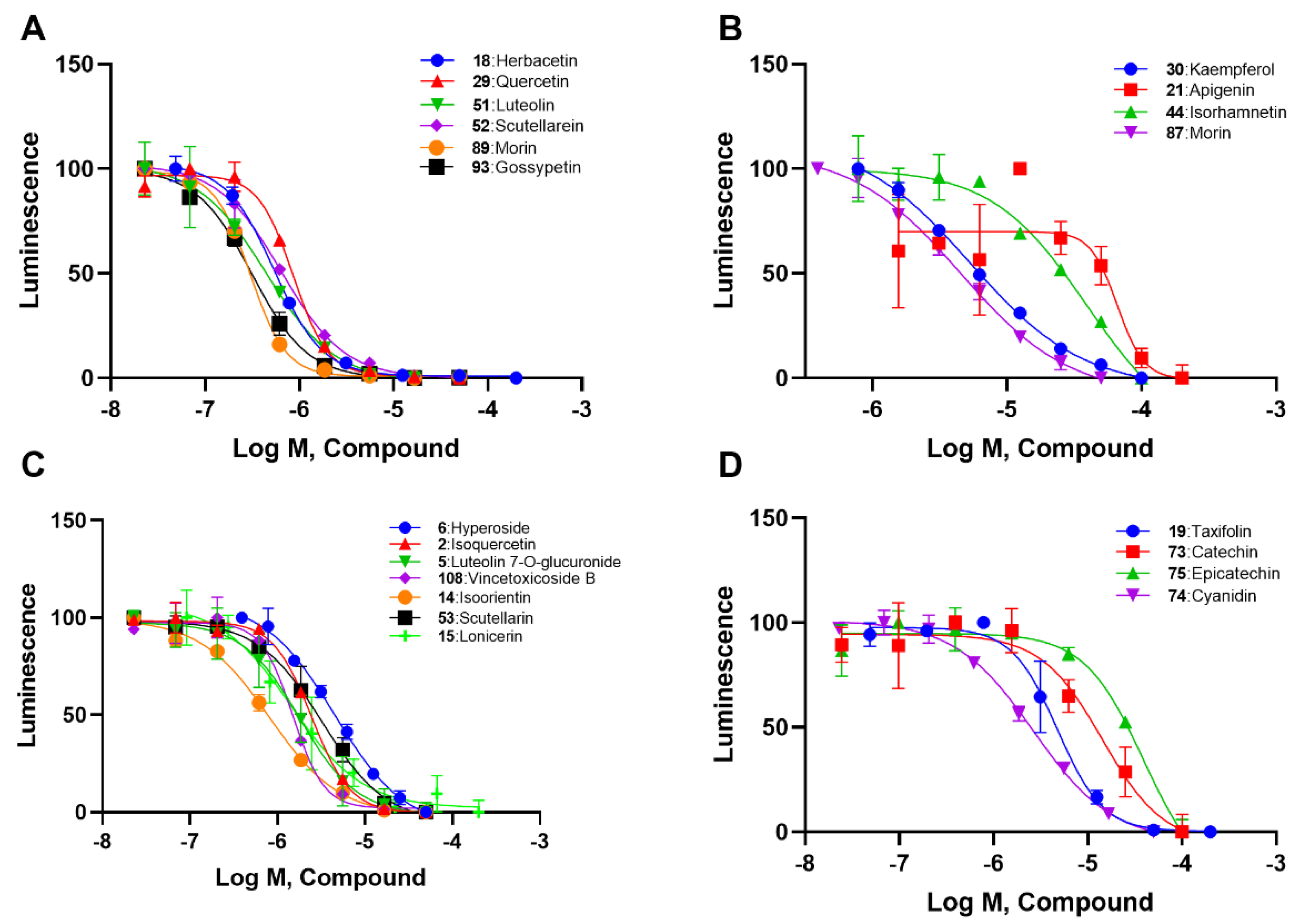
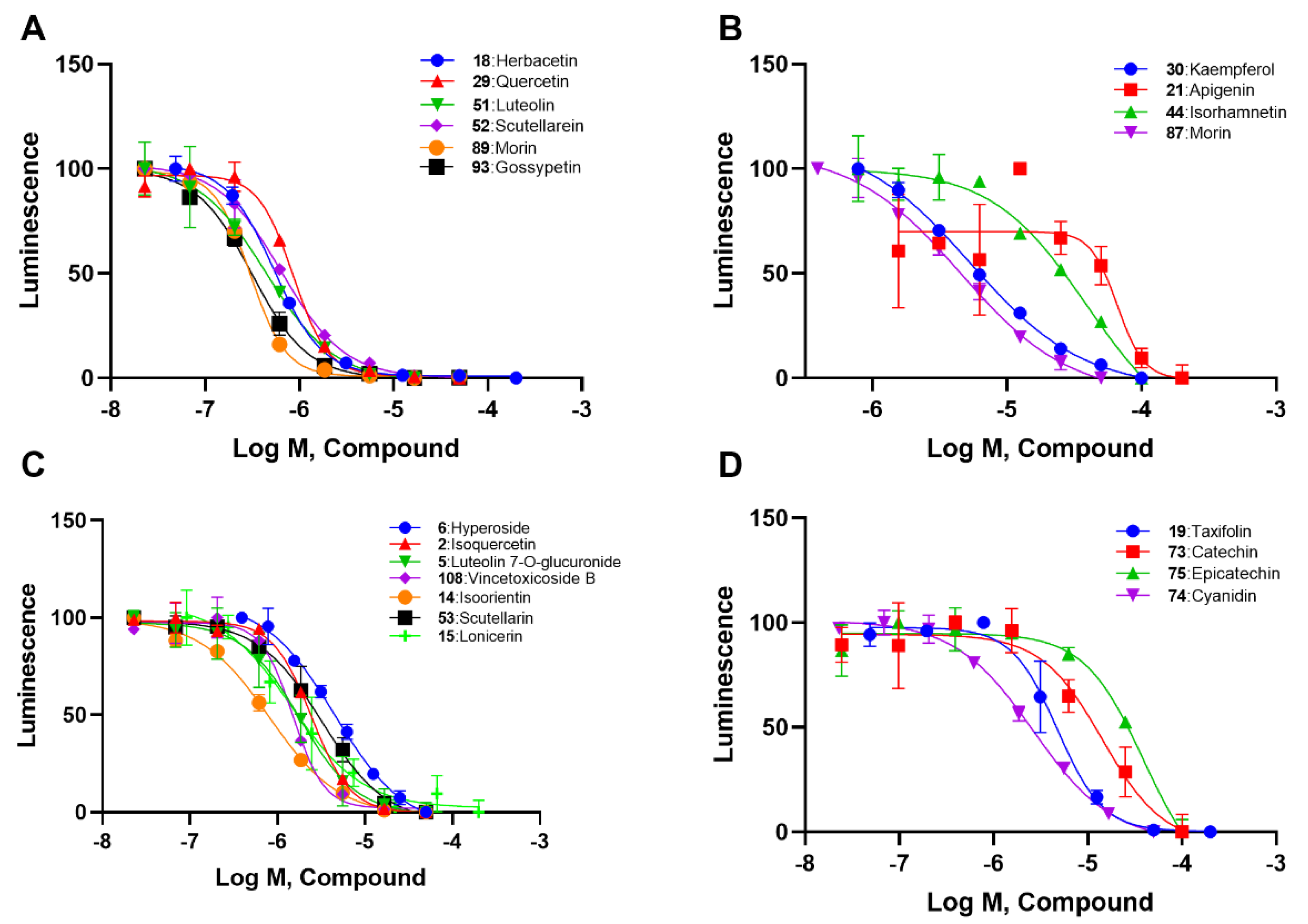
2.2. SAR Analysis of Flavonoids
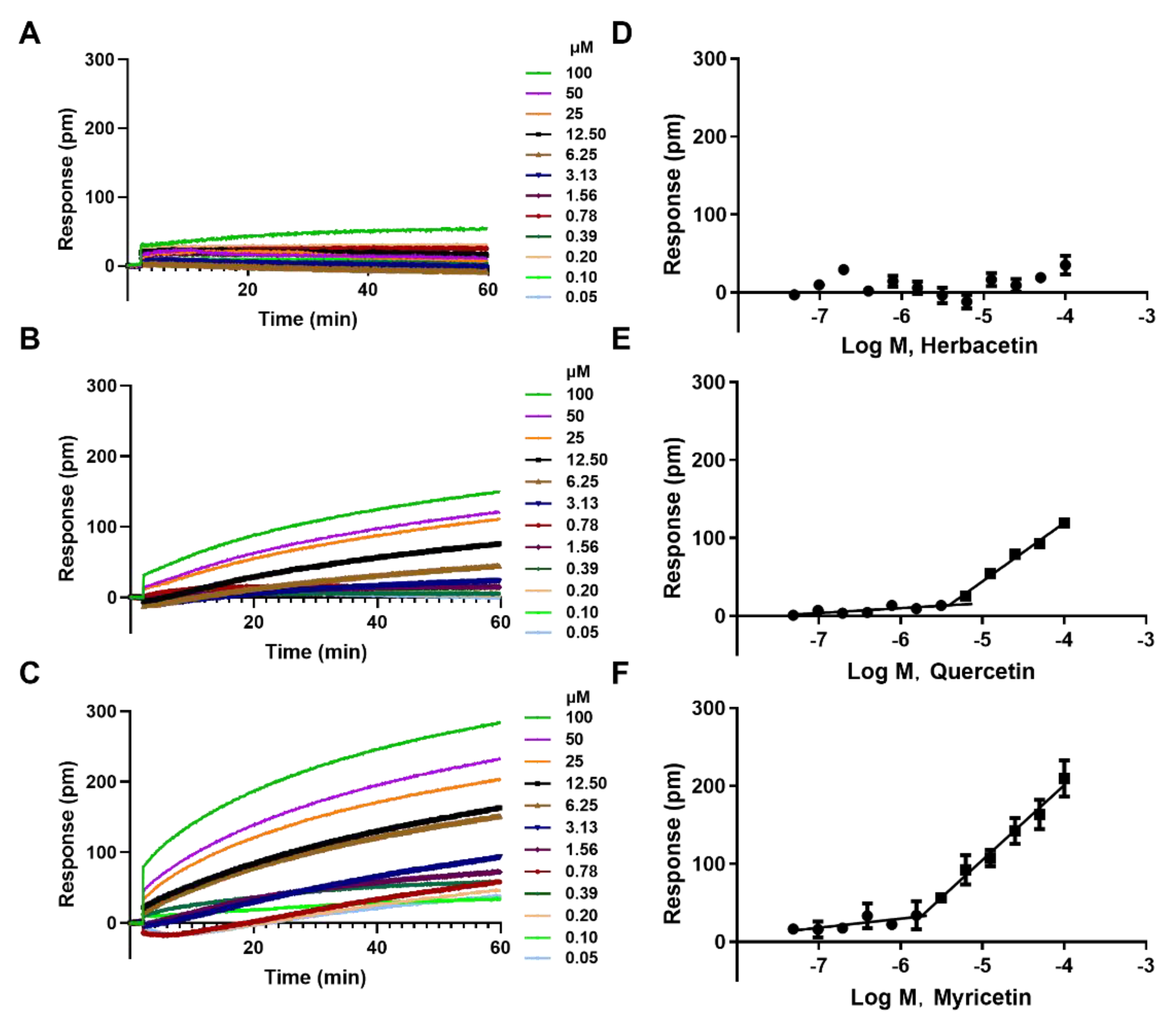
2.3. The Aggregation Analysis of 3 Ortho–Dihydroxyphenyl Flavonoids
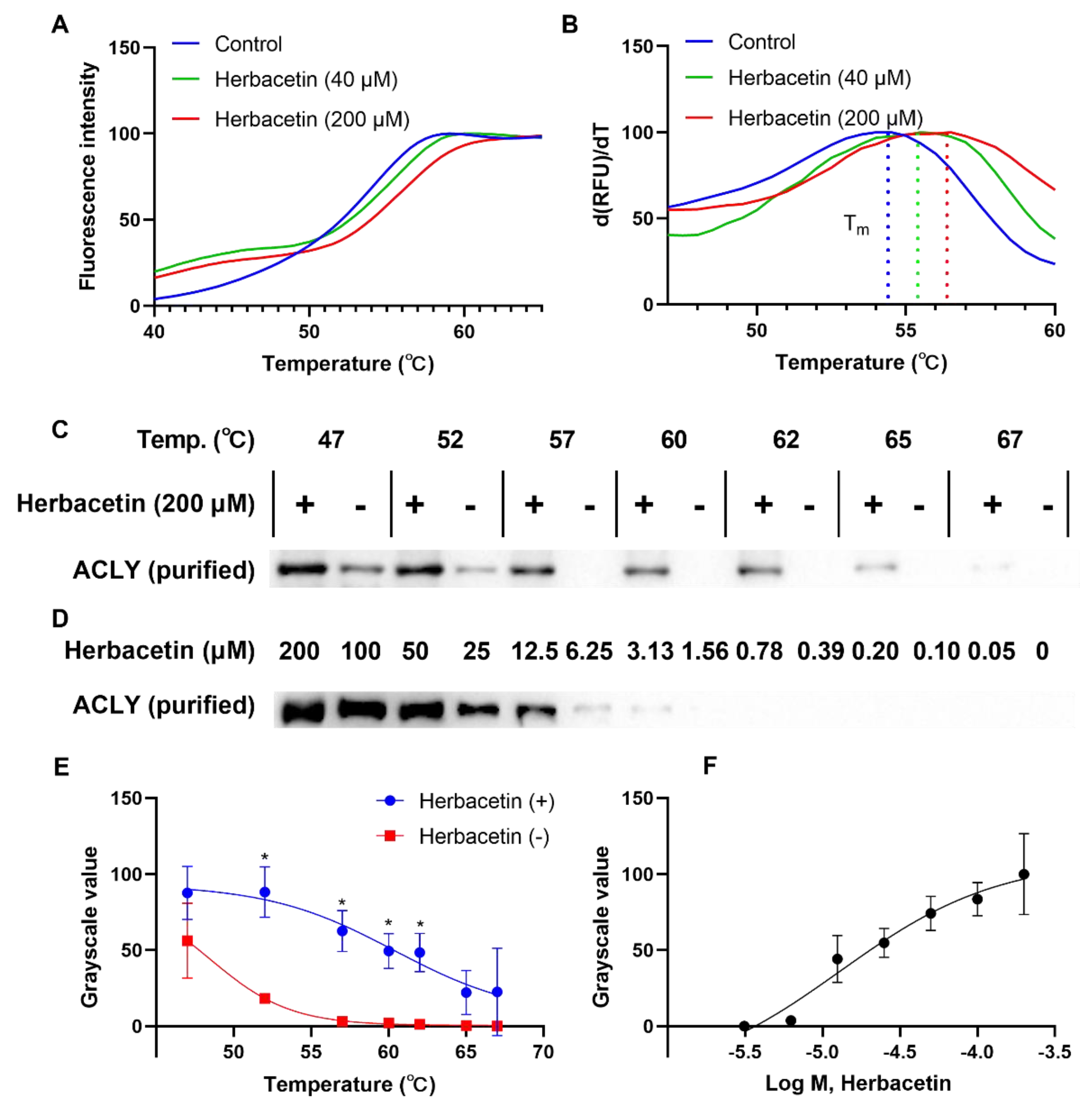
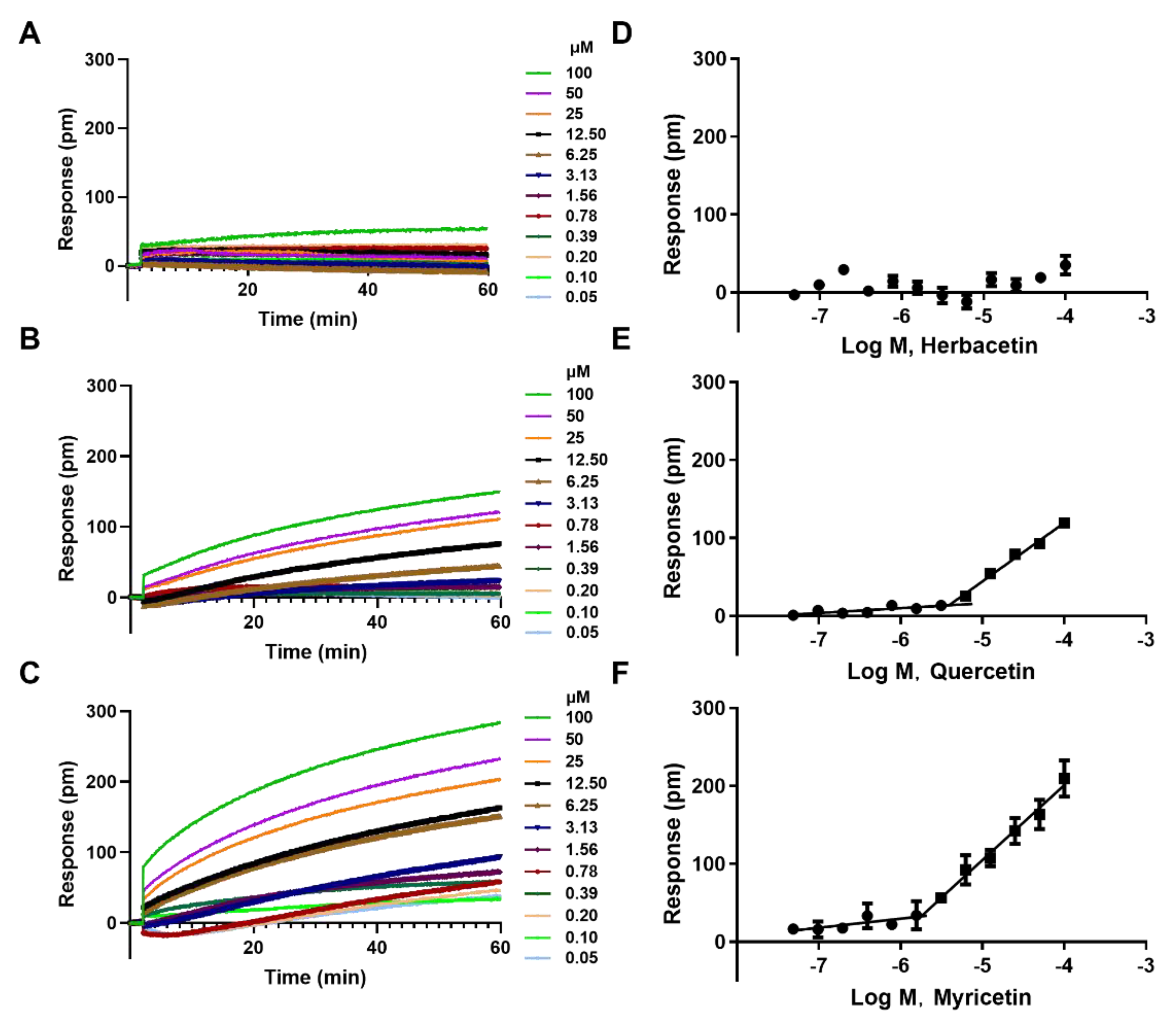
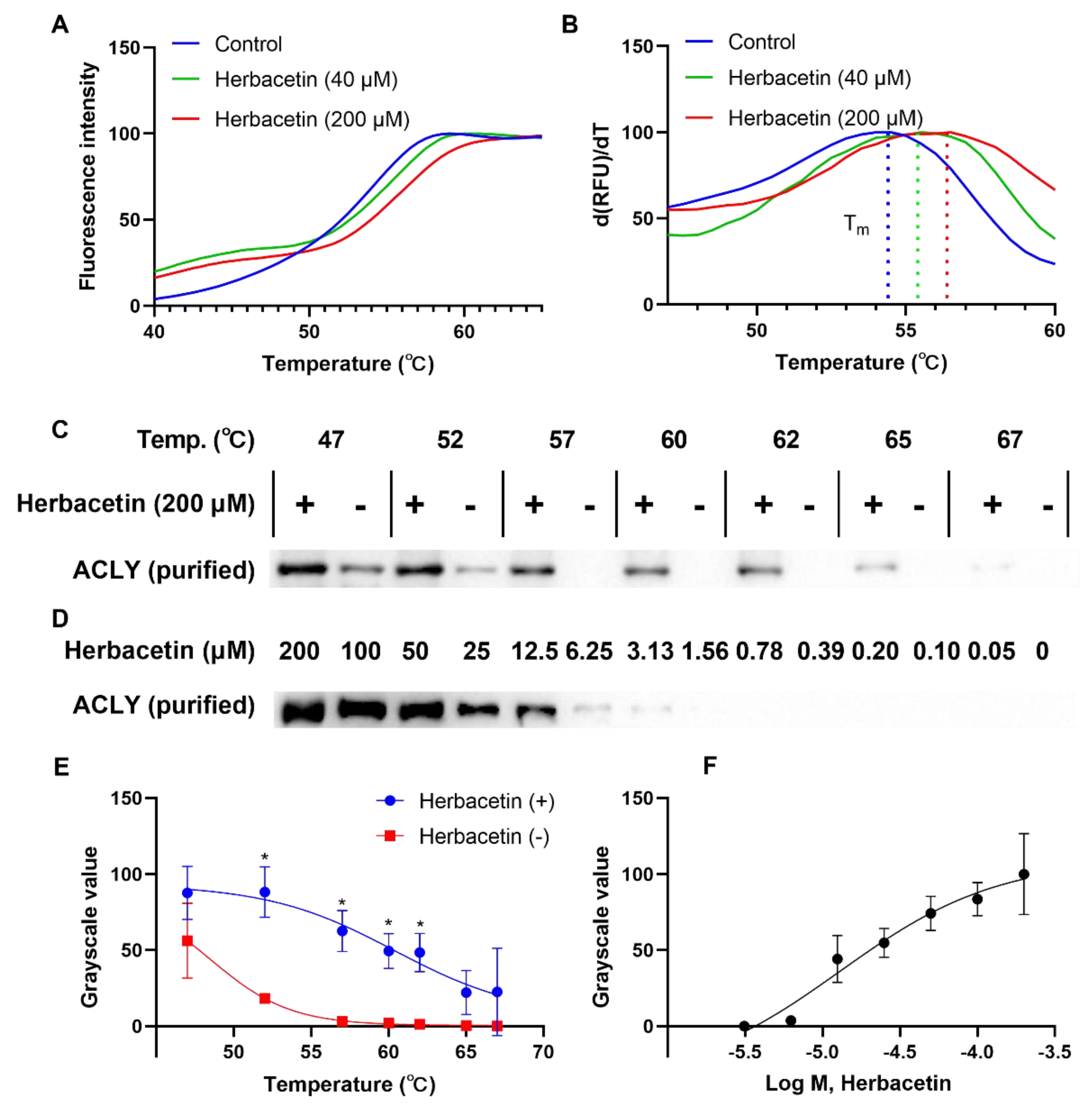
2.4. Direct Interaction of Herbacetin with ACLY
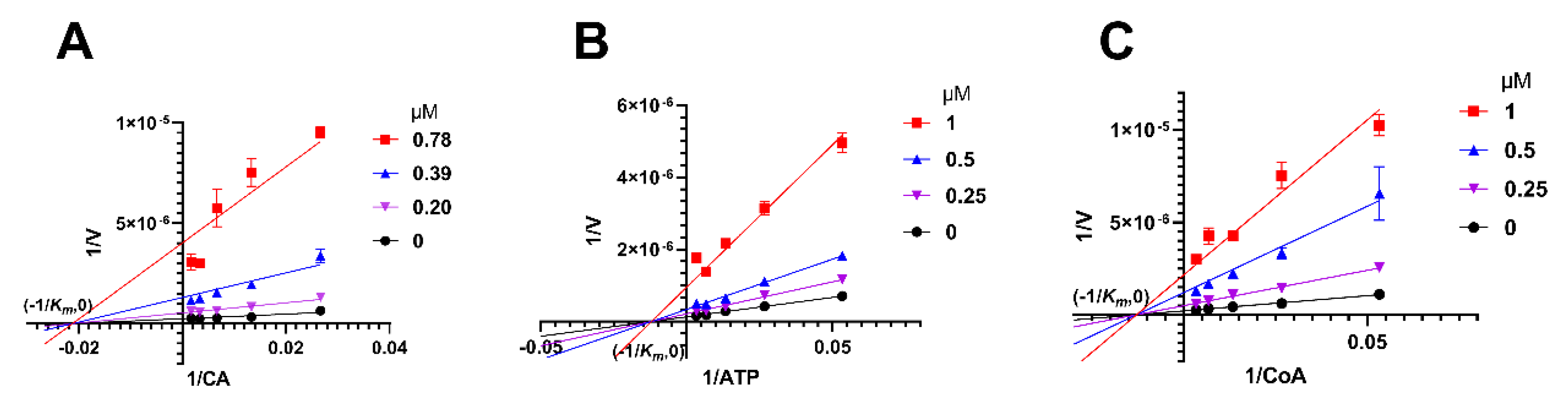
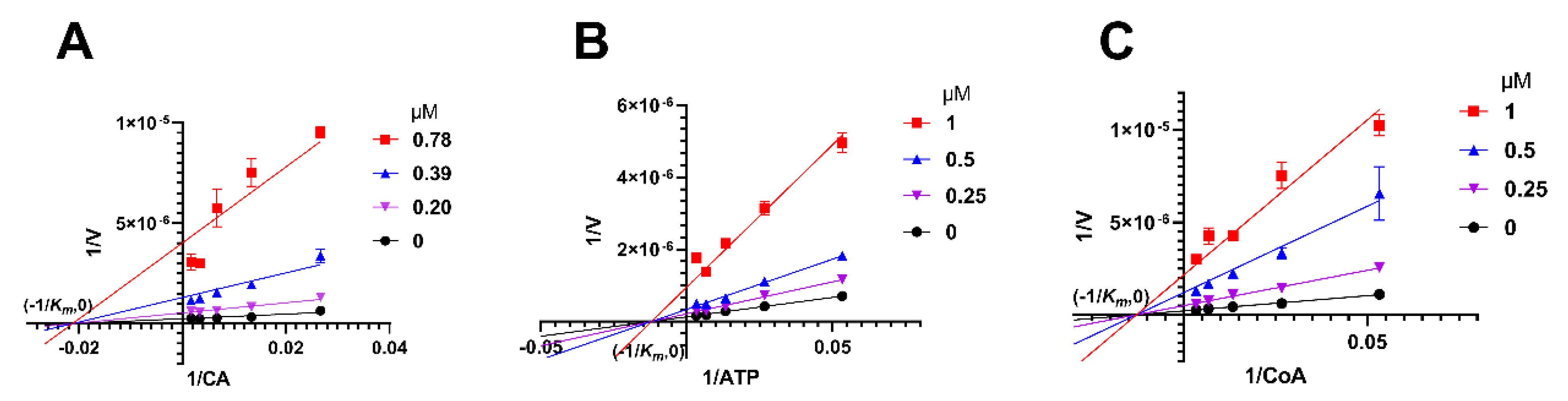
2.5. Herbacetin Was a Noncompetitive Inhibitor of ACLY
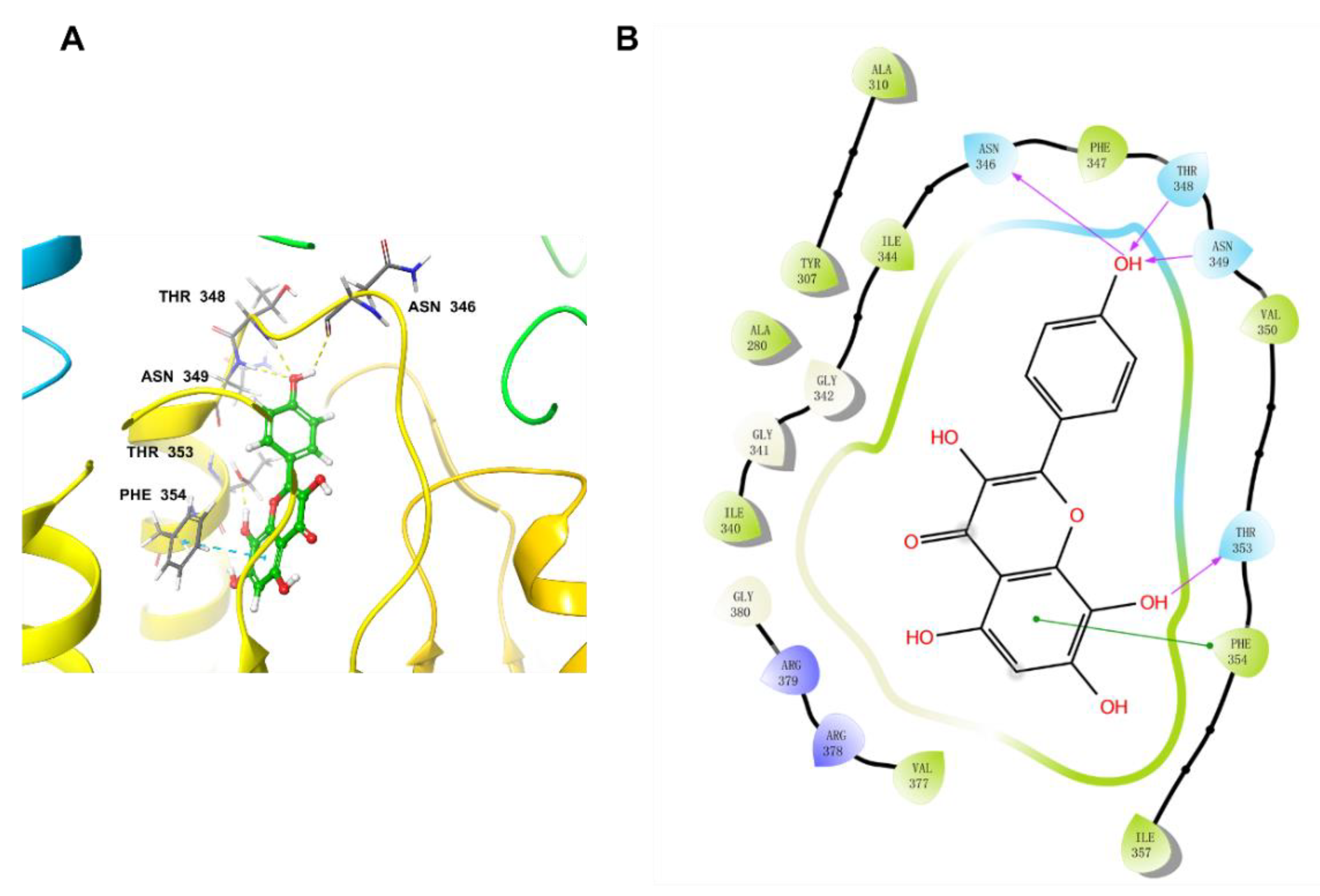
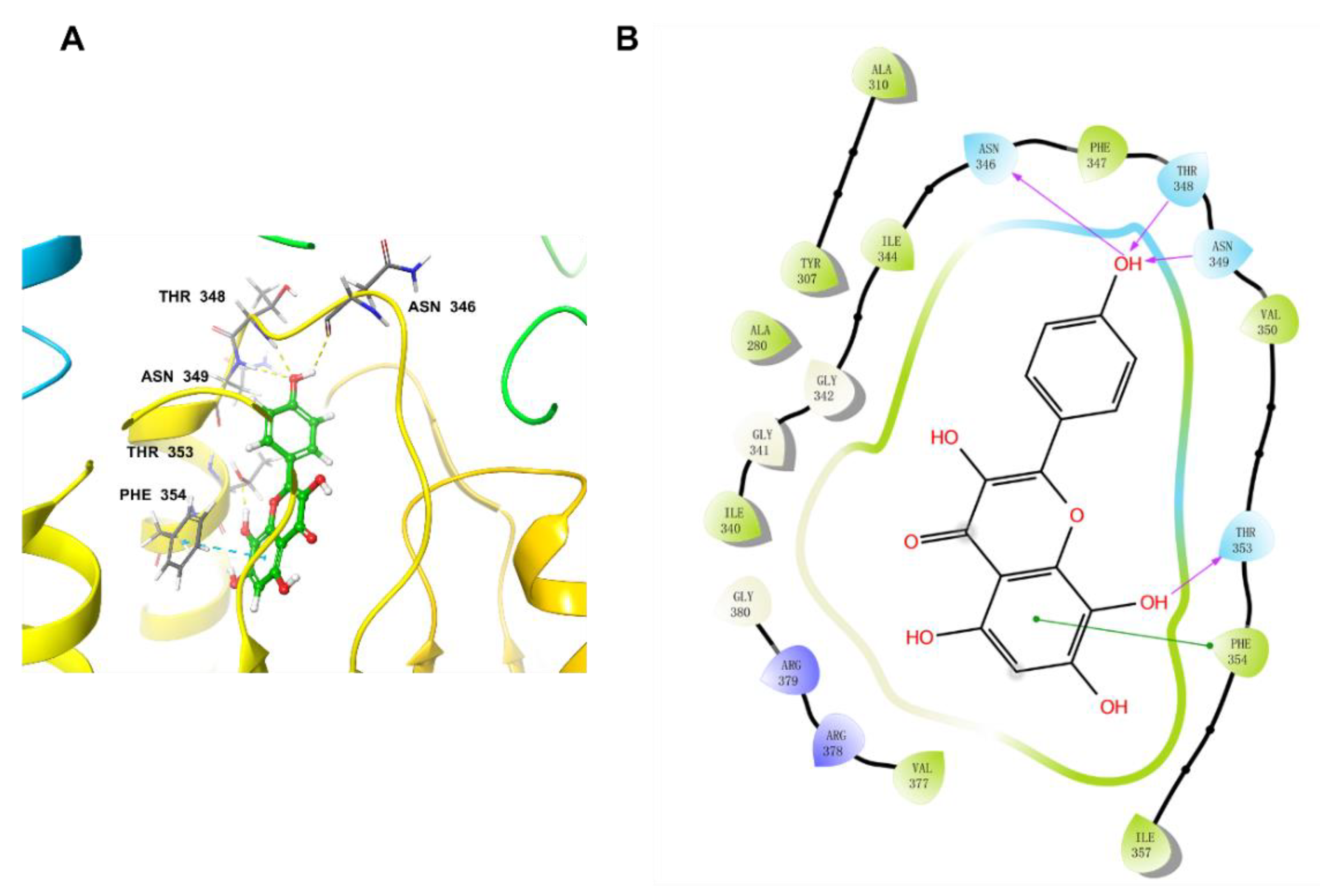
2.6. Docking Studies
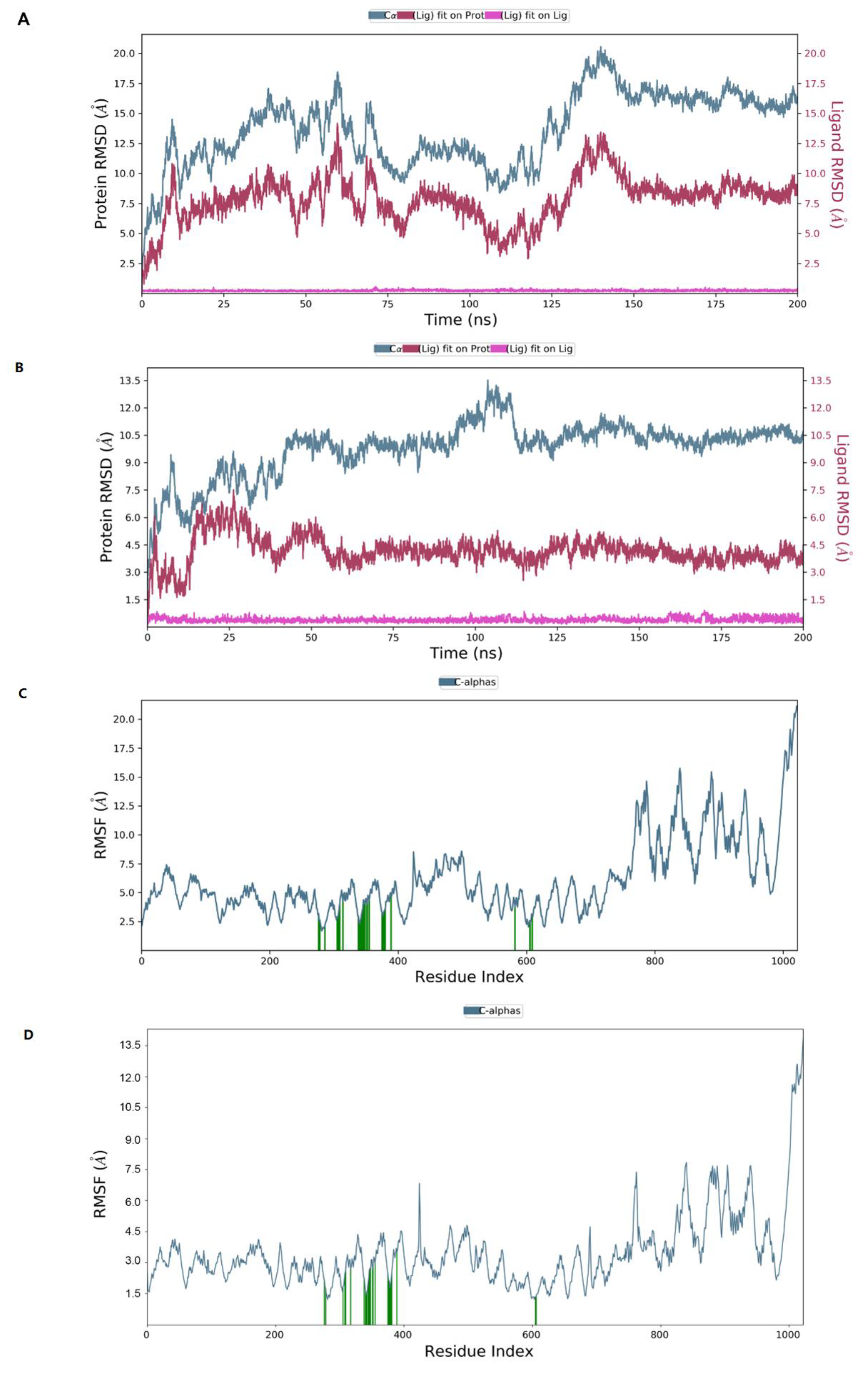
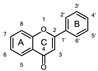
2.7. Molecular Dynamics Simulation
3. Discussion
4. Materials and Methods
4.1. Reagents and Materials
4.2. Enzymatic Inhibition Assay
4.3. Cell–Free Aggregation Assay
4.4. Thermal Shift Assay
4.5. Western Blotting Analysis
4.6. Analysis of Inhibitory Mechanism of Herbacetin
4.7. Molecular Docking
4.8. Molecular Dynamics Simulation
4.9. Data Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Srere, P.A. CitryI-CoA. An substrate for the citrate-cleavage enzyme. BBA-Mol. Cell Biol. L 1963, 73, 523–525. [Google Scholar]
- Fan, F.; Williams, H.J.; Boyer, J.G.; Graham, T.L.; Zhao, H.Z.; Lehr, R.; Qi, H.W.; Schwartz, B.; Raushel, F.M.; Meek, T.D. On the catalytic mechanism of human ATP citrate lyase. Biochemistry 2012, 51, 5198–5211. [Google Scholar] [CrossRef] [PubMed]
- Chypre, M.; Zaidi, N.; Smans, K. ATP-citrate lyase: A mini-review. Biochem. Biophys. Res. Commun. 2012, 422, 1–4. [Google Scholar] [CrossRef] [PubMed]
- Zu, X.Y.; Zhang, Q.H.; Liu, J.H.; Cao, R.X.; Zhong, J.; Yi, G.H.; Quan, Z.H.; Pizzorno, G. ATP citrate lyase inhibitors as novel cancer therapeutic agents. Recent Pat. Anti-Cancer Drug Discov. 2012, 7, 154–167. [Google Scholar] [CrossRef]
- Szutowicz, A.; Kwiatkowski, J.; Angielski, S. Lipogenetic and glycolytic enzyme activities in carcinoma and nonmalignant diseases of the human breast. Br. J. Cancer 1979, 39, 681–687. [Google Scholar] [CrossRef]
- Gao, Y.J.; Islam, M.S.; Tian, J.; Lui, V.W.Y.; Xiao, D. Inactivation of ATP citrate lyase by Cucurbitacin B: A bioactive compound from cucumber, inhibits prostate cancer growth. Cancer Lett. 2014, 349, 15–25. [Google Scholar] [CrossRef]
- Granchi, C. ATP citrate lyase (ACLY) inhibitors: An anti-cancer strategy at the crossroads of glucose and lipid metabolism. Eur. J. Med. Chem. 2018, 157, 1276–1291. [Google Scholar] [CrossRef]
- Abramson, H.N. The lipogenesis pathway as a cancer target. J. Med. Chem. 2011, 54, 5615–5638. [Google Scholar] [CrossRef]
- Xin, M.; Qiao, Z.; Li, J.; Liu, J.; Song, S.; Zhao, X.; Miao, P.; Tang, T.; Wang, L.; Liu, W.; et al. miR-22 inhibits tumor growth and metastasis by targeting ATP citrate lyase evidence in osteosarcoma, prostate cancer, cervical cancer and lung cancer. Oncotarget 2016, 7, 44252–44265. [Google Scholar] [CrossRef]
- Gribble, A.D.; Dolle, R.E.; Shaw, A.; McNair, D.; Novelli, R.; Novelli, C.E.; Slingsby, B.P.; Shah, V.P.; Tew, D.; Saxty, B.A.; et al. ATP-citrate lyase as a target for hypolipidemic intervention. Design and synthesis of 2-substituted butanedioic acids as novel, potent inhibitors of the enzyme. J. Med. Chem. 1996, 39, 3569–3584. [Google Scholar] [CrossRef]
- Cramer, C.T.; Goetz, B.; Hopson, K.L.; Fici, G.J.; Ackermann, R.M.; Brown, S.C.; Bisgaier, C.L.; Rajeswaran, W.G.; Oniciu, D.C.; Pape, M.E. Effects of a novel dual lipid synthesis inhibitor and its potential utility in treating dyslipidemia and metabolic syndrome. J. Lipid Res. 2004, 45, 1289–1301. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feng, X.J.; Zhang, L.; Xu, S.W.; Shen, A.Z. ATP-citrate lyase (ACLY) in lipid metabolism and atherosclerosis: An updated review. Prog. Lipid Res. 2020, 77, 101006. [Google Scholar] [CrossRef] [PubMed]
- Ray, K.K.; Bays, H.E.; Catapano, A.L.; Lalwani, N.D.; Bloedon, L.T.; Sterling, L.R.; Robinson, P.L.; Ballantyne, C.M.; Trial, C.H. Safety and efficacy of bempedoic acid to reduce LDL cholesterol. N. Engl. J. Med. 2019, 380, 1022–1032. [Google Scholar] [CrossRef] [PubMed]
- Pinkosky, S.L.; Filippov, S.; Srivastava, R.A.K.; Hanselman, J.C.; Bradshaw, C.D.; Hurley, T.R.; Cramer, C.T.; Spahr, M.A.; Brant, A.F.; Houghton, J.L.; et al. AMP-activated protein kinase and ATP-citrate lyase are two distinct molecular targets for ETC-1002, a novel small molecule regulator of lipid and carbohydrate metabolism. J. Lipid Res. 2013, 54, 134–151. [Google Scholar] [CrossRef]
- Watson, J.A.; Fang, M.; Lowenstein, J.M. Tricarballylate and hydroxycitrate: Substrate and inhibitor of ATP citrate oxaloacetate lyase. Arch. Biochem. Biophys. 1969, 135, 209–217. [Google Scholar] [CrossRef]
- Tana, J.B.; Shoshan, S.B.; Blum, J.; Migron, Y.; Hertz, R.; Rill, J. Synthesis and hypolipidemic and antidiabetogenic activities of beta,beta,beta’,beta’-tetrasubstituted, long-chain dioic acids. J. Med. Chem. 1989, 32, 2072–2084. [Google Scholar] [CrossRef]
- Pearce, N.J.; Yates, J.W.; Berkhout, T.A.; Jackson, B.; Tew, D.; Boyd, H.; Camilleri, P.; Sweeney, P.; Gribble, A.D.; Shaw, A.; et al. The role of ATP citrate-lyase in the metabolic regulation of plasma lipids. Hypolipidaemic effects of SB-204990, a lactone prodrug of the potent ATP citrate-lyase inhibitor SB-201076. Biochem. J. 1998, 334, 113–119. [Google Scholar] [CrossRef]
- Hatzivassiliou, G.; Zhao, F.P.; Bauer, D.E.; Andreadis, C.; Shaw, A.N.; Dhanak, D.; Hingorani, S.R.; Tuveson, D.A.; Thompson, C.B. ATP citrate lyase inhibition can suppress tumor cell growth. Cancer Cell 2005, 8, 311–321. [Google Scholar] [CrossRef]
- Li, J.J.; Wang, H.X.; Tino, J.A.; Robl, J.A.; Herpin, T.F.; Lawrence, R.M.; Biller, S.; Jamil, H.; Ponticiello, R.; Chen, L.P.; et al. 2-Hydroxy-N-arylbenzenesulfonamides as ATP-citrate lyase inhibitors. Bioorg. Med. Chem. Lett. 2007, 17, 3208–3211. [Google Scholar] [CrossRef]
- Wei, J.; Leit, S.; Kuai, J.; Therrien, E.; Rafi, S.; Harwood, H.J.; DeLaBarre, B.; Tong, L. An allosteric mechanism for potent inhibition of human ATP-citrate lyase. Nature 2019, 568, 566–570. [Google Scholar] [CrossRef]
- Guo, Q.; Kang, H.; Wang, J.; Dong, Y.; Peng, R.; Zhao, H.; Wu, W.; Guan, H.; Li, F. Inhibition of ACLY leads to suppression of osteoclast differentiation and function via regulation of histone acetylation. J. Bone Miner. Res. 2021, 36, 2065–2080. [Google Scholar] [CrossRef] [PubMed]
- Imran, M.; Rauf, A.; Abu-Izneid, T.; Nadeem, M.; Shariati, M.A.; Khan, I.A.; Imran, A.; Orhan, I.E.; Rizwan, M.; Atif, M.; et al. Luteolin, a flavonoid, as an anticancer agent: A review. Biomed. Pharmacother. 2019, 112, 108612. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.J.; Lee, M.H.; Liu, K.; Lim, D.Y.; Roh, E.; Chen, H.; Kim, S.H.; Shim, J.H.; Kim, M.O.; Li, W.; et al. Herbacetin suppresses cutaneous squamous cell carcinoma and melanoma cell growth by targeting AKT and ODC. Carcinogenesis 2017, 38, 1136–1146. [Google Scholar] [CrossRef] [PubMed]
- Al-Numair, K.S.; Chandramohan, G.; Alsaif, M.A.; Veeramani, C.; El Newehy, A.S. Morin, a flavonoid, on lipid peroxidation and antioxidant status in experimental myocardial ischemic rats. Afr. J. Tradit. Complement. Altern. Med. 2014, 11, 14–20. [Google Scholar] [CrossRef]
- Deng, Q.; Li, X.X.; Fang, Y.; Chen, X.; Xue, J. Therapeutic Potential of Quercetin as an Antiatherosclerotic Agent in Atherosclerotic Cardiovascular Disease: A Review. Evid. Based Complement. Alternat. Med. 2020, 2020, 5926381. [Google Scholar] [CrossRef]
- Wan, J.; Jiang, C.X.; Tang, Y.; Ma, G.L.; Tong, Y.P.; Jin, Z.X.; Zang, Y.; Osman, E.E.; Li, J.; Xiong, J.; et al. Structurally diverse glycosides of secoiridoid, bisiridoid, and triterpene-bisiridoid conjugates from the flower buds of two Caprifoliaceae plants and their ATP-citrate lyase inhibitory activities. Bioorg. Chem. 2022, 120, 105630. [Google Scholar] [CrossRef]
- Wang, R.; Wang, J.; Liu, Y.; Zhang, X.; Liang, X. Resonant waveguide grating based assays for colloidal aggregate detection and promiscuity characterization in natural products. RSC Adv. 2019, 9, 38055–38064. [Google Scholar] [CrossRef]
- Xiong, Y.; Zhu, G.-H.; Zhang, Y.-N.; Hu, Q.; Wang, H.-N.; Yu, H.-N.; Qin, X.-Y.; Guan, X.-Q.; Xiang, Y.-W.; Tang, H.; et al. Flavonoids in Ampelopsis grossedentata as covalent inhibitors of SARS-CoV-2 3CL(pro): Inhibition potentials, covalent binding sites and inhibitory mechanisms. Int. J. Biol. Macromol. 2021, 187, 976–987. [Google Scholar] [CrossRef]
- Koerner, S.K.; Hanai, J.I.; Bai, S.; Jernigan, F.E.; Oki, M.W.; Komaba, C.; Shuto, E.; Sukhatme, V.P.; Sun, L. Design and synthesis of emodin derivatives as novel inhibitors of ATP-citrate lyase. Eur. J. Med. Chem. 2017, 126, 920–928. [Google Scholar] [CrossRef]
- Weng, G.; Wang, E.; Wang, Z.; Liu, H.; Zhu, F.; Li, D.; Hou, T. HawkDock: A web server to predict and analyze the protein-protein complex based on computational docking and MM/GBSA. Nucleic Acids Res. 2019, 47, W322–W330. [Google Scholar] [CrossRef]
- Ylilauri, M.; Pentikäinen, O.T. MMGBSA as a tool to understand the binding affinities of filamin-peptide interactions. J. Chem. Inf. Modeling 2013, 53, 2626–2633. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Wang, Y.; Yu, M.; Shang, Y.; Chang, Y.; Zhao, H.; Kang, Y.; Zhao, L.; Xu, L.; Zhao, X.; et al. Discovery of herbacetin as a novel SGK1 inhibitor to alleviate myocardial hypertrophy. Adv. Sci. 2022, 9, 2101485. [Google Scholar] [CrossRef] [PubMed]
- Veeramani, C.; Alsaif, M.A.; Al-Numair, K.S. Herbacetin, a flaxseed flavonoid, ameliorates high percent dietary fat induced insulin resistance and lipid accumulation through the regulation of hepatic lipid metabolizing and lipid-regulating enzymes. Chem. Biol. Interact. 2018, 288, 49–56. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.J.; Roh, E.; Lee, M.H.; Oi, N.; Lim, D.Y.; Kim, M.O.; Cho, Y.Y.; Pugliese, A.; Shim, J.H.; Chen, H.; et al. Herbacetin is a novel allosteric inhibitor of ornithine decarboxylase with antitumor activity. Cancer Res. 2016, 76, 1146–1157. [Google Scholar] [CrossRef] [PubMed]
- Friesner, R.A.; Murphy, R.B.; Repasky, M.P.; Frye, L.L.; Greenwood, J.R.; Halgren, T.A.; Sanschagrin, P.C.; Mainz, D.T. Extra precision glide: Docking and scoring incorporating a model of hydrophobic enclosure for protein-ligand complexes. J. Med. Chem. 2006, 49, 6177–6196. [Google Scholar] [CrossRef]
- Halgren, T.A.; Murphy, R.B.; Friesner, R.A.; Beard, H.S.; Frye, L.L.; Pollard, W.T.; Banks, J.L. Glide: A new approach for rapid, accurate docking and scoring. 2. Enrichment factors in database screening. J. Med. Chem. 2004, 47, 1750–1759. [Google Scholar] [CrossRef]
- Søndergaard, C.R.; Olsson, M.H.; Rostkowski, M.; Jensen, J.H. Improved Treatment of Ligands and Coupling Effects in Empirical Calculation and Rationalization of pKa Values. J. Chem. Theory Comput. 2011, 7, 2284–2295. [Google Scholar] [CrossRef]
- Olsson, M.H.; Søndergaard, C.R.; Rostkowski, M.; Jensen, J.H. PROPKA3: Consistent Treatment of Internal and Surface Residues in Empirical pKa Predictions. J. Chem. Theory Comput. 2011, 7, 525–537. [Google Scholar] [CrossRef]
- Sastry, G.M.; Adzhigirey, M.; Day, T.; Annabhimoju, R.; Sherman, W. Protein and ligand preparation: Parameters, protocols, and influence on virtual screening enrichments. J. Comput.-Aided Mol. Des. 2013, 27, 221–234. [Google Scholar] [CrossRef]
- Greenwood, J.R.; Calkins, D.; Sullivan, A.P.; Shelley, J.C. Towards the comprehensive, rapid, and accurate prediction of the favorable tautomeric states of drug-like molecules in aqueous solution. J. Comput.-Aided Mol. Des. 2010, 24, 591–604. [Google Scholar] [CrossRef]
- Shelley, J.C.; Cholleti, A.; Frye, L.L.; Greenwood, J.R.; Timlin, M.R.; Uchimaya, M. Epik: A software program for pK(a) prediction and protonation state generation for drug-like molecules. J. Comput.-Aided Mol. Des. 2007, 21, 681–691. [Google Scholar] [CrossRef] [PubMed]
- Jacobson, M.P.; Pincus, D.L.; Rapp, C.S.; Day, T.J.; Honig, B.; Shaw, D.E.; Friesner, R.A. A hierarchical approach to all-atom protein loop prediction. Proteins 2004, 55, 351–367. [Google Scholar] [CrossRef] [PubMed]
- Gohlke, H.; Case, D.A. Converging free energy estimates: MM-PB(GB)SA studies on the protein-protein complex Ras-Raf. J. Comput. Chem. 2004, 25, 238–250. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Abel, R.; Zhu, K.; Cao, Y.; Zhao, S.; Friesner, R.A. The VSGB 2.0 model: A next generation energy model for high resolution protein structure modeling. Proteins 2011, 79, 2794–2812. [Google Scholar] [CrossRef]
- Mahanta, S.; Naiya, T.; Biswas, K.; Changkakoti, L.; Mohanta, Y.K.; Tanti, B.; Mishra, A.K.; Mohanta, T.K.; Sharma, N. Plant Source Derived Compound Exhibited In Silico Inhibition of Membrane Glycoprotein In SARS-CoV-2: Paving the Way to Discover a New Class of Compound For Treatment of COVID-19. Front. Pharmacol. 2022, 13, 805344. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Entry | Group | Compound | A Ring | B Ring | C Ring | Classification | IC50 (μM) |
|---|---|---|---|---|---|---|---|
| 18 | A | Herbacetin | 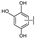 |  |  | Flavonol | 0.50 ± 0.08 |
| 29 | A | Quercetin | 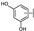 |  | 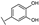 | Flavonol | 0.86 ± 0.05 |
| 51 | A | Luteolin | 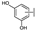 |  | 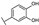 | Flavone | 0.64 ± 0.13 |
| 52 | A | Scutellarein |  |  |  | Flavone | 0.71 ± 0.06 |
| 89 | A | Myricetin | 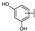 |  |  | Flavonol | 0.57 ± 0.03 |
| 93 | A | Gossypetin | 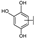 | 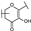 |  | Flavonol | 0.31 ± 0.02 |
| 30 | B | Kaempferol |  | 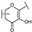 | 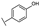 | Flavonol | 10.16 ± 1.07 |
| 21 | B | Apigenin | 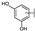 | 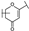 | 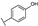 | Flavone | 19.11 ± 11.63 |
| 44 | B | Isorhamnetin | 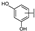 | 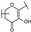 |  | Flavonol | 49.74 ± 14.76 |
| 87 | B | Morin | 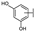 |  | 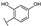 | Flavonol | 4.07 ± 0.69 |
| 6 | C | Hyperoside | 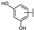 |  |  | Flavonol | 1.53 ± 0.08 |
| 20 | C | Isoquercetin | 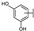 |  | 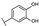 | Flavonol | 2.14 ± 0.12 |
| 5 | C | Luteolin 7-O-glucuronide |  |  | 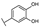 | Flavone | 1.47 ± 0.20 |
| 108 | C | Vincetoxicoside B |  | 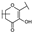 |  | Flavonol | 0.57 ± 0.09 |
| 14 | C | Isoorientin | 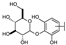 | 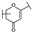 | 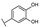 | Flavone | 1.86 ± 0.29 |
| 53 | C | Scutellarin | 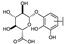 | 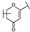 |  | Flavone | 3.77 ± 0.26 |
| 15 | C | Lonicerin | 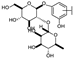 | 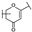 |  | Flavone | 1.43 ± 0.44 |
| 19 | D | Taxifolin |  | 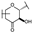 | 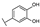 | Flavononol | 2.54 ± 0.25 |
| 73 | D | Catechin | 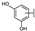 | 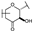 | 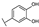 | Flavononol | 23.64 ± 19.19 |
| 75 | D | Epicatechin |  | 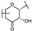 |  | Flavononol | 31.63 ± 9.45 |
| 74 | D | Cyanidin | 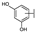 |  | 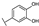 | Anthocyanin | 1.29 ± 0.14 |
| Types | Equations | Vmax | Km |
|---|---|---|---|
| No inhibitor | Vmax | Km | |
| Competitive | constant | decrease | |
| Noncompetitive | decrease | constant | |
| Uncompetitive | decrease | decrease |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, P.; Hou, T.; Xu, F.; Luo, F.; Zhou, H.; Liu, F.; Xie, X.; Liu, Y.; Wang, J.; Guo, Z.; et al. Discovery of Flavonoids as Novel Inhibitors of ATP Citrate Lyase: Structure–Activity Relationship and Inhibition Profiles. Int. J. Mol. Sci. 2022, 23, 10747. https://doi.org/10.3390/ijms231810747
Wang P, Hou T, Xu F, Luo F, Zhou H, Liu F, Xie X, Liu Y, Wang J, Guo Z, et al. Discovery of Flavonoids as Novel Inhibitors of ATP Citrate Lyase: Structure–Activity Relationship and Inhibition Profiles. International Journal of Molecular Sciences. 2022; 23(18):10747. https://doi.org/10.3390/ijms231810747
Chicago/Turabian StyleWang, Pan, Tao Hou, Fangfang Xu, Fengbin Luo, Han Zhou, Fan Liu, Xiaomin Xie, Yanfang Liu, Jixia Wang, Zhimou Guo, and et al. 2022. "Discovery of Flavonoids as Novel Inhibitors of ATP Citrate Lyase: Structure–Activity Relationship and Inhibition Profiles" International Journal of Molecular Sciences 23, no. 18: 10747. https://doi.org/10.3390/ijms231810747
APA StyleWang, P., Hou, T., Xu, F., Luo, F., Zhou, H., Liu, F., Xie, X., Liu, Y., Wang, J., Guo, Z., & Liang, X. (2022). Discovery of Flavonoids as Novel Inhibitors of ATP Citrate Lyase: Structure–Activity Relationship and Inhibition Profiles. International Journal of Molecular Sciences, 23(18), 10747. https://doi.org/10.3390/ijms231810747