
In Vitro Rescue of the Bile Acid Transport Function of ABCB11 Variants by CFTR Potentiators

 ,
,  , ,
, ,  ,
,
Abstract
:1. Introduction
2. Results
2.1. Patients
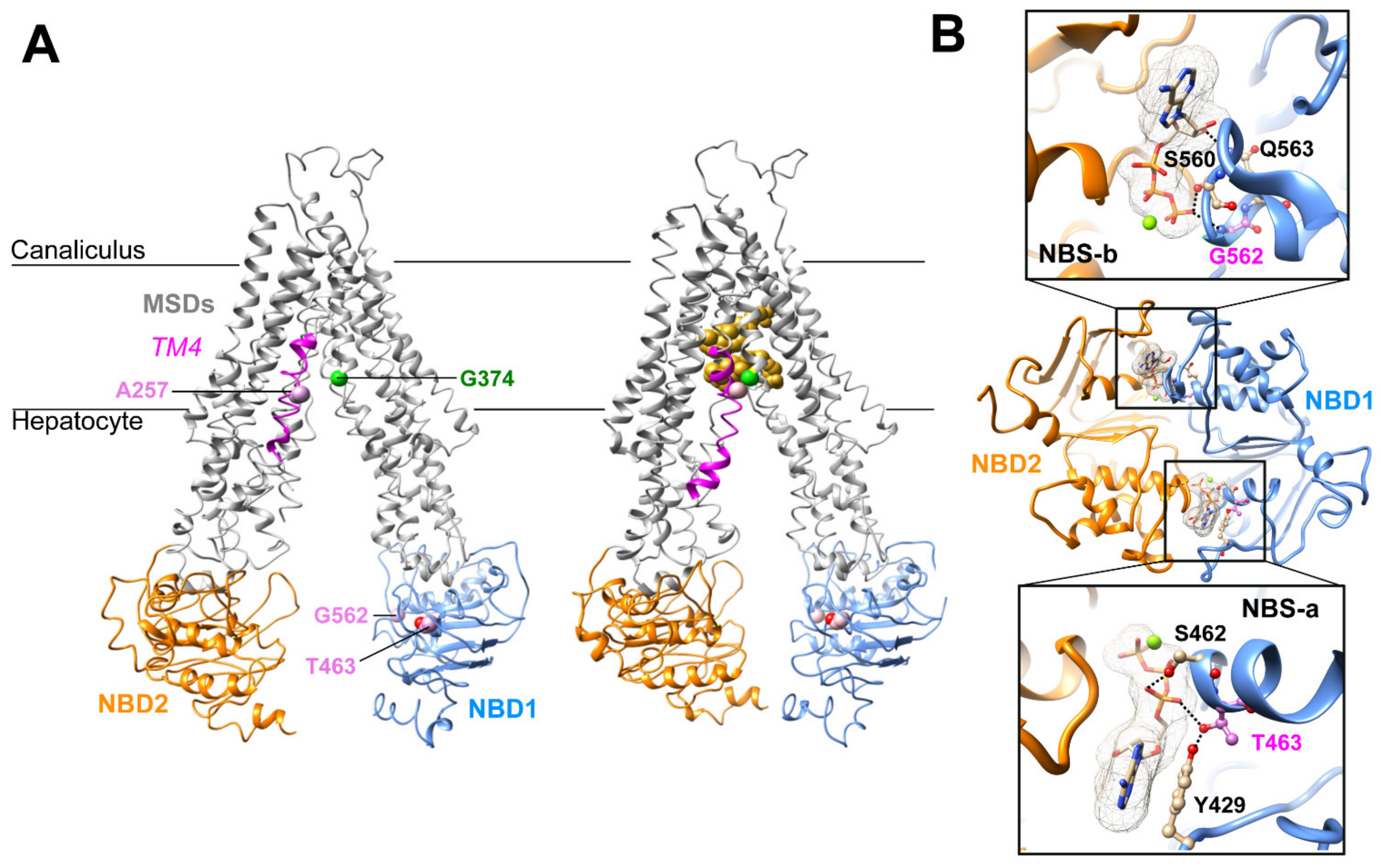
2.2. Three-Dimensional Structure Analysis Predicts a Functional Defect of ABCB11 Variants
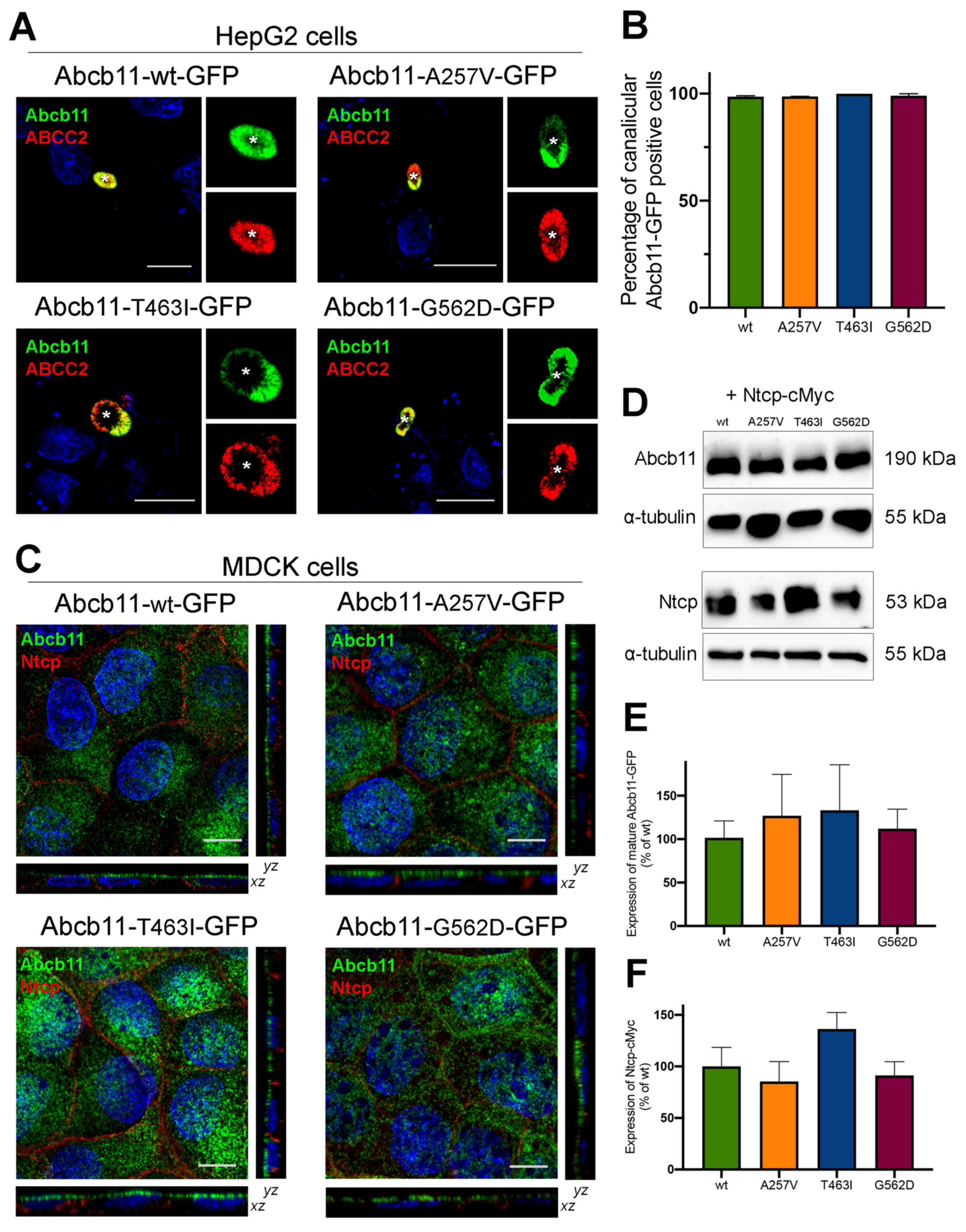
2.3. Abcb11 Variants Are Correctly Targeted to the Canalicular/Apical Membrane
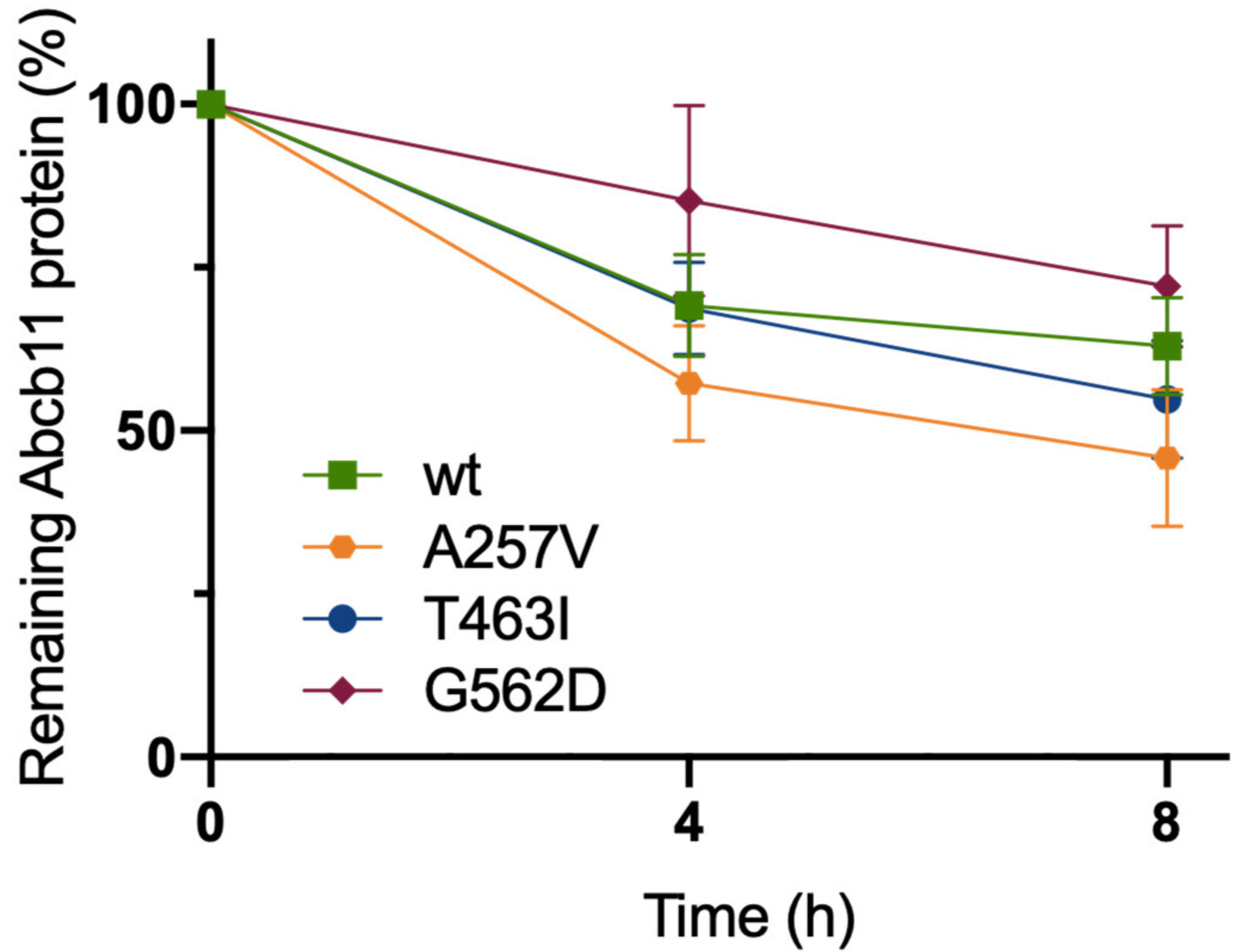
2.4. The Three Variations of Abcb11 Do Not Impact the Stability of the Transporter
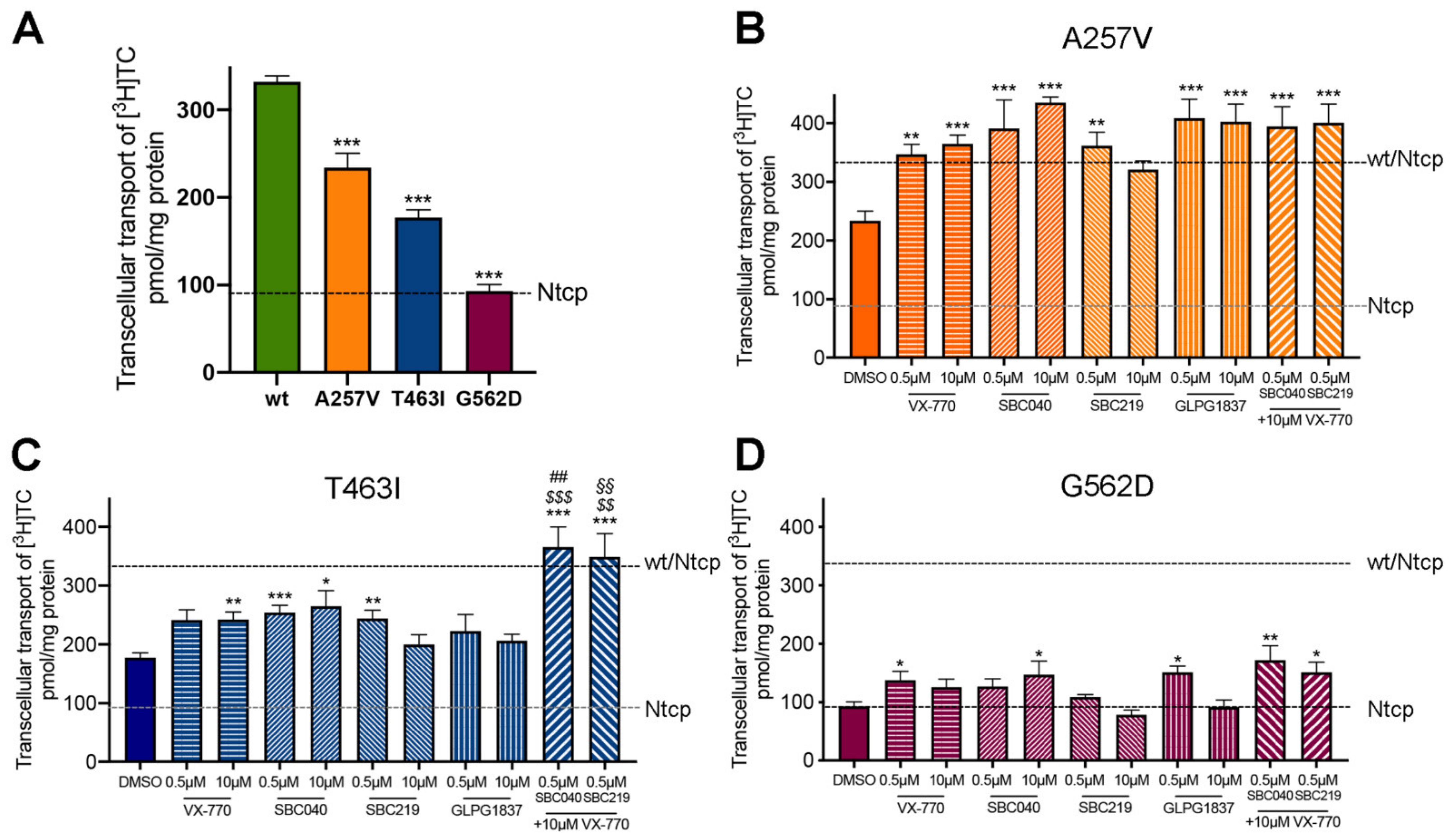
2.5. CFTR Potentiators Rescue the Functional Defect Due to Abcb11 Variations
3. Discussion
4. Materials and Methods
4.1. Patients
4.2. Three-Dimensional Analysis
4.3. DNA Constructs and Mutagenesis
4.4. Cell Culture, Transfection, Lentiviral Infection and Immunoanalyses
4.5. Chemicals and Cell Treatments
4.6. Analysis of Abcb11 Protein Stability
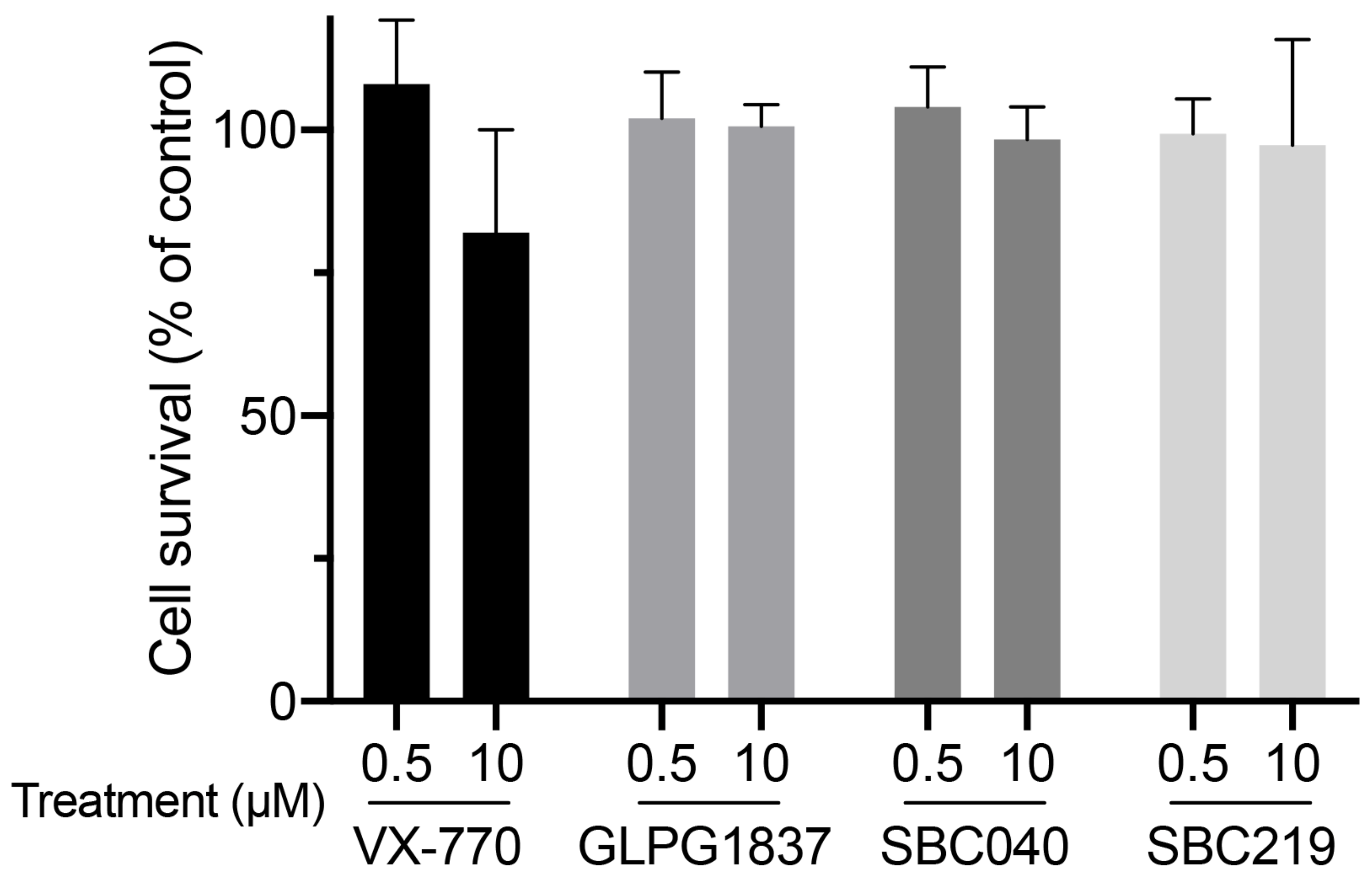
4.7. Cytotoxicity Assays
4.8. Taurocholate Transport Assays
4.9. Statistical Analyses
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| ABC | ATP-binding cassette |
| BA | Bile acid |
| BSEP | Bile salt export pump |
| CFTR | Cystic fibrosis transmembrane conductance regulator |
| DMSO | Dimethylsulfoxide |
| MDCK | Madin–Darby canine kidney |
| MSD | Membrane-spanning domain |
| NBD | Nucleotide-binding domain |
| NBS | Nucleotide-binding site |
| NTCP | Na+-taurocholate cotransporting polypeptide |
| PFIC | Progressive familial intrahepatic cholestasis |
| SBC | Small binder of CFTR |
| TC | Taurocholate |
| TM | Transmembrane helix |
| WT | Wild type |
References
- Davit-Spraul, A.; Gonzales, E.; Baussan, C.; Jacquemin, E. Progressive familial intrahepatic cholestasis. Orphanet J. Rare Dis. 2009, 4, 1. [Google Scholar] [CrossRef] [PubMed]
- Dean, M.; Rzhetsky, A.; Allikmets, R. The human ATP-binding cassette (ABC) transporter superfamily. Genome Res. 2001, 11, 1156–1166. [Google Scholar] [CrossRef] [PubMed]
- Soroka, C.J.; Boyer, J.L. Biosynthesis and trafficking of the bile salt export pump, BSEP: Therapeutic implications of BSEP mutations. Mol. Asp. Med. 2014, 37, 3–14. [Google Scholar] [CrossRef] [PubMed]
- Sohail, M.I.; Schmid, D.; Wlcek, K.; Spork, M.; Szakacs, G.; Trauner, M.; Stockner, T.; Chiba, P. Molecular Mechanism of Taurocholate Transport by the Bile Salt Export Pump, an ABC Transporter Associated with Intrahepatic Cholestasis. Mol. Pharm. 2017, 92, 401–413. [Google Scholar] [CrossRef]
- van Wessel, D.B.E.; Thompson, R.J.; Gonzales, E.; Jankowska, I.; Sokal, E.; Grammatikopoulos, T.; Kadaristiana, A.; Jacquemin, E.; Spraul, A.; Lipinski, P.; et al. Genotype correlates with the natural history of severe bile salt export pump deficiency. J. Hepatol. 2020, 73, 84–93. [Google Scholar] [CrossRef]
- Strautnieks, S.S.; Byrne, J.A.; Pawlikowska, L.; Cebecauerova, D.; Rayner, A.; Dutton, L.; Meier, Y.; Antoniou, A.; Stieger, B.; Arnell, H.; et al. Severe bile salt export pump deficiency: 82 different ABCB11 mutations in 109 families. Gastroenterology 2008, 134, 1203–1214. [Google Scholar] [CrossRef]
- Byrne, J.A.; Strautnieks, S.S.; Ihrke, G.; Pagani, F.; Knisely, A.S.; Linton, K.J.; Mieli-Vergani, G.; Thompson, R.J. Missense mutations and single nucleotide polymorphisms in ABCB11 impair bile salt export pump processing and function or disrupt pre-messenger RNA splicing. Hepatology 2009, 49, 553–567. [Google Scholar] [CrossRef]
- Davit-Spraul, A.; Fabre, M.; Branchereau, S.; Baussan, C.; Gonzales, E.; Stieger, B.; Bernard, O.; Jacquemin, E. ATP8B1 and ABCB11 analysis in 62 children with normal gamma-glutamyl transferase progressive familial intrahepatic cholestasis (PFIC): Phenotypic differences between PFIC1 and PFIC2 and natural history. Hepatology 2010, 51, 1645–1655. [Google Scholar] [CrossRef]
- Droge, C.; Bonus, M.; Baumann, U.; Klindt, C.; Lainka, E.; Kathemann, S.; Brinkert, F.; Grabhorn, E.; Pfister, E.D.; Wenning, D.; et al. Sequencing of FIC1, BSEP and MDR3 in a large cohort of patients with cholestasis revealed a high number of different genetic variants. J. Hepatol. 2017, 67, 1253–1264. [Google Scholar] [CrossRef]
- Mareux, E.; Lapalus, M.; Amzal, R.; Almes, M.; Ait-Slimane, T.; Delaunay, J.L.; Adnot, P.; Collado-Hilly, M.; Davit-Spraul, A.; Falguieres, T.; et al. Functional rescue of an ABCB11 mutant by ivacaftor: A new targeted pharmacotherapy approach in bile salt export pump deficiency. Liver Int. 2020, 40, 1917–1925. [Google Scholar] [CrossRef]
- Mareux, E.; Lapalus, M.; Ben-Saad, A.; Callebaut, I.; Falguieres, T.; Gonzales, E.; Jacquemin, E. In vitro functional rescue by ivacaftor of an ABCB11 variant involved in PFIC2 and intrahepatic cholestasis of pregnancy. Orphanet J. Rare Dis. 2021, 16, 484. [Google Scholar] [CrossRef]
- Van Goor, F.; Hadida, S.; Grootenhuis, P.D.; Burton, B.; Cao, D.; Neuberger, T.; Turnbull, A.; Singh, A.; Joubran, J.; Hazlewood, A.; et al. Rescue of CF airway epithelial cell function in vitro by a CFTR potentiator, VX-770. Proc. Natl. Acad. Sci. USA 2009, 106, 18825–18830. [Google Scholar] [CrossRef] [PubMed]
- Yeh, H.I.; Sohma, Y.; Conrath, K.; Hwang, T.C. A common mechanism for CFTR potentiators. J. Gen. Physiol. 2017, 149, 1105–1118. [Google Scholar] [CrossRef]
- Froux, L.; Elbahnsi, A.; Boucherle, B.; Billet, A.; Baatallah, N.; Hoffmann, B.; Alliot, J.; Zelli, R.; Zeinyeh, W.; Haudecoeur, R.; et al. Targeting different binding sites in the CFTR structures allows to synergistically potentiate channel activity. Eur. J. Med. Chem. 2020, 190, 112116. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.; Chen, J. Molecular structure of human P-glycoprotein in the ATP-bound, outward-facing conformation. Science 2018, 359, 915–919. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Hou, W.T.; Wang, J.; Xu, D.; Guo, C.; Sun, L.; Ruan, K.; Zhou, C.Z.; Chen, Y. Structures of human bile acid exporter ABCB11 reveal a transport mechanism facilitated by two tandem substrate-binding pockets. Cell Res. 2022, 32, 501–504. [Google Scholar] [CrossRef]
- Gonzales, E.; Grosse, B.; Schuller, B.; Davit-Spraul, A.; Conti, F.; Guettier, C.; Cassio, D.; Jacquemin, E. Targeted pharmacotherapy in progressive familial intrahepatic cholestasis type 2: Evidence for improvement of cholestasis with 4-phenylbutyrate. Hepatology 2015, 62, 558–566. [Google Scholar] [CrossRef]
- Wang, L.; Hou, W.T.; Chen, L.; Jiang, Y.L.; Xu, D.; Sun, L.; Zhou, C.Z.; Chen, Y. Cryo-EM structure of human bile salts exporter ABCB11. Cell Res. 2020, 30, 623–625. [Google Scholar] [CrossRef]
- Stockner, T.; Gradisch, R.; Schmitt, L. The role of the degenerate nucleotide binding site in type I ABC exporters. FEBS Lett. 2020, 594, 3815–3838. [Google Scholar] [CrossRef]
- Tsui, L.C. The cystic fibrosis transmembrane conductance regulator gene. Am. J. Respir. Crit. Care Med. 1995, 151, S47–S53. [Google Scholar] [CrossRef] [Green Version]
- Li, C.; Ramjeesingh, M.; Wang, W.; Garami, E.; Hewryk, M.; Lee, D.; Rommens, J.M.; Galley, K.; Bear, C.E. ATPase activity of the cystic fibrosis transmembrane conductance regulator. J. Biol. Chem. 1996, 271, 28463–28468. [Google Scholar] [CrossRef] [PubMed]
- Delaunay, J.L.; Durand-Schneider, A.M.; Dossier, C.; Falguieres, T.; Gautherot, J.; Davit-Spraul, A.; Ait-Slimane, T.; Housset, C.; Jacquemin, E.; Maurice, M. A functional classification of ABCB4 variations causing progressive familial intrahepatic cholestasis type 3. Hepatology 2016, 63, 1620–1631. [Google Scholar] [CrossRef] [PubMed]
- De Boeck, K.; Munck, A.; Walker, S.; Faro, A.; Hiatt, P.; Gilmartin, G.; Higgins, M. Efficacy and safety of ivacaftor in patients with cystic fibrosis and a non-G551D gating mutation. J. Cyst. Fibros. 2014, 13, 674–680. [Google Scholar] [CrossRef] [PubMed]
- Csanady, L.; Torocsik, B. Cystic fibrosis drug ivacaftor stimulates CFTR channels at picomolar concentrations. Elife 2019, 8, e46450. [Google Scholar] [CrossRef] [PubMed]
- Liu, F.; Zhang, Z.; Levit, A.; Levring, J.; Touhara, K.K.; Shoichet, B.K.; Chen, J. Structural identification of a hotspot on CFTR for potentiation. Science 2019, 364, 1184–1188. [Google Scholar] [CrossRef]
- Eckford, P.D.; Li, C.; Ramjeesingh, M.; Bear, C.E. Cystic fibrosis transmembrane conductance regulator (CFTR) potentiator VX-770 (ivacaftor) opens the defective channel gate of mutant CFTR in a phosphorylation-dependent but ATP-independent manner. J. Biol. Chem. 2012, 287, 36639–36649. [Google Scholar] [CrossRef]
- Jih, K.Y.; Hwang, T.C. Vx-770 potentiates CFTR function by promoting decoupling between the gating cycle and ATP hydrolysis cycle. Proc. Natl. Acad. Sci. USA 2013, 110, 4404–4409. [Google Scholar] [CrossRef]
- Laselva, O.; Qureshi, Z.; Zeng, Z.W.; Petrotchenko, E.V.; Ramjeesingh, M.; Hamilton, C.M.; Huan, L.J.; Borchers, C.H.; Pomes, R.; Young, R.; et al. Identification of binding sites for ivacaftor on the cystic fibrosis transmembrane conductance regulator. iScience 2021, 24, 102542. [Google Scholar] [CrossRef]
- Barbieri, A.; Thonghin, N.; Shafi, T.; Prince, S.M.; Collins, R.F.; Ford, R.C. Structure of ABCB1/P-glycoprotein bound to the CFTR potentiator ivacaftor. bioRxiv 2021. [Google Scholar] [CrossRef]
- Lingam, S.; Thonghin, N.; Ford, R.C. Investigation of the effects of the CFTR potentiator ivacaftor on human P-glycoprotein (ABCB1). Sci. Rep. 2017, 7, 17481. [Google Scholar] [CrossRef] [Green Version]
- Davies, J.C.; Van de Steen, O.; van Koningsbruggen-Rietschel, S.; Drevinek, P.; Derichs, N.; McKone, E.F.; Kanters, D.; Allamassey, L.; Namour, F.; de Kock, H.; et al. GLPG1837, a CFTR potentiator, in p.Gly551Asp (G551D)-CF patients: An open-label, single-arm, phase 2a study (SAPHIRA1). J. Cyst. Fibros. 2019, 18, 693–699. [Google Scholar] [CrossRef] [PubMed]
- Van der Plas, S.E.; Kelgtermans, H.; De Munck, T.; Martina, S.L.X.; Dropsit, S.; Quinton, E.; De Blieck, A.; Joannesse, C.; Tomaskovic, L.; Jans, M.; et al. Discovery of N-(3-Carbamoyl-5,5,7,7-tetramethyl-5,7-dihydro-4H-thieno[2,3-c]pyran-2-yl)-lH-pyr azole-5-carboxamide (GLPG1837), a Novel Potentiator Which Can Open Class III Mutant Cystic Fibrosis Transmembrane Conductance Regulator (CFTR) Channels to a High Extent. J. Med. Chem. 2018, 61, 1425–1435. [Google Scholar] [PubMed]
- Webb, B.; Sali, A. Protein Structure Modeling with MODELLER. Methods Mol. Biol. 2017, 1654, 39–54. [Google Scholar] [PubMed]
- Amzal, R.; Thebaut, A.; Lapalus, M.; Almes, M.; Grosse, B.; Mareux, E.; Collado-Hilly, M.; Davit-Spraul, A.; Bidou, L.; Namy, O.; et al. Pharmacological Premature Termination Codon Readthrough of ABCB11 in Bile Salt Export Pump Deficiency: An In Vitro Study. Hepatology 2021, 73, 1449–1463. [Google Scholar] [CrossRef] [PubMed]
- Sormunen, R.; Eskelinen, S.; Lehto, V.P. Bile canaliculus formation in cultured HEPG2 cells. Lab. Investig. 1993, 68, 652–662. [Google Scholar]
- van Meerloo, J.; Kaspers, G.J.; Cloos, J. Cell sensitivity assays: The MTT assay. Methods Mol. Biol. 2011, 731, 237–245. [Google Scholar]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Patient (No./Sex) | First Allele | Domain | Second Allele | Domain | Biliary BA (N < 10 µM) | IHC ABCB11 | Reference |
|---|---|---|---|---|---|---|---|
| 1/F | c.770C > T p.A257V | TM4 | c.2944G > A p.G982R | TM11 | na | na | [17] |
| 2/F | c.1388C > T p.T463I | NBD1 NBS-a | c.3169C > T p.R1057X | Linker TM6-NBD2 | 1 mM | Faint | [10] |
| 3/na | c.1685G > A p.G562D | NBD1 NBS-b | c.1445A_G p.D482G | NBD1 | na | Normal | [6] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mareux, E.; Lapalus, M.; Ben Saad, A.; Zelli, R.; Lakli, M.; Riahi, Y.; Almes, M.; Banet, M.; Callebaut, I.; Decout, J.-L.; et al. In Vitro Rescue of the Bile Acid Transport Function of ABCB11 Variants by CFTR Potentiators. Int. J. Mol. Sci. 2022, 23, 10758. https://doi.org/10.3390/ijms231810758
Mareux E, Lapalus M, Ben Saad A, Zelli R, Lakli M, Riahi Y, Almes M, Banet M, Callebaut I, Decout J-L, et al. In Vitro Rescue of the Bile Acid Transport Function of ABCB11 Variants by CFTR Potentiators. International Journal of Molecular Sciences. 2022; 23(18):10758. https://doi.org/10.3390/ijms231810758
Chicago/Turabian StyleMareux, Elodie, Martine Lapalus, Amel Ben Saad, Renaud Zelli, Mounia Lakli, Yosra Riahi, Marion Almes, Manon Banet, Isabelle Callebaut, Jean-Luc Decout, and et al. 2022. "In Vitro Rescue of the Bile Acid Transport Function of ABCB11 Variants by CFTR Potentiators" International Journal of Molecular Sciences 23, no. 18: 10758. https://doi.org/10.3390/ijms231810758
APA StyleMareux, E., Lapalus, M., Ben Saad, A., Zelli, R., Lakli, M., Riahi, Y., Almes, M., Banet, M., Callebaut, I., Decout, J.-L., Falguières, T., Jacquemin, E., & Gonzales, E. (2022). In Vitro Rescue of the Bile Acid Transport Function of ABCB11 Variants by CFTR Potentiators. International Journal of Molecular Sciences, 23(18), 10758. https://doi.org/10.3390/ijms231810758