
Role of Biomarkers for the Diagnosis of Prion Diseases: A Narrative Review



Abstract
1. Introduction
2. Neurophysiological Biomarkers
2.1. Electroencephalogram (EEG)
2.2. Polysomnogram (PSG)
3. Neuroimaging Biomarkers
3.1. Brain Magnetic Resonance (MR)
3.2. Fluorodesoxyglucose Positron Emission Tomography (PET-FDG)
4. Genetics
5. Cerebrospinal Fluid (CSF) Biomarkers
5.1. Biochemical Analysis
5.2. CSF Surrogate Biomarkers
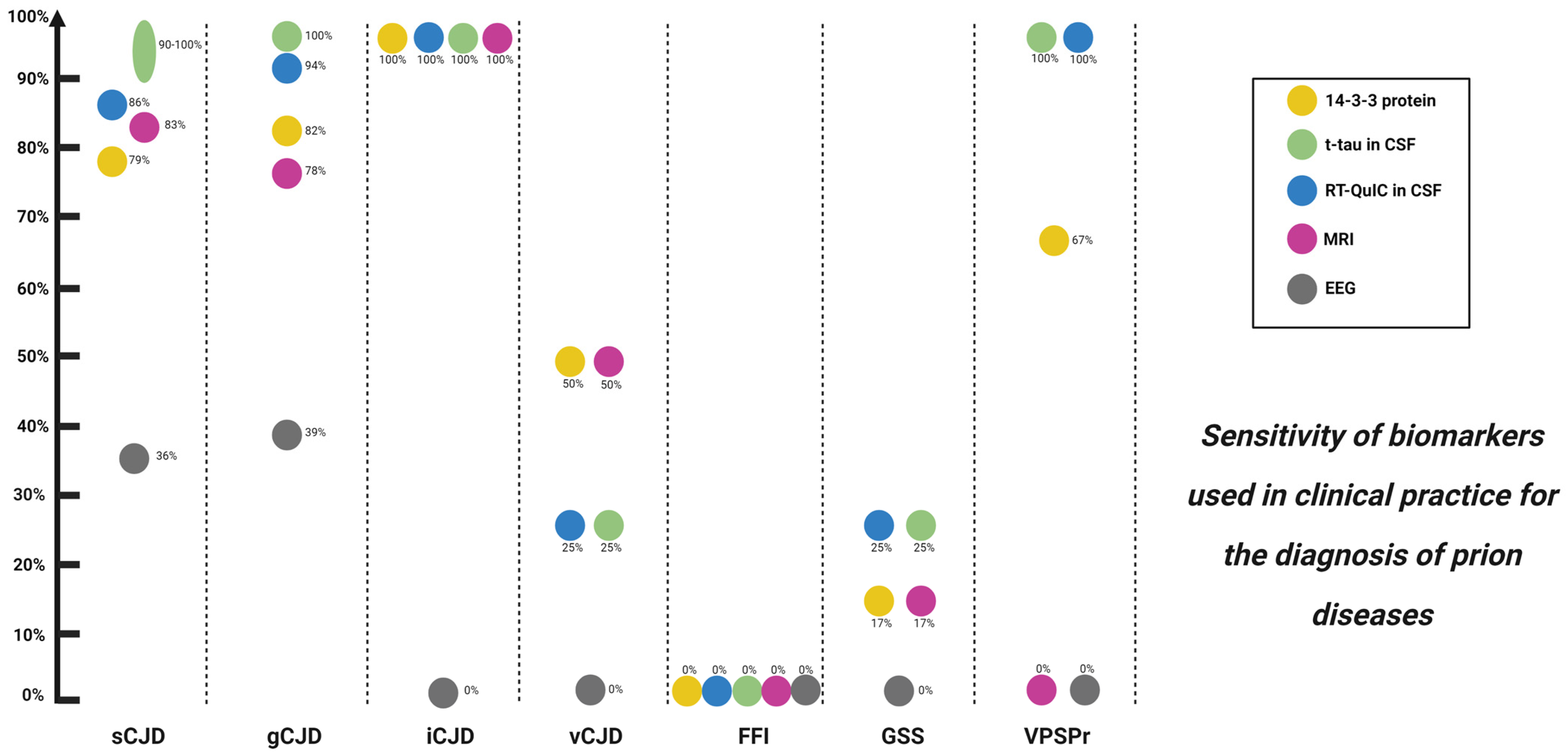
5.2.1. 14-3-3 Protein
5.2.2. Total Tau (t-tau) and Total Tau/Phosphorylated Tau (t-tau/p-tau) Ratio
5.2.3. Neurofilament Light Chain Protein (NfL)
5.2.4. Other Biomarkers
5.3. CSF Prion Proteins
5.3.1. Total PrP
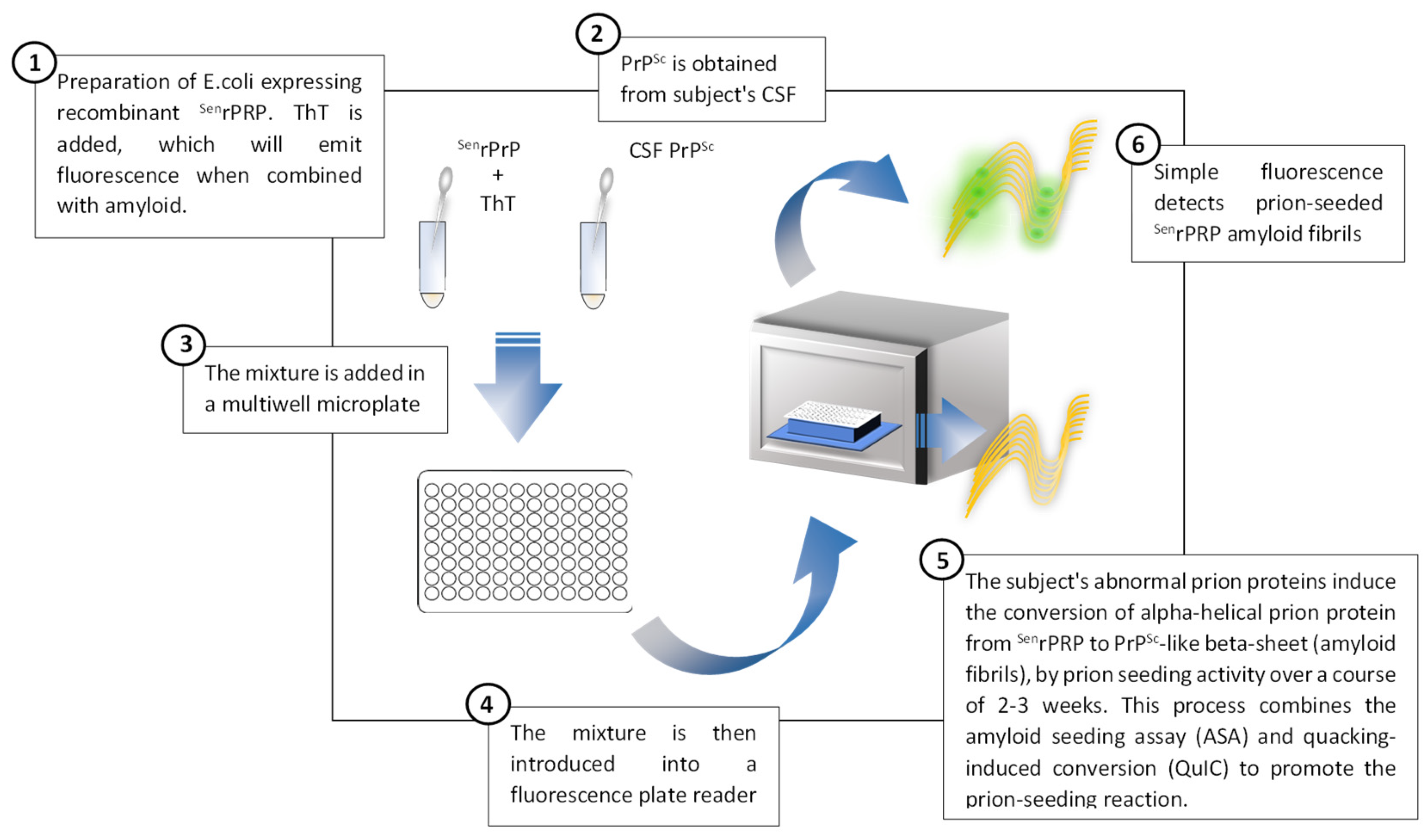
5.3.2. Prion Real-Time Quaking-Induced Conversion (RT-QuIC)
6. Plasma Biomarkers
6.1. NfL
6.2. t-tau
6.3. YKL-40
6.4. MicroRNA
6.5. Total Prion Protein (t-PrP)
7. Nasal Mucosa
8. Future Directions
9. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Llorens, F.; Rübsamen, N.; Hermann, P.; Schmitz, M.; Villar-Piqué, A.; Goebel, S.; Karch, A.; Zerr, I. A Prognostic Model for Overall Survival in Sporadic Creutzfeldt-Jakob Disease. Alzheimer’s Dement. 2020, 16, 1438–1447. [Google Scholar] [CrossRef] [PubMed]
- Ladogana, A.; Kovacs, G.G. Genetic Creutzfeldt-Jakob Disease. Handb. Clin. Neurol. 2018, 153, 219–242. [Google Scholar] [CrossRef] [PubMed]
- Hermann, P.; Appleby, B.; Brandel, J.-P.; Caughey, B.; Collins, S.; Geschwind, M.D.; Green, A.; Haïk, S.; Kovacs, G.G.; Ladogana, A.; et al. Biomarkers and Diagnostic Guidelines for Sporadic Creutzfeldt-Jakob Disease. Lancet Neurol. 2021, 20, 235–246. [Google Scholar] [CrossRef]
- Atkinson, C.J.; Zhang, K.; Munn, A.L.; Wiegmans, A.; Wei, M.Q. Prion Protein Scrapie and the Normal Cellular Prion Protein. Prion 2016, 10, 63–82. [Google Scholar] [CrossRef]
- Blennow, K.; Diaz-Lucena, D.; Zetterberg, H.; Villar-Pique, A.; Karch, A.; Vidal, E.; Hermann, P.; Schmitz, M.; Ferrer Abizanda, I.; Zerr, I.; et al. CSF Neurogranin as a Neuronal Damage Marker in CJD: A Comparative Study with AD. J. Neurol. Neurosurg. Psychiatry 2019, 90, 846–853. [Google Scholar] [CrossRef]
- Marino, S.; Morabito, R.; De Salvo, S.; Bonanno, L.; Bramanti, A.; Pollicino, P.; Giorgianni, R.; Bramanti, P. Quantitative, Functional MRI and Neurophysiological Markers in a Case of Gerstmann-Sträussler-Scheinker Syndrome. Funct. Neurol. 2017, 32, 153–158. [Google Scholar] [CrossRef] [PubMed]
- Moore, R.C.; Xiang, F.; Monaghan, J.; Han, D.; Zhang, Z.; Edström, L.; Anvret, M.; Prusiner, S.B. Huntington Disease Phenocopy Is a Familial Prion Disease. Am. J. Hum. Genet. 2001, 69, 1385–1388. [Google Scholar] [CrossRef] [PubMed]
- Brandel, J.-P.; Knight, R. Variant Creutzfeldt-Jakob Disease. Handb. Clin. Neurol. 2018, 153, 191–205. [Google Scholar] [CrossRef] [PubMed]
- Spencer, M.D.; Knight, R.S.G.; Will, R.G. First Hundred Cases of Variant Creutzfeldt-Jakob Disease: Retrospective Case Note Review of Early Psychiatric and Neurological Features. BMJ 2002, 324, 1479–1482. [Google Scholar] [CrossRef]
- Mok, T.H.; Mead, S. Preclinical Biomarkers of Prion Infection and Neurodegeneration. Curr. Opin. Neurobiol. 2020, 61, 82–88. [Google Scholar] [CrossRef]
- Hill, A.F.; Joiner, S.; Wadsworth, J.D.F.; Sidle, K.C.L.; Bell, J.E.; Budka, H.; Ironside, J.W.; Collinge, J. Molecular Classification of Sporadic Creutzfeldt–Jakob Disease. Brain 2003, 126, 1333–1346. [Google Scholar] [CrossRef] [PubMed]
- Safar, J.G. Molecular Pathogenesis of Sporadic Prion Diseases in Man. Prion 2012, 6, 108–115. [Google Scholar] [CrossRef] [PubMed]
- Ascari, L.M.; Rocha, S.C.; Gonçalves, P.B.; Vieira, T.C.R.G.; Cordeiro, Y. Challenges and Advances in Antemortem Diagnosis of Human Transmissible Spongiform Encephalopathies. Front. Bioeng. Biotechnol. 2020, 8, 585896. [Google Scholar] [CrossRef] [PubMed]
- Abu-Rumeileh, S.; Baiardi, S.; Polischi, B.; Mammana, A.; Franceschini, A.; Green, A.; Capellari, S.; Parchi, P. Diagnostic Value of Surrogate CSF Biomarkers for Creutzfeldt-Jakob Disease in the Era of RT-QuIC. J. Neurol. 2019, 266, 3136–3143. [Google Scholar] [CrossRef] [PubMed]
- Wieser, H.G.; Schindler, K.; Zumsteg, D. EEG in Creutzfeldt-Jakob Disease. Clin. Neurophysiol. Off. J. Int. Fed. Clin. Neurophysiol. 2006, 117, 935–951. [Google Scholar] [CrossRef]
- Skillbäck, T.; Rosén, C.; Asztely, F.; Mattsson, N.; Blennow, K.; Zetterberg, H. Diagnostic Performance of Cerebrospinal Fluid Total Tau and Phosphorylated Tau in Creutzfeldt-Jakob Disease: Results from the Swedish Mortality Registry. JAMA Neurol. 2014, 71, 476–483. [Google Scholar] [CrossRef]
- Figgie, M.P.J.; Appleby, B.S. Clinical Use of Improved Diagnostic Testing for Detection of Prion Disease. Viruses 2021, 13, 789. [Google Scholar] [CrossRef] [PubMed]
- Franko, E.; Wehner, T.; Joly, O.; Lowe, J.; Porter, M.-C.; Kenny, J.; Thompson, A.; Rudge, P.; Collinge, J.; Mead, S. Quantitative EEG Parameters Correlate with the Progression of Human Prion Diseases. J. Neurol. Neurosurg. Psychiatry 2016, 87, 1061–1067. [Google Scholar] [CrossRef]
- Matsubayashi, T.; Akaza, M.; Hayashi, Y.; Hamaguchi, T.; Yamada, M.; Shimohata, T.; Yokota, T.; Sanjo, N. Focal Sharp Waves Are a Specific Early-Stage Marker of the MM2-Cortical Form of Sporadic Creutzfeldt-Jakob Disease. Prion 2020, 14, 207–213. [Google Scholar] [CrossRef]
- Wu, L.-Y.; Zhan, S.-Q.; Huang, Z.-Y.; Zhang, B.; Wang, T.; Liu, C.-F.; Lu, H.; Dong, X.-P.; Wu, Z.-Y.; Zhang, J.-W.; et al. Expert Consensus on Clinical Diagnostic Criteria for Fatal Familial Insomnia. Chin. Med. J. 2018, 131, 1613–1617. [Google Scholar] [CrossRef]
- Kang, P.; de Bruin, G.S.; Wang, L.H.; Ward, B.A.; Ances, B.M.; Lim, M.M.; Bucelli, R.C. Sleep Pathology in Creutzfeldt-Jakob Disease. J. Clin. Sleep Med. JCSM Off. Publ. Am. Acad. Sleep Med. 2016, 12, 1033–1039. [Google Scholar] [CrossRef] [PubMed]
- Dai, Y.; Shao, J.; Lang, Y.; Lv, Y.; Cui, L. Clinical Manifestations and Polysomnography-Based Analysis in Nine Cases of Probable Sporadic Creutzfeldt-Jakob Disease. Neurol. Sci. Off. J. Ital. Neurol. Soc. Ital. Soc. Clin. Neurophysiol. 2021, 42, 4209–4219. [Google Scholar] [CrossRef] [PubMed]
- Macfarlane, R.G.; Wroe, S.J.; Collinge, J.; Yousry, T.A.; Jäger, H.R. Neuroimaging Findings in Human Prion Disease. J. Neurol. Neurosurg. Psychiatry 2007, 78, 664–670. [Google Scholar] [CrossRef] [PubMed]
- Sacco, S.; Paoletti, M.; Staffaroni, A.M.; Kang, H.; Rojas, J.; Marx, G.; Goh, S.-Y.; Luisa Mandelli, M.; Allen, I.E.; Kramer, J.H.; et al. Multimodal MRI Staging for Tracking Progression and Clinical-Imaging Correlation in Sporadic Creutzfeldt-Jakob Disease. NeuroImage. Clin. 2021, 30, 102523. [Google Scholar] [CrossRef] [PubMed]
- Baiardi, S.; Magherini, A.; Capellari, S.; Redaelli, V.; Ladogana, A.; Rossi, M.; Tagliavini, F.; Pocchiari, M.; Giaccone, G.; Parchi, P. Towards an Early Clinical Diagnosis of Sporadic CJD VV2 (Ataxic Type). J. Neurol. Neurosurg. Psychiatry 2017, 88, 764–772. [Google Scholar] [CrossRef]
- Ioannides, P.; Karacostas, D. Neuroimaging in Human Prion Disease: Searching in the Mist. World J. Radiol. 2009, 1, 45–49. [Google Scholar] [CrossRef]
- Divya, K.P.; Menon, R.N.; Thomas, B.; Nair, M. A Hospital-Based Registry of Creutzfeldt-Jakob Disease: Can Neuroimaging Serve as a Surrogate Biomarker? Neurol. India 2016, 64, 411–418. [Google Scholar] [CrossRef]
- Rudge, P.; Hyare, H.; Green, A.; Collinge, J.; Mead, S. Imaging and CSF Analyses Effectively Distinguish CJD from Its Mimics. J. Neurol. Neurosurg. Psychiatry 2018, 89, 461–466. [Google Scholar] [CrossRef]
- Kim, J.H.; Choi, B.S.; Jung, C.; Chang, Y.; Kim, S. Diffusion-Weighted Imaging and Magnetic Resonance Spectroscopy of Sporadic Creutzfeldt-Jakob Disease: Correlation with Clinical Course. Neuroradiology 2011, 53, 939–945. [Google Scholar] [CrossRef]
- Shiga, Y.; Miyazawa, K.; Sato, S.; Fukushima, R.; Shibuya, S.; Sato, Y.; Konno, H.; Doh-ura, K.; Mugikura, S.; Tamura, H.; et al. Diffusion-Weighted MRI Abnormalities as an Early Diagnostic Marker for Creutzfeldt-Jakob Disease. Neurology 2004, 63, 443–449. [Google Scholar] [CrossRef]
- Lee, H.; Rosenmann, H.; Chapman, J.; Kingsley, P.B.; Hoffmann, C.; Cohen, O.S.; Kahana, E.; Korczyn, A.D.; Prohovnik, I. Thalamo-Striatal Diffusion Reductions Precede Disease Onset in Prion Mutation Carriers. Brain 2009, 132, 2680–2687. [Google Scholar] [CrossRef] [PubMed]
- Kim, E.-J.; Cho, S.-S.; Jeong, B.-H.; Kim, Y.-S.; Seo, S.W.; Na, D.L.; Geschwind, M.D.; Jeong, Y. Glucose Metabolism in Sporadic Creutzfeldt-Jakob Disease: A Statistical Parametric Mapping Analysis of (18) F-FDG PET. Eur. J. Neurol. 2012, 19, 488–493. [Google Scholar] [CrossRef] [PubMed]
- Green, A.J.E.; Ramljak, S.; Müller, W.E.G.; Knight, R.S.G.; Schröder, H.C. 14-3-3 in the Cerebrospinal Fluid of Patients with Variant and Sporadic Creutzfeldt-Jakob Disease Measured Using Capture Assay Able to Detect Low Levels of 14-3-3 Protein. Neurosci. Lett. 2002, 324, 57–60. [Google Scholar] [CrossRef]
- Matsui, Y.; Satoh, K.; Miyazaki, T.; Shirabe, S.; Atarashi, R.; Mutsukura, K.; Satoh, A.; Kataoka, Y.; Nishida, N. High Sensitivity of an ELISA Kit for Detection of the Gamma-Isoform of 14-3-3 Proteins: Usefulness in Laboratory Diagnosis of Human Prion Disease. BMC Neurol. 2011, 11, 120. [Google Scholar] [CrossRef] [PubMed]
- Castellani, R.J.; Colucci, M.; Xie, Z.; Zou, W.; Li, C.; Parchi, P.; Capellari, S.; Pastore, M.; Rahbar, M.H.; Chen, S.G.; et al. Sensitivity of 14-3-3 Protein Test Varies in Subtypes of Sporadic Creutzfeldt-Jakob Disease. Neurology 2004, 63, 436–442. [Google Scholar] [CrossRef] [PubMed]
- Leitão, M.J.; Baldeiras, I.; Almeida, M.R.; Ribeiro, M.H.; Santos, A.C.; Ribeiro, M.; Tomás, J.; Rocha, S.; Santana, I.; Oliveira, C.R. CSF Tau Proteins Reduce Misdiagnosis of Sporadic Creutzfeldt-Jakob Disease Suspected Cases with Inconclusive 14-3-3 Result. J. Neurol. 2016, 263, 1847–1861. [Google Scholar] [CrossRef] [PubMed]
- Torres, M.; Cartier, L.; Matamala, J.M.; Hernández, N.; Woehlbier, U.; Hetz, C. Altered Prion Protein Expression Pattern in CSF as a Biomarker for Creutzfeldt-Jakob Disease. PLoS ONE 2012, 7, e36159. [Google Scholar] [CrossRef]
- Ladogana, A.; Sanchez-Juan, P.; Mitrová, E.; Green, A.; Cuadrado-Corrales, N.; Sánchez-Valle, R.; Koscova, S.; Aguzzi, A.; Sklaviadis, T.; Kulczycki, J.; et al. Cerebrospinal Fluid Biomarkers in Human Genetic Transmissible Spongiform Encephalopathies. J. Neurol. 2009, 256, 1620–1628. [Google Scholar] [CrossRef]
- Higuma, M.; Sanjo, N.; Satoh, K.; Shiga, Y.; Sakai, K.; Nozaki, I.; Hamaguchi, T.; Nakamura, Y.; Kitamoto, T.; Shirabe, S.; et al. Relationships between Clinicopathological Features and Cerebrospinal Fluid Biomarkers in Japanese Patients with Genetic Prion Diseases. PLoS ONE 2013, 8, e60003. [Google Scholar] [CrossRef]
- Leitão, M.J.; Baldeiras, I.; Almeida, M.R.; Ribeiro, M.H.; Santos, A.C.; Ribeiro, M.; Tomás, J.; Rocha, S.; Santana, I.; Oliveira, C.R. Sporadic Creutzfeldt-Jakob Disease Diagnostic Accuracy Is Improved by a New CSF ELISA 14-3-3γ Assay. Neuroscience 2016, 322, 398–407. [Google Scholar] [CrossRef]
- Lattanzio, F.; Abu-Rumeileh, S.; Franceschini, A.; Kai, H.; Amore, G.; Poggiolini, I.; Rossi, M.; Baiardi, S.; McGuire, L.; Ladogana, A.; et al. Prion-Specific and Surrogate CSF Biomarkers in Creutzfeldt-Jakob Disease: Diagnostic Accuracy in Relation to Molecular Subtypes and Analysis of Neuropathological Correlates of p-Tau and Aβ42 Levels. Acta Neuropathol. 2017, 133, 559–578. [Google Scholar] [CrossRef] [PubMed]
- Zanusso, G.; Fiorini, M.; Ferrari, S.; Gajofatto, A.; Cagnin, A.; Galassi, A.; Richelli, S.; Monaco, S. Cerebrospinal Fluid Markers in Sporadic Creutzfeldt-Jakob Disease. Int. J. Mol. Sci. 2011, 12, 6281–6292. [Google Scholar] [CrossRef] [PubMed]
- Hyeon, J.W.; Kim, S.Y.; Lee, J.; Park, J.S.; Hwang, K.J.; Lee, S.M.; An, S.A.; Lee, M.K.; Ju, Y.R. Alternative Application of Tau Protein in Creutzfeldt-Jakob Disease Diagnosis: Improvement for Weakly Positive 14-3-3 Protein in the Laboratory. Sci. Rep. 2015, 5, 15283. [Google Scholar] [CrossRef] [PubMed]
- Chohan, G.; Pennington, C.; Mackenzie, J.M.; Andrews, M.; Everington, D.; Will, R.G.; Knight, R.S.G.; Green, A.J.E. The Role of Cerebrospinal Fluid 14-3-3 and Other Proteins in the Diagnosis of Sporadic Creutzfeldt-Jakob Disease in the UK: A 10-Year Review. J. Neurol. Neurosurg. Psychiatry 2010, 81, 1243–1248. [Google Scholar] [CrossRef] [PubMed]
- Lemstra, A.W.; van Meegen, M.T.; Vreyling, J.P.; Meijerink, P.H.; Jansen, G.H.; Bulk, S.; Baas, F.; van Gool, W.A. 14-3-3 Testing in Diagnosing Creutzfeldt-Jakob Disease: A Prospective Study in 112 Patients. Neurology 2000, 55, 514–516. [Google Scholar] [CrossRef]
- Schmitz, M.; Ebert, E.; Stoeck, K.; Karch, A.; Collins, S.; Calero, M.; Sklaviadis, T.; Laplanche, J.-L.; Golanska, E.; Baldeiras, I.; et al. Validation of 14-3-3 Protein as a Marker in Sporadic Creutzfeldt-Jakob Disease Diagnostic. Mol. Neurobiol. 2016, 53, 2189–2199. [Google Scholar] [CrossRef]
- Stoeck, K.; Sanchez-Juan, P.; Gawinecka, J.; Green, A.; Ladogana, A.; Pocchiari, M.; Sanchez-Valle, R.; Mitrova, E.; Sklaviadis, T.; Kulczycki, J.; et al. Cerebrospinal Fluid Biomarker Supported Diagnosis of Creutzfeldt-Jakob Disease and Rapid Dementias: A Longitudinal Multicentre Study over 10 Years. Brain 2012, 135, 3051–3061. [Google Scholar] [CrossRef]
- Llorens, F.; Karch, A.; Golanska, E.; Schmitz, M.; Lange, P.; Sikorska, B.; Liberski, P.P.; Zerr, I. Cerebrospinal Fluid Biomarker-Based Diagnosis of Sporadic Creutzfeldt-Jakob Disease: A Validation Study for Previously Established Cutoffs. Dement. Geriatr. Cogn. Disord. 2017, 43, 71–80. [Google Scholar] [CrossRef]
- Hamlin, C.; Puoti, G.; Berri, S.; Sting, E.; Harris, C.; Cohen, M.; Spear, C.; Bizzi, A.; Debanne, S.M.; Rowland, D.Y. A Comparison of Tau and 14-3-3 Protein in the Diagnosis of Creutzfeldt-Jakob Disease. Neurology 2012, 79, 547–552. [Google Scholar] [CrossRef]
- Coulthart, M.B.; Jansen, G.H.; Olsen, E.; Godal, D.L.; Connolly, T.; Choi, B.C.K.; Wang, Z.; Cashman, N.R. Diagnostic Accuracy of Cerebrospinal Fluid Protein Markers for Sporadic Creutzfeldt-Jakob Disease in Canada: A 6-Year Prospective Study. BMC Neurol. 2011, 11, 133. [Google Scholar] [CrossRef]
- Foucault-Fruchard, L.; Delaye, J.B.; Morange, V.; Beaufils, E.; Duwicquet, C.; Quadrio, I.; Balageas, A.C.; Dufour-Rainfray, D. An Automated Alert System Based on the P-Tau/Tau Ratio to Quickly Inform Health Professionals upon a Suspected Case of Sporadic Creutzfeldt-Jakob Disease. J. Neurol. Sci. 2020, 415, 116971. [Google Scholar] [CrossRef] [PubMed]
- Llorens, F.; Villar-Piqué, A.; Hermann, P.; Schmitz, M.; Goebel, S.; Waniek, K.; Lachmann, I.; Zerr, I. Cerebrospinal Fluid Non-Phosphorylated Tau in the Differential Diagnosis of Creutzfeldt-Jakob Disease: A Comparative Prospective Study with 14-3-3. J. Neurol. 2020, 267, 543–550. [Google Scholar] [CrossRef] [PubMed]
- Meiner, Z.; Kahana, E.; Baitcher, F.; Korczyn, A.D.; Chapman, J.; Cohen, O.S.; Milo, R.; Aharon-Perez, J.; Abramsky, O.; Gabizon, R.; et al. Tau and 14-3-3 of Genetic and Sporadic Creutzfeldt-Jakob Disease Patients in Israel. J. Neurol. 2011, 258, 255–262. [Google Scholar] [CrossRef]
- Gmitterová, K.; Heinemann, U.; Krasnianski, A.; Gawinecka, J.; Zerr, I. Cerebrospinal Fluid Markers in the Differentiation of Molecular Subtypes of Sporadic Creutzfeldt-Jakob Disease. Eur. J. Neurol. 2016, 23, 1126–1133. [Google Scholar] [CrossRef] [PubMed]
- Karch, A.; Hermann, P.; Ponto, C.; Schmitz, M.; Arora, A.; Zafar, S.; Llorens, F.; Müller-Heine, A.; Zerr, I. Cerebrospinal Fluid Tau Levels Are a Marker for Molecular Subtype in Sporadic Creutzfeldt-Jakob Disease. Neurobiol. Aging 2015, 36, 1964–1968. [Google Scholar] [CrossRef] [PubMed]
- Karch, A.; Llorens, F.; Schmitz, M.; Arora, A.S.; Zafar, S.; Lange, P.; Schmidt, C.; Zerr, I. Stratification by Genetic and Demographic Characteristics Improves Diagnostic Accuracy of Cerebrospinal Fluid Biomarkers in Rapidly Progressive Dementia. J. Alzheimer’s Dis. 2016, 54, 1385–1393. [Google Scholar] [CrossRef]
- Satoh, K.; Shirabe, S.; Eguchi, H.; Tsujino, A.; Motomura, M.; Satoh, A.; Tsujihata, M.; Eguchi, K. Chronological Changes in MRI and CSF Biochemical Markers in Creutzfeldt-Jakob Disease Patients. Dement. Geriatr. Cogn. Disord. 2007, 23, 372–381. [Google Scholar] [CrossRef]
- Cohen, O.S.; Chapman, J.; Korczyn, A.D.; Siaw, O.L.; Warman-Alaluf, N.; Nitsan, Z.; Appel, S.; Kahana, E.; Rosenmann, H.; Hoffmann, C. CSF Tau Correlates with the Degree of Cortical Involvement in E200K Familial Creutzfeldt-Jakob Disease. Neurosci. Lett. 2016, 634, 76–78. [Google Scholar] [CrossRef]
- Van Everbroeck, B.; Quoilin, S.; Boons, J.; Martin, J.J.; Cras, P. A Prospective Study of CSF Markers in 250 Patients with Possible Creutzfeldt-Jakob Disease. J. Neurol. Neurosurg. Psychiatry 2003, 74, 1210–1214. [Google Scholar] [CrossRef]
- Wang, G.-R.; Gao, C.; Shi, Q.; Zhou, W.; Chen, J.-M.; Dong, C.-F.; Shi, S.; Wang, X.; Wei, Y.; Jiang, H.-Y.; et al. Elevated Levels of Tau Protein in Cerebrospinal Fluid of Patients with Probable Creutzfeldt-Jakob Disease. Am. J. Med. Sci. 2010, 340, 291–295. [Google Scholar] [CrossRef]
- Thompson, A.G.B.; Mead, S.H. Review: Fluid Biomarkers in the Human Prion Diseases. Mol. Cell. Neurosci. 2019, 97, 81–92. [Google Scholar] [CrossRef]
- Zerr, I.; Schmitz, M.; Karch, A.; Villar-Piqué, A.; Kanata, E.; Golanska, E.; Díaz-Lucena, D.; Karsanidou, A.; Hermann, P.; Knipper, T.; et al. Cerebrospinal Fluid Neurofilament Light Levels in Neurodegenerative Dementia: Evaluation of Diagnostic Accuracy in the Differential Diagnosis of Prion Diseases. Alzheimer’s Dement. 2018, 14, 751–763. [Google Scholar] [CrossRef] [PubMed]
- Kanata, E.; Golanska, E.; Villar-Piqué, A.; Karsanidou, A.; Dafou, D.; Xanthopoulos, K.; Schmitz, M.; Ferrer, I.; Karch, A.; Sikorska, B.; et al. Cerebrospinal Fluid Neurofilament Light in Suspected Sporadic Creutzfeldt-Jakob Disease. J. Clin. Neurosci. Off. J. Neurosurg. Soc. Australas. 2019, 60, 124–127. [Google Scholar] [CrossRef] [PubMed]
- Abu-Rumeileh, S.; Capellari, S.; Stanzani-Maserati, M.; Polischi, B.; Martinelli, P.; Caroppo, P.; Ladogana, A.; Parchi, P. The CSF Neurofilament Light Signature in Rapidly Progressive Neurodegenerative Dementias. Alzheimer’s Res. Ther. 2018, 10, 3. [Google Scholar] [CrossRef] [PubMed]
- Llorens, F.; Kruse, N.; Schmitz, M.; Shafiq, M.; da Cunha, J.E.G.; Gotzman, N.; Zafar, S.; Thune, K.; de Oliveira, J.R.M.; Mollenhauer, B.; et al. Quantification of CSF Biomarkers Using an Electrochemiluminescence-Based Detection System in the Differential Diagnosis of AD and SCJD. J. Neurol. 2015, 262, 2305–2311. [Google Scholar] [CrossRef] [PubMed]
- Llorens, F.; Kruse, N.; Schmitz, M.; Gotzmann, N.; Golanska, E.; Thüne, K.; Zejneli, O.; Kanata, E.; Knipper, T.; Cramm, M.; et al. Evaluation of α-Synuclein as a Novel Cerebrospinal Fluid Biomarker in Different Forms of Prion Diseases. Alzheimer’s Dement. 2017, 13, 710–719. [Google Scholar] [CrossRef] [PubMed]
- Oeckl, P.; Metzger, F.; Nagl, M.; von Arnim, C.A.F.; Halbgebauer, S.; Steinacker, P.; Ludolph, A.C.; Otto, M. Alpha-, Beta-, and Gamma-Synuclein Quantification in Cerebrospinal Fluid by Multiple Reaction Monitoring Reveals Increased Concentrations in Alzheimer’s and Creutzfeldt-Jakob Disease but No Alteration in Synucleinopathies. Mol. Cell. Proteom. 2016, 15, 3126–3138. [Google Scholar] [CrossRef] [PubMed]
- Mastrangelo, A.; Baiardi, S.; Zenesini, C.; Poleggi, A.; Mammana, A.; Polischi, B.; Ladogana, A.; Capellari, S.; Parchi, P. Diagnostic and Prognostic Performance of CSF α-Synuclein in Prion Disease in the Context of Rapidly Progressive Dementia. Alzheimer’s Dement. 2021, 13, e12214. [Google Scholar] [CrossRef]
- Abu-Rumeileh, S.; Oeckl, P.; Baiardi, S.; Halbgebauer, S.; Steinacker, P.; Capellari, S.; Otto, M.; Parchi, P. CSF Ubiquitin Levels Are Higher in Alzheimer’s Disease than in Frontotemporal Dementia and Reflect the Molecular Subtype in Prion Disease. Biomolecules 2020, 10, 497. [Google Scholar] [CrossRef]
- Steinacker, P.; Rist, W.; Swiatek-de-Lange, M.; Lehnert, S.; Jesse, S.; Pabst, A.; Tumani, H.; von Arnim, C.A.F.; Mitrova, E.; Kretzschmar, H.A.; et al. Ubiquitin as Potential Cerebrospinal Fluid Marker of Creutzfeldt-Jakob Disease. Proteomics 2010, 10, 81–89. [Google Scholar] [CrossRef]
- Chen, C.; Hu, C.; Zhou, W.; Chen, J.; Shi, Q.; Xiao, K.; Wang, Y.; Dong, X.-P. Calmodulin Level Is Significantly Increased in the Cerebrospinal Fluid of Patients with Sporadic Creutzfeldt-Jakob Disease. Eur. J. Neurol. 2021, 28, 1134–1141. [Google Scholar] [CrossRef] [PubMed]
- Schmitz, M.; Llorens, F.; Pracht, A.; Thom, T.; Correia, Â.; Zafar, S.; Ferrer, I.; Zerr, I. Regulation of Human Cerebrospinal Fluid Malate Dehydrogenase 1 in Sporadic Creutzfeldt-Jakob Disease Patients. Aging 2016, 8, 2927–2935. [Google Scholar] [CrossRef] [PubMed]
- Zerr, I.; Villar-Piqué, A.; Schmitz, V.E.; Poleggi, A.; Pocchiari, M.; Sánchez-Valle, R.; Calero, M.; Calero, O.; Baldeiras, I.; Santana, I.; et al. Evaluation of Human Cerebrospinal Fluid Malate Dehydrogenase 1 as a Marker in Genetic Prion Disease Patients. Biomolecules 2019, 9, 800. [Google Scholar] [CrossRef] [PubMed]
- Abu-Rumeileh, S.; Steinacker, P.; Polischi, B.; Mammana, A.; Bartoletti-Stella, A.; Oeckl, P.; Baiardi, S.; Zenesini, C.; Huss, A.; Cortelli, P.; et al. CSF Biomarkers of Neuroinflammation in Distinct Forms and Subtypes of Neurodegenerative Dementia. Alzheimer’s Res. Ther. 2019, 12, 2. [Google Scholar] [CrossRef] [PubMed]
- Herbst, A.; McIlwain, S.; Schmidt, J.J.; Aiken, J.M.; Page, C.D.; Li, L. Prion Disease Diagnosis by Proteomic Profiling. J. Proteome Res. 2009, 8, 1030–1036. [Google Scholar] [CrossRef][Green Version]
- Foutz, A.; Appleby, B.S.; Hamlin, C.; Liu, X.; Yang, S.; Cohen, Y.; Chen, W.; Blevins, J.; Fausett, C.; Wang, H.; et al. Diagnostic and Prognostic Value of Human Prion Detection in Cerebrospinal Fluid. Ann. Neurol. 2017, 81, 79–92. [Google Scholar] [CrossRef]
- Sano, K.; Satoh, K.; Atarashi, R.; Takashima, H.; Iwasaki, Y.; Yoshida, M.; Sanjo, N.; Murai, H.; Mizusawa, H.; Schmitz, M.; et al. Early Detection of Abnormal Prion Protein in Genetic Human Prion Diseases Now Possible Using Real-Time QUIC Assay. PLoS ONE 2013, 8, e54915. [Google Scholar] [CrossRef]
- Dorey, A.; Tholance, Y.; Vighetto, A.; Perret-Liaudet, A.; Lachman, I.; Krolak-Salmon, P.; Wagner, U.; Struyfs, H.; De Deyn, P.P.; El-Moualij, B.; et al. Association of Cerebrospinal Fluid Prion Protein Levels and the Distinction between Alzheimer Disease and Creutzfeldt-Jakob Disease. JAMA Neurol. 2015, 72, 267–275. [Google Scholar] [CrossRef]
- Vallabh, S.M.; Minikel, E.V.; Williams, V.J.; Carlyle, B.C.; McManus, A.J.; Wennick, C.D.; Bolling, A.; Trombetta, B.A.; Urick, D.; Nobuhara, C.K.; et al. Cerebrospinal Fluid and Plasma Biomarkers in Individuals at Risk for Genetic Prion Disease. BMC Med. 2020, 18, 140. [Google Scholar] [CrossRef]
- Villar-Piqué, A.; Schmitz, M.; Lachmann, I.; Karch, A.; Calero, O.; Stehmann, C.; Sarros, S.; Ladogana, A.; Poleggi, A.; Santana, I.; et al. Cerebrospinal Fluid Total Prion Protein in the Spectrum of Prion Diseases. Mol. Neurobiol. 2019, 56, 2811–2821. [Google Scholar] [CrossRef]
- Rhoads, D.D.; Wrona, A.; Foutz, A.; Blevins, J.; Glisic, K.; Person, M.; Maddox, R.A.; Belay, E.D.; Schonberger, L.B.; Tatsuoka, C.; et al. Diagnosis of Prion Diseases by RT-QuIC Results in Improved Surveillance. Neurology 2020, 95, e1017–e1026. [Google Scholar] [CrossRef] [PubMed]
- Connor, A.; Wang, H.; Appleby, B.S.; Rhoads, D.D. Clinical Laboratory Tests Used to Aid in Diagnosis of Human Prion Disease. J. Clin. Microbiol. 2019, 57, e00769-19. [Google Scholar] [CrossRef] [PubMed]
- Atarashi, R.; Satoh, K.; Sano, K.; Fuse, T.; Yamaguchi, N.; Ishibashi, D.; Matsubara, T.; Nakagaki, T.; Yamanaka, H.; Shirabe, S.; et al. Ultrasensitive Human Prion Detection in Cerebrospinal Fluid by Real-Time Quaking-Induced Conversion. Nat. Med. 2011, 17, 175–178. [Google Scholar] [CrossRef] [PubMed]
- Piconi, G.; Peden, A.H.; Barria, M.A.; Green, A.J.E. Epitope Mapping of the Protease Resistant Products of RT-QuIC Does Not Allow the Discrimination of SCJD Subtypes. PLoS ONE 2019, 14, e0218509. [Google Scholar] [CrossRef]
- Llorens, F.; Kruse, N.; Karch, A.; Schmitz, M.; Zafar, S.; Gotzmann, N.; Sun, T.; Köchy, S.; Knipper, T.; Cramm, M.; et al. Validation of α-Synuclein as a CSF Biomarker for Sporadic Creutzfeldt-Jakob Disease. Mol. Neurobiol. 2018, 55, 2249–2257. [Google Scholar] [CrossRef]
- Orrù, C.D.; Groveman, B.R.; Hughson, A.G.; Manca, M.; Raymond, L.D.; Raymond, G.J.; Campbell, K.J.; Anson, K.J.; Kraus, A.; Caughey, B. RT-QuIC Assays for Prion Disease Detection and Diagnostics. Methods Mol. Biol. 2017, 1658, 185–203. [Google Scholar] [CrossRef]
- Zerr, I.; Villar-Piqué, A.; Hermann, P.; Schmitz, M.; Varges, D.; Ferrer, I.; Riggert, J.; Zetterberg, H.; Blennow, K.; Llorens, F. Diagnostic and Prognostic Value of Plasma Neurofilament Light and Total-Tau in Sporadic Creutzfeldt-Jakob Disease. Alzheimer’s Res. Ther. 2021, 13, 86. [Google Scholar] [CrossRef]
- Steinacker, P.; Blennow, K.; Halbgebauer, S.; Shi, S.; Ruf, V.; Oeckl, P.; Giese, A.; Kuhle, J.; Slivarichova, D.; Zetterberg, H.; et al. Neurofilaments in Blood and CSF for Diagnosis and Prediction of Onset in Creutzfeldt-Jakob Disease. Sci. Rep. 2016, 6, 38737. [Google Scholar] [CrossRef]
- Abu-Rumeileh, S.; Baiardi, S.; Ladogana, A.; Zenesini, C.; Bartoletti-Stella, A.; Poleggi, A.; Mammana, A.; Polischi, B.; Pocchiari, M.; Capellari, S.; et al. Comparison between Plasma and Cerebrospinal Fluid Biomarkers for the Early Diagnosis and Association with Survival in Prion Disease. J. Neurol. Neurosurg. Psychiatry 2020, 91, 1181–1188. [Google Scholar] [CrossRef]
- Thompson, A.G.B.; Anastasiadis, P.; Druyeh, R.; Whitworth, I.; Nayak, A.; Nihat, A.; Mok, T.H.; Rudge, P.; Wadsworth, J.D.F.; Rohrer, J.; et al. Evaluation of Plasma Tau and Neurofilament Light Chain Biomarkers in a 12-Year Clinical Cohort of Human Prion Diseases. Mol. Psychiatry 2021, 26, 5955–5966. [Google Scholar] [CrossRef]
- Abu-Rumeileh, S.; Parchi, P. Cerebrospinal Fluid and Blood Neurofilament Light Chain Protein in Prion Disease and Other Rapidly Progressive Dementias: Current State of the Art. Front. Neurosci. 2021, 15, 648743. [Google Scholar] [CrossRef] [PubMed]
- Thompson, A.G.B.; Luk, C.; Heslegrave, A.J.; Zetterberg, H.; Mead, S.H.; Collinge, J.; Jackson, G.S. Neurofilament Light Chain and Tau Concentrations Are Markedly Increased in the Serum of Patients with Sporadic Creutzfeldt-Jakob Disease, and Tau Correlates with Rate of Disease Progression. J. Neurol. Neurosurg. Psychiatry 2018, 89, 955–961. [Google Scholar] [CrossRef] [PubMed]
- Noguchi-Shinohara, M.; Hamaguchi, T.; Nozaki, I.; Sakai, K.; Yamada, M. Serum Tau Protein as a Marker for the Diagnosis of Creutzfeldt-Jakob Disease. J. Neurol. 2011, 258, 1464–1468. [Google Scholar] [CrossRef] [PubMed]
- Villar-Piqué, A.; Schmitz, M.; Hermann, P.; Goebel, S.; Bunck, T.; Varges, D.; Ferrer, I.; Riggert, J.; Llorens, F.; Zerr, I. Plasma YKL-40 in the Spectrum of Neurodegenerative Dementia. J. Neuroinflamm. 2019, 16, 145. [Google Scholar] [CrossRef]
- Norsworthy, P.J.; Thompson, A.G.B.; Mok, T.H.; Guntoro, F.; Dabin, L.C.; Nihat, A.; Paterson, R.W.; Schott, J.M.; Collinge, J.; Mead, S.; et al. A Blood MiRNA Signature Associates with Sporadic Creutzfeldt-Jakob Disease Diagnosis. Nat. Commun. 2020, 11, 3960. [Google Scholar] [CrossRef]
- Llorens, F.; Villar-Piqué, A.; Schmitz, M.; Diaz-Lucena, D.; Wohlhage, M.; Hermann, P.; Goebel, S.; Schmidt, I.; Glatzel, M.; Hauw, J.-J.; et al. Plasma Total Prion Protein as a Potential Biomarker for Neurodegenerative Dementia: Diagnostic Accuracy in the Spectrum of Prion Diseases. Neuropathol. Appl. Neurobiol. 2020, 46, 240–254. [Google Scholar] [CrossRef]
- Behaeghe, O.; Mangelschots, E.; De Vil, B.; Cras, P. A Systematic Review Comparing the Diagnostic Value of 14-3-3 Protein in the Cerebrospinal Fluid, RT-QuIC and RT-QuIC on Nasal Brushing in Sporadic Creutzfeldt-Jakob Disease. Acta Neurol. Belg. 2018, 118, 395–403. [Google Scholar] [CrossRef]
- Di Fede, G.; Giaccone, G.; Salmona, M.; Tagliavini, F. Translational Research in Alzheimer’s and Prion Diseases. J. Alzheimer’s Dis. 2018, 62, 1247–1259. [Google Scholar] [CrossRef]
- López-Pérez, Ó.; Sanz-Rubio, D.; Hernaiz, A.; Betancor, M.; Otero, A.; Castilla, J.; Andréoletti, O.; Badiola, J.J.; Zaragoza, P.; Bolea, R.; et al. Cerebrospinal Fluid and Plasma Small Extracellular Vesicles and MiRNAs as Biomarkers for Prion Diseases. Int. J. Mol. Sci. 2021, 22, 6822. [Google Scholar] [CrossRef]
- Vassileff, N.; Cheng, L.; Hill, A.F. Extracellular Vesicles—Propagators of Neuropathology and Sources of Potential Biomarkers and Therapeutics for Neurodegenerative Diseases. J. Cell Sci. 2020, 133. [Google Scholar] [CrossRef]
- Minikel, E.V.; Vallabh, S.M.; Orseth, M.C.; Brandel, J.-P.; Haïk, S.; Laplanche, J.-L.; Zerr, I.; Parchi, P.; Capellari, S.; Safar, J.; et al. Age at Onset in Genetic Prion Disease and the Design of Preventive Clinical Trials. Neurology 2019, 93, e125–e134. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Probable Disease | Definitive Disease | ||||||
|---|---|---|---|---|---|---|---|
| CJD | sCJD | Rapidly progressive cognitive impairment AND typical EEG (generalized periodic complexes) OR typical MRI (high signal in caudate/putamen or at least two cortical regions either on DWI or FLAIR) OR positive 14-3-3 AND two of the following: -Myoclonus -Visual or cerebellar problems -Pyramidal or extrapyramidal features -Akinetic mutism | OR | Progressive neurological syndrome AND positive RT-QuIC in CSF or other tissues. | Progressive neurological syndrome AND neuropathologically OR immunocytochemically OR biochemically confirmed. | ||
| gCJD | Progressive neuropsychiatric disorder AND definite or probable CJD in 1st degree relative | OR | Progressive neuropsychiatric disorder AND pathogenic PRNP mutation | Definitive CJD AND definitive or probable CJD in 1st degree relative | OR | Definitive CJD AND pathogenic PRNP mutation | |
| vCJD | Progressive neuropsychiatric disorder with duration of illness >6 months, no history of potential iatrogenic exposure, no evidence of gCJD and no alternative diagnosis suggested by routine investigations AND positive tonsil biopsy OR bilateral pulvinar high signal on MRI AND atypical appearance of sCJD on EEG in the early stages AND four of the following: -Early psychiatric symptoms (depression, anxiety, apathy, withdrawal, delusions) -Persistent painful sensory symptoms (frank pain or dysaesthesia) -Ataxia -Myoclonus, chorea or dystonia -Dementia | Progressive neuropsychiatric disorder AND neuropathological confirmation (spongiform change and extensive PrP deposition with florid plaques throughout the cerebrum and cerebellum) | |||||
| iCJD | Progressive cerebellar syndrome in a recipient of human cadaveric-derived pituitary hormone OR sporadic CJD with a recognized exposure risk | Progressive cerebellar syndrome or sporadic CJD with a recognized exposure risk AND neuropathological confirmation | |||||
| VSPr | Cognitive impairment and/or two of the following: -Psychiatric symptoms -Parkinsonism -Aphasia -Ataxia -Myoclonus | AND | Less than 8 years duration AND absence of alternative etiology or phenotype divergence from atypical neurodegenerative dementias | Progressive neurological syndrome AND neuropathological confirmation | |||
| sFI and FFI | Organic sleep disturbances. If not yet clinically apparent, a polysomnography has to be performed. At least two of the following: -Psychiatric symptoms (visual hallucinations, personality changes, depression, anxiety, aggressiveness, disinhibition, listlessness) -Ataxia -Visual symptoms -Myoclonus -Cognitive/mnesic deficits | AND | One of the following: -Loss of >10 kg during the last 6 months -Vegetative signs (hyperhidrosis, newly diagnosed arterial hypertonia, tachycardia, constipation, hyperthermia) -Husky voice | Progressive neurological syndrome AND neuropathological confirmation | |||
| GSS | Progressive cerebellar syndrome, cognitive impairment and/or sensory symptoms AND pathogenic PRNP mutation | Progressive neurological syndrome AND neuropathological confirmation (amyloid deposits immunoreactive for PrP are the morphological hallmark of GSS) | |||||
| HDL-1 | Abnormal involuntary movements, coordination difficulty, dementia, personality changes and psychiatric symptoms AND pathogenic PRNP mutation | Kuru and multicentric plaques that stain with anti-prion antibodies | |||||
| PRNP Variants | DNA Nucleotide Change | Predicted Protein Change | Related Prionopathy | Phenotype | Expected Survival (Median) |
|---|---|---|---|---|---|
| P102L | c.305C>T | Proline to leucine substitution at codon 102 | GSS | Ataxia (100%), pyramidal (75%), dementia (62%), extrapyramidal (50%), myoclonus (25%). Others: dysarthria, sleep and sensory disturbances. | 40 months. |
| D178N | c.532G>A | aspartic acid to asparagine substitution at codon 178 | FFI and genetic CJD (Depends on the allele on codon 129. M allele: FFI, and V allele: genetic CJD) | Dementia (96%), myoclonus (89%), ataxia (82%), extrapyramidal (82%), pyramidal (79%), cortical blindness (79%). Others: sleep disturbances, dysarthria, weight loss and hyperhidrosis. | 15 months (earlier onset and shorter duration of symptomatic disease in genetic CJD). |
| V180I | c.538G>A | Valine to isoleucine change at codon 180 | Genetic CJD | Dementia (100%), extrapyramidal (54%), pyramidal (46%). Others: akinetic mutism (57%) and psychiatric (50%). Cortical blindness, myoclonus and ataxia are more infrequent. | 16,4 months (wide range of survival). |
| E200K | c.598G>A | Glutamic acid to lysine substitution at codon 200 | Genetic CJD | Ataxia (100%), dementia (95%), myoclonus (85%), pyramidal (70%), cortical blindness (70%), extrapyramidal (65%). Others: dysarthria, sleep disturbances and weight loss. | 5 months (wide range of survival 1–74 months). |
| V210I | c.628G>A | Valine to isoleucine substitution at codon 210 | Genetic CJD | Ataxia (100%), dementia (92%), myoclonus (92%), extrapyramidal (92%), cortical blindness (85%), pyramidal (72%). Others: dysarthria and sensory symptoms. | 4 months (wide range of survival). |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Altuna, M.; Ruiz, I.; Zelaya, M.V.; Mendioroz, M. Role of Biomarkers for the Diagnosis of Prion Diseases: A Narrative Review. Medicina 2022, 58, 473. https://doi.org/10.3390/medicina58040473
Altuna M, Ruiz I, Zelaya MV, Mendioroz M. Role of Biomarkers for the Diagnosis of Prion Diseases: A Narrative Review. Medicina. 2022; 58(4):473. https://doi.org/10.3390/medicina58040473
Chicago/Turabian StyleAltuna, Miren, Iñigo Ruiz, María Victoria Zelaya, and Maite Mendioroz. 2022. "Role of Biomarkers for the Diagnosis of Prion Diseases: A Narrative Review" Medicina 58, no. 4: 473. https://doi.org/10.3390/medicina58040473
APA StyleAltuna, M., Ruiz, I., Zelaya, M. V., & Mendioroz, M. (2022). Role of Biomarkers for the Diagnosis of Prion Diseases: A Narrative Review. Medicina, 58(4), 473. https://doi.org/10.3390/medicina58040473