Abstract
Catecholaminergic polymorphic ventricular tachycardia (CPVT) is a fatal rare inherited cardiac channelopathy. Affected patients are susceptible to develop deadly ventricular arrythmias after physical or emotional stress. The typical arrhythmia presents as bidirectional and/or polymorphic ventricular tachycardias. To illustrate the clinical challenges in properly diagnosing this disease, we report two cases of CPVT together with a brief literature review.
Introduction
A sudden cardiac arrest in the young is a rare but catastrophic event. Catecholaminergic polymorphic ventricular tachycardia (CPVT) is one possible cause. An early diagnosis can prevent a sudden cardiac death (SCD). Symptoms of CPVT range from palpitations during exercise or emotional stress to exercise induced syncopes or cardiac arrest [1]. Because the resting 12-lead electrocardiography (ECG) is commonly inconspicuous and different imaging modalities do not show abnormalities, the diagnosis must be made through additional examinations including exercise stress test, Holter monitor and genetic testing [2]. In very young children, an adrenaline test can help to make the diagnosis. The following two cases highlight the young age of the patients at presentation and the unfortunate fact that the correct diagnosis is often only made after a deadly event occurs.
Case Reports
- Case 1
Seven-year-old identical twin brothers both had episodes of syncopes while playing. The initial pediatric evaluation did not detect any abnormalities. At the age of ten, after a syncope with seizure-like episodes, epilepsy was diagnosed in both boys. Although epileptic treatment was started, the syncopal episodes persisted. At the age of 15, one of the boys died suddenly. The surviving boy had a normal resting ECG (Figure 1A). During a stress test, polymorphic ventricular ectopy/bigeminy was recorded (Figure 1B).
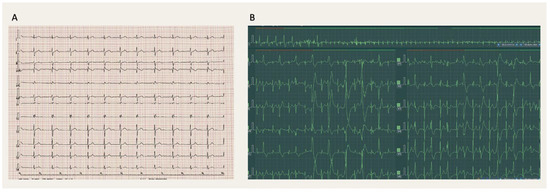
Figure 1.
(A) Patient 1: Resting 12-lead electrocardiogram (ECG, 25 mm/s) with sinus rhythm at a rate of 68 bpm, normal PR and QRS duration, normal QRS axis and no ST-segment changes. There is an rsR pattern in V1-V2 typical for a right bundle branch block, although not meeting the other criteria. (B) Patient 1: Exercise ECG (25 mm/s) revealing polymorphic ventricular ectopy.
After obtaining informed consent, genetic testing was performed. The pathogenic mutation NM_001035.3(RYR2):c.1259G>A (p.Arg420Gln) was detected (Figure 2A) and the diagnose of CPVT was made.
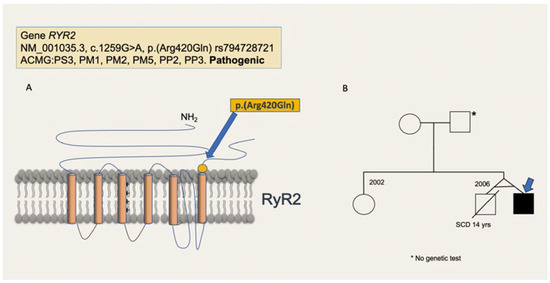
Figure 2.
(A) Pathogenic variant of patient 1. The variant RYR2: c.1259G>A (p.Arg420Gln) replaces an arginine to a glutamine at codon 420 and localizes to the hotspot I (amino acids 44-466) of the RYR2 protein in which an important cluster of pathogenic variants has been described. The variant is not present in the Genome Aggregation Database, but has been reported in individuals with clinical features of catecholaminergic polymorphic ventricular tachycardia. Experimental studies have shown that this missense change affects the RYR2 function [7,8]. There are other pathogenic variants at position Arg420 that have been described. Following the American College of Medical Genetics (ACMG) criteria it is classified as pathogenic. (B) Family tree of patient 1. The pathogenic variant RYR2: NM_001035.3, c.1259G>A, p.(Arg420Gln) was not found in the mother. The father did not want to be tested.
No genetic testing was performed in the deceased twin. The variant was not found in the mother and the asymptomatic father did not want to be tested (Figure 2B).
- Case 2
The second patient had a family history of SCD in his elder brother (aged 17). An autopsy was performed, but the data was not available. Our index patient also experienced a cardiac arrest during exercise at the age of 16 but fortunately survived. Shortly thereafter, an implantable cardioverter defibrillator (ICD) was implanted as secondary prevention, even though the diagnosis was not yet clear. First, Long QT syndrome (LQTS) was suspected, since at the time when the patient had had the event, the clinical and genetic causes of CPVT were not yet described. The resting ECG was normal (Figure 3A), though during the exercise stress test polymorphic ventricular triplets occurred (Figure 3B).
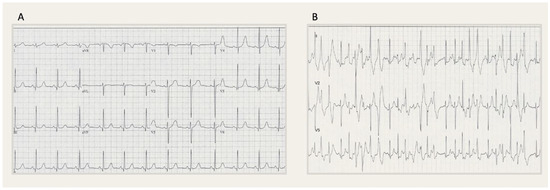
Figure 3.
(A) Patient 2: Resting 12-lead electrocardiogram (ECG, 25 mm/s) showing a sinus rhythm at a rate of 71 bpm, normal PR and QRS duration, a normal QRS axis and no ST-segment changes. (B) Patient 2: Exercise ECG (25 mm/s) showing polymorphic ventricular triplets.
During follow-up, the patient had several appropriate shocks. After initiating propranolol, the number and frequency of shocks was dramatically reduced. Fourteen years after his cardiac arrest, genetic testing was performed. The homozygous, likely pathogenic variant in the CASQ2 gene NM_001232.4:c. 838G>A (p.Asp280Asn) was detected (Figure 4A) and the most common recessive form of CPVT was diagnosed. The patient’s parents were consanguineous, both asymptomatic and heterozygous carriers of the variant. The patient had two sisters and a brother. One sister was found to be an asymptomatic heterozygous carrier of the same CASQ2 variant whereas the other sister was not. The patient’s deceased brother was not tested since no genetic material was available (Figure 4B).
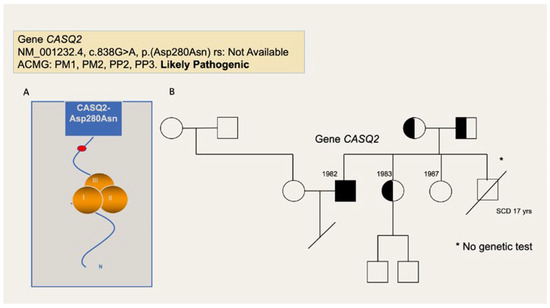
Figure 4.
(A) Pathogenic variant of patient 2. The variant CASQ2:c.838G>A (p.Asp280Asn) replaces an aspartic acid with an asparagine at position 280 of the CASQ2 protein product. There are important physicochemical differences between both residues and the aspartic acid at position 280 is conserved across species. This variant localizes to the calsequestrin-2 C-terminus, a region known to regulate Ca-binding capacity and aggregation state. In silico software programs, designed to assess the effect of the missense variants in the protein product, agree in its the deleterious effect. The variant has not been described in the literature nor is it listed in the Genome Aggregation Data-base or in the clinical database ClinVar. It was found homozygous in the patient, who previously showed typical signs and symptoms of catecholaminergic polymorphic ventricular tachycardia. Following the American College of Medical Genetics (ACMG) criteria, the variant is classified as likely pathogenic. (B) Family tree of patient 2. The patient was homozygous for the pathogenic variant in the CASQ2-gene. The patient’s parents and one of the patient’s sisters were asymptomatic heterozygous carriers of the variant. The other sister was not a carrier and the patient´s deceased brother was not tested. SCD: Sudden cardiac death.
Genetic Testing Methods
A Cardio Panel containing 173 genes (SureSelectQXT Target Enrichment, Agilent Technologies, Santa Clara, US) and next generation sequencing (MiSeq™-System, Illumina, San Diego, US) was used. Alignment of the sequences and local realignment against the human reference genome (GRCh37-hg19) was performed with lllumina Alignment Software v2.5.42.7 (Burrows-Wheeler algorithm and Genome Analysis Toolkit for variant calling). Variants with an allele frequency <1% in the coding regions including the flanking intronic regions (±8 base pairs) were evaluated. We reported variants classified by the American College of Medical Genetics criteria as pathogenic, likely pathogenic or variants of uncertain significance based on current knowledge. VariantStudio Software v3.0 (Illumina), VarSome Clinical (Saphetor SA, Lausanne, Switzerland), JSI medical systems software 5.3 (JSI medical systems Corp. New York, US), Single Nucleotide Polymorphism database release 153, Genome Aggregation Database, PubMed and ClinVar were used for data interpretation. Following the European Heart Rhythm Association (EHRA) expert consensus statement from 2022 on genetic testing of inherited heart diseases, in the context of CPVT, a genetic test, including the genes RYR2, CASQ2, CALM1-3, TRDN and TECRL should be performed when the criteria for CPVT (score >3.5) are fulfilled (Table 1) [3]. Initially, there is no indication to expand the testing to other cardiomyopathy genes. Therefore, our analysis included only the genes indicated by the EHRA expert consensus statement. Only if the results come back negative in patients otherwise fulfilling the criteria for CPVT, genetic testing could be expanded, then including for example pathogenetic variants in the KCNJ2, which causes phenocopies [3].

Table 1.
Catecholaminergic Polymorphic Ventricular Tachycardia (CPVT) Score; Adapted from [9].
Discussion
CPVT is a highly lethal, inherited cardiac channelopathy with an approximated prevalence of 1 in 10,000 [2]. The adrenergic induced arrythmias can lead to dizziness, palpitation, syncope and SCD, in the worst case [1]. Unfortunately, the diagnosis of CPVT is repeatedly missed, even though it is often already symptomatic during childhood [2]. The fact that palpitations during exercise in the young are usually benign and that patients with CPVT commonly have a normal resting ECG and a structurally normal heart [1], may contribute to the disease being missed. The hallmark arrhythmias can usually be seen-during an exercise stress test or on a Holter monitor. During the stress test, ectopic ventricular beats followed by ventricular bigeminy can often be detected. With ongoing adrenergic stimulation, bidirectional and polymorphic ventricular tachycardias can occur [1,4].
The incomplete penetrance and variable expressivity further complicate the diagnosis [1]. Genetic testing is recommended for all patients with clinically suspected or diagnosed CPVT [2]. A diagnostic score >3.5 is enough to proceed with genetic testing (Table 1) [3].
CPVT has been recognized as a disease in which the intracellular calcium in the cardiomyocytes is not handled properly [1]. As of today, seven genes have been clearly associated with the disease (Table 2).
Table 2.
Table of genes implicated in catecholaminergic polymorphic ventricular tachycardia (CPVT) or CPVT phenotypes.
The two most common genes in CPVT are RYR2 and CASQ2 [2]. RYR2 encodes the ryanodine receptor calcium release channel and CASQ2 encodes cardiac calsequestrin, which plays an important role in calcium storage and release inside the cell. Both proteins are involved in the sarcoplasmic reticulum calcium release complex (or excitation-contraction coupling) [5]. Genetic defects of the RYR2 gene are autosomal dominant and are found in the majority of genetically confirmed CPVT cases [5]. Genetic defects in the CASQ2 gene are autosomal recessive and represents the second most common genetic cause for CPVT [2,5]. In recent years, additional, although very rare genes have been described (Table 2). All these genes are part of the calcium signal regulation [4]. Importantly, an overlap syndrome with long QT phenotype has been detected for genes TRDN and CALM1-3 [4]. The precise diagnosis can only be made through genetic testing.
Differential Diagnosis
Many cases of CPVT are initially diagnosed with LQTS because the arrhythmic triggers are identical. Patients with LQTS, particularly with type 1 LQTS, can exhibit syncopes under exercise [2]. Moreover, LQTS can be concealed since the QT interval might not be obviously prolonged [6]. For the differential diagnosis a stress test is necessary, as the QT interval in patients with LQTS will prolong in the recovery phase [6].
Mutations in KCNJ2, which are associated with Andersen-Tawil syndrome (LQTS type 7), can mimic the CPVT phenotype (Table 2). Affected patients can exhibit bidirectional or polymorphic ventricular tachycardias (VT) during exercise. The expression of these mutations is thus considered a phenocopy [4]. However, these patients can have other syndromic features and muscular disorders characterized by periodic paralysis [2].
Further, other idiopathic VTs should be considered as potential differential diagnosis, although these arrhythmias are commonly monomorphic, not polymorphic [2].
CPVT is usually diagnosed in the first decade of life [2], during which cardiac ischemia is rare. Therefore, routinary exclusion of ischemic heart disease is not recommended, although, if ischemic symptoms are present, it is important to rule out coronary malformation, muscular bridges and spasms.
Cardiac magnetic resonance imaging (CMR) is indicated if cardiac anomalies are detected during echocardiography. CMR may also be useful in distinguishing CPVT from arrhythmogenic right ventricular cardiomyopathy (ARVC). The main difference between those two being the age at presentation. While the symptoms of CPVT usually occur in the first decade of life, the symptoms of ARVC commonly appear after the third decade. Depending on the clinical presentation, ARVC with its characteristic predominantly right ventricular fibrofatty replacement of myocardial tissue, can be difficult to discriminate from CPVT. During early stages of the disease, the associated structural changes may be lacking or may not visible on echocardiography, however, they might be seen on CMR [2].
The cornerstone of the medical treatment of CPVT are beta blockers (preferably nadolol or propranolol). Even asymptomatic mutation carriers without ventricular arrythmias should be prescribed a beta blocker [1,2]. Although beta blockers have shown to reduce the rate of SCD in CPVT patients, the incidence of ventricular arrythmias can still remain significant. Accordingly, the addition of flecainide should be considered in cases were the arrythmias are not adequately controlled under beta blockers [1,2]. In patients with CPVT and persistent syncopal episodes or ventricular arrythmias despite maximal pharmacological treatment, an ICD should be considered. An ICD is also indicated in CPVT patients already treated with beta blockers after surviving an aborted cardiac arrest [1,2]. Because the stress of appropriate or inappropriate shock therapy itself can trigger additional arrythmias, the time from arrythmia onset to defibrillation should be programmed as long as possible with high tracking rates. In addition, ventricular fibrillation or polymorphic ventricular tachycardias respond better to defibrillation than the initially occurring bidirectional ventricular tachycardia [2].
Left cardiac sympathetic denervation has shown a reduction of arrhythmic episodes and should therefore be considered in addition to ICD in patients with persistent symptoms despite maximal pharmacological therapy [1,2].
Finally, CPVT patients are recommended to avoid exhausting exercise and pressured environments [2].
Conclusion
A missed or delayed precise diagnosis of CPVT can have fatal consequences. Recurrent syncopes during exercise in the first decade of life should be considered an important symptom for suspected CPVT. In the evaluation of unexplained syncopes, it is important to perform an exercise stress test and Holter monitoring to rule out exercise induced arrhythmias, even during childhood.
Author Contributions
LR: Concept, design and construction of the manuscript, implementation of reviewers comments, production of figures, literature research. AMD: Data acquisition, idea of the manuscript, interpretation of patient data, proofreading and correction of the manuscript, production of figures. NBS: Data acquisition, interpretation of patient data, proofreading and correction of the manuscript.
Ethics Statement
The families reported in this manuscript accepted to anonymously participate in this study. Written informed consent was obtained.
Conflicts of Interest
LR and NBS have no potential conflicts of interest to declare. AMD is the medical director of Swiss DNAlysis.
References
- Baltogiannis, G.G.; Lysitsas, D.N.; di Giovanni, G.; Ciconte, G.; Sieira, J.; Conte, G.; et al. CPVT: Arrhythmogenesis, Therapeutic Management, and Future Perspectives. A Brief Review of the Literature. Front Cardiovasc Med. 2019, 6, 92. [Google Scholar] [CrossRef] [PubMed]
- Zeppenfeld, K.; Tfelt-Hansen, J.; de Riva, M.; Winkel, B.G.; Behr, E.R.; Blom, N.A.; et al.; ESC Scientific Document Group 2022 ESC Guidelines for the management of patients with ventricular arrhythmias and the prevention of sudden cardiac death. Eur Heart J. 2022, 43, 3997–4126. [Google Scholar] [CrossRef] [PubMed]
- Wilde, A.A.; Semsarian, C.; Márquez, M.F.; Shamloo, A.S.; Ackerman, M.J.; Ashley, E.A.; et al. Document Reviewers; Developed in partnership with and endorsed by the European Heart Rhythm Association (EHRA), a branch of the European Society of Cardiology 8ESC), the Heart Rhythm Society (HRS), the Asia Pacific Heart Rhythm Society (APHRS), and the Latin American Heart Rhythm Society (LAHRS). European Heart Rhythm Association (EHRA)/Heart Rhythm Society (HRS)/Asia Pacific Heart Rhythm Society (APHRS)/ Latin American Heart Rhythm Society (LAHRS) Expert Consensus Statement on the state of genetic testing for cardiac diseases. Europace 2022, 24, 1307–1367. [Google Scholar] [PubMed]
- Walsh, R.; Adler, A.; Amin, A.S.; Abiusi, E.; Care, M.; Bikker, H.; et al. Evaluation of gene validity for CPVT and short QT syndrome in sudden arrhythmic death. Eur Heart J. 2022, 43, 1500–1510. [Google Scholar] [CrossRef] [PubMed]
- Wleklinski, M.J.; Kannankeril, P.J.; Knollmann, B.C. Molecular and tissue mechanisms of catecholaminergic polymorphic ventricular tachycardia. J Physiol. 2020, 598, 2817–2834. [Google Scholar] [CrossRef] [PubMed]
- Horner, J.M.; Horner, M.M.; Ackerman, M.J. The diagnostic utility of recovery phase QTc during treadmill exercise stress testing in the evaluation of long QT syndrome. Heart Rhythm. 2011, 8, 1698–1704. [Google Scholar] [CrossRef] [PubMed]
- Novak, A.; Barad, L.; Lorber, A.; Gherghiceanu, M.; Reiter, I.; Eisen, B.; et al. Functional abnormalities in iPSC-derived cardiomyocytes generated from CPVT1 and CPVT2 patients carrying ryanodine or calsequestrin mutations. J Cell Mol Med. 2015, 19, 2006–2018. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.Y.; Mesirca, P.; Marqués-Sulé, E.; Zahradnikova, A., Jr.; Villejoubert, O.; D’Ocon, P.; et al. RyR2R420Q catecholaminergic polymorphic ventricular tachycardia mutation induces bradycardia by disturbing the coupled clock pacemaker mechanism. JCI Insight. 2017, 2, e91872. [Google Scholar] [CrossRef] [PubMed]
- Giudicessi, J.R.; Lieve, K.V.; Rohatgi, R.K.; Koca, F.; Tester, D.J.; van der Werf, C.; et al. Assessment and Validation of a Phenotype-Enhanced Variant Classification Frame-work to Promote or Demote RYR2 Missense Variants of Uncertain Significance. Circ Genom Precis Med. 2019, 12, e002510. [Google Scholar] [CrossRef] [PubMed]


© 2024 by the author. Attribution - Non-Commercial - NoDerivatives 4.0