
Fucoxanthin Loaded in Palm Stearin- and Cholesterol-Based Solid Lipid Nanoparticle-Microcapsules, with Improved Stability and Bioavailability In Vivo

 ,
,  ,
,
Abstract
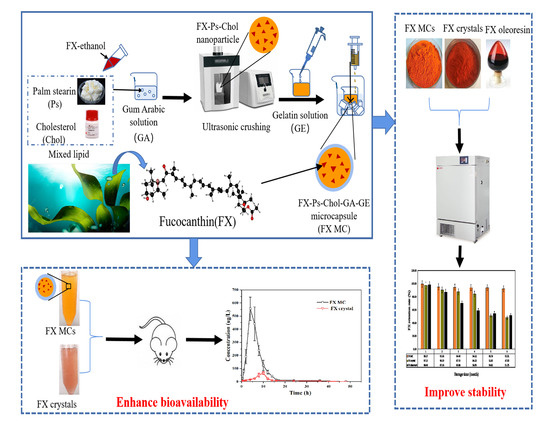
1. Introduction
2. Results and Discussion
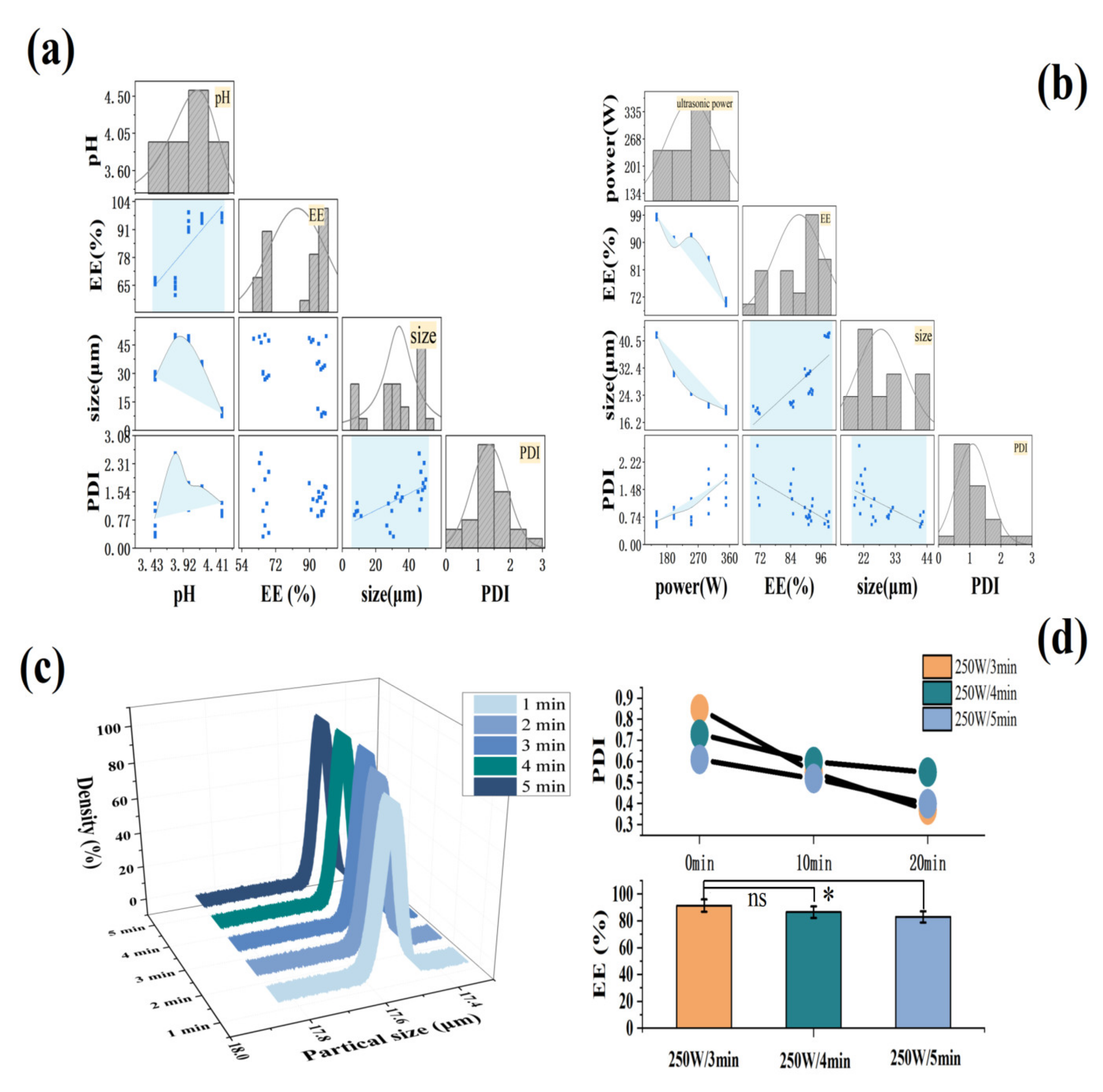
2.1. Optimization of FX MC Preparation Process
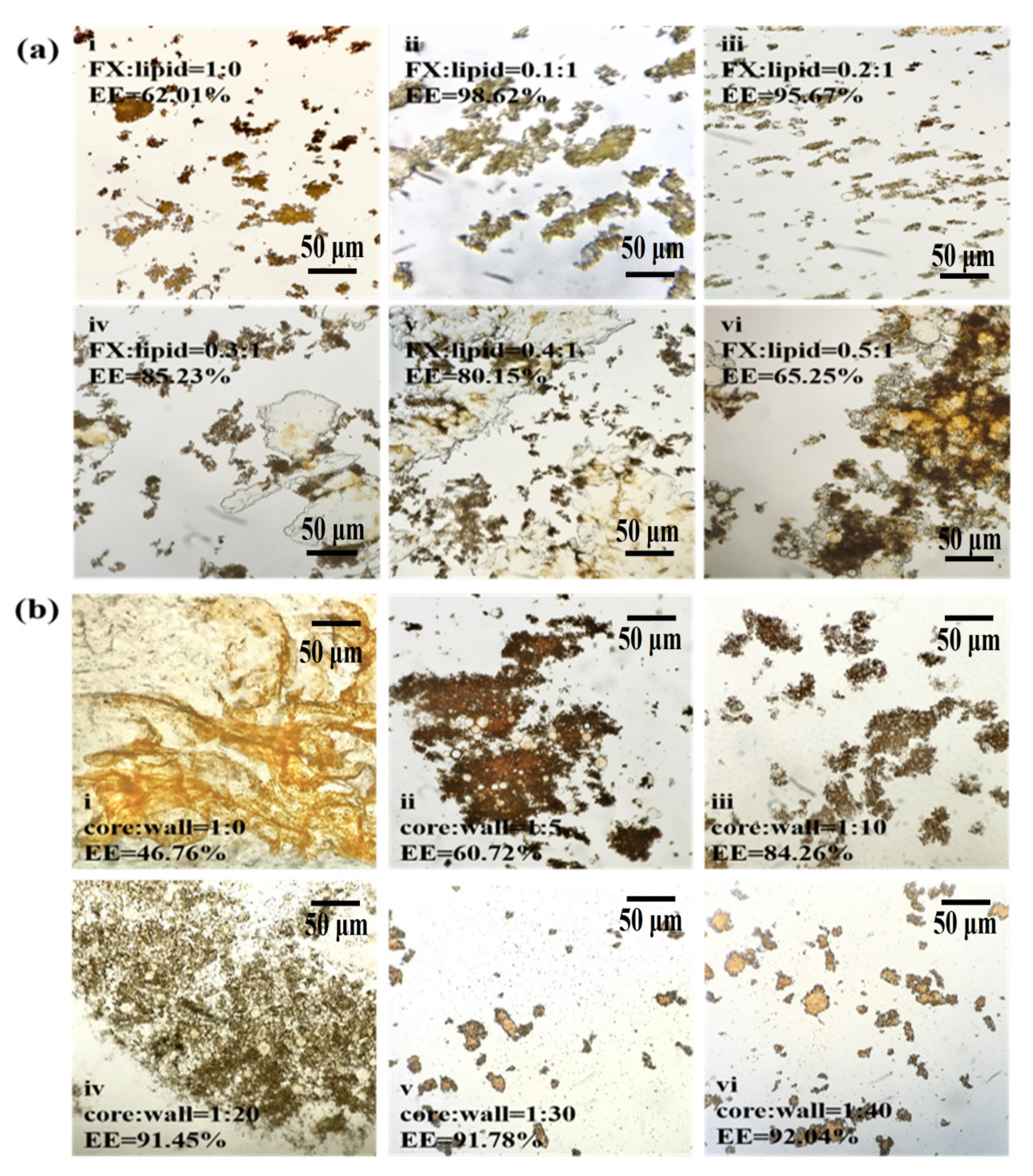
2.1.1. Effect of the Ratio of FX to Lipid and the Ratio of Core to Wall on FX MCs
2.1.2. Effect of Aggregation pH and Ultrasonic Conditions on FX MCs
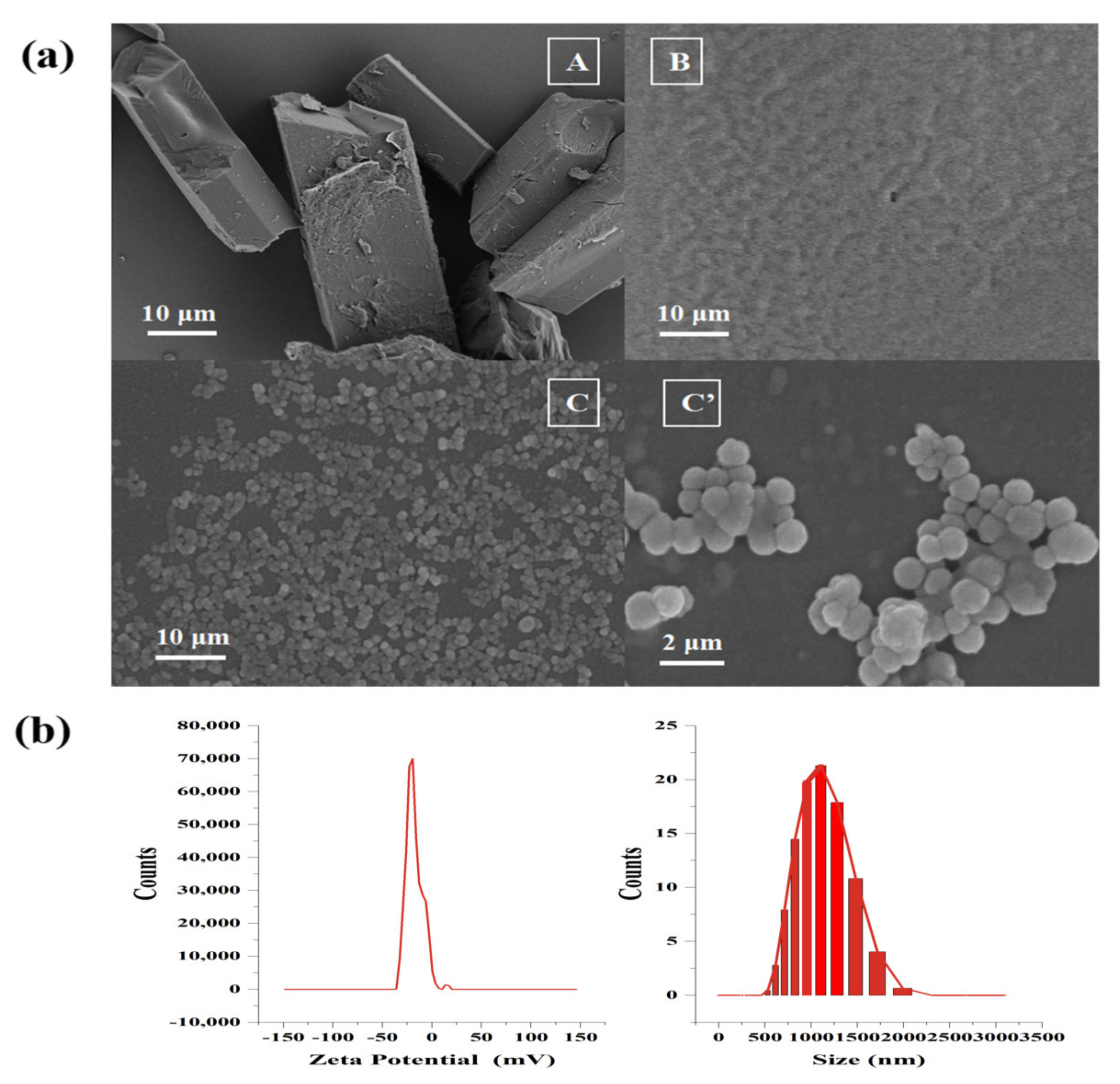
2.2. Characterization of FX MCs
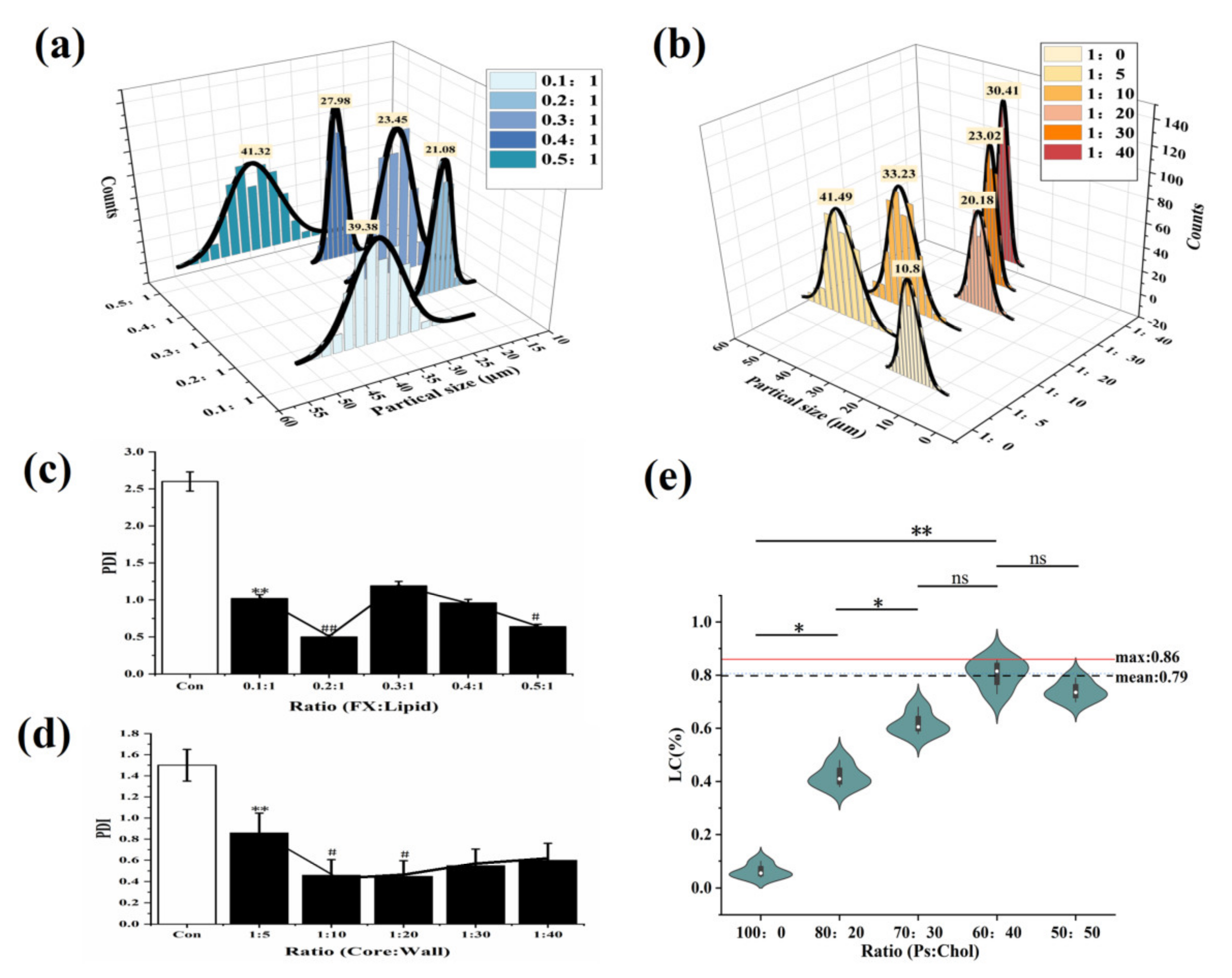
2.2.1. Surface Morphology, Particle Size, and Zeta Potential Analysis
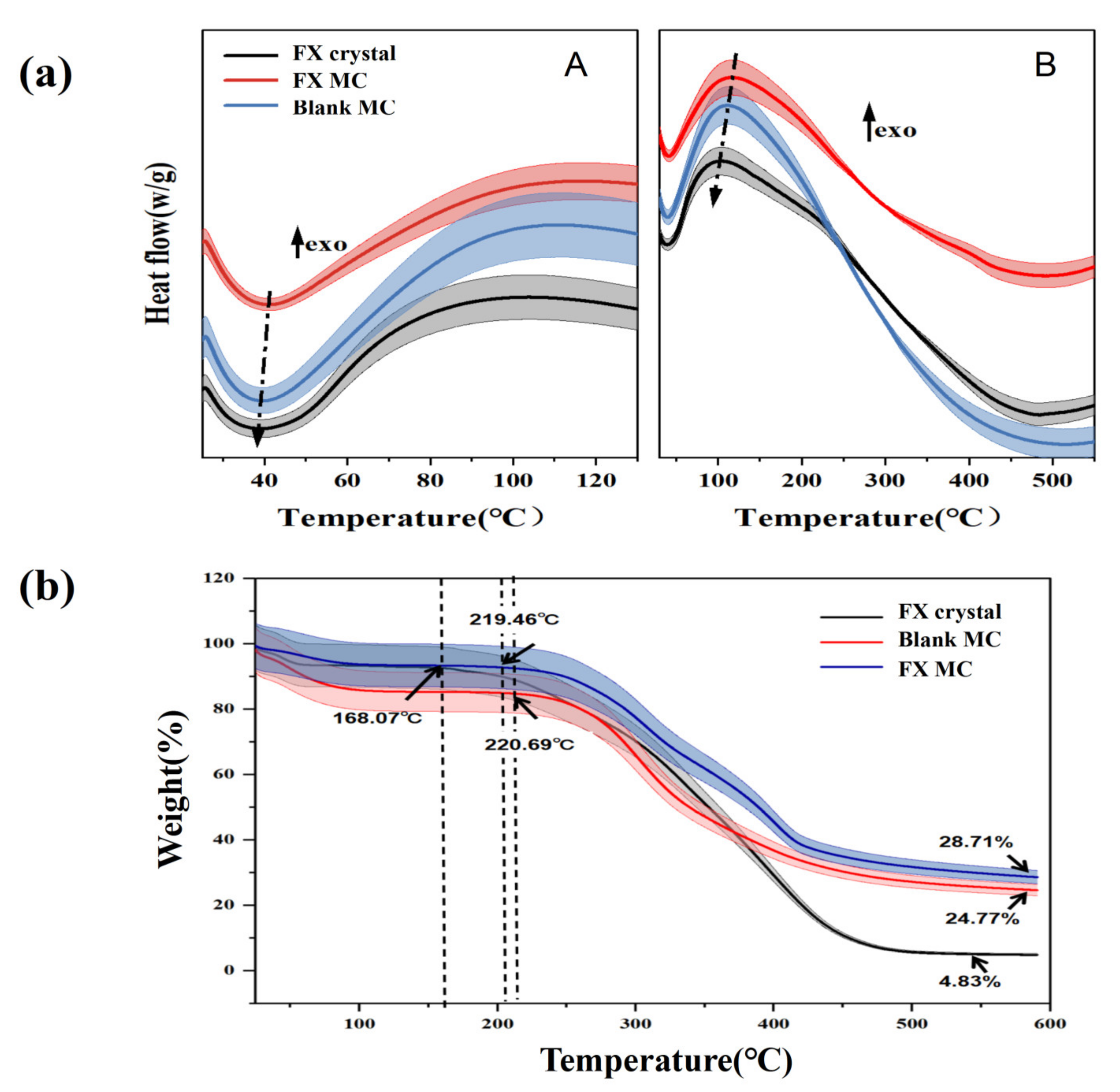
2.2.2. DSC Analysis of Materials
2.2.3. Thermogravimetric (TG) Analysis of Materials
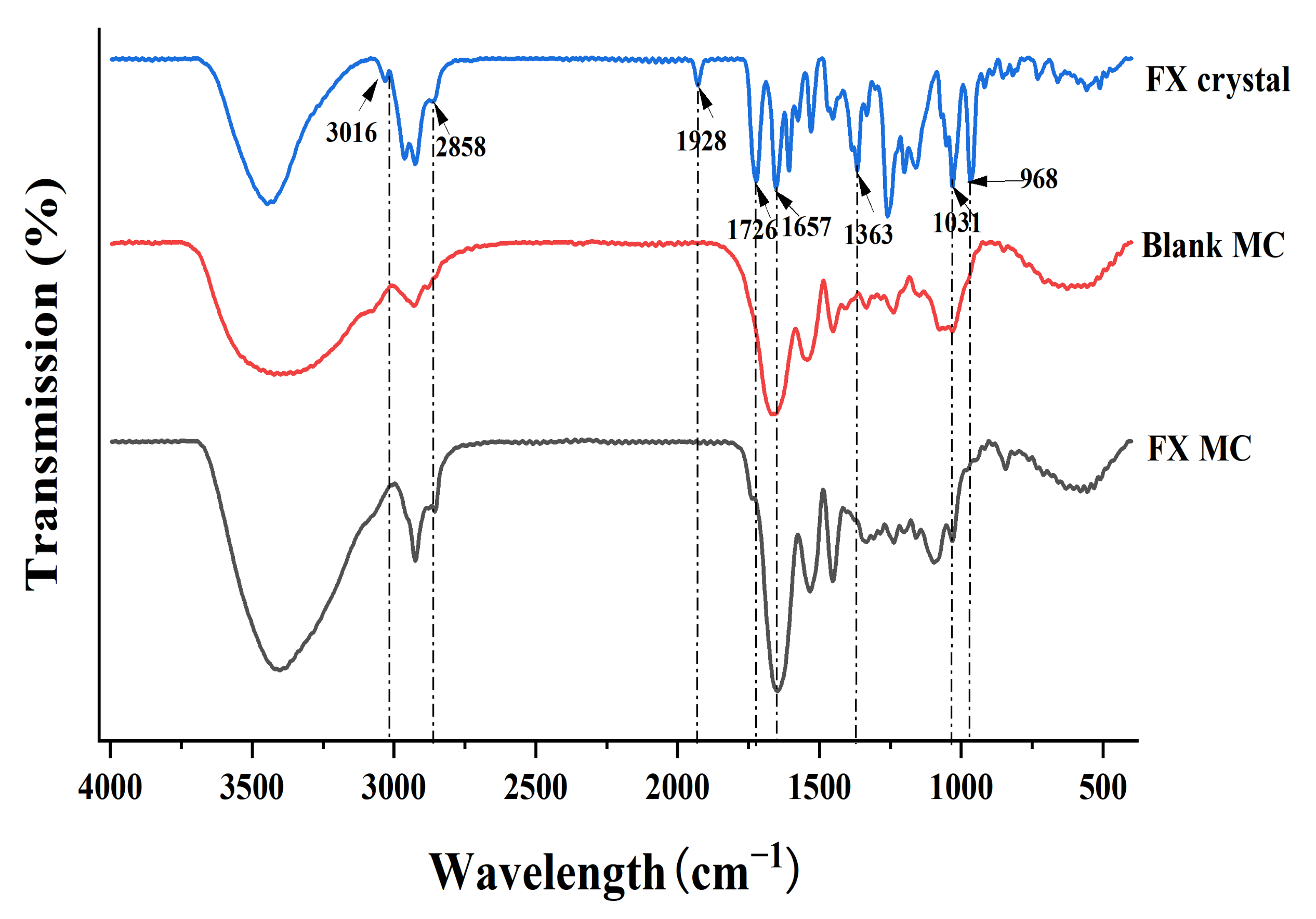
2.2.4. FTIR Analysis of Materials
2.3. Performance Analysis of FX MCs
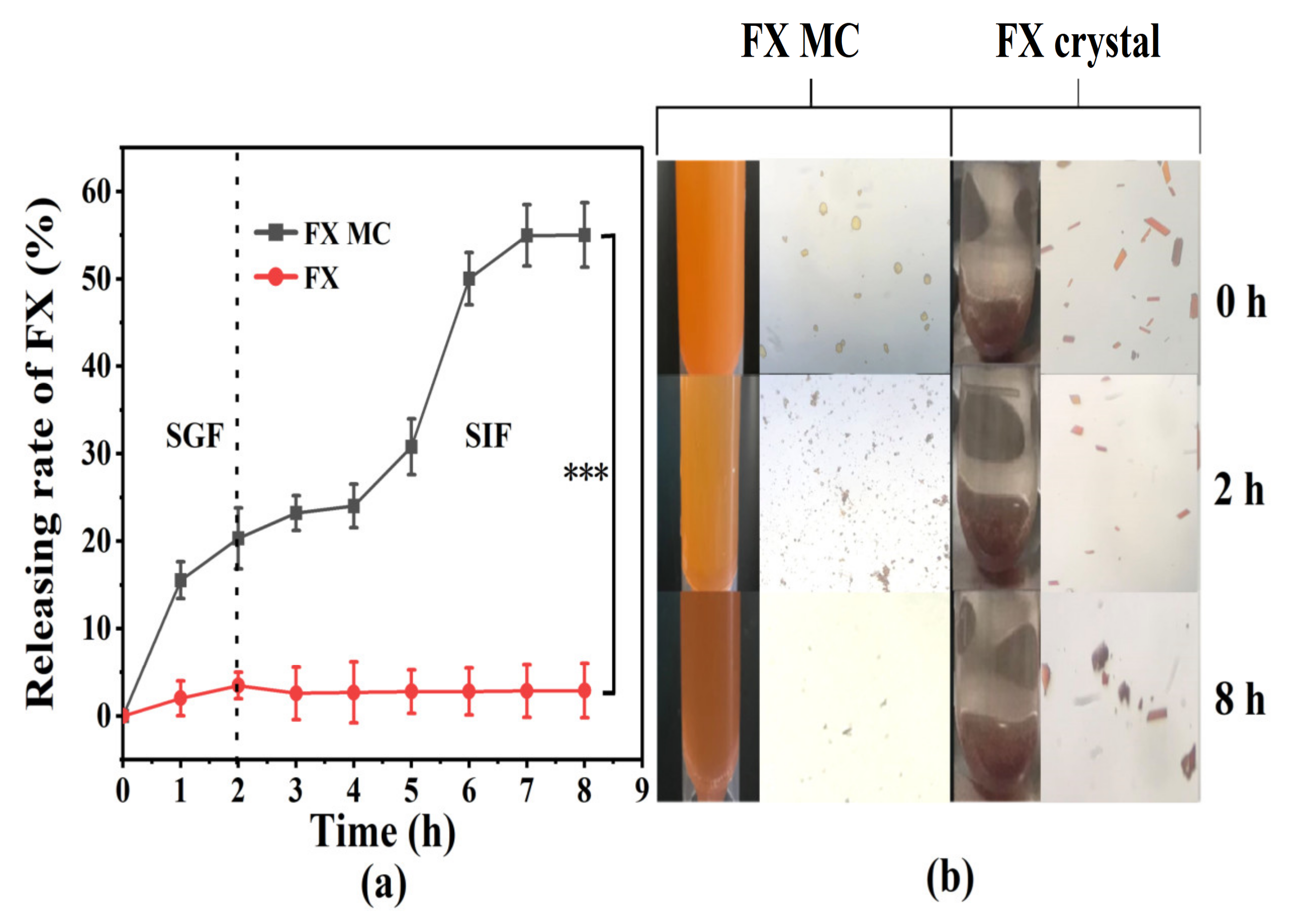
2.3.1. In Vitro Release Study
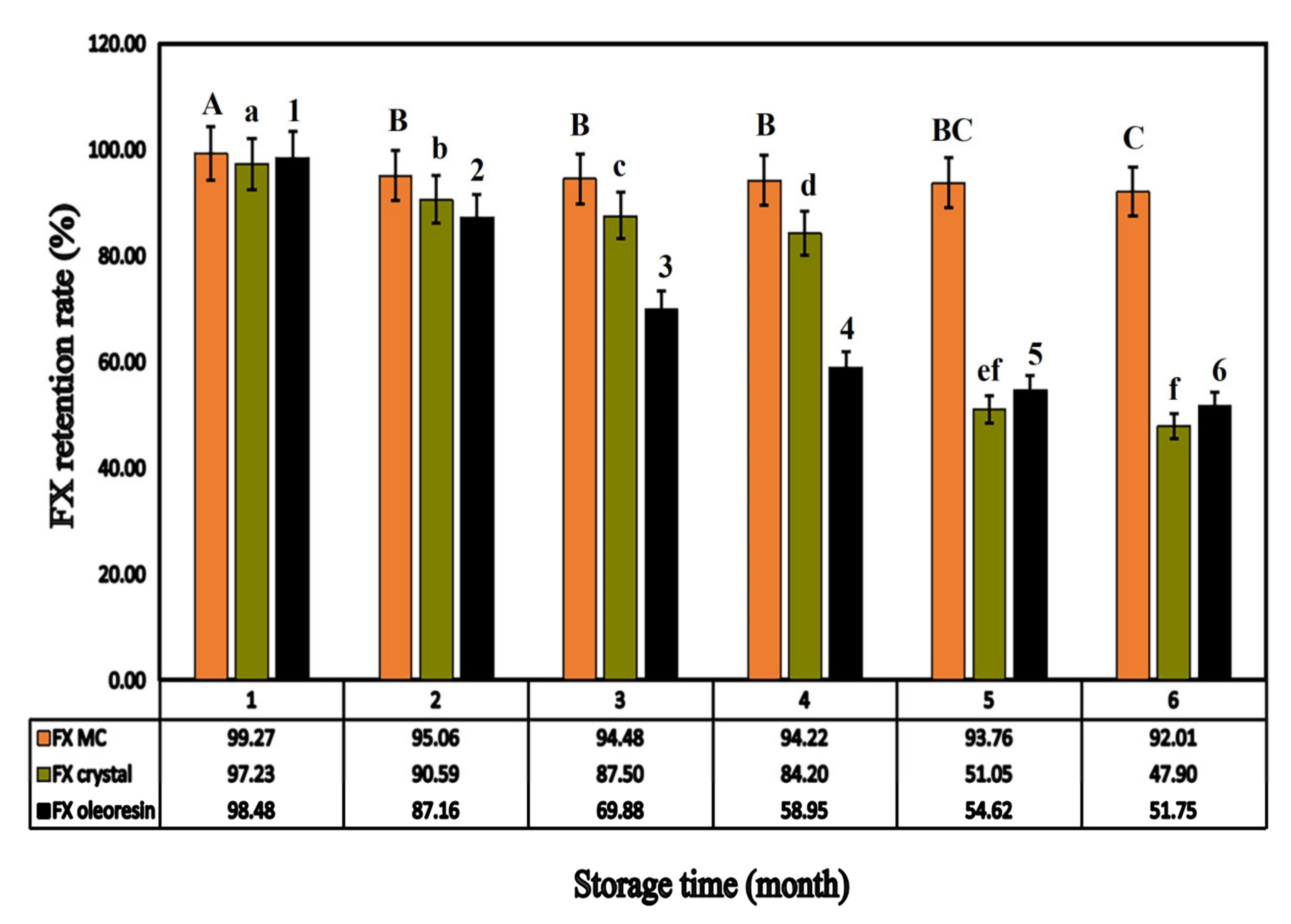
2.3.2. Accelerated Stability Study
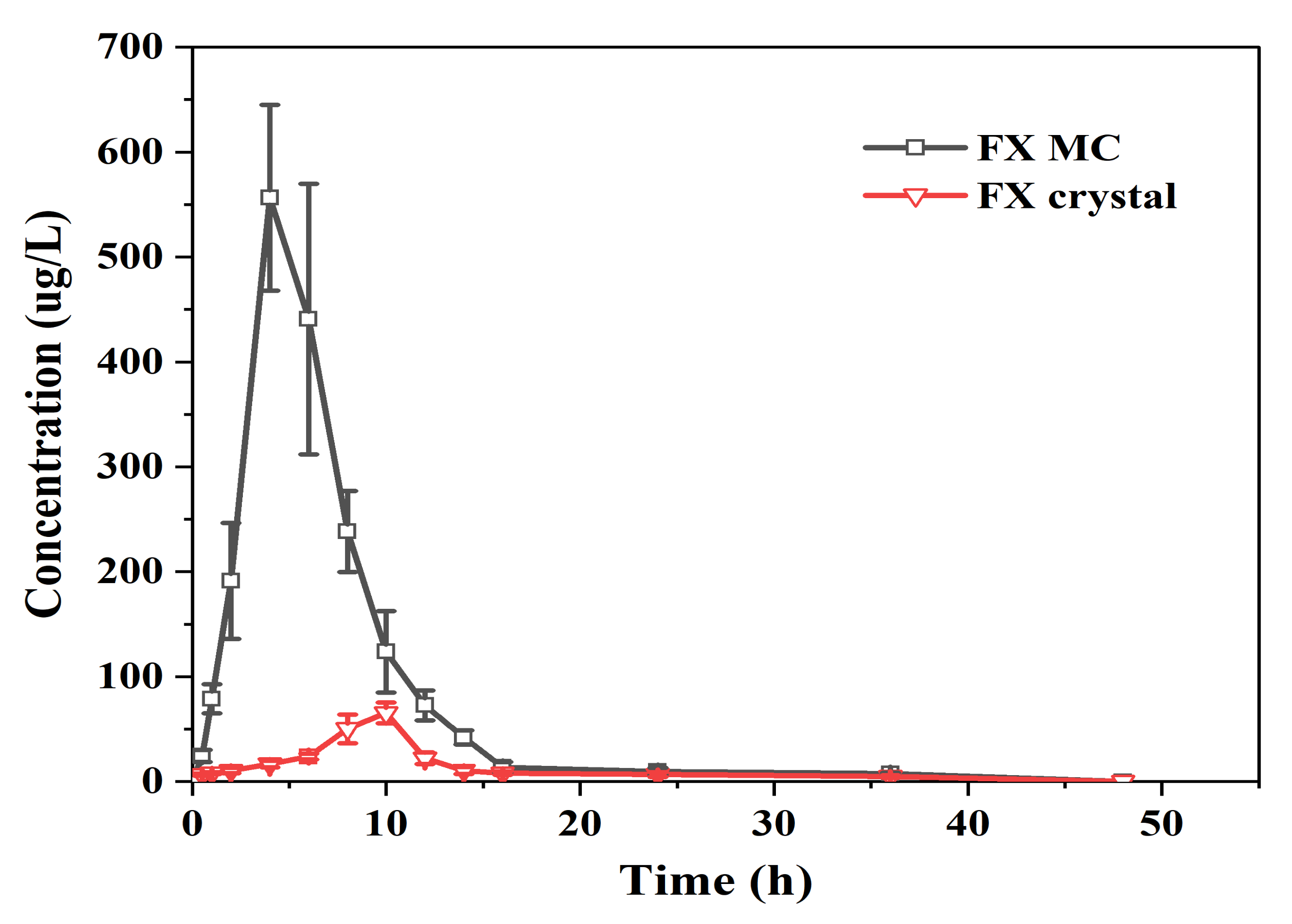
2.3.3. In Vivo Bioavailability Studies of FX MCs Compared with FX Crystals
3. Materials and Methods
3.1. Materials and Animals
3.2. Preparation of FX MCs
3.3. Optimization of the Preparation
3.3.1. Effect of Ps-to-Chol Ratios on FX MCs
3.3.2. Effect of FX-to-Lipid Ratios on FX MCs
3.3.3. Effect of Core-to-Wall Ratios on FX MCs
3.3.4. Effect of Aggregation pH on FX MCs
3.3.5. Effect of Ultrasound Conditions on FX MCs
3.4. HPLC Analysis of FX
3.5. Determination of EE
3.6. Determination of Drug LC
3.7. Characterization of FX MCs
3.7.1. Surface Morphology Analysis
3.7.2. Particle Size, PDI, and Zeta Potential Analysis
3.7.3. Thermodynamic Properties Analysis
3.7.4. Functional Group Analysis
3.8. Performance Evaluation of FX MCs
3.8.1. In Vitro Release Study
3.8.2. Accelerated Stability Study
3.8.3. In Vivo Bioavailability Study
3.9. LC-MS Analysis of FX Metabolites
3.10. Pharmacokinetic Data Analysis
3.11. Statistical Analysis
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Gille, A.; Neumann, U.; Louis, S.; Bischoff, S.C.; Briviba, K. Microalgae as a potential source of carotenoids: Comparative results of an in vitro digestion method and a feeding experiment with C57BL/6J mice. J. Funct. Foods 2018, 49, 285–294. [Google Scholar] [CrossRef]
- Obluchinskaya, E.D.; Pozharitskaya, O.N.; Zakharova, L.V.; Daurtseva, A.V.; Flisyuk, E.V.; Shikov, A.N. Efficacy of natural deep eutectic solvents for extraction of hydrophilic and lipophilic compounds from Fucus vesiculosus. Molecules 2021, 49, 4198. [Google Scholar] [CrossRef] [PubMed]
- Lourenço-Lopes, C.; Fraga-Corral, M.; Jimenez-Lopez, C.; Carpena, A.G.M.; Garcia-Oliveira, P.; Prieto, M.A.; Simal-Gandara, J. Biological action mechanisms of fucoxanthin extracted from algae for application in food and cosmetic industries. Trends Food Sci. Technol. 2021, 117, 163–181. [Google Scholar]
- Minkyung, B.; Mi-Bo, K.; Young-Ki, P.; Ji-Young, L. Health benefits of fucoxanthin in the prevention of chronic diseases. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2020, 11, 158618. [Google Scholar]
- Leong, Y.K.; Chen, C.-Y.; Varjani, S.; Chang, J.-S. Producing fucoxanthin from algae—Recent advances in cultivation strategies and downstream processing. Bioresour. Technol. 2022, 344, 126170. [Google Scholar]
- Li, Y.; Liu, Y.; Wang, Y.; Yu, Y.; Zeng, Y.; Li, L.; Wang, L. The Bioactivity of Fucoxanthin from Undaria pinnatifida in Vitro. Am. J. Biochem. Biotechnol. 2016, 12, 139–148. [Google Scholar] [CrossRef]
- Hii, S.; Choong, P.; Woo, K.; Wong, C.; Sciences, A. Stability studies of fucoxanthin from Sargassum binderi. J. Basic Appl. Sci. 2010, 4, 4580–4584. [Google Scholar]
- Huang, Z.; Xu, L.; Zhu, X.; Hu, J.; Peng, H.; Zeng, Z.; Xiong, H. Stability and bioaccessibility of fucoxanthin in nanoemulsions prepared from pinolenic acid-contained structured lipid. Int. J. Food Eng. 2017, 13, 0273. [Google Scholar] [CrossRef]
- Li, H.; Xu, Y.; Sun, X.; Wang, S.; Wang, J.; Zhu, J.; Wang, D.; Zhao, L. Stability, bioactivity, and bioaccessibility of fucoxanthin in zein-caseinate composite nanoparticles fabricated at neutral pH by antisolvent precipitation. Food Hydrocoll. 2018, 84, 379–388. [Google Scholar] [CrossRef]
- Rehman, A.Q.T.; Jafari, S.M.; Assadpour, E.Q.S.; Aadil, R.M.; Iqbal, M.W.; Rashed, M.M.A.; Sajid, B.; Mushtaq, W.A. Carotenoid-loaded nanocarriers: A comprehensive review. Colloid Interface Sci. 2019, 275, 102048. [Google Scholar] [CrossRef] [PubMed]
- Asai, A.; Yonekura, L.; Nagao, A. Low bioavailability of dietary epoxyxanthophylls in humans. Br. J. Nutr. 2008, 100, 273–277. [Google Scholar] [CrossRef] [PubMed]
- Yiping, Z.; Hao, W.; Hongmei, W.; Hua, F.; Zhuan, H.; Ruizao, Y.; Rui, L. Simultaneous Determination of Fucoxanthin and Its Deacetylated Metabolite Fucoxanthinol in Rat Plasma by Liquid Chromatography-Tandem Mass Spectrometry. Mar. Drugs 2015, 13, 6521–6536. [Google Scholar]
- Hashimoto, T.; Ozaki, Y.; Mizuno, M.; Yoshida, M.; Nishitani, Y.; Azuma, T.; Komoto, A.; Maoka, T.; Tanino, Y.; Kanazawa, K. Pharmacokinetics of fucoxanthinol in human plasma after the oral administration of kombu extract. Br. J. Nutr. 2012, 107, 1566–1569. [Google Scholar] [CrossRef] [PubMed]
- Rizvi, S.Z.H.; Shah, F.A.; Khan, N.; Muhammad, I.; Ali, K.H.; Ansari, M.M.; Din, F.U.; Qureshi, O.S.; Kim, K.-W.; Choe, Y.-H.; et al. Simvastatin-loaded solid lipid nanoparticles for enhanced anti-hyperlipidemic activity in hyperlipidemia animal model. Int. J. Pharm. 2019, 560, 136–143. [Google Scholar] [CrossRef]
- Ruktanonchai, U.; Limpakdee, S.; Meejoo, S.; Sakulkhu, U.; Bunyapraphatsara, N.; Junyaprasert, V.; Puttipipatkhachorn, S. The effect of cetyl palmitate crystallinity on physical properties of gamma-oryzanol encapsulated in solid lipid nanoparticles. Nanotechnology 2008, 19, 095701. [Google Scholar] [CrossRef]
- Koo, S.Y.; Hwang, J.H.; Yang, S.H.; Um, J.I.; Hong, K.W.; Kang, K.; Pan, C.H.; Hwang, K.T.; Kim, S.M. Anti-Obesity Effect of Standardized Extract of Microalga Phaeodactylum tricornutum Containing Fucoxanthin. Mar. Drugs 2019, 17, 15. [Google Scholar] [CrossRef]
- Wang, Y.; Ye, H.; Zhou, C.; Lv, F.; Bie, X.; Lu, Z. Study on the spray-drying encapsulation of lutein in the porous starch and gelatin mixture. Eur. Food Res. Technol. 2012, 234, 157–163. [Google Scholar] [CrossRef]
- Wang, X.; Li, H.; Wang, F.; Xia, G.; Liu, H.; Cheng, X.; Kong, M.; Liu, Y.; Feng, C.; Chen, X. Isolation of fucoxanthin from Sargassum thunbergii and preparation of microcapsules based on palm stearin solid lipid core. Front. Mater. Sci. 2017, 11, 66–74. [Google Scholar] [CrossRef]
- Quan, J.; Kim, S.-M.; Pan, C.-H.; Chung, D. Characterization of fucoxanthin-loaded microspheres composed of cetyl palmitate-based solid lipid core and fish gelatin–gum arabic coacervate shell. Food Res. Int. 2013, 50, 31–37. [Google Scholar] [CrossRef]
- Li, Y.; Dou, X.; Pang, J.; Liang, M.; Feng, C.; Kong, M.; Liu, Y.; Cheng, X.; Wang, Y.; Chen, X. Improvement of fucoxanthin oral efficacy via vehicles based on gum Arabic, gelatin and alginate hydrogel: Delivery system for oral efficacy enhancement of functional food ingredients. J. Funct. Foods 2019, 63, 103573. [Google Scholar] [CrossRef]
- Zhang, Y.; Lin, J.; Yan, G.; Jin, W.; Chen, W.; Sun, J.; Yang, L.; Huang, M.; Hong, Z. Determination of fucoxanthinol in rat plasma by liquid chromatography-tandem mass spectrometry. J. Pharm. Biomed. Anal. 2019, 164, 155–163. [Google Scholar] [CrossRef] [PubMed]
- Xiaodong, C.; Rong, L.; Fang, Z.; Yokoyama, W.H. Effect of Beta-carotene status in Microcapsules on Its in vivo Bioefficacy and in vitro Bioaccessibility. Food Hydrocoll. 2020, 106, 105848. [Google Scholar]
- Lemmens, L.; De Vleeschouwer, K.; Moelants, K.R.; Colle, I.J.; Van Loey, A.M.; Hendrickx, M.E. Beta-carotene isomerization kinetics during thermal treatments of carrot puree. J. Agric. Food Chem. 2010, 58, 6816–6824. [Google Scholar] [CrossRef] [PubMed]
- Yi, J.; Fan, Y.; Yokoyama, W.; Zhang, Y.; Zhao, L. Thermal Degradation and Isomerization of beta-Carotene in Oil-in-Water Nanoemulsions Supplemented with Natural Antioxidants. J. Agric. Food Chem. 2016, 64, 1970–1976. [Google Scholar] [CrossRef]
- Seung-Rak, S.; Jongil, A.; Jin-Wook, C.; Jisung, P.; Beom, P.C.; Hyup, L.J. Hierarchical self-constructed biomolecular nanolayers comprising cholesterol and cholesteryl hemisuccinate for automatic alignment of liquid crystals. J. Mol. Liq. 2021, 340, 116842. [Google Scholar]
- Ma, Z.; Khalid, N.; Shu, G.; Zhao, Y.; Kobayashi, I.; Neves, M.A.; Tuwo, A.; Nakajima, M. Fucoxanthin-Loaded Oil-in-Water Emulsion-Based Delivery Systems: Effects of Natural Emulsifiers on the Formulation, Stability, and Bioaccessibility. ACS Omega 2019, 4, 10502–10509. [Google Scholar] [CrossRef]
- Macdougall, L.J.; Wechsler, M.E.; Culver, H.R.; Benke, E.H.; Broerman, A.; Bowman, C.N.; Anseth, K.S. Charged Poly(N-isopropylacrylamide) Nanogels for the Stabilization of High Isoelectric Point Proteins. ACS Biomater. Sci. Eng. 2021, 7, 4282–4292. [Google Scholar] [CrossRef]
- Huang, Y.-I.; Cheng, Y.-H.; Yu, C.-C.; Tsai, T.-R.; Cham, T.-M. Microencapsulation of extract containing shikonin using gelatin–acacia coacervation method: A formaldehyde-free approach. Colloids Surf. 2007, 58, 290–297. [Google Scholar] [CrossRef]
- Zhu, J.; Li, H.; Xu, Y.; Wang, D. Construction of Fucoxanthin Vector Based on Binding of Whey Protein Isolate and Its Subsequent Complex Coacervation with Lysozyme. J. Agric. Food Chem. 2019, 67, 2980–2990. [Google Scholar] [CrossRef]
- Sébastien, B.; Claire, D.; Olivier, R.; Nathalie, B.-M. Effect of constituent particle polydispersion on VSSA-based equivalent particle diameter: Theoretical rationale and application to a set of eight powders with constituent particle median diameters ranging from 9 to 130 nm. Adv. Powder Technol. 2021, 32, 1369–1379. [Google Scholar]
- Vicente, J.; Pereira, L.J.B.; Bastos, L.P.H.; Carvalho, M.G.; Garcia-Rojas, E.E. Effect of xanthan gum or pectin addition on Sacha Inchi oil-in-water emulsions stabilized by ovalbumin or tween 80: Droplet size distribution, rheological behavior, and stability. Int. J. Biol. Macromol. 2018, 120, 339–345. [Google Scholar] [CrossRef] [PubMed]
- Jamile, S.; Hadi, A.; Hamed, H.; Sajed, A. Sodium caseinate-coated and β-cyclodextrin/vitamin E inclusion complex-loaded nanoliposomes: A novel stabilized nanocarrier. LWT-Food Sci. Technol. 2021, 151, 112174. [Google Scholar]
- Mandana, S.-K.; Saeed, J.; Mohammad, R.; Amir, R. Ionic strength and zeta potential effects on colloid transport and retention processes. Colloid Interface Sci. Commun. 2021, 42, 100389. [Google Scholar]
- Noviendri, D.; Jaswir, I.; Taher, M.; Mohamed, F.; Salleh, H.M.; Noorbatcha, I.A.; Octavianti, F.; Lestari, W.; Hendri, R.; Ahmad, H. Fabrication of fucoxanthin-loaded microsphere (F-LM) by two steps double-emulsion solvent evaporation method and characterization of fucoxanthin before and after microencapsulation. J. Oleo Sci. 2016, 65, 641–653. [Google Scholar] [CrossRef] [PubMed]
- Emami, J.; Hamishehkar, H.; Najafabadi, A.R.; Gilani, K.; Minaiyan, M.; Mahdavi, H.; Mirzadeh, H.; Fakhari, A.; Nokhodchi, A. Particle size design of PLGA microspheres for potential pulmonary drug delivery using response surface methodology. J. Microencapsul. 2009, 26, 1–8. [Google Scholar] [CrossRef]
- Gabriela, O.; Lukáš, V.; Jan, Z.; Johannes, B.; Thomas, L.; Dominik, H. Using Excimeric Fluorescence to Study How the Cooling Rate Determines the Behavior of Naphthalenes in Freeze-Concentrated Solutions: Vitrification and Crystallization. J. Phys. Chem. B 2020, 124, 10556–10566. [Google Scholar]
- Tao, Y.; Wu, Y.; Yang, J.; Jiang, N.; Wang, Q.; Chu, D.-T.; Han, Y.; Zhou, J. Thermodynamic sorption properties, water plasticizing effect and particle characteristics of blueberry powders produced from juices, fruits, and pomaces. Powder Technol. 2018, 323, 208–218. [Google Scholar] [CrossRef]
- Milanovic, J.; Manojlovic, V.; Levic, S.; Rajic, N.; Nedovic, V.; Bugarski, B. Microencapsulation of Flavors in Carnauba Wax. Sensors 2010, 10, 901–912. [Google Scholar] [CrossRef]
- Gannimani, R.; Walvekar, P.; Naidu, V.R.; Aminabhavi, T.M.; Govender, T. Acetal containing polymers as pH-responsive nano-drug delivery systems. J. Control. Release 2020, 328, 736–761. [Google Scholar] [CrossRef]
- Sahil, K.; Akanksha, M. Microsphere: A Review. Int. J. Res. Pharm. Chem. 2011, 1, 1184–1198. [Google Scholar]
- Zhao, D.; Yu, D.; Kim, M.; Gu, M.Y.; Kim, S.M.; Pan, C.H.; Kim, G.H.; Chung, D. Effects of temperature, light, and pH on the stability of fucoxanthin in an oil-in-water emulsion. Food Chem. 2019, 291, 87–93. [Google Scholar] [CrossRef] [PubMed]
- Maeda, H.; Hosokawa, M.; Sashima, T.; Takahashi, N.; Kawada, T.; Miyashita, K. Fucoxanthin and its metabolite, fucoxanthinol, suppress adipocyte differentiation in 3T3-L1 cells. Int. J. Mol. Med. 2006, 18, 147–152. [Google Scholar] [CrossRef] [PubMed]
- Beppu, F.; Niwano, Y.; Sato, E.; Kohno, M.; Tsukui, T.; Hosokawa, M.; Miyashita, K. In vitro and in vivo evaluation of mutagenicity of fucoxanthin (FX) and its metabolite fucoxanthinol (FXOH). J. Toxicol. Sci. 2009, 34, 693–698. [Google Scholar] [CrossRef] [PubMed]
- Guo, B.; Oliviero, T.; Fogliano, V.; Ma, Y.; Chen, F.; Capuano, E. Gastrointestinal Bioaccessibility and Colonic Fermentation of Fucoxanthin from the Extract of the Microalga Nitzschia laevis. J. Agric. Food Chem. 2020, 68, 1844–1850. [Google Scholar] [CrossRef] [PubMed]
- Foo, S.C.; Khong, N.M.H.; Yusoff, F. Physicochemical, microstructure and antioxidant properties of microalgae-derived fucoxanthin rich microcapsules. Algal Res. 2020, 51, 102061. [Google Scholar] [CrossRef]
- Jaswir, I.; Noviendri, D.; Taher, M.; Mohamed, F.; Octavianti, F.; Lestari, W.; Mukti, A.G.; Nirwandar, S.; Hamad Almansori, B.B. Optimization and Formulation of Fucoxanthin-Loaded Microsphere (F-LM) Using Response Surface Methodology (RSM) and Analysis of Its Fucoxanthin Release Profile. Molecules 2019, 24, 947. [Google Scholar] [CrossRef] [PubMed]
- Aditya, N.P.; Macedo, A.S.; Doktorovova, S.; Souto, E.B.; Kim, S.; Chang, P.-S.; Ko, S. Development and evaluation of lipid nanocarriers for quercetin delivery: A comparative study of solid lipid nanoparticles (SLN), nanostructured lipid carriers (NLC), and lipid nanoemulsions (LNE). LWT-Food Sci. Technol. 2014, 59, 115–121. [Google Scholar] [CrossRef]
- Ruben, G.S.; Stephen, B.; Zoe, Z.; Young, J.Y. Method Development and Application of an Accelerated Solution Stability Screen for Drug Discovery. SLAS Discov. 2020, 25, 1191–1196. [Google Scholar]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameters | FX Cystal | FX MC |
|---|---|---|
| AUC0-∞ [(ng/mL)·h] | 856.14 ± 43.28 | 3871.07 ± 47.54 ** |
| AUC0-t [(ng/mL)·h] | 493.89 ± 40.12 | 3517.89 ± 272.53 ** |
| CL (L/h/kg) | 34364.14 ± 1330.66 | 6515.23 ± 652.06 ** |
| Cmax (ng/mL) | 65.42 ± 9.08 | 556.60 ± 46.20 ** |
| T1/2 (h) | 11.30 ± 2.29 | 7.80 ± 0.75 ** |
| MRT0-t (h) | 12.87 ± 0.53 | 7.01 ± 0.41 ** |
| Tmax (h) | 9.33 ± 0.81 | 4.67 ± 1.03 ** |
| Vd (L) | 1562721.16 ± 79156.37 | 304910.07 ± 24075.80 ** |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, Y.; He, N.; Yang, T.; Cai, S.; Zhang, Y.; Lin, J.; Huang, M.; Chen, W.; Zhang, Y.; Hong, Z. Fucoxanthin Loaded in Palm Stearin- and Cholesterol-Based Solid Lipid Nanoparticle-Microcapsules, with Improved Stability and Bioavailability In Vivo. Mar. Drugs 2022, 20, 237. https://doi.org/10.3390/md20040237
Chen Y, He N, Yang T, Cai S, Zhang Y, Lin J, Huang M, Chen W, Zhang Y, Hong Z. Fucoxanthin Loaded in Palm Stearin- and Cholesterol-Based Solid Lipid Nanoparticle-Microcapsules, with Improved Stability and Bioavailability In Vivo. Marine Drugs. 2022; 20(4):237. https://doi.org/10.3390/md20040237
Chicago/Turabian StyleChen, Yaxin, Niaoniao He, Ting Yang, Shuyun Cai, Yi Zhang, Jinjing Lin, Mingqing Huang, Weizhu Chen, Yiping Zhang, and Zhuan Hong. 2022. "Fucoxanthin Loaded in Palm Stearin- and Cholesterol-Based Solid Lipid Nanoparticle-Microcapsules, with Improved Stability and Bioavailability In Vivo" Marine Drugs 20, no. 4: 237. https://doi.org/10.3390/md20040237
APA StyleChen, Y., He, N., Yang, T., Cai, S., Zhang, Y., Lin, J., Huang, M., Chen, W., Zhang, Y., & Hong, Z. (2022). Fucoxanthin Loaded in Palm Stearin- and Cholesterol-Based Solid Lipid Nanoparticle-Microcapsules, with Improved Stability and Bioavailability In Vivo. Marine Drugs, 20(4), 237. https://doi.org/10.3390/md20040237