Abstract
Although experimental evidence indicates that mitochondrial collapse is a common effect of both Chagas disease and post-ischemic heart failure and that cardiac anatomy and function are partially restored by stem cell therapy, the responsible molecular mechanisms are still under debate. Gene expression data from our publicly accessible transcriptomic dataset obtained by profiling the left ventricle myocardia of mouse models of Chagas disease and post-ischemic heart failure were re-analyzed from the perspective of the Genomic Fabric Paradigm. In addition to the regulation of the gene expression levels, we determined the changes in the strength of the homeostatic control of transcript abundance and the remodeling of the gene networks responsible for the mitochondrial respiration. The analysis revealed that most of the mitochondrial genes assigned to the five complexes of the respiratory chain were significantly downregulated by both Chagas disease and ischemia but exhibited outstanding recovery of the normal expression levels following direct injection of bone-marrow-derived stem cells. However, instead of regaining the original expression control and gene networking, the treatment induced novel mitochondrial arrangements, suggesting that multiple transcriptomic topologies might be compatible with any given physiological or pathological state. This study confirmed several established mechanisms and identified novel gene expression signals, especially Cox4i2, Cox6b1, Cox7b, Ndufb11, and Tmem186, that warrant further investigations. Their broad rescue with cell therapy underscores mitochondria as a convergent, tractable target for cardiac repair.
1. Introduction
Mitochondria are critical organelles that regulate cell metabolism and survival, especially in the heart, where mitochondria comprise 30–40% of cardiomyocyte volume and play a primary role in energy production, calcium homeostasis, and regulation of cellular apoptosis. Mitochondrial dysfunction leads to impaired cardiomyocyte function, a hallmark of heart failure [1]. Since mitochondria are essential for the cellular energy-demanding cardiac contractile function, a decline in mitochondrial biogenesis and function [2] was associated with the development of ventricular dysfunction caused by myocardial infarction [3], type 2 diabetes [4], Chagas disease [5], or anticancer-drug-related cardiotoxicity [6] among several cardiovascular afflictions.
Decades after initial infection with the parasitic euglenoid Trypanosoma cruzi [7], transmitted by the so-called “kissing bug” [8], ~30% of individuals can develop chronic Chagas disease (CCC), a congestive heart failure and dilated cardiomyopathy [9,10,11]. It is estimated that CCC affects about 7 million people worldwide, most of them in Latin America [12], and it recently became endemic even in the United States [13]. Although the pathogenesis of CCC remains a matter of debate [14,15], the involvement of cardiac mitochondria was first demonstrated by Garg et al. in 2003 [16] and confirmed by Báez et al. in 2011 [17]. It is known that a year post-infection parasite persistence and inflammation are associated with structural and functional alterations in mouse cardiac mitochondria in a parasite-strain-dependent manner [18]. Common comorbidities include dyslipidemia and hypertension [19] and might lead to cryptogenic stroke [20]. As expected for any infectious disease, CCC triggers the immune response [21] that changes during the development of the disease [22].
Myocardial infarction [23], described as the cardiomyocytes’ death caused by insufficient oxygen supply, whose definition and management are still under debate [24,25], is directly related to mitochondrial dysfunction [26] and affects several functional pathways [27,28].
Previous studies on mouse models have shown that myocardial infarction induced by interruption of blood supply [29] activates a strong inflammatory response and leads to ventricular remodeling and ischemic heart failure (IHF) [30]. These effects were reversed by injecting bone-marrow-derived mononuclear stem cells into the cardiac scar tissue [31]. Whatever the cause, heart failure has severe consequences on all organs, including triggering various mental disorders [32].
Transcriptomic studies on cardiomyocytes and the left heart ventricle of CCC mice revealed remodeling of the immune response [33], extracellular matrix, cell adhesion [34], intercellular communication via gap junction channels [35,36], mitochondrial oxidative phosphorylation [37], JAK/STAT signaling, and cell cycle [38] functional pathways.
The progenitor cells are well recognized for their therapeutic potential in cardiac regeneration [39]. We found that injecting bone-marrow-derived stem cells restores most of the heart function and recovers most of the normal heart gene expression profile [40,41].
This research investigated the mitochondria-related genomic alterations in failing hearts due to CCC and IHF before and after stem cell treatment. We analyzed the KEGG-constructed oxidative phosphorylation functional pathway [42] as arranged in the mitochondrial module of the KEGG-constructed cardiovascular disease pathway of diabetic cardiomyopathy [43]. This study did not stop at the alteration of the gene expression profile but extended to encompass the entire mitochondrial transcriptomic topology, i.e., to include the control of transcripts’ abundances and the transcriptomic networks, as common in our Genomic Fabric Paradigm [41].
2. Materials and Methods
2.1. Experimental Data
We re-analyzed publicly accessible gene expression data generated in former Iacobas’ lab by profiling the transcriptomes of the left heart ventricle myocardium of adult, age-matched male C57Bl/6j mouse models of CCC [44] and IHF [45] before and after the treatment with bone-marrow-derived mononuclear cells [46,47]. The treated stem cells were collected from femurs and tibiae of adult C57Bl/6 mice and purified by centrifugation in a Ficoll gradient [30]. Chagas disease was induced by infecting adult male C57Bl/6j mice with trypomastigotes of the Colombian Trypanosoma cruzi strain [48], while the permanent myocardial infarcts were obtained by ligating the descending branch of the left coronary artery [39]. The experimental protocols and raw and normalized expression data are fully described and publicly accessible in the Gene Expression Omnibus (GEO) of the (USA) National Center for Biotechnology Information (NCBI) [44,45,46,47].
If in a microarray a spot had corrupted or saturated pixels, or the background exceeded half of the foreground fluorescence signal, that spot was eliminated from the analysis for all microarrays in the experiment. The background-subtracted foreground fluorescence signal from each spot was normalized to the median for that channel of the microarray block printed with the same pin and then averaged for all biological replicas of the corresponding condition. This “multiple yellow strategy” adopted by us in hundreds of transcriptomic experiments is theoretically the least affected by the bias between the two laser channels of the scanner and the potential differences among the spotting pins of the microarray printer.
For each cardiomyopathy, we examined two conditions: IN (infected/infarcted—not treated) and IT (infected/infarcted—treated) and compared them with CN (healthy/control—not treated), all in four biological replicates.
2.2. Primary Independent Characteristics of Individual Genes
Background-subtracted foreground fluorescence signals were averaged for spots redundantly probing the same gene. According to our standard procedure [41] (flowchart in Figure 1), each properly quantified gene was characterized in each condition by three independent characteristics: AVE, REV, and COR.
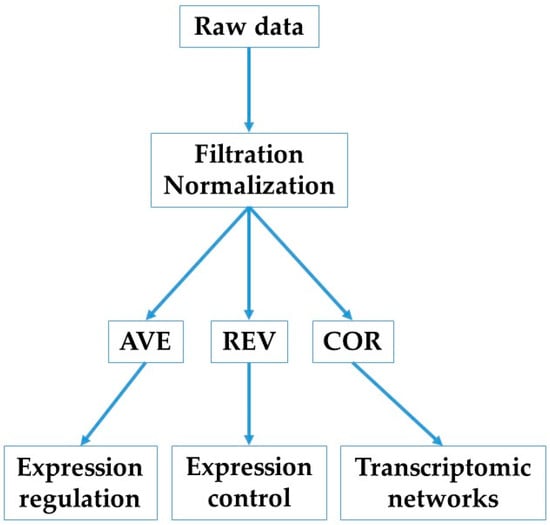
Figure 1.
Flowchart of this study.
AVE is the average expression level across biological replicas. Comparing AVE values of a gene in two conditions indicates whether expression of that gene was altered.
REV (relative expression variation) is defined as the mid-interval of the chi-square estimate of the coefficient of variation of the expression levels on all spots redundantly probing the same gene across biological replicas of one condition. REV is used to calculate the REC (relative expression control) and RCS (relative control strength) as
where ⟨REV⟩(c) is the median REV of all genes quantified in the condition c = CN, IN, IT. A positive/negative REC value indicates that the expression of that gene is more/less controlled (i.e., more/less stably expressed across biological replicas) than the median in the condition. The control level of a gene indicates the importance of that gene for the cell survival and phenotypic expression, with the top positive value pointing to the most critical mitochondrial gene for the respective condition.
COR is the Pearson pairwise correlation coefficient of the (log2) expression levels of two genes. There are three (p < 0.05) statistically significant cases when each of the two genes “i” and “j” in condition “c” are probed by single microarray spots:
- (1)
- synergistically expressed, i.e., their expression levels oscillate in phase across biological replicates.
- (2)
- antagonistically expressed, i.e., their expression levels oscillate in antiphase across biological replicates.
- (3)
- independently expressed genes, i.e., the expression of one gene has no influence on the expression of the other gene.
COR analysis was used to identify the (p < 0.05) significant transcriptomic networks based on the “principle of transcriptomic stoichiometry” [49], requiring coordinated expressions of genes whose encoded products are linked in a functional pathway. The overall expression correlation of the mitochondrial genes responsible for adjacent complexes (Cx) of the respiratory chain was measured by their coordination (COORD) percentage:
where SYN, ANT, and IND are the percentages of the p < 0.05 significant synergistically, antagonistically, and independently expressed gene pairs out of all pairs that can be formed between the two complexes.
REC and COR values were used to hierarchize the genes according to their Gene Commanding Height (CGH) score [50]:
2.3. Transcriptomic Changes in Individual Genes and Functional Pathways
Our standard protocol [51] considers the expression of a gene as statistically significantly regulated (here in the left heart ventricle of the diseased mouse without and with cell treatment (d = IN, IT) with respect to the control counterpart CN) if the absolute expression fold-change
exceeds the appropriate cut-off and the p-value of the heteroscedastic t-test of the means equality is less than 0.05. The absolute fold-change cutoff (CUT) is computed for each gene by considering both the expression variabilities in the compared conditions and the technical noises of the probing microarray spots in the two sets of biological replicas.
Contributions of individual genes to the transcriptomic alteration in condition “d” (CCC, IHF) with respect to their healthy counterparts were measured by their WIR (weighted individual (gene) regulation) score:
The overall alteration of a functional pathway Φ in a particular condition (CCC or IHF) was measured by the median WIR of the pathway genes. A positive median WIR indicates that the total number of (significantly and not significantly) upregulated genes in the pathway exceeded the number of the downregulated genes, and vice versa.
The alteration of expression control in CCC and IHF mice without cell treatment with respect to the condition CN was measured by the fold-change (FC) defined as
The fold-change alteration (negative for downregulation) of GCH in a treated (IT) or untreated (IN) Chagas and post-ischemic heart failure is computed as
3. Results
3.1. Both Chagasic Disease and Post-Ischemic Heart Failure Are Characterized by Substantial Downregulation of Mitochondrial Genes
Figure 2 presents the mitochondrial genes that were significantly regulated (according to the composite criterion of the absolute fold-change and p-value (condition 5)) in Chagas disease and post-ischemic heart failure. Thus, in the untreated CCC, four of the five complexes contain only downregulated genes, with Cx4 making the exception by having one upregulated gene (Cox6a1), although two other genes (Cox5a, Cox6b1) were downregulated. The percentage of downregulated genes is even larger in the post-ischemic heart despite the upregulation of Ndufa9.
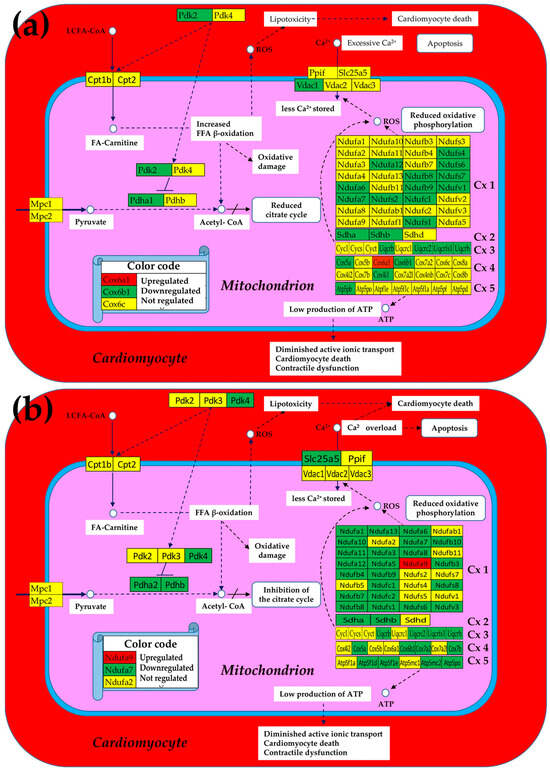
Figure 2.
Significantly regulated mitochondrial genes in (a) Chagas disease and in (b) post-ischemic heart failure. A red/green background of the gene symbol indicates significant up/downregulation, while a yellow background indicates that the expression change was not statistically significant. Regulated genes: Atp5f1d/e (ATP synthase, H+ transporting, mitochondrial F1 complex, delta/epsilon subunit), Atp5mc2 (ATP synthase membrane subunit c locus 2), Atp5pb/o (ATP synthase peripheral Uqcrh stalk-membrane subunit b/OSCP), Cox4i1 (cytochrome c oxidase subunit IV isoform 1), Cox17 (cytochrome c oxidase, subunit XVII assembly protein homolog (yeast)), Cox5a (cytochrome c oxidase, subunit Va), Cox6a1/6b1 (cytochrome c oxidase, subunit VI a/b, polypeptide 1), Cox7a2/b (cytochrome c oxidase, subunit VIIa 2/VIIb), Cpt2 (carnitine palmitoyltransferase 2), Cyc1 (cytochrome c-1), Ndufa6/7/12 (NADH:ubiquinone oxidoreductase subunit A6/7/12), Ndufb8/9/10 (NADH: ubiquinone oxidoreductase subunit beta8/9/10), Ndufc1/2 (NADH:ubiquinone oxidoreductase subunit C1/C2), Ndufs1/4/6/7/8 (NADH dehydrogenase (ubiquinone) Fe-S protein 1/4/6/7/8), Ndufv3 (NADH:ubiquinone oxidoreductase core subunit V3), Pdha1 (pyruvate dehydrogenase E1 alpha 1), Pdk2/4 (pyruvate dehydrogenase kinase, isoenzyme 2/4), Sdha/b (succinate dehydrogenase complex, subunit A/B), Slc25a5 (solute carrier family 25 (mitochondrial carrier, adenine nucleotide translocator), member 5), Uqcrc2 (ubiquinol-cytochrome c reductase core protein 2), Uqcrfs1 (ubiquinol-cytochrome c reductase, Rieske iron–sulfur polypeptide 1), Uqcrh (ubiquinol-cytochrome c reductase hinge protein), Uqcrb/h (ubiquinol-cytochrome c reductase binding/hinge protein), and Vdac1 (voltage-dependent anion channel 1).
The large numbers of the downregulated genes in the five complexes indicate a significant reduction in oxidative phosphorylation and, by consequence, less production of ATP. Remarkably, in both cardiomyopathies, Ca2+ storage in the mitochondrion was diminished by downregulation of one of the involved genes (Vdac1 in CCC and Slc25a5 in IHF mice). The downregulation of these transporters consistently led to Ca2+ accumulation in the cardiomyocyte that affected the contractility function of this heart-muscle cell.
3.2. Cell Treatment Restores the Normal Expression of Most Mitochondrial Genes Altered in Both Chagasic Disease and Post-Ischemic Heart Failure
As illustrated in the Figure 3, we found that the expressions of most mitochondrial genes that have been altered in both Chagasic and ischemic mice were markedly restored following the cell treatment.
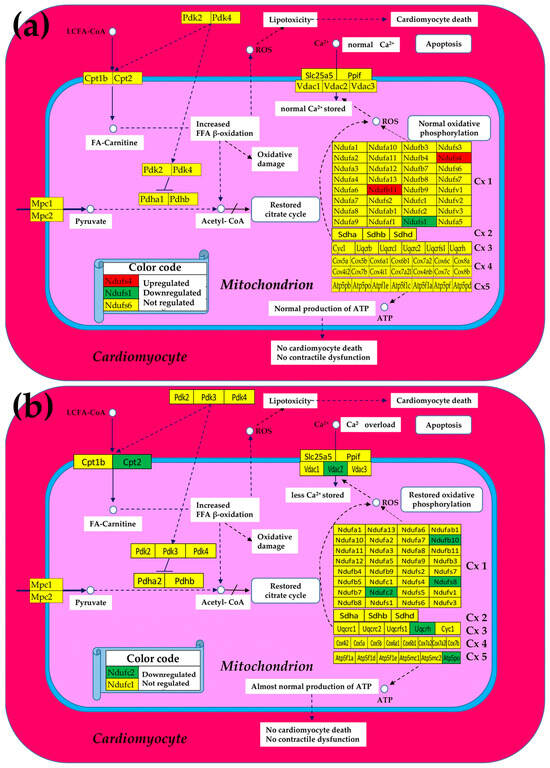
Figure 3.
Remaining significantly regulated mitochondrial genes after stem cell treatment in (a) Chagas disease cardiomyopathy and in (b) post-ischemic heart failure. A red/green background of the gene symbol indicates significant up/downregulation, while a yellow background indicates that the expression change was not statistically significant. Regulated genes: Atp5po (ATP synthase peripheral Uqcrh stalk-membrane subunit OSCP), Cpt2 (carnitine palmitoyltransferase 2), Ndufb1/10/11 (NADH: ubiquinone oxidoreductase subunit beta1/10/11), Ndufc2 (NADH:ubiquinone oxidoreductase subunit C2), Ndufs1/4/8 (NADH dehydrogenase (ubiquinone) Fe-S protein 1/4/8), Uqcrh (ubiquinol-cytochrome c reductase hinge protein), and Vdac2 (voltage-dependent anion channel 2).
Moreover, in the case of CCC mice, the initially not affected Ndufb11 was upregulated by treatment, while the originally downregulated Ndufs4 was even switched to upregulated. Of note is that normal Ca2+ storage in the mitochondrion was restored in the treated CCC mice but remained diminished in the treated IHF now because of the downregulation of Vdac2 despite recovering the normal expression of initially downregulated Slc25a5.
3.3. The Largest Mitochondrial Gene Contributors to the Transcriptomic Alterations in Both Chagasic and Post-Ischemic Mice
Figure 2 and Figure 3 implicitly consider significantly regulated genes as equal contributors to the transcriptomic alteration, presenting the overall change as percentages of up- and downregulated genes, which is an oversimplified description of the biological reality. A better representation of the contribution would be by the pair (expression ratio, p-value) of each affected gene. However, we think that a more comprehensive characterization is by the weighted individual (gene) regulation (WIR), computed with Formula (6), that incorporates the total expression change AVE(CN)(|x| − 1) and the statistical confidence (1 − p-value) of this change.
Table 1 presents the largest five contributors (as absolute value of WIR) in Chagasic (CCC) and ischemic (IHF) mice, together with their normal average expression levels (AVE), expression ratios (X), and p-values of their regulation with respect to the normal expression. As expected, all top contributors had negative expression ratios.

Table 1.
The largest mitochondrial (MITO) gene contributors to the heart transcriptome alteration in Chagasic (CCC) and ischemic (IHF) mice. AVE = average expression in the control mice, X-IN = expression ratio in untreated infected/infarcted with respect to control (negative for downregulation), X-IT = expression ratio in treated infected/infarcted with respect to control (negative for downregulation), |WIR| = absolute value of the weighted individual (gene) regulation. The gray background indicates the most important values in each condition.
The effectiveness of the cell treatment in restoring the expression profiles of the mitochondrial genes is evident by the change in the median WIR from −18.74 in infected not treated to −4.50 after treatment in CCC mice and from −16.83 to −1.81 in IHF mice. These results indicate both substantial reduction in mitochondrial function caused by parasitic infection and partial recovery following cell treatment. While in CCC mice, the non-mitochondrial gene Pln was the largest contributor (WIR = − 222), in the ischemic mice, one mitochondrial gene, Cox5a, tops the list with the impressive WIR = −2603(!).
Interestingly, the median WIR value for the entire transcriptome was positive in all conditions, meaning that overall, the contributions of the upregulated genes (like those involved in the inflammatory response as previously reported [23,39]) exceeded those of the downregulated. As expected, the median WIRs of the entire transcriptome were larger for both untreated CCC (0.67 vs. 0.13) and IHF (1.63 vs. 0.70) mice, pointing to an overall reduction in the alteration of the expression profile.
Figure 4 is a graphical representation of the fold-change (negative for downregulation) with respect to the healthy condition of the five largest mitochondrial gene contributors to the transcriptomic alterations in the two treated and untreated cardiomyopathies studied (data from Table 1).
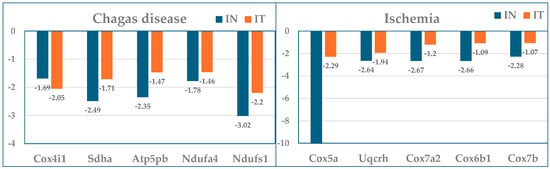
Figure 4.
Expression ratios with respect to control (healthy mice) of the five largest mitochondrial gene contributors to the transcriptomic alterations in the treated (IT) and untreated (IN) Chagas disease and post-ischemic heart failure.
3.4. The Most and the Least Controlled Mitochondrial Genes in CCC and IHF Mice
The experimental design with four biological replicates allowed us to estimate the relative expression control (REC) of individual genes in each condition and how much both treated and untreated cardiomyopathies affected them. Table 2 presents the REC values (computed according to Equation (1)) of the most (REC > 0) and the least (REC < 0) controlled mitochondrial genes in all profiled conditions (healthy, treated, and untreated CCC and IHF mice). We also listed the most and the least controlled genes in the entire transcriptome and the fold-change (negative for downregulation) of diseased states with respect to the healthy mice.
Table 2.
The most and the least controlled mitochondrial genes in Chagasic and ischemic mice. Abbreviations: CN = control (reference); IN = infected/ischemic not treated; IT = infected/ischemic treated; MITO = mitochondrial; RCS-FC, fold-change of the relative control strength in IN/IT with respect to CN. For reference, the table includes the RECs and the RCS-FCs of the most and least controlled genes in the entire profiled transcriptome in control, untreated, and treated CCC mice. The gray background indicates the most important values in each condition.
3.5. Both CCC and IHF Alter the Transcriptomic Networks of the Mitochondrial Genes by Partially Decoupling the Oxidative Phosphorylation Complexes
Figure 5 presents the (p < 0.05) significant transcriptomic networks coupling the five complexes of the oxidative phosphorylation in the left heart ventricles of healthy and untreated CCC and IHF mice. One observes that the adjacent complexes are coupled in all conditions mostly by synergistic expression correlations and that the coordination degree (computed with Formula (2)) is significantly lower in the untreated cardiomyopathies, indicating a partial decoupling of the complexes. The coordinated percentages were computed for the 80 possible gene pairs between Cx1 and Cx2, 27 between Cx2 and Cx3, 84 between Cx3 and Cx4, and 140 between Cx4 and Cx5.
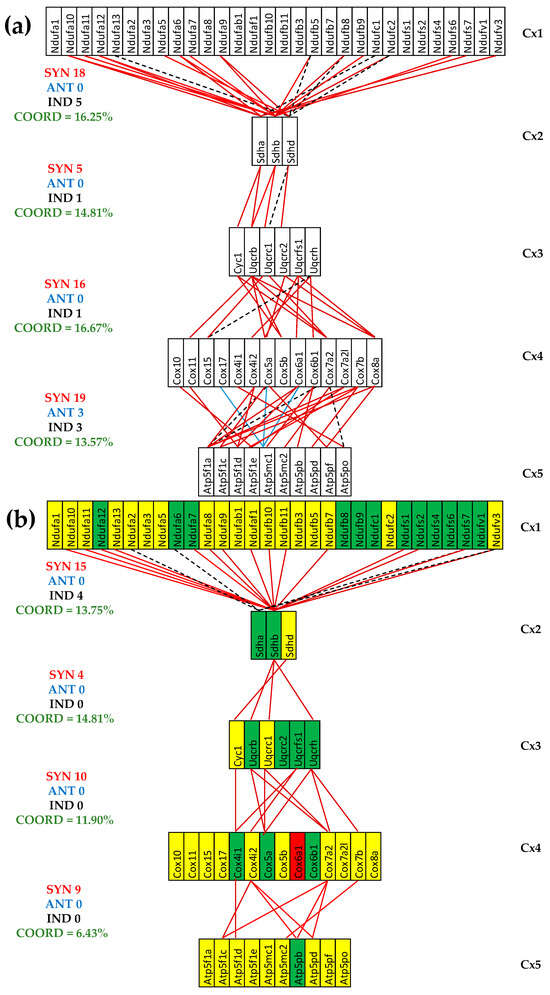
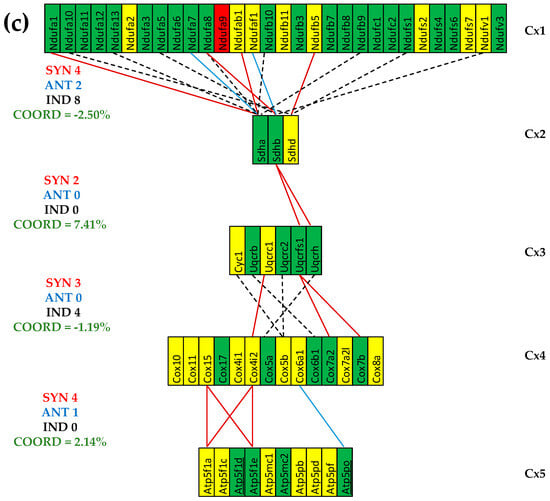
Figure 5.
The (p < 0.05) significant transcriptomic networks coupling the five complexes of the oxidative phosphorylation in the left heart ventricles of healthy and untreated CCC and IHF mice. (a) Healthy mice, (b) untreated CCC mice, (c) untreated IHF mice. Continuous red/blue lines indicate (p < 0.05) significant synergistic/antagonistic expression correlations of the paired genes, while dashed black lines point to significantly independently expressed gene pairs. Missing lines mean that the expression correlation was not (p < 0.05) statistically significant. A red/green background of the gene symbols indicates significant up/downregulation, while a yellow background means no significant change in the expression level with respect to the healthy mice.
The reduction was much more considerable for the untreated IHF mice, going to even negative values (COORD(Cx1-Cx2) = −2.50%, COORD(Cx3-Cx4) = −1.19%), which means more significantly independently that synergistically + antagonistically expressed gene pairs were identified. For instance, the number of synergistically + antagonistically expressed gene pairs between complexes Cx1 and Cx2 decreased from 18 (=18 + 0) in CN to 15 (=15 + 0) in CCC and to 6 (4 + 2) in IHF, while the number of independently expressed pairs goes from five in CN to four in CCC and eight in IHF. In general, there are fewer antagonistically than independently expressed gene pairs, explaining the synchrony of the respiratory chain.
3.6. Stem Cell Treatment Benefits for the Transcriptomic Coupling of the Oxidative Phosphorylation Complexes
Figure 6 presents the (p < 0.05) significant gene expression correlations in treated CCC and IHF mice and the coordination degrees in all five conditions. As summarized in Figure 6c, both cardiomyopathies reduced the expression coordination degrees among all pairs of coupled complexes. However, the stem cell treatment partially restored the coordination degree in the infarcted mice while even substantially increasing it in the CCC mice.
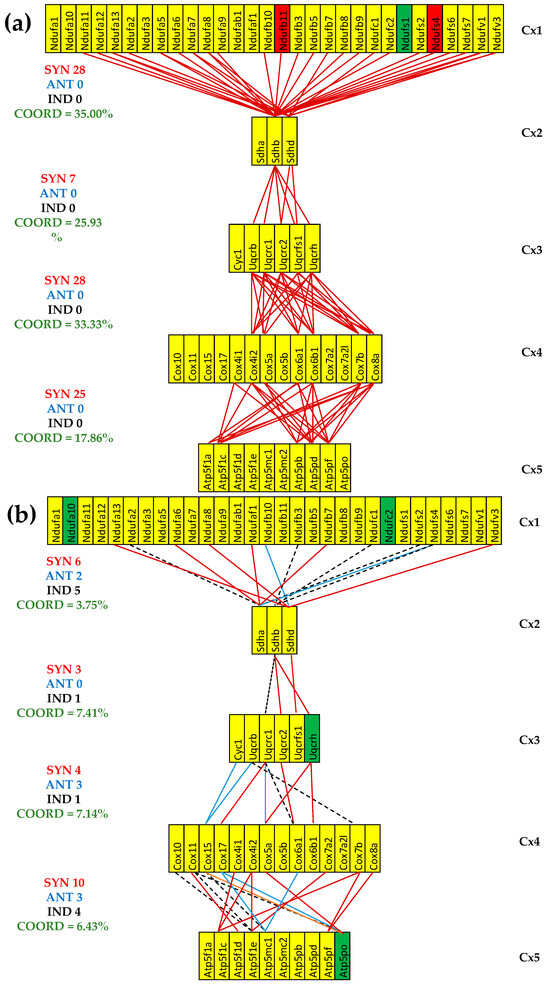
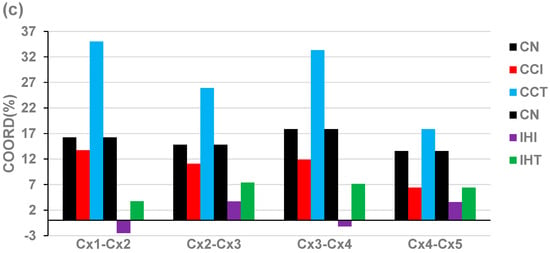
Figure 6.
The (p < 0.05) significant transcriptomic networks coupling the five complexes of the oxidative phosphorylation in the left heart ventricles of treated CCC and IHF mice. (a) CCC-treated mice, (b) IHF-treated mice, (c) coordination degrees between adjacent complexes of the oxidative phosphorylation functional pathway in the left heart ventricle of healthy (denoted by CN) untreated CCC (CCI) and IHF (IHI) and treated CCC (CCT) and IHF (IHT) mice. Continuous red/blue lines indicate (p < 0.05) significant synergistic/antagonistic expression correlations of the paired genes, while dashed black lines point to independently expressed gene pairs. Missing lines indicate that the expression correlation of the two genes was not (p < 0.05) statistically significant. A red/green background of the gene symbols indicates significant up/downregulation, while a yellow background means no significant change in the expression level with respect to the healthy mice. Note the coordination reduction in both untreated cardiomyopathies, reaching even negative values for two pairs of complexes (Cx1-Cx2, Cx3-Cx4) in IHF mice. (c) IHF-treated mice.
One important observation is that although the cell treatment restored the normal expression levels for most mitochondrial genes, the transcriptomic coupling of the complexes exhibits different than normal patterns. Importantly, the patterns of treated CCC and IHF are also largely distinct from one another.
3.7. Both CCC and IHF Alter the Hierarchy of Mitochondrial Genes
Table 3 presents the most prominent five mitochondrial genes in treated and untreated Chagasic and ischemic mice according to their Gene Commanding Height (GCH) scores computed with Formula (3). It also contains the fold-change (GCH-FC, negative for downregulation) of the GCHs in disease states with respect to the healthy condition, computed with Formula (8). For comparison, the table also presents the GCH scores of the top five mitochondrial genes in healthy mice and their GCHs in the untreated CCC and IHF mice.
Table 3.
The Gene Commanding Heights (GCHs) of the five most prominent mitochondrial genes in control, treated, and untreated Chagasic and ischemic mice. Abbreviations: IN = infected/ischemic not treated; IT = infected/ischemic treated; MITO = mitochondrial; GCH-FC = fold-change of the Gene Commanding Height; CCC = Chagasic mice; IHF = ischemic mice. For reference, the table includes the GCHs of the most prominent MITO genes in control mice and their scores in untreated CCC and IHF mice. The gray background indicates the most important values in each condition.
4. Discussion
Alteration of the mitochondrial respiration and excessive generation of reactive oxygen species (ROS) are regarded as the main causes of heart failure [52,53], given that mitochondria provide about 90% of the energy used by the heart during normal functioning [54,55].
Using the Genomic Fabric Paradigm [51], which provides the most theoretically possible comprehensive transcriptomic characterization, our present study has brought new insights into the current understanding of mitochondrial gene involvement underlying Chagas disease [56] and post-ischemic heart failure [57]. Although several previous studies specifically analyzed alteration of the expression of genes/proteins that regulate mitochondrial oxidative phosphorylation [16,17], none of them studied the alteration and recovery of the gene expression control and transcriptomic networking.
Our transcriptomic characterization of untreated CCC and IHF hearts indicated a unified failure of mitochondrial energetics that was substantially reversed by bone-marrow-derived cell therapy. At the level of Complex 1 [58], the downregulation of 11/32 Nduf genes in untreated CCC and 22/32 in untreated IHF mice likely predicts a severe block in NADH oxidation, a surge in reactive oxygen species, and consequent energy depletion. Complex 1 (NADH ubiquinone oxidoreductase [59]) is responsible for NADH oxidation and proton pumping along the electron transport chain [60]. For instance, the downregulation of Ndufa7 found by us (x = −1.92 in the untreated CCC and x = −1.72 in untreated IHF) was also reported in cardiac hypertrophy [61]. Although Ndufa10, known for its role in spontaneous hypertension [62,63], was equally downregulated (x = −1.60) in both untreated cardiomyopathies, its downregulation was statistically significant only in IHF mice (p(IHF) = 0.009), while in untreated CCC mice it was not because of larger biological variability (p(CCC) = 0.142). Interestingly, while Ndufa9 was (even if not statistically significant) downregulated in untreated CCC, it was upregulated (x = 4.18) in untreated IHF, as happens in high-intensity training [62].
The collapse of 2/3 of the quantified Complex 2 genes, critical for the Krebs cycle [64,65,66,67], in both untreated heart diseases investigated by us, resulted from the significant downregulation of Sdha (x(CCC) = −2.49, x(IHF) = −2.29) and Sdhb (x(CCC) = −2.07, x(IHF) = −1.59).
Electron flow stops at Complex 3: the hinge protein Uqcrch shows one of the strongest negative WIR scores (WIR = −44.38), consistent with knockout data in which its loss alone causes contractile failure [68], thereby supporting and broadening those functional observations to two distinct heart failure models. Complex 3 (CoQ: cytochrome c-oxidoreductase [69]) deficiency is responsible for fewer mitochondrial disorders [70], although carbonylation of genes like Uqcrc2 (found as downregulated in both untreated CCC and IHF) is a direct response to the stress of Trypanosoma cruzi infection [71].
Complex 4 is compromised by suppression of the ATP/ADP sensor Cox4i1 in CCC, which matches the marked reduction in state-3 respiration and cytochrome-c-oxidase deficits clinically documented [72]. The downregulation (x = −2.3, WIR = −80) in untreated IHF mice of Cox7b likely undermines Complex 4 assembly, highlighting a novel candidate for future functional probing. Cox5a, the largest contributor listed in Table 1 to the downregulation of Cx4 in IHF (WIR = −2603), is considered a biomarker for the blood stasis syndrome [67], cyanotic heart disease [73], and acute myocardial infarction [74].
Adaptive changes in energy preservation were evident at Complex 5, where the ATP synthase ε-subunit Atp5f1e was downregulated (x = −2.22) in the ischemic hearts and then normalized after therapy. This not only agrees with but also now quantifies the prior enzymology-based findings [75,76]. Our results about the downregulation of Cx 5 contradict the report of its upregulation found by another group [77] through meta-analysis of 13 patients with heart failure subjected to heart transplantation compared to 10 healthy counterparts. However, Cpt2 (Carnitine palmitoyltransferase 2), not affected by either CCC or IHF, is downregulated in treated IHF, which, according to some research [78], might result in rapamycin-resistant, acetylation-independent hypertrophy.
By comparing Figure 2 and Figure 3, one observes that the cell treatment triggered marked recovery of the expression levels of almost all mitochondrial genes in both CCC and IHF mice. The recovery happens in all five oxidative phosphorylation complexes. While the commonly decreased expression of Ndufs1 in the infarcted heart [79] is restored to the normal level by the cell treatment in our model, it remains (even less but still) downregulated in the treated CCC mice (from x(IN) = −3.02 to x(IT) = −2.20). However, the downregulation of Ndufs4 in T. cruzi-infected mice was reversed to upregulation by the cell treatment, confirming the cardioprotection role of its upregulation [80].
A very important part of this study was devoted to the control of the transcripts’ abundances by the cellular homeostatic mechanisms and its modification during the disease development and following the treatment. Among the most controlled genes (Table 2), there were five subunits of NADH:ubiquinone oxidoreductase (i.e., mitochondrial complex I), including Ndufa10, Ndufb10, Ndufb5, Ndufb11, and Ndufaf4. These genes can affect mitochondria function profoundly. Especially Ndufa10, the mitochondrial gene with the largest REC (relative expression control) in healthy mice whose RCS (relative control strength) was markedly reduced in both treated (by 32.80×) and untreated (by 22.70×) CCC, meaning a high expression flexibility to adapt to changeable environmental conditions that might eventually be used to alleviate diabetic cardiomyopathy [81]. Ndufa10 has also been implicated in diabetic cardiomyopathy [81] and ischemic injury [72]. Note also the substantial reduction in the expression control in both treated and untreated CCC mice for other stably expressed mitochondrial genes in the healthy mice: Cox7b and Ndufb10. To our best knowledge, this is the first report revealing the significant alteration of the expression control of these genes in Chagasic hearts. Ndufb10 observed destabilization agrees with its known role in holoenzyme assembly failure [82].
The high RCS of Tmem186 in the entire normal transcriptome was significantly reduced (RCS-FC = −39.13) in untreated IHF that persisted (RCS-FC = −22.50) after treatment, but the role of this gene is much less understood. Tmem186 is a component of the mitochondrial Complex 1 intermediate assembly [58]. In contrast, IHF increased by 36.56× the control strength of the antigen Cd164, but the cell treatment restores the normal transcription control.
There are several other interesting data in Table 2. For instance, while the RCS of Vdac2, whose downregulation is one of the main responsible factors for heart failure [83], increased by 2.89× in the not-treated CCC mice (IN) with respect to the healthy animals (CN), it decreased in treated mice (IT) by 3.23×. The control of Cox6b1, known to relieve hypoxia/reoxygenation injury [83] increased by 8.31× in the untreated IHF but decreased by 2.48 in treated IHF. The situation is opposite for Ndufaf4, whose expression control decreased in untreated CCC but increased in treated CCC. We found substantial changes in ischemic mice, with some genes exhibiting more expression control (e.g., Atp5f1e, Cox6b1). The control of Atp13a2 decreased in untreated IHF to considerably increase (by 14.82×) in treated IHF.
All these differences indicate that the homeostatic control mechanisms to keep the expression levels within certain intervals are neither uniform among genes nor similarly altered by each of the two cardiomyopathies nor restored following the same therapeutic approach. A very important observation comparing data from Table 2 with Figure 2 and Figure 3 is that, even though the cell treatment restored the normal expression of several genes, their expression control was not. For instance, in the ischemic mice, the expression of the significantly downregulated Cox6b1 (x(IN) = −2.87) was restored (x(IT) = 1.1) by the treatment, but its RCS was decreased by 2.48×, while the recovered downregulation of Atp5f1e (x(IN) = −2.22) was accompanied by an increase in the RCS by 2.77×). This observation indicates that whereas the treatment recovered the expression profile of most mitochondrial genes, it did not restore the initial control of the transcripts’ abundances.
Recent work in acute experimental models of Chagas disease showed that an imbalance between ROS and NO creates a strongly proarrhythmic substrate independent of major structural remodeling and directly links mitochondrial redox output to electrical instability [2,84]. The same study demonstrates that NO inhibition reverses early after-depolarizations and action potential alternans, underscoring redox–calcium crosstalk as a therapeutic lever. Interestingly, the parasite developed a system of antioxidant enzymes by which to resist the high ROS concentration in the infected cardiomyocyte [85]. Our findings of Complexes 1–5 suppression and Vdac1 dysregulation align with this mechanism and suggest that transcriptomic loss of the ETC (electron transfer chain) control may be an upstream driver of the ROS/NO disequilibrium [86]. There is no contradiction between our finding of downregulation of Slc25a5, one of the genes responsible for pumping in Ca2+, and recent reports of mitochondrial Ca2+ overload during the acute phase of the myocardial ischemia-reperfusion [87] because in the post-ischemic failing heart (our case), mitochondrial Ca2+ content is lower [88].
Additional analysis highlighted that mitochondrial injury is shaped by both inflammatory environment and genotype [89]. This supports our observation that nuclear-encoded ETC subunits (Ndufb11, Cox7b) are vulnerable nodes that could synergize with inflammatory signaling to depress ATP output. It also strengthens the rationale to stratify future cohorts by mitochondrial genotype and inflammatory profile when validating control metrics [81,90]. There was evidence for adipose tissue as a chronic reservoir for Trypanosoma cruzi and a source of adipokine-associated mitochondrial stress, implying that systemic metabolic inputs can burden cardiac bioenergetics [86]. This concept aligns with our Cpt2 signal finding that reduced fatty acid oxidation capacity in the heart may be compounded by parasite-driven adipose dysfunction and argues for integrating peripheral metabolic tissues into future mechanistic and therapeutic designs [91]. Moreover, the downregulation of the two pyruvate dehydrogenase modules (PDK and PDH) generating lipotoxicity by reducing the oxidation of the fatty acids [92] in both untreated cardiomyopathies was corrected by the cell treatment (Figure 3).
Figure 5 presents for the first time to our knowledge the transcriptomic networks that couple the respiratory chain complexes. It also shows how infection with Trypanosoma cruzi and ischemia alter the transcriptomic coupling, indicating profound remodeling of the oxidative phosphorylation functional pathway. The most dramatic alteration occurred between Cx1 and Cx2 in the IHF mice, where the coordination degree decreased from +16.25% to −2.50%, meaning a significant decoupling of the paired genes between the two complexes. The marked coupling decrease between the adjacent complexes is an additional explanation of the mitochondrial failure in the two investigated cardiomyopathies.
Understanding the importance of some of these expression correlations for normal heart function and what pathological consequences their alterations bring might open a very interesting and challenging field of research. For instance, Chagas disease turned the significant synergism of the downregulated Nfdufa6 and Sdha, whose simultaneous upregulation is an indication of acute myeloid leukemia [93], into statistically significant independence. We found that the pairs Uqcrb and Coxcb, and Atp5pf and Cox5b, reported as simultaneously upregulated in paradoxical low transvalvular flow and low gradient patients who develop advanced heart failure symptoms [94], are synergistically expressed in normal conditions. The synergism of Sdhb and Uqcrffs1 downregulated by ischemia supports the efforts to improve the diagnosis and gene-targeted therapies of Alzheimer’s disease [95].
It will be very interesting (and the object of a future study of our group) to investigate the interaction between the oxidative phosphorylation pathway and the mTOR pathway to understand their implication in the observed mitophagy in T. cruzi-infected cardiomyocytes [96]. It will also be of interest to study the interplay between the oxidative phosphorylation pathway and the PINK1-PRKN-mediated mitophagy [97]. Interesting future research should also investigate how Chagas disease and myocardial infarction affect the mitochondrial network plasticity [98].
As illustrated in Figure 6c, cell therapy recovered part of the decrease in the four coordination degrees in the IHF mice and even increased them over the original levels in the CCC mice. In the CCC mice, the cell therapy not only restored the normal expressions of the downregulated Ndufv1 and Sdhb (also downregulated in diabetic patients caused by a high-fat diet [99]) but also made significant their synergistic expression. To prove this assertion, one can use an online available calculator [100] to determine the p-values of the statistical significance of the expression correlations in each of the three conditions: COR(CN) = 0.832 (p-value = 0.168), COR(IN) = 0.397 (p-value = 0.603), and COR(IT) = 0.996 (p-value = 0.004). Interestingly, Ndufa13 and Sdhd, which are independently expressed in healthy mice housed in normal atmospheric conditions, maintained their independence in untreated IHF but became synergistically expressed in both treated CCC and IHF mice. The treatment turned the practically neutral correlations: COR(CN) = 0.320 (p-value = 0.680) and COR(IN) = 0.222 (0.778) between the downregulated Cox17 and Atp5mc1 in ischemic mice into a significant antagonistic expression: COR(IT) = −0.952 (p-value = 0.048). However, together with NDUFB1, COX17, and ATP5MC1, they were reported as upregulated in patients with Parkinson’s, Alzheimer’s, and Huntington’s diseases [101].
Nevertheless, we must recognize that the correlation analysis cannot determine which gene of the pair is the master, i.e., whose gene expression level commands the expression level of the other, nor about the energy coupling [102] and the electric charge transfer within or between the respiratory complexes [103,104,105].
The prominence analysis ranked the mitochondrial genes according to their importance for the functioning of the cell powerhouse. From Table 3 one may learn the following:
- (i)
- Gene hierarchy is altered in cardiomyopathy, as revealed by the differences between the top five genes in healthy mice compared to the sets of five in the other heart conditions.
- (ii)
- Each type of cardiomyopathy induces distinct alteration of the gene hierarchy (see GCH differences between CCC and IHF for the most prominent genes in healthy mice).
- (iii)
- Restoration of the normal expression level is not accompanied by the reinstatement of the genes in their right hierarchy, indicated by the non-unit GCH-FC. For instance, Uqcrh, found by us as downregulated in both untreated CCC and IHF mice, was reported as upregulated in patients with hypertrophic cardiomyopathy [106]. Cell treatment restored the normal expression in the CCC but not in the IHF mice. However, the restored expression level is not followed by the full restoration of its GCH after the treatment. Instead, its RCS became 6.67× stronger than in the normal condition (even stronger than in the untreated CCC, where it was 4.66×).
- (iv)
- Cell treatment has different effects on the two types of heart afflictions. For instance, the somehow low GCH of Cox6b1 healthy mice that was considerably raised in both untreated cardiomyopathies (GCH-FC(IN-CCC) = 4.81, GCH-FC(IN-IHF) = 5.53) is raised even more in treated CCC (GCH-FC) = 7.34) but downgraded in treated IHF (GCH-FC = −2.10).
In previous publications, we have shown that the most prominent gene of a condition might be the most effective target for the gene therapy of that condition [50,107]. Therefore, this analysis may open new therapeutic avenues for both ischemic failure and Chagas disease.
There are several reports from our former collaborators about the beneficial effects of stem cell therapy on the same mouse model of Chagas disease (e.g., [108,109]) in recovering the normal cardiac anatomy and functions that correlate with recovery of the gene expression profile. Also, our previous report on transcriptomic recovery of cell-treated IHF mice [31] mentioned that after 25 days of therapy, the mice demonstrated absence of the pathological Q wave in the electrocardiogram, improved systolic performance, and much less ventricular dilatation. However, a quantitative gene expression study like the present one cannot explain the complex interaction between the damaged cardiomyocytes and the stem cells [110,111] or the molecular mechanisms involved in the rescue of the mitochondrial gene expression profile.
5. Conclusions and Future Directions
Taken together, the findings of this study establish an exhaustive profile of mitochondrial collapse in Chagas disease and post-ischemic heart failure. This investigation demonstrated that targeted cell therapy can partially restore the functional integrity of key ETC components. It further confirmed several established mechanisms and introduced new signals. Their broad rescue with cell therapy underscores mitochondria as a convergent, tractable target for cardiac repair.
Heart malfunction affects all organs, brain included, and myocardial infarction is often followed by depression and other mental disorders [32]. Therefore, one major task of clinical management of the post-infarction mental problems is the restoration of the normal cardiac function. As such, cell treatment might be a solution, and clinical trials are underway [112,113].
Interestingly, although cell treatment recovers most of the normal gene expression profile, it does not restore either the normal expression control or the inter-complex transcriptomic coupling. Moreover, both treated cardiomyopathies present different expression control and correlation patterns. Since the treated animals displayed almost normal electrophysiological parameters and excitatory and contractility properties of the ventricular myocardium, our results suggest that a pathophysiological state is compatible with several transcriptomic topologies. The transcriptomic topology was mathematically defined by us ([114], Appendix A) as the weighted superposition of virtual transcriptomes where the expressions of the genes are controlled and correlated in groups of two, three, four, …. Given the stochastic nature of the gene transcription, transcript abundances can fluctuate within intervals subject to relative control strength. Therefore, each pathophysiological condition might be associated with a family of transcriptomic topologies, each defined by a distinct configuration function. This conclusion points to the need of defining a kind of “transcriptomic entropy of a state” that might be useful to assess the “transcriptomic health” of that state.
Author Contributions
Conceptualization, D.A.I., L.X., and D.D.; methodology, S.I. and D.A.I.; software, D.A.I.; validation, S.I. and D.A.I.; formal analysis, D.A.I.; investigation, S.I. and D.A.I.; resources, D.A.I.; data curation, L.X.; writing—original draft preparation, D.A.I. and L.X.; writing—review and editing, S.M., L.X., and D.D.; visualization, D.A.I.; supervision, D.A.I. and L.X.; project administration, D.A.I.; funding acquisition, D.A.I. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Microarray protocol and raw data can be found in the databases https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE17363, https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE18703, https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE24088, https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE29769 (accessed on 15 September 2025).
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Hinton, A., Jr.; Claypool, S.M.; Neikirk, K.; Senoo, N.; Wanjalla, C.N.; Kirabo, A.; Williams, C.R. Mitochondrial Structure and Function in Human Heart Failure. Circ. Res. 2024, 135, 372–396. [Google Scholar] [CrossRef] [PubMed]
- Murphy, E.; Ardehali, H.; Balaban, R.S.; DiLisa, F.; Dorn, G.W., 2nd; Kitsis, R.N.; Otsu, K.; Ping, P.; Rizzuto, R.; Sack, M.N.; et al. Mitochondrial Function, Biology, and Role in Disease: A Scientific Statement from the American Heart Association. Circ. Res. 2016, 118, 1960–1991. [Google Scholar] [CrossRef]
- Jia, D.; Hou, L.; Lv, Y.; Xi, L.; Tian, Z. Postinfarction exercise training alleviates cardiac dysfunction and adverse remodeling via mitochondrial biogenesis and SIRT1/PGC-1alpha/PI3K/Akt signaling. J. Cell Physiol. 2019, 234, 23705–23718. [Google Scholar] [CrossRef]
- Koka, S.; Aluri, H.S.; Xi, L.; Lesnefsky, E.J.; Kukreja, R.C. Chronic inhibition of phosphodiesterase 5 with tadalafil attenuates mitochondrial dysfunction in type 2 diabetic hearts: Potential role of NO/SIRT1/PGC-1alpha signaling. Am. J. Physiol. Heart Circ. Physiol. 2014, 306, H1558–H1568. [Google Scholar] [CrossRef]
- Nunes, J.P.S.; Roda, V.M.P.; Andrieux, P.; Kalil, J.; Chevillard, C.; Cunha-Neto, E. Inflammation and mitochondria in the pathogenesis of chronic Chagas disease cardiomyopathy. Exp. Biol. Med. 2023, 248, 2062–2071. [Google Scholar] [CrossRef]
- Zhu, S.G.; Kukreja, R.C.; Das, A.; Chen, Q.; Lesnefsky, E.J.; Xi, L. Dietary nitrate supplementation protects against Doxorubicin-induced cardiomyopathy by improving mitochondrial function. J. Am. Coll. Cardiol. 2011, 57, 2181–2189. [Google Scholar] [CrossRef] [PubMed]
- Schaub, G.A. Trypanosoma cruzi/Triatomine Interactions—A Review. Pathogens 2025, 14, 392. [Google Scholar] [CrossRef]
- Ouali, R.; Bousbata, S. Rhodnius prolixus (kissing bug). Trends Parasitol. 2025, 41, 1064–1065. [Google Scholar] [CrossRef]
- Mukherjee, S.; Belbin, T.J.; Spray, D.C.; Iacobas, D.A.; Weiss, L.M.; Kitsis, R.N.; Wittner, M.; Jelicks, L.A.; Scherer, P.E.; Ding, A.; et al. Microarray analysis of changes in gene expression in a murine model of chronic chagasic cardiomyopathy. Parasitol. Res. 2003, 91, 187–196. [Google Scholar] [CrossRef] [PubMed]
- Lopez, M.; Tanowitz, H.B.; Garg, N.J. Pathogenesis of Chronic Chagas Disease: Macrophages, Mitochondria, and Oxidative Stress. Curr. Clin. Microbiol. Rep. 2018, 5, 45–54. [Google Scholar] [CrossRef]
- Hernandez, S.; Srikanth, K.K.; Bommireddi, A.; Leong, T.K.; Miller, D.A.; Ambrosy, A.P.; Zaroff, J. Chagas Disease in Northern California: Observed Prevalence, Clinical Characteristics, and Outcomes Within an Integrated Health Care Delivery System. Perm. J. 2025, 29, 40–48. [Google Scholar] [CrossRef] [PubMed]
- da Silva, J.C.; da Silva, M.T.S.; Bezerra Dos Santos, L.; Abdala, M.G.G.; Lopes, A.B.O.; de Oliveira, G.A.; Guedes-da-Silva, F.H.; Rigoni, T.D.S.; Damasceno, F.S.; Barros-Neto, J.A.; et al. Epidemiological assessment of the first year of mandatory notification of chronic Chagas disease in Alagoas, Northeast Brazil. Acta Trop. 2025, 270, 107805. [Google Scholar] [CrossRef]
- Beatty, N.L.; Hamer, G.L.; Moreno-Peniche, B.; Mayes, B.; Hamer, S.A. Chagas Disease, an Endemic Disease in the United States. Emerg. Infect. Dis. 2025, 31, 1691–1697. [Google Scholar] [CrossRef]
- Bunkofske, M.E.; Sanchez-Valdez, F.J.; Tarleton, R.L. The importance of persistence and dormancy in Trypanosoma cruzi infection and Chagas disease. Curr. Opin. Microbiol. 2025, 86, 102615. [Google Scholar] [CrossRef]
- Telleria, J.; Costales, J.A. An Overview of Trypanosoma cruzi Biology Through the Lens of Proteomics: A Review. Pathogens 2025, 14, 337. [Google Scholar] [CrossRef]
- Garg, N.; Popov, V.L.; Papaconstantinou, J. Profiling gene transcription reveals a deficiency of mitochondrial oxidative phosphorylation in Trypanosoma cruzi-infected murine hearts: Implications in chagasic myocarditis development. Biochim. Biophys. Acta 2003, 1638, 106–120. [Google Scholar] [CrossRef] [PubMed]
- Báez, A.; Lo Presti, M.S.; Rivarola, H.W.; Mentesana, G.G.; Pons, P.; Fretes, R.; Paglini-Oliva, P. Mitochondrial involvement in chronic chagasic cardiomyopathy. Trans. R. Soc. Trop. Med. Hyg. 2011, 105, 239–246. [Google Scholar] [CrossRef] [PubMed]
- Serrano-Coll, H. Chagas disease: Host responses, parasite evasion and vaccine advances. Trans. R. Soc. Trop. Med. Hyg. 2025, traf109. [Google Scholar] [CrossRef] [PubMed]
- Costa, C.J.D.N.; da Silva, P.S.; Saraiva, R.M.; Sangenis, L.H.C.; de Holanda, M.T.; Sperandio da Silva, G.M.; Mendes, F.S.N.S.; Xavier, I.G.G.; Costa, H.S.; Gonçalves, T.R.; et al. Food insecurity is associated with decreased quality of life in patients with chronic Chagas disease. PLoS ONE 2025, 20, e0328466. [Google Scholar] [CrossRef]
- Echeverría, L.E.; Serrano-García, A.Y.; Rojas, L.Z.; Silva-Sieger, F.; Navarro, M.; Aguilera, L.; Gómez-Ochoa, S.A.; Morillo, C.A. Beyond cardiac embolism and cryptogenic stroke: Unveiling the mechanisms of cerebrovascular events in Chagas disease. Lancet Reg. Health Am. 2025, 50, 101203. [Google Scholar] [CrossRef]
- Souza-Silva, T.G.; Figueiredo, A.; Morais, K.L.P.; Apostólico, J.; Pantaleao, A.; Mutarelli, A.; Araújo, S.S.; Nunes, M.D.C.P.; Gollob, K.J.; Dutra, W.O. Single-cell targeted transcriptomics reveals subset-specific immune signatures differentiating asymptomatic and cardiac patients with chronic Chagas disease. J. Infect. Dis. 2025, jiaf269. [Google Scholar] [CrossRef] [PubMed]
- Duque, C.; So, J.; Castro-Sesquen, Y.E.; DeToy, K.; Gutierrez Guarnizo, S.A.; Jahanbakhsh, F.; Malaga Machaca, E.; Miranda-Schaeubinger, M.; Chakravarti, I.; Cooper, V.; et al. Immunologic changes in the peripheral blood transcriptome of individuals with early-stage chronic Chagas cardiomyopathy: A cross-sectional study. Lancet Reg. Health Am. 2025, 45, 101090. [Google Scholar] [CrossRef] [PubMed]
- Saha, T.; Soliman-Aboumarie, H. Review of Current Management of Myocardial Infarction. J. Clin. Med. 2025, 14, 6241. [Google Scholar] [CrossRef]
- Mueller, C.; White, H.D.; Lopez-Ayala, P.; de Silva, R.; Kaski, J.C. Great debate: The universal definition of myocardial infarction is flawed and should be put to rest. Eur. Heart J. 2025, ehaf641. [Google Scholar] [CrossRef]
- Taggart, C.; Ferry, A.V.; Chapman, A.R.; Schulberg, S.D.; Bularga, A.; Wereski, R.; Boeddinghaus, J.; Kimenai, D.M.; Lowry, M.T.H.; Chew, D.P.; et al. The assessment and management of patients with type 2 myocardial infarction: An international Delphi study. Eur. Heart J. Qual. Care Clin. Outcomes 2025, qcaf069. [Google Scholar] [CrossRef]
- Sun, S.; Zhang, Z.; Li, Y.; Zhang, H.; Guo, H.; Chen, G.; Wei, P.; Lin, F.; Zhao, G. Mitochondrial dysfunction as a central hub linking Na+/Ca2+ homeostasis and inflammation in ischemic arrhythmias: Therapeutic implications. Front. Cardiovasc. Med. 2025, 12, 1506501. [Google Scholar] [CrossRef]
- Meco, M.; Giustiniano, E.; Nisi, F.; Zulli, P.; Agosteo, E. MAPK, PI3K/Akt Pathways, and GSK-3β Activity in Severe Acute Heart Failure in Intensive Care Patients: An Updated Review. J. Cardiovasc. Dev. Dis. 2025, 12, 266. [Google Scholar] [CrossRef]
- Yang, Q.; Ji, H.; Modarresi Chahardehi, A. JAK/STAT pathway in myocardial infarction: Crossroads of immune signaling and cardiac remodeling. Mol. Immunol. 2025, 186, 206–217. [Google Scholar] [CrossRef]
- Deng, G.; Yang, Y.; Qing, O.; Linhui, J.; Haotao, S.; Liu, C.; Li, G.; Nasser, M.I. Chrysin Attenuates Myocardial Cell Apoptosis in Mice. Cardiovasc. Toxicol. 2025, 25, 1791–1806. [Google Scholar] [CrossRef]
- Lachtermacher, S.; Esporcatte, B.L.; Montalvao, F.; Costa, P.C.; Rodrigues, D.C.; Belem, L.; Rabischoffisky, A.; Faria Neto, H.C.; Vasconcellos, R.; Iacobas, S.; et al. Cardiac gene expression and systemic cytokine profile are complementary in a murine model of post-ischemic heart failure. Braz. J. Med. Biol. Res. 2010, 43, 377–389. [Google Scholar] [CrossRef] [PubMed]
- Lachtermacher, S.; Esporcatte, B.L.; da Silva de Azevedo Fortes, F.; Rocha, N.N.; Montalvao, F.; Costa, P.C.; Belem, L.; Rabischoffisky, A.; Faria Neto, H.C.; Vasconcellos, R.; et al. Functional and transcriptomic recovery of infarcted mouse myocardium treated with bone marrow mononuclear cells. Stem Cell Rev. Rep. 2012, 8, 251–261. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Patel, P.; Yang, F.; Iacobas, D.A.; Xi, L. Mental disorders after myocardial infarction: Potential mediator role for chemokines in heart-brain interaction? J. Geriatr. Cardiol. 2024, 21, 913–926. [Google Scholar] [CrossRef] [PubMed]
- Soares, M.B.; de Lima, R.S.; Rocha, L.L.; Vasconcelos, J.F.; Rogatto, S.R.; dos Santos, R.R.; Iacobas, S.; Goldenberg, R.C.; Iacobas, D.A.; Tanowitz, H.B.; et al. Gene expression changes associated with myocarditis and fibrosis in hearts of mice with chronic chagasic cardiomyopathy. J. Infect. Dis. 2010, 202, 416–426. [Google Scholar] [CrossRef]
- Goldenberg, R.C.; Iacobas, D.A.; Iacobas, S.; Rocha, L.L.; da Silva de Azevedo Fortes, F.; Vairo, L.; Nagajyothi, F.; Campos de Carvalho, A.C.; Tanowitz, H.B.; Spray, D.C. Transcriptomic alterations in Trypanosoma cruzi-infected cardiac myocytes. Microbes Infect. 2009, 11, 1140–1149. [Google Scholar] [CrossRef]
- Adesse, D.; Goldenberg, R.C.; Fortes, F.S.; Jasmin; Iacobas, D.A.; Iacobas, S.; Campos de Carvalho, A.C.; de Narareth Meirelles, M.; Huang, H.; Soares, M.B.; et al. Gap junctions and chagas disease. Adv. Parasitol. 2011, 76, 63–81. [Google Scholar] [CrossRef]
- Xinxin, Z.; Pan, H.; Qiao, L. Research progress of connexin 43 in cardiovascular diseases. Front. Cardiovasc. Med. 2025, 12, 1650548. [Google Scholar] [CrossRef]
- Caetano-da-Silva, J.E.; Gonçalves-Santos, E.; Domingues, E.L.B.C.; Caldas, I.S.; Lima, G.D.A.; Diniz, L.F.; Gonçalves, R.V.; Novaes, R.D. The mitochondrial uncoupler 2,4-dinitrophenol modulates inflammatory and oxidative responses in Trypanosoma cruzi-induced acute myocarditis in mice. Cardiovasc. Pathol. 2024, 72, 107653. [Google Scholar] [CrossRef]
- Nisimura, L.M.; Coelho, L.L.; de Melo, T.G.; Vieira, P.C.; Victorino, P.H.; Garzoni, L.R.; Spray, D.C.; Iacobas, D.A.; Iacobas, S.; Tanowitz, H.B.; et al. Trypanosoma cruzi Promotes Transcriptomic Remodeling of the JAK/STAT Signaling and Cell Cycle Pathways in Myoblasts. Front. Cell Infect. Microbiol. 2020, 10, 255. [Google Scholar] [CrossRef]
- Narimani, S.; Rahbarghazi, R.; Salehipourmehr, H.; Taghavi Narmi, M.; Lotfimehr, H.; Mehdipour, R. Therapeutic Potential of Endothelial Progenitor Cells in Angiogenesis and Cardiac Regeneration: A Systematic Review and Meta-Analysis of Rodent Models. Adv Pharm Bull. 2025, 15, 268–283. [Google Scholar] [CrossRef]
- Soares, M.B.; Lima, R.S.; Souza, B.S.; Vasconcelos, J.F.; Rocha, L.L.; Dos Santos, R.R.; Iacobas, S.; Goldenberg, R.C.; Lisanti, M.P.; Iacobas, D.A.; et al. Reversion of gene expression alterations in hearts of mice with chronic chagasic cardiomyopathy after transplantation of bone marrow cells. Cell Cycle 2011, 10, 1448–1455. [Google Scholar] [CrossRef] [PubMed]
- Iacobas, D.A.; Iacobas, S.; Tanowitz, H.B.; Campos de Carvalho, A.; Spray, D.C. Functional genomic fabrics are remodeled in a mouse model of Chagasic cardiomyopathy and restored following cell therapy. Microbes Infect. 2018, 20, 185–195. [Google Scholar] [CrossRef]
- Oxidative Phosphorylation. Available online: https://www.kegg.jp/pathway/mmu00190 (accessed on 1 September 2025).
- Diabetic Cardiomyopathy. Available online: https://www.kegg.jp/pathway/mmu05415 (accessed on 1 September 2025).
- Genomic Data: Gene Expression Changes Associated with Myocarditis and Fibrosis in Hearts of Mice with Chronic Chagasic Cardiomyopathy. Available online: https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE17363 (accessed on 1 September 2025).
- Genomic Data: Cardiac Gene Expression and Systemic Cytokine Profile Are Complementary in a Murine Model of Post Ischemic Heart Failure. Available online: https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE18703 (accessed on 1 September 2025).
- Genomic Data: Therapy with Bone Marrow Cells Recovers Gene Expression Alterations in Hearts of Mice with Chronic Chagasic Cardiomyopathy. Available online: https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE24088 (accessed on 1 September 2025).
- Genomic Data: Functional and Transcriptomic Recovery of Infarcted Mouse Myocardium Treated with Bone Marrow Mononuclear Cells. Available online: https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE29769 (accessed on 1 September 2025).
- Adesse, D.; Iacobas, D.A.; Iacobas, S.; Garzoni, L.R.; Meirelles, M.d.N.; Tanowitz, H.B.; Spray, D.C. Transcriptomic signatures of alterations in a myoblast cell line infected with four distinct strains of Trypanosoma cruzi. Am. J. Trop. Med. Hyg. 2010, 82, 846–854. [Google Scholar] [CrossRef]
- Iacobas, D.A.; Iacobas, S.; Lee, P.R.; Cohen, J.E.; Fields, R.D. Coordinated Activity of Transcriptional Networks Responding to the Pattern of Action Potential Firing in Neurons. Genes 2019, 10, 754. [Google Scholar] [CrossRef] [PubMed]
- Iacobas, D.A. Biomarkers, Master Regulators and Genomic Fabric Remodeling in a Case of Papillary Thyroid Carcinoma. Genes 2020, 11, 1030. [Google Scholar] [CrossRef]
- Iacobas, D.A.; Xi, L. Theory and Applications of the (Cardio) Genomic Fabric Approach to Post-Ischemic and Hypoxia-Induced Heart Failure. J. Pers. Med. 2022, 12, 1246. [Google Scholar] [CrossRef]
- Parker, A.M.; Lees, J.G.; Murray, A.J.; Velagic, A.; Lim, S.Y.; De Blasio, M.J.; Ritchie, R.H. Precision Medicine: Therapeutically Targeting Mitochondrial Alterations in Heart Failure. JACC Basic Transl. Sci. 2025, 10, 101345. [Google Scholar] [CrossRef]
- Mongelli, A.; Mengozzi, A.; Geiger, M.; Gorica, E.; Mohammed, S.A.; Paneni, F.; Ruschitzka, F.; Costantino, S. Mitochondrial epigenetics in aging and cardiovascular diseases. Front. Cardiovasc. Med. 2023, 10, 1204483. [Google Scholar] [CrossRef]
- Pietrangelo, D.; Lopa, C.; Litterio, M.; Cotugno, M.; Rubattu, S.; Lombardi, A. Metabolic Disturbances Involved in Cardiovascular Diseases: The Role of Mitochondrial Dysfunction, Altered Bioenergetics and Oxidative Stress. Int. J. Mol. Sci. 2025, 26, 6791. [Google Scholar] [CrossRef]
- Dhalla, N.S.; Ostadal, P.; Tappia, P.S. Involvement of Oxidative Stress in Mitochondrial Abnormalities During the Development of Heart Disease. Biomedicines 2025, 13, 1338. [Google Scholar] [CrossRef] [PubMed]
- Mueller, M.; Blandino, A.; Scherer, D.; Zulantay, I.; Apt, W.; Varela, N.M.; Llancaqueo, M.; Garcia, L.; Ortiz, L.; Nicastri, E.; et al. Small-RNA sequencing identifies serum microRNAs associated with abnormal electrocardiography findings in patients with Chagas disease. J. Infect. 2025, 91, 106613. [Google Scholar] [CrossRef] [PubMed]
- Ma, H.; Xu, H. Diagnosis and treatment of post-acute myocardial infarction ventricular aneurysm: A review. Medicine 2025, 8, e43696. [Google Scholar] [CrossRef] [PubMed]
- Formosa, L.E.; Muellner-Wong, L.; Reljic, B.; Sharpe, A.J.; Jackson, T.D.; Beilharz, T.H.; Stojanovski, D.; Lazarou, M.; Stroud, D.A.; Ryan, M.T. Dissecting the Roles of Mitochondrial Complex I Intermediate Assembly Complex Factors in the Biogenesis of Complex I. Cell Rep. 2020, 31, 107541. [Google Scholar] [CrossRef]
- Yu, F.; Zhao, H.; Luo, L.; Wu, W. Nicotinamide Adenine Dinucleotide Supplementation to Alleviate Heart Failure: A Mitochondrial Dysfunction Perspective. Nutrients 2025, 17, 1855. [Google Scholar] [CrossRef] [PubMed]
- Bozdemir, N.; Cakir, C.; Topcu, U.; Uysal, F. A Comprehensive Review of Mitochondrial Complex I During Mammalian Oocyte Maturation. Genesis 2025, 63, e70017. [Google Scholar] [CrossRef] [PubMed]
- Shi, X.; Zhang, Y.; Chen, R.; Gong, Y.; Zhang, M.; Guan, R.; Rotstein, O.D.; Liu, X.; Wen, X.Y. ndufa7 plays a critical role in cardiac hypertrophy. J. Cell Mol. Med. 2020, 24, 13151–13162. [Google Scholar] [CrossRef]
- Wyckelsma, V.L.; Levinger, I.; McKenna, M.J.; Formosa, L.E.; Ryan, M.T.; Petersen, A.C.; Anderson, M.J.; Murphy, R.M. Preservation of skeletal muscle mitochondrial content in older adults: Relationship between mitochondria, fibre type and high-intensity exercise training. J. Physiol. 2017, 595, 3345–3359. [Google Scholar] [CrossRef]
- Meng, C.; Jin, X.; Xia, L.; Shen, S.M.; Wang, X.L.; Cai, J.; Chen, G.Q.; Wang, L.S.; Fang, N.Y. Alterations of mitochondrial enzymes contribute to cardiac hypertrophy before hypertension development in spontaneously hypertensive rats. J. Proteome Res. 2009, 8, 2463–2475. [Google Scholar] [CrossRef]
- Huang, L.; Jin, X.; Xia, L.; Wang, X.; Yu, Y.; Liu, C.; Shao, D.; Fang, N.; Meng, C. Characterization of mitochondrial NADH dehydrogenase 1alpha subcomplex 10 variants in cardiac muscles from normal Wistar rats and spontaneously hypertensive rats: Implications in the pathogenesis of hypertension. Mol. Med. Rep. 2016, 13, 961–966. [Google Scholar] [CrossRef][Green Version]
- Iverson, T.M.; Singh, P.K.; Cecchini, G. An evolving view of complex II-noncanonical complexes, megacomplexes, respiration, signaling, and beyond. J. Biol. Chem. 2023, 299, 104761. [Google Scholar] [CrossRef]
- Lin, S.; Fasham, J.; Al-Hijawi, F.; Qutob, N.; Gunning, A.; Leslie, J.S.; McGavin, L.; Ubeyratna, N.; Baker, W.; Zeid, R.; et al. Consolidating biallelic SDHD variants as a cause of mitochondrial complex II deficiency. Eur. J. Hum. Genet. 2021, 29, 1570–1576. [Google Scholar] [CrossRef]
- Zhang, Y.; Wei, J.; Qiao, L.; Yu, R.; Ren, H.; Zhao, A.; Sun, Y.; Wang, A.; Li, B.; Wang, X.; et al. Exploring the biological basis for the identification of different syndromes in ischemic heart failure based on joint multi-omics analysis. Front. Pharmacol. 2025, 16, 1641422. [Google Scholar] [CrossRef] [PubMed]
- Spielmann, N.; Schenkl, C.; Komlodi, T.; da Silva-Buttkus, P.; Heyne, E.; Rohde, J.; Amarie, O.V.; Rathkolb, B.; Gnaiger, E.; Doenst, T.; et al. Knockout of the Complex III subunit Uqcrh causes bioenergetic impairment and cardiac contractile dysfunction. Mamm. Genome 2023, 34, 229–243. [Google Scholar] [CrossRef]
- Čunátová, K.; Fernández-Vizarra, E. Pathological variants in nuclear genes causing mitochondrial complex III deficiency: An update. J. Inherit. Metab. Dis. 2024, 47, 1278–1291. [Google Scholar] [CrossRef]
- Sinkler, C.A.; Kalpage, H.; Shay, J.; Lee, I.; Malek, M.H.; Grossman, L.I.; Huttemann, M. Tissue- and Condition-Specific Isoforms of Mammalian Cytochrome c Oxidase Subunits: From Function to Human Disease. Oxidative Med. Cell Longev. 2017, 2017, 1534056. [Google Scholar] [CrossRef]
- Wen, J.J.; Garg, N. Oxidative modification of mitochondrial respiratory complexes in response to the stress of Trypanosoma cruzi infection. Free Radic. Biol. Med. 2004, 37, 2072–2081. [Google Scholar] [CrossRef]
- Pandey, R.; Velasquez, S.; Durrani, S.; Jiang, M.; Neiman, M.; Crocker, J.S.; Benoit, J.B.; Rubinstein, J.; Paul, A.; Ahmed, R.P. MicroRNA-1825 induces proliferation of adult cardiomyocytes and promotes cardiac regeneration post-ischemic injury. Am. J. Transl. Res. 2017, 9, 3120–3137. [Google Scholar] [PubMed]
- Elbatarny, M.; Lu, Y.T.; Hu, M.; Coles, J.; Mital, S.; Ross-White, A.; Honjo, O.; Barron, D.J.; Gramolini, A.O. Systems biology approaches investigating mitochondrial dysfunction in cyanotic heart disease: A systematic review. EBioMedicine 2025, 118, 105839. [Google Scholar] [CrossRef]
- Qiu, J.; Gu, Y. Analysis of the prognostic value of mitochondria-related genes in patients with acute myocardial infarction. BMC Cardiovasc. Disord. 2024, 24, 408. [Google Scholar] [CrossRef] [PubMed]
- Hejzlarova, K.; Mracek, T.; Vrbacky, M.; Kaplanova, V.; Karbanova, V.; Nuskova, H.; Pecina, P.; Houstek, J. Nuclear genetic defects of mitochondrial ATP synthase. Physiol. Res. 2014, 63, S57–S71. [Google Scholar] [CrossRef]
- Mayr, J.A.; Havlickova, V.; Zimmermann, F.; Magler, I.; Kaplanova, V.; Jesina, P.; Pecinova, A.; Nuskova, H.; Koch, J.; Sperl, W.; et al. Mitochondrial ATP synthase deficiency due to a mutation in the ATP5E gene for the F1 epsilon subunit. Hum. Mol. Genet. 2010, 19, 3430–3439. [Google Scholar] [CrossRef]
- Pereyra, A.S.; Hasek, L.Y.; Harris, K.L.; Berman, A.G.; Damen, F.W.; Goergen, C.J.; Ellis, J.M. Loss of cardiac carnitine palmitoyltransferase 2 results in rapamycin-resistant, acetylation-independent hypertrophy. J. Biol. Chem. 2017, 292, 18443–18456. [Google Scholar] [CrossRef] [PubMed]
- Giménez-Escamilla, I.; Benedicto, C.; Pérez-Carrillo, L.; Delgado-Arija, M.; González-Torrent, I.; Vilchez, R.; Martínez-Dolz, L.; Portolés, M.; Tarazón, E.; Roselló-Lletí, E. Alterations in Mitochondrial Oxidative Phosphorylation System: Relationship of Complex V and Cardiac Dysfunction in Human Heart Failure. Antioxidants 2024, 13, 285. [Google Scholar] [CrossRef]
- Zhang, Q.; Jin, A.; Cheng, H.; Zheng, Y.; Li, B. PCBP2 alleviates myocardial infarction by inhibiting cardiomyocyte ferroptosis via the NDUFS1/NRF2 pathway. Mol. Immunol. 2025, 186, 13–25. [Google Scholar] [CrossRef] [PubMed]
- Shao, M.; Li, L.; Ma, L.; Song, C.; Li, W.; Zhang, Y.; Cheng, W.; Chen, Y.; Yang, Y.; Wang, Q.; et al. Activation of RXRα exerts cardioprotection through transcriptional upregulation of Ndufs4 in heart failure. Sci. Bull. 2024, 69, 1202–1207. [Google Scholar] [CrossRef]
- Guo, P.; Hu, S.; Liu, X.; He, M.; Li, J.; Ma, T.; Huang, M.; Fang, Q.; Wang, Y. CAV3 alleviates diabetic cardiomyopathy via inhibiting NDUFA10-mediated mitochondrial dysfunction. J. Transl. Med. 2024, 22, 390. [Google Scholar] [CrossRef] [PubMed]
- Friederich, M.W.; Erdogan, A.J.; Coughlin, C.R., 2nd; Elos, M.T.; Jiang, H.; O’Rourke, C.P.; Lovell, M.A.; Wartchow, E.; Gowan, K.; Chatfield, K.C.; et al. Mutations in the accessory subunit NDUFB10 result in isolated complex I deficiency and illustrate the critical role of intermembrane space import for complex I holoenzyme assembly. Hum. Mol. Genet. 2017, 26, 702–716. [Google Scholar] [CrossRef]
- Zhang, W.; Wang, Y.; Wan, J.; Zhang, P.; Pei, F. COX6B1 relieves hypoxia/reoxygenation injury of neonatal rat cardiomyocytes by regulating mitochondrial function. Biotechnol. Lett. 2019, 41, 59–68. [Google Scholar] [CrossRef]
- Nogueira, S.S.; Souza, M.A.; Santos, E.C.; Caldas, I.S.; Gonçalves, R.V.; Novaes, R.D. Oxidative stress, cardiomyocytes premature senescence and contractile dysfunction in in vitro and in vivo experimental models of Chagas disease. Acta Trop. 2023, 244, 106950. [Google Scholar] [CrossRef]
- Gomez, J.; Coll, M.; Guarise, C.; Cifuente, D.; Masone, D.; Tello, P.F.; Piñeyro, M.D.; Robello, C.; Reta, G.; Sosa, M.Á.; et al. New insights into the pro-oxidant mechanism of dehydroleucodine on Trypanosoma cruzi. Sci. Rep. 2024, 14, 18875. [Google Scholar] [CrossRef]
- Santos-Miranda, A.; Joviano-Santos, J.V.; Ribeiro, G.A.; Botelho, A.F.M.; Rocha, P.; Vieira, L.Q.; Cruz, J.S.; Roman-Campos, D. Reactive oxygen species and nitric oxide imbalances lead to in vivo and in vitro arrhythmogenic phenotype in acute phase of experimental Chagas disease. PLoS Pathog. 2020, 16, e1008379, Erratum in PLoS Pathog. 2020, 16, e1009049. [Google Scholar] [CrossRef]
- Paillard, M.; Abdellatif, M.; Andreadou, I.; Bär, C.; Bertrand, L.; Brundel, B.J.J.; Chiva-Blanch, G.; Davidson, S.M.; Dawson, D.; Di Lisa, F.; et al. Mitochondrial targets in ischaemic heart disease and heart failure, and their potential for a more efficient clinical translation. A scientific statement of the ESC Working Group on Cellular Biology of the Heart and the ESC Working Group on Myocardial Function. Eur. J. Heart Fail. 2025, 27, 1720–1736. [Google Scholar] [CrossRef]
- Kohlhaas, M.; Nickel, A.G.; Maack, C. Mitochondrial energetics and calcium coupling in the heart. J. Physiol. 2017, 595, 3753–3763. [Google Scholar] [CrossRef]
- Frade, A.F.; Guerin, H.; Nunes, J.P.S.; Silva, L.; Roda, V.M.P.; Madeira, R.P.; Brochet, P.; Andrieux, P.; Kalil, J.; Chevillard, C.; et al. Cardiac and Digestive Forms of Chagas Disease: An Update on Pathogenesis, Genetics, and Therapeutic Targets. Mediat. Inflamm. 2025, 2025, 8862004. [Google Scholar] [CrossRef]
- Yang, Y.; Li, J.H.; Yao, B.C.; Chen, Q.L.; Jiang, N.; Wang, L.Q.; Guo, Z.G. NDUFB11 and NDUFS3 play a role in atherosclerosis and chronic stress. Aging 2023, 15, 8026–8043. [Google Scholar] [CrossRef]
- Nagajyothi, J.F.; Weiss, L.M. Advances in understanding the role of adipose tissue and mitochondrial oxidative stress in Trypanosoma cruzi infection. F1000Research 2019, 8, 1152. [Google Scholar] [CrossRef] [PubMed]
- Schulze, P.C.; Drosatos, K.; Goldberg, I.J. Lipid Use and Misuse by the Heart. Circ. Res. 2016, 118, 1736–1751. [Google Scholar] [CrossRef]
- Abdel-Aziz, A.K. OXPHOS mediators in acute myeloid leukemia patients: Prognostic biomarkers and therapeutic targets for personalized medicine. World J. Surg. Oncol. 2024, 22, 298. [Google Scholar] [CrossRef] [PubMed]
- Elkenani, M.; Barallobre-Barreiro, J.; Schnelle, M.; Mohamed, B.A.; Beuthner, B.E.; Jacob, C.F.; Paul, N.B.; Yin, X.; Theofilatos, K.; Fischer, A.; et al. Cellular and extracellular proteomic profiling of paradoxical low-flow low-gradient aortic stenosis myocardium. Front. Cardiovasc. Med. 2024, 11, 1398114. [Google Scholar] [CrossRef]
- Chen, F.; Bai, J.; Zhong, S.; Zhang, R.; Zhang, X.; Xu, Y.; Zhao, M.; Zhao, C.; Zhou, Z. Molecular Signatures of Mitochondrial Complexes Involved in Alzheimer’s Disease via Oxidative Phosphorylation and Retrograde Endocannabinoid Signaling Pathways. Oxidative Med. Cell Longev. 2022, 2022, 9565545. [Google Scholar] [CrossRef]
- Alvim, J.M.; Venturini, G.; Oliveira, T.G.M.; Seidman, J.G.; Seidman, C.E.; Krieger, J.E.; Pereira, A.C. mTOR signaling inhibition decreases lysosome migration and impairs the success of Trypanosoma cruzi infection and replication in cardiomyocytes. Acta Trop. 2023, 240, 106845. [Google Scholar] [CrossRef] [PubMed]
- Nàger, M.; Calvoli, M.; Larsen, K.B.; Birgisdottir, A.B. The multifaceted role of autophagy and mitophagy in cardiovascular health and disease. Autophagy Rep. 2025, 4, 2572511. [Google Scholar] [CrossRef]
- Piquereau, J.; Veksler, V.; Novotova, M.; Ventura-Clapier, R. Energetic Interactions Between Subcellular Organelles in Striated Muscles. Front. Cell Dev. Biol. 2020, 8, 581045. [Google Scholar] [CrossRef]
- Sparks, L.M.; Xie, H.; Koza, R.A.; Mynatt, R.; Hulver, M.W.; Bray, G.A.; Smith, S.R. A high-fat diet coordinately downregulates genes required for mitochondrial oxidative phosphorylation in skeletal muscle. Diabetes 2005, 54, 1926–1933. [Google Scholar] [CrossRef]
- Social Science Statistics. Available online: https://www.socscistatistics.com/pvalues/pearsondistribution.aspx (accessed on 1 September 2025).
- Ahmad, B.; Dumbuya, J.S.; Li, W.; Tang, J.X.; Chen, X.; Lu, J. Evaluation of GFM1 mutations pathogenicity through in silico tools, RNA sequencing and mitophagy pahtway in GFM1 knockout cells. Int. J. Biol. Macromol. 2025, 304 Pt 2, 140970, Erratum in Int. J. Biol. Macromol. 2025, 318 Pt 1, 145547. [Google Scholar] [CrossRef] [PubMed]
- Lemeshko, V.V. Mechanism of Na+ ions contribution to the generation and maintenance of a high inner membrane potential in mitochondria. Biochim. Biophys. Acta Bioenerg. 2025, 1867, 149571. [Google Scholar] [CrossRef] [PubMed]
- Fedor, J.G.; Jones, A.J.Y.; Di Luca, A.; Kaila, V.R.I.; Hirst, J. Correlating kinetic and structural data on ubiquinone binding and reduction by respiratory complex I. Proc. Natl. Acad. Sci. USA 2017, 114, 12737–12742. [Google Scholar] [CrossRef]
- Galemou Yoga, E.; Haapanen, O.; Wittig, I.; Siegmund, K.; Sharma, V.; Zickermann, V. Mutations in a conserved loop in the PSST subunit of respiratory complex I affect ubiquinone binding and dynamics. Biochim. Biophys. Acta Bioenerg. 2019, 1860, 573–581. [Google Scholar] [CrossRef]
- Röpke, M.; Saura, P.; Riepl, D.; Pöverlein, M.C.; Kaila, V.R.I. Functional Water Wires Catalyze Long-Range Proton Pumping in the Mammalian Respiratory Complex I. J. Am. Chem. Soc. 2020, 142, 21758–21766. [Google Scholar] [CrossRef]
- Chen, P.; Yawar, W.; Farooqui, A.R.; Ali, S.; Lathiya, N.; Ghous, Z.; Sultan, R.; Alhomrani, M.; Alghamdi, S.A.; Almalki, A.A.; et al. Transcriptomics data integration and analysis to uncover hallmark genes in hypertrophic cardiomyopathy. Am. J. Transl. Res. 2024, 16, 637–653. [Google Scholar] [CrossRef]
- Iacobas, S.; Ede, N.; Iacobas, D.A. The Gene Master Regulators (GMR) Approach Provides Legitimate Targets for Personalized, Time-Sensitive Cancer Gene Therapy. Genes 2019, 10, 560. [Google Scholar] [CrossRef] [PubMed]
- Soares, M.B.; Lima, R.S.; Rocha, L.L.; Takyia, C.M.; Pontes-de-Carvalho, L.; de Carvalho, A.C.; Ribeiro-dos-Santos, R. Transplanted bone marrow cells repair heart tissue and reduce myocarditis in chronic chagasic mice. Am. J. Pathol. 2004, 164, 441–447. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Jasmin; Jelicks, L.A.; Tanowitz, H.B.; Peters, V.M.; Mendez-Otero, R.; de Carvalho, A.C.C.; Spray, D.C. Molecular imaging, biodistribution and efficacy of mesenchymal bone marrow cell therapy in a mouse model of Chagas disease. Microbes Infect. 2014, 16, 923–935. [Google Scholar] [CrossRef]
- Ma, Z.; Yang, H.; Liu, H.; Xu, M.; Runyan, R.B.; Eisenberg, C.A.; Markwald, R.R.; Borg, T.K.; Gao, B.Z. Mesenchymal stem cell-cardiomyocyte interactions under defined contact modes on laser-patterned biochips. PLoS ONE 2013, 8, e56554. [Google Scholar] [CrossRef]
- He, L.; Nguyen, N.B.; Ardehali, R.; Zhou, B. Heart Regeneration by Endogenous Stem Cells and Cardiomyocyte Proliferation: Controversy, Fallacy, and Progress. Circulation 2020, 142, 275–291. [Google Scholar] [CrossRef]
- Mello, D.B.; Mesquita, F.C.P.; Silva dos Santos, D.; Asensi, K.D.; Dias, M.L.; Campos de Carvalho, A.C.; Goldenberg, R.C.d.S.; Kasai-Brunswick, T.H. Mesenchymal Stromal Cell-Based Products: Challenges and Clinical Therapeutic Options. Int. J. Mol. Sci. 2024, 25, 6063. [Google Scholar] [CrossRef]
- Carvalho, A.B.; Kasai-Brunswick, T.H.; Campos de Carvalho, A.C. Advanced cell and gene therapies in cardiology. EBioMedicine 2024, 103, 105125. [Google Scholar] [CrossRef] [PubMed]
- Iacobas, D.A.; Obiomon, E.A.; Iacobas, S. Genomic Fabrics of the Excretory System’s Functional Pathways Remodeled in Clear Cell Renal Cell Carcinoma. Curr. Issues Mol. Biol. 2023, 45, 9471–9499. [Google Scholar] [CrossRef] [PubMed]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).