A Molecular Docking Approach to Evaluate the Pharmacological Properties of Natural and Synthetic Treatment Candidates for Use against Hypertension

 ,
, 

 , , and
, , and
Abstract
1. Introduction
2. Materials and Methods
2.1. Physiochemical Properties
2.2. Secondary Structure Predictions
2.3. Domain and Motif Analysis
2.4. Selection of Ligand, Receptor, and Active Site Prediction
2.5. Preparation of Ligands and Receptor
2.6. Molecular Docking
2.7. Lipinski’s Rule of Five for Drug-Likeness or ADME (Absorption, Distribution, Metabolism, and Excretion) Analysis
3. Results and Discussion
3.1. Physiochemical Properties of ACE
3.2. Membrane Topology of ACE
3.3. Reported Inhibitors of ACE
3.4. Molecular Docking
3.5. Drug-Likeness and ADME Predictions of Our Inhibitors
4. Conclusions
Author Contributions
Acknowledgments
Conflicts of Interest
References
- Mendis, S.; Puska, P.; Norrving, B.; World Health Organization. Global Atlas on Cardiovascular Disease Prevention and Control; World Health Organization: Geneva, Switzerland, 2011. [Google Scholar]
- Catalá López, F.; Tabarés Seisdedos, R. Global, regional, and national life expectancy, all-cause mortality, and cause-specific mortality for 249 causes of death, 1980–2015: A systematic analysis for the Global Burden of Disease Study 2015. Lancet 2016, 8, 1459–1544. [Google Scholar]
- Santulli, G. Epidemiology of cardiovascular disease in the 21st century: Updated numbers and updated facts. J. Cardiovasc. Dis. 2013, 1, 1–2. [Google Scholar]
- Bundy, J.D.; He, J. Hypertension and Related Cardiovascular Disease Burden in China. Ann. Glob. Health 2016, 82, 227–233. [Google Scholar] [CrossRef]
- Campbell, N.R.; Lackland, D.T.; Lisheng, L.; Niebylski, M.L.; Nilsson, P.M.; Zhang, X.H. Using the Global Burden of Disease study to assist development of nation-specific fact sheets to promote prevention and control of hypertension and reduction in dietary salt: A resource from the World Hypertension League. J. Clin. Hypertens. 2015, 17, 165–167. [Google Scholar] [CrossRef] [PubMed]
- Ruppert, V.; Maisch, B. Genetics of human hypertension. Herz 2003, 28, 655–662. [Google Scholar] [CrossRef] [PubMed]
- Newhouse, S.J.; Wallace, C.; Dobson, R.; Mein, C.; Pembroke, J.; Farrall, M.; Clayton, D.; Brown, M.; Samani, N.; Dominiczak, A. Haplotypes of the WNK1 gene associate with blood pressure variation in a severely hypertensive population from the British Genetics of Hypertension study. Hum. Mol. Genet. 2005, 14, 1805–1814. [Google Scholar] [CrossRef] [PubMed]
- Brand, E.; Chatelain, N.; Paillard, F.; Tiret, L.; Visvikis, S.; Lathrop, M.; Soubrier, F.; Demenais, F. Detection of putative functional angiotensinogen (AGT) gene variants controlling plasma AGT levels by combined segregation-linkage analysis. Eur. J. Hum. Genet. 2002, 10, 715. [Google Scholar] [CrossRef]
- Jimsheena, V.; Gowda, L.R. Angiotensin I-converting enzyme (ACE) inhibitory peptides derived from arachin by simulated gastric digestion. Food Chem. 2011, 125, 561–569. [Google Scholar] [CrossRef]
- Riordan, J.F. Angiotensin-I-converting enzyme and its relatives. Genome Biol. 2003, 4, 225. [Google Scholar] [CrossRef][Green Version]
- Passos-Silva, D.G.; Brandan, E.; Santos, R.A.S. Angiotensins as therapeutic targets beyond heart disease. Trends Pharmacol. Sci. 2015, 36, 310–320. [Google Scholar] [CrossRef]
- Kouranov, A.; Xie, L.; de la Cruz, J.; Chen, L.; Westbrook, J.; Bourne, P.E.; Berman, H.M. The RCSB PDB information portal for structural genomics. Nucleic Acids Res. 2006, 34, D302–D305. [Google Scholar] [CrossRef] [PubMed]
- Edelhoch, H.J.B. Spectroscopic determination of tryptophan and tyrosine in proteins. Biochemistry 1967, 6, 1948–1954. [Google Scholar] [CrossRef] [PubMed]
- McGuffin, L.J.; Bryson, K.; Jones, D.T.J.B. The PSIPRED protein structure prediction server. Bioinformatics 2000, 16, 404–405. [Google Scholar] [CrossRef]
- Bailey, T.L.; Johnson, J.; Grant, C.E.; Noble, W.S. The MEME suite. Nucleic Acids Res. 2015, 43, W39–W49. [Google Scholar] [CrossRef] [PubMed]
- Bailey, T.L.; Boden, M.; Buske, F.A.; Frith, M.; Grant, C.E.; Clementi, L.; Ren, J.; Li, W.W.; Noble, W.S. MEME SUITE: Tools for motif discovery and searching. Nucleic Acids Res. 2009, 37, W202–W208. [Google Scholar] [CrossRef]
- Kim, S.; Thiessen, P.A.; Bolton, E.E.; Chen, J.; Fu, G.; Gindulyte, A.; Han, L.; He, J.; He, S.; Shoemaker, B.A. PubChem substance and compound databases. Nucleic Acids Res. 2015, 44, D1202–D1213. [Google Scholar] [CrossRef] [PubMed]
- Scholz, C.; Knorr, S.; Hamacher, K.; Schmidt, B. DOCKTITE—A Highly Versatile Step-by-Step Workflow for Covalent Docking and Virtual Screening in the Molecular Operating Environment. J. Chem. Inf. Model. 2015, 55, 398–406. [Google Scholar] [CrossRef]
- Bajorath, J. Integration of virtual and high-throughput screening. Nat. Rev. Drug Discov. 2002, 1, 882. [Google Scholar] [CrossRef]
- Jorgensen, W.L. The many roles of computation in drug discovery. Science 2004, 303, 1813–1818. [Google Scholar] [CrossRef]
- Kuntz, I.D.; Blaney, J.M.; Oatley, S.J.; Langridge, R.; Ferrin, T.E. A geometric approach to macromolecule-ligand interactions. J. Mol. Biol. 1982, 161, 269–288. [Google Scholar] [CrossRef]
- Kalyaanamoorthy, S.; Chen, Y.-P.P. Structure-based drug design to augment hit discovery. Drug Discov. Today 2011, 16, 831–839. [Google Scholar] [CrossRef]
- Chemical Computing Group Inc. Molecular Operating Environment (MOE); H3A 2R7; Chemical Computing Group Inc.: Montreal, QC, Canada, 2016. [Google Scholar]
- Clark, A.M.; Labute, P. 2D depiction of protein–ligand complexes. J. Chem. Inf. Model. 2007, 47, 1933–1944. [Google Scholar] [CrossRef] [PubMed]
- Vilar, S.; Cozza, G.; Moro, S. Medicinal chemistry and the molecular operating environment (MOE): Application of QSAR and molecular docking to drug discovery. Curr. Top. Med. Chem. 2008, 8, 1555–1572. [Google Scholar] [CrossRef]
- Yuliana, D.; Bahtiar, F.I.; Najib, A. In silico screening of chemical compounds from roselle (Hibiscus Sabdariffa) as angiotensin-I converting enzyme inhibitor used PyRx program. ARPN J. Sci. Technol. 2013, 3, 1158–1160. [Google Scholar]
- Friesner, R.A.; Banks, J.L.; Murphy, R.B.; Halgren, T.A.; Klicic, J.J.; Mainz, D.T.; Repasky, M.P.; Knoll, E.H.; Shelley, M.; Perry, J.K. Glide: A new approach for rapid, accurate docking and scoring. 1. Method and assessment of docking accuracy. J. Med. Chem. 2004, 47, 1739–1749. [Google Scholar] [CrossRef] [PubMed]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef] [PubMed]
- Verdonk, M.L.; Cole, J.C.; Hartshorn, M.J.; Murray, C.W.; Taylor, R.D. Improved protein–ligand docking using GOLD. Proteins Struct. Funct. Bioinform. 2003, 52, 609–623. [Google Scholar] [CrossRef]
- Jain, A.N. Surflex: Fully automatic flexible molecular docking using a molecular similarity-based search engine. J. Med. Chem. 2003, 46, 499–511. [Google Scholar] [CrossRef] [PubMed]
- Schellhammer, I.; Rarey, M. FlexX-Scan: Fast, structure-based virtual screening. Proteins Struct. Funct. Bioinform. 2004, 57, 504–517. [Google Scholar] [CrossRef] [PubMed]
- Abagyan, R.; Totrov, M.; Kuznetsov, D. ICM—A new method for protein modeling and design: applications to docking and structure prediction from the distorted native conformation. J. Comput. Chem. 1994, 15, 488–506. [Google Scholar] [CrossRef]
- Molegro, A. MVD 5.0 Molegro Virtual Docker; CLC bio: Aarhus, Denmark, 2011. [Google Scholar]
- Kellenberger, E.; Rodrigo, J.; Muller, P.; Rognan, D. Comparative evaluation of eight docking tools for docking and virtual screening accuracy. Proteins Struct. Funct. Bioinform. 2004, 57, 225–242. [Google Scholar] [CrossRef] [PubMed]
- Venkatachalam, C.M.; Jiang, X.; Oldfield, T.; Waldman, M. LigandFit: A novel method for the shape-directed rapid docking of ligands to protein active sites. J. Mol. Graph. Model. 2003, 21, 289–307. [Google Scholar] [CrossRef]
- Corbeil, C.R.; Englebienne, P.; Moitessier, N. Docking ligands into flexible and solvated macromolecules. 1. Development and validation of FITTED 1.0. J. Chem. Inf. Model. 2007, 47, 435–449. [Google Scholar] [CrossRef] [PubMed]
- Tietze, S.; Apostolakis, J. GlamDock: Development and validation of a new docking tool on several thousand protein−ligand complexes. J. Chem. Inf. Model. 2007, 47, 1657–1672. [Google Scholar] [CrossRef] [PubMed]
- Hsu, K.-C.; Chen, Y.-F.; Lin, S.-R.; Yang, J.-M. iGEMDOCK: A graphical environment of enhancing GEMDOCK using pharmacological interactions and post-screening analysis. BMC Bioinform. 2011, 12, S33. [Google Scholar] [CrossRef]
- Daina, A.; Michielin, O.; Zoete, V. SwissADME: A free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci. Rep. 2017, 7, 42717. [Google Scholar] [CrossRef]
- Daina, A.; Zoete, V. A BOILED-Egg To Predict Gastrointestinal Absorption and Brain Penetration of Small Molecules. ChemMedChem 2016, 11, 1117–1121. [Google Scholar] [CrossRef]
- Jamerson, K.; Weber, M.A.; Bakris, G.L.; Dahlöf, B.; Pitt, B.; Shi, V.; Hester, A.; Gupte, J.; Gatlin, M.; Velazquez, E.J. Benazepril plus amlodipine or hydrochlorothiazide for hypertension in high-risk patients. N. Engl. J. Med. 2008, 359, 2417–2428. [Google Scholar] [CrossRef] [PubMed]
- Nagaoka, S.; Kawasaki, S.; Kawasaki, H.; Kamei, K. The angiotensin converting enzyme (ACE) inhibitor, captopril disrupts the motility activation of sperm from the silkworm, Bombyx mori. J. Insect Physiol. 2017, 103, 18–28. [Google Scholar] [CrossRef]
- Narayanam, M.; Sahu, A.; Singh, S. Use of LC–MS/TOF, LC–MSn, NMR and LC–NMR in characterization of stress degradation products: Application to cilazapril. J. Pharm. Biomed. Anal. 2015, 111, 190–203. [Google Scholar] [CrossRef]
- Younas, F.; Aslam, B.; Muhammad, F.; Mohsin, M.; Raza, A.; Faisal, M.N.; Hassan, S.-U.; Majeed, W. Haematopoietic effects of Angelica sinensis root cap polysaccharides against lisinopril-induced anaemia in albino rats. Pharm. Biol. 2017, 55, 108–113. [Google Scholar] [CrossRef]
- Reddy, C.P.; Ramakrishna, K.; Narayana Rao, K. Structural identification of degradants of moexipril by LC-MS/MS. Biomed. Chromatogr. 2017, 31, e4004. [Google Scholar]
- Elgendy, I.Y.; Bavry, A.A.; Gong, Y.; Handberg, E.M.; Cooper-DeHoff, R.M.; Pepine, C.J. Long-term mortality in hypertensive patients with coronary artery disease: Results from the US cohort of the International Verapamil (SR)/Trandolapril Study. Hypertension 2016, 68, 1110–1114. [Google Scholar] [CrossRef]
- Shaddy, R.; Canter, C.; Halnon, N.; Kochilas, L.; Rossano, J.; Bonnet, D.; Bush, C.; Zhao, Z.; Kantor, P.; Burch, M. Design for the sacubitril/valsartan (LCZ696) compared with enalapril study of pediatric patients with heart failure due to systemic left ventricle systolic dysfunction (PANORAMA-HF study). Am. Heart J. 2017, 193, 23–34. [Google Scholar] [CrossRef]
- Araiza-Saldaña, C.I.; Pedraza-Priego, E.F.; Torres-López, J.E.; Rocha-González, H.I.; Castañeda-Corral, G.; Hong-Chong, E.; Granados-Soto, V. Fosinopril Prevents the Development of Tactile Allodynia in a Streptozotocin-Induced Diabetic Rat Model. Drug Dev. Res. 2015, 76, 442–449. [Google Scholar] [CrossRef]
- El-Bagary, R.I.; Elkady, E.F.; Mowaka, S.; Attallah, M.A. A Validated HPLC Method for Simultaneous Determination of Perindopril Arginine, Amlodipine, and Indapamide: Application in Bulk and in Different Pharmaceutical Dosage Forms. J. AOAC Int. 2017, 100, 992–999. [Google Scholar] [CrossRef]
- Wojewodzka-Zelezniakowicz, M.; Kisiel, W.; Kramkowski, K.; Gromotowicz-Poplawska, A.; Zakrzeska, A.; Stankiewicz, A.; Kolodziejczyk, P.; Szemraj, J.; Ladny, J.R.; Chabielska, E. Quinapril decreases antifibrinolytic and prooxidative potential of propofol in arterial thrombosis in hypertensive rats. J. Renin-Angiotensin-Aldosterone Syst. 2016, 17, 1470320316647239. [Google Scholar] [CrossRef]
- Mancia, G.; Schumacher, H.; Böhm, M.; Redon, J.; Schmieder, R.E.; Verdecchia, P.; Sleight, P.; Teo, K.; Yusuf, S. Relative and Combined Prognostic Importance of On-Treatment Mean and Visit-to-Visit Blood Pressure Variability in ONTARGET and TRANSCEND PatientsNovelty and Significance. Hypertension 2017, 70, 938–948. [Google Scholar] [CrossRef]
- Reuter, H.; Koch, H.; Lawson, L. The Science and Therapeutic Application of Allium sativum L. and Related Species; Williams and Wilkins: Baltimore, MD, USA, 1996. [Google Scholar]
- Cragg, G.M.; Newman, D.J.; Snader, K.M. Natural products in drug discovery and development. J. Nat. Prod. 1997, 60, 52–60. [Google Scholar] [CrossRef]
- El-Sheakh, A.R.; Ghoneim, H.A.; Suddek, G.M.; Ammar, E.S. Attenuation of oxidative stress, inflammation, and endothelial dysfunction in hypercholesterolemic rabbits by allicin. Can. J. Physiol. Pharmacol. 2015, 94, 216–224. [Google Scholar] [CrossRef]
- Ford, N.F.; Fulmor, I.E.; Nichola, P.S.; Alpin, P.G.; Herron, J.M. Fosinopril monotherapy: Relationship between blood pressure reduction and time of administration. Clin. Cardiol. 1993, 16, 324–330. [Google Scholar] [CrossRef]
- Hayek, T.; Attias, J.; Coleman, R.; Brodsky, S.; Smith, J.; Breslow, J.L.; Keidar, S.J. The angiotensin-converting enzyme inhibitor, fosinopril, and the angiotensin II receptor antagonist, losartan, inhibit LDL oxidation and attenuate atherosclerosis independent of lowering blood pressure in apolipoprotein E deficient mice. Cardiovasc. Res. 1999, 44, 579–587. [Google Scholar] [CrossRef]
- Heart Outcomes Prevention Evaluation Study Investigators. Effects of an angiotensin-converting–enzyme inhibitor, ramipril, on cardiovascular events in high-risk patients. N. Engl. J. Med. 2000, 342, 145–153. [Google Scholar] [CrossRef]
- Lipinski, C.A. Lead-and drug-like compounds: The rule-of-five revolution. Drug Discov. Today Technol. 2004, 1, 337–341. [Google Scholar] [CrossRef]
- Lipinski, C.A. Rule of five in 2015 and beyond: Target and ligand structural limitations, ligand chemistry structure and drug discovery project decisions. Adv. Drug Deliv. Rev. 2016, 101, 34–41. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
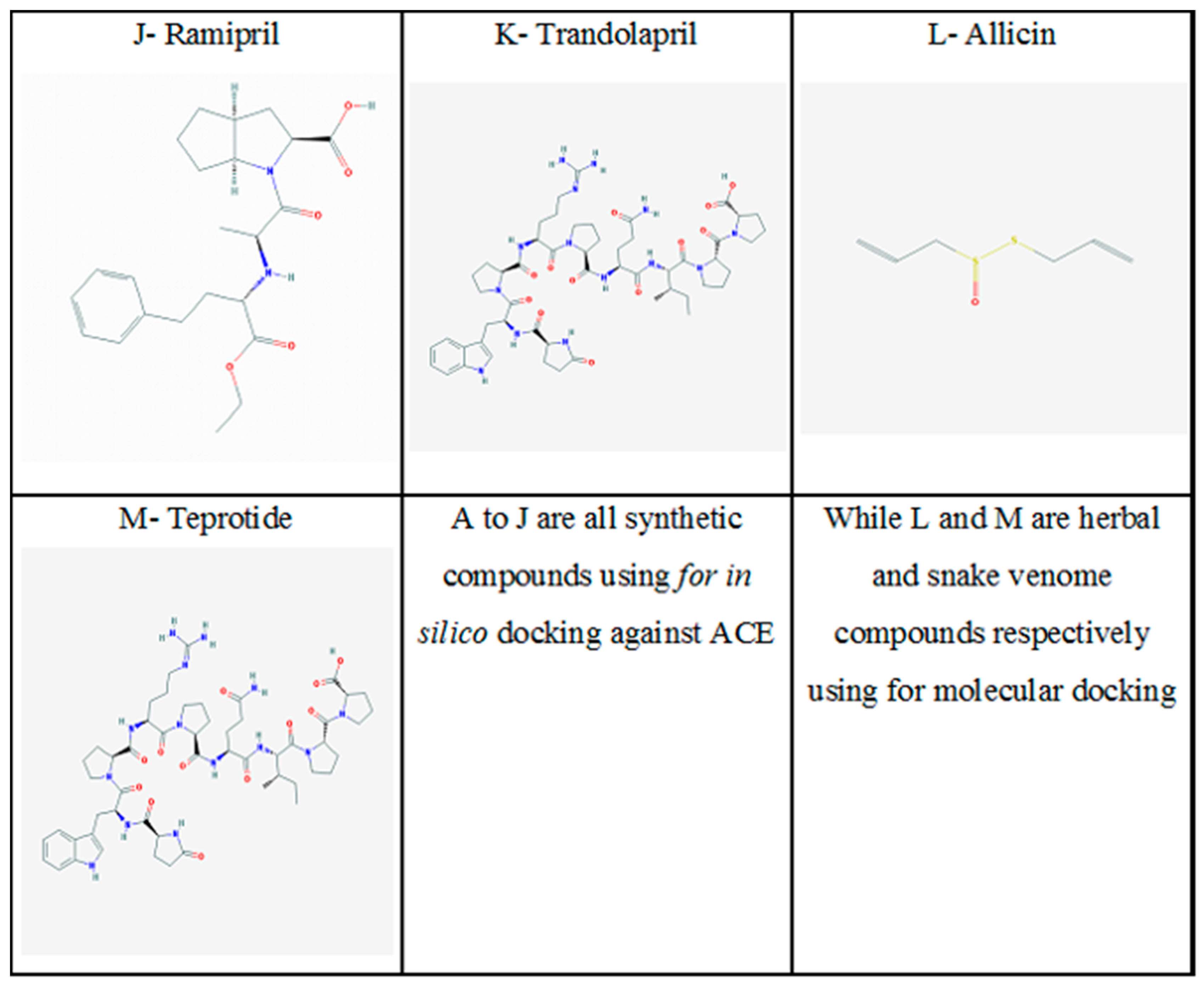{kind=link}
{kind=link}
| No. | Software/Tools | Algorithm | Scoring Term | Advantages | Ref. |
|---|---|---|---|---|---|
| 1. | Molecular Operating Environment (MOE) | High-Speed α shapes algorithms | London dG, FlexX, DrugScore, Mcdock | Customizable, available source-code, gives binding affinity score, shows interacting amino acids with position, and is user-friendly. | [25] |
| 2. | PyRx | Lamarckian genetic algorithm | Binding energy, Internal energy, Internal energy, Unbound energy | Temperature Resistance. Pyrex’s excellent thermal properties at both high and low temperatures are one of its key features. | [26] |
| 3. | Glide (Grid-based Ligand Docking with Energetics) | Monte Carlo | Glide score | Lead discovery and lead optimization | [27] |
| 4. | AutoDock | Lamarckian genetic algorithm | Empirical free-energy function | Adaptability to user-defined input | [28] |
| 5. | GOLD (Genetic Optimization for Ligand Docking) | Genetic algorithm | GoldScore, ChemScore, ASP (Astex Statistical Potential), CHEMPLP (Piecewise Linear Potential), User-defined | Allows atomic overlapping between protein and ligand | [29] |
| 6. | Surflex | Surflex-Dock search Algorithm | Bohm’s scoring function | High accuracy level by extending force fields | [30] |
| 7. | FlexX | Incremental reconstruction | Modified Bohm scoring function | Provides a large number of conformations | [31] |
| 8. | ICM (Internal Coordinate Modeling) | Monte Carlo minimization | Virtual library screening scoring function | Allows side chain flexibility to find a parallel arrangement of two rigid helixes | [32] |
| 9. | MVD (Molegro Virtual Docker) | Evolutionary algorithm | MolDock score | High accuracy level of predicting binding mode | [33] |
| 10. | Fred (Fast Rigid Exhaustive Docking) | Exhaustive search algorithm | Gaussian scoring function | Nonstochastic approach to examine all possible poses within a protein active site | [34] |
| 11. | LigandFit | Monte Carlo method | LigScore, Piecewise Linear Potential (PLP), Potential of Mean Force (PMF) | Generates good hit rates based on LigScore | [35] |
| 12. | FITTED (Flexibility Induced Through Targeted Evolutionary Description) | Genetic algorithm | Potential of Mean Force (PMF), Drug Score | Analyzes the effect of water molecules on protein–ligand complexes | [36] |
| 13. | GlamDock | Monte Carlo method | ChillScore | Provides provision of two-dimensional analysis to screen ligands by targeting protein | [37] |
| 14. | iGEMDOCK | Genetic algorithm | Empirical scoring function | Integrates the structure-based virtual screening and post-screening analysis. Provides a graphical integrated environment for virtual screening | [38] |
| No. | Ligand | Features | Source | Function | Citation |
|---|---|---|---|---|---|
| 1. | Benazepril | 97% protein binding, a half-life of 10–11 h, pregnancy category: D | Synthetic | Cures hypertension | [41] |
| 2. | Captopril | 25–30% protein binding, a half-life of 2 h, pregnancy category: D | Synthetic | Controls blood pressure | [42] |
| 3. | Cilazapril | A half-life of 1 to 4 h | Synthetic | ACE inhibition | [43] |
| 4. | Lisinopril | Pregnancy category: D, does not bind serum proteins other than ACE | Synthetic | Inhibition of ACE | [44] |
| 5. | Moexipril | Pregnancy category: D, <90% protein binding and 1 h half-life | Synthetic | Treatment of hypertension and congestive heart failure | [45] |
| 6. | Trandolapril | Half-life 6 to 10 h, pregnancy category: D | Synthetic | Controls high blood pressure | [46] |
| 7. | Enalapril | Pregnancy category: D, half-life of 11 h | Synthetic | ACE inhibition to control hypertension | [47] |
| 8. | Fosinopril | 12 h half-life, pregnancy category: D, ≥95% protein-binding capacity | Synthetic | Normalizes blood pressure | [48] |
| 9. | Perindopril | 20% protein binding, pregnancy category: D and 1–2 h half life | Synthetic | Controls blood pressure | [49] |
| 10. | Quinapril | 97% protein binding, 2 h biological half-life, pregnancy category: D | Synthetic | Inhibition of ACE | [50] |
| 11. | Ramipril | Protein binding 73% (ramipril), 56% (ramiprilat), half-life of 2–4 h | Synthetic | Congestive heart failure control | [51] |
| 12. | Allicin | Has water solubility of 24 mg/mL at 10 °C, solid, melting point >25 °C | Garlic and onion | Inhibition of ACE | [52] |
| 13. | Teprotide | Has 10 hydrogen bond donors, 13 hydrogen bond acceptors, and 79 heavy atoms | Snake venom | Antihypertensive agent | [53] |
| Serial Number | Property | Value |
|---|---|---|
| 1. | Number of amino acids | 589 |
| 2. | Total number of atoms | 9457 |
| 3. | Molecular weight | 67,993.20 Dalton |
| 4. | Theoretical pI | 5.82 |
| 5. | Extinction coefficient * | 143,240 at Abs 0.1% 2.112, assuming all pairs of Cys residues form cystines |
| 6. | Instability index | 39.46 |
| 7. | Aliphatic index | 78.86 |
| 8. | Grand average of hydropathicity (GRAVY) | −0.441 |
| 9. | Chemical Formula | C3076H4656N818O883S24 |
| 10. | Charge | Negative |
| Amino Acid | Position | Amino Acid | Position |
|---|---|---|---|
| Histidine | 317 | Alanine | 318 |
| Serine | 319 | Histidine | 347 |
| Glutamic Acid | 348 | Histidine | 351 |
| Glutamic Acid | 375 | Phenylalanine | 421 |
| Lysine | 475 | Phenylalanine | 476 |
| Histidine | 477 | Valine | 482 |
| Tyrosine | 484 | Tyrosine | 487 |
| No. | Name | S-Values |
|---|---|---|
| 1. | Teprotide | −20.1163 |
| 2. | Fosinopril | −18.9225 |
| 3. | Moexipril | −16.816 |
| 4. | Quinapril | −13.456 |
| 5. | Lisinopril | −12.502 |
| 6. | Cilazapril | −12.493 |
| 7. | Trandolapril | −12.2673 |
| 8. | Enalapril | −11.7516 |
| 9. | Ramipril | −11.3562 |
| 10. | Captopril | −10.8282 |
| 11. | Benazepril | −9.3245 |
| 12. | Perindopril | −8.105 |
| 13. | Allicin | −5.5448 |
| No. | Name | Lipinski’s Rule of Five | Drug-Likeness | ||||
|---|---|---|---|---|---|---|---|
| Molecular Weight (g/mol) | Lipophilicity (MLog P) | Hydrogen Bond Donors | Hydrogen Bond Acceptors | No. of Rule Violations | |||
| Less than 500 Dalton | Less than 5 | Less than 5 | Less than 10 | Less than 2 Violations | Lipinski’s Rule Follows | ||
| 1. | Teprotide | 1101.26 | −3.11 | 10 | 13 | 3: MW > 500, NH or OH > 5, N or O > 10, 1: MW > 500 | No |
| 2. | Fosinopril | 563.66 | 3.74 | 1 | 7 | 0 | Yes |
| 3. | Moexipril | 498.57 | 1.54 | 2 | 8 | 0 | Yes |
| 4. | Quinapril | 438.52 | 2.17 | 2 | 6 | 0 | Yes |
| 5. | Lisinopril | 405.49 | −1.46 | 4 | 7 | 0 | Yes |
| 6. | Cilizapril | 417.50 | 1.79 | 2 | 7 | 0 | Yes |
| 7. | Trandolapril | 430.54 | 2.19 | 2 | 6 | 0 | Yes |
| 8. | Enalapril | 376.45 | 1.32 | 2 | 6 | 0 | Yes |
| 9. | Ramipril | 416.51 | 1.98 | 2 | 6 | 0 | Yes |
| 10. | Caprtopril | 217.29 | 0.45 | 1 | 3 | 0 | Yes |
| 11. | Benzapril | 424.49 | 2.23 | 2 | 6 | 0 | Yes |
| 12. | Perindopril | 368.47 | 1.36 | 2 | 6 | 0 | Yes |
| 13. | Allicin | 162.27 | 1.18 | 0 | 1 | 0 | Yes |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Attique, S.A.; Hassan, M.; Usman, M.; Atif, R.M.; Mahboob, S.; Al-Ghanim, K.A.; Bilal, M.; Nawaz, M.Z. A Molecular Docking Approach to Evaluate the Pharmacological Properties of Natural and Synthetic Treatment Candidates for Use against Hypertension. Int. J. Environ. Res. Public Health 2019, 16, 923. https://doi.org/10.3390/ijerph16060923
Attique SA, Hassan M, Usman M, Atif RM, Mahboob S, Al-Ghanim KA, Bilal M, Nawaz MZ. A Molecular Docking Approach to Evaluate the Pharmacological Properties of Natural and Synthetic Treatment Candidates for Use against Hypertension. International Journal of Environmental Research and Public Health. 2019; 16(6):923. https://doi.org/10.3390/ijerph16060923
Chicago/Turabian StyleAttique, Syed Awais, Muhammad Hassan, Muhammad Usman, Rana Muhammad Atif, Shahid Mahboob, Khalid A. Al-Ghanim, Muhammad Bilal, and Muhammad Zohaib Nawaz. 2019. "A Molecular Docking Approach to Evaluate the Pharmacological Properties of Natural and Synthetic Treatment Candidates for Use against Hypertension" International Journal of Environmental Research and Public Health 16, no. 6: 923. https://doi.org/10.3390/ijerph16060923
APA StyleAttique, S. A., Hassan, M., Usman, M., Atif, R. M., Mahboob, S., Al-Ghanim, K. A., Bilal, M., & Nawaz, M. Z. (2019). A Molecular Docking Approach to Evaluate the Pharmacological Properties of Natural and Synthetic Treatment Candidates for Use against Hypertension. International Journal of Environmental Research and Public Health, 16(6), 923. https://doi.org/10.3390/ijerph16060923