
Using AlphaFold Predictions in Viral Research


Abstract
1. Introduction
2. Application of AF2 for Research on Eukaryotic Viruses
2.1. Application of AlphaFold for SARS-CoV-2 Research
2.2. Application of AlphaFold to Study Eukaryotic Viruses
3. Application of AlphaFold for Research on Bacteriophages
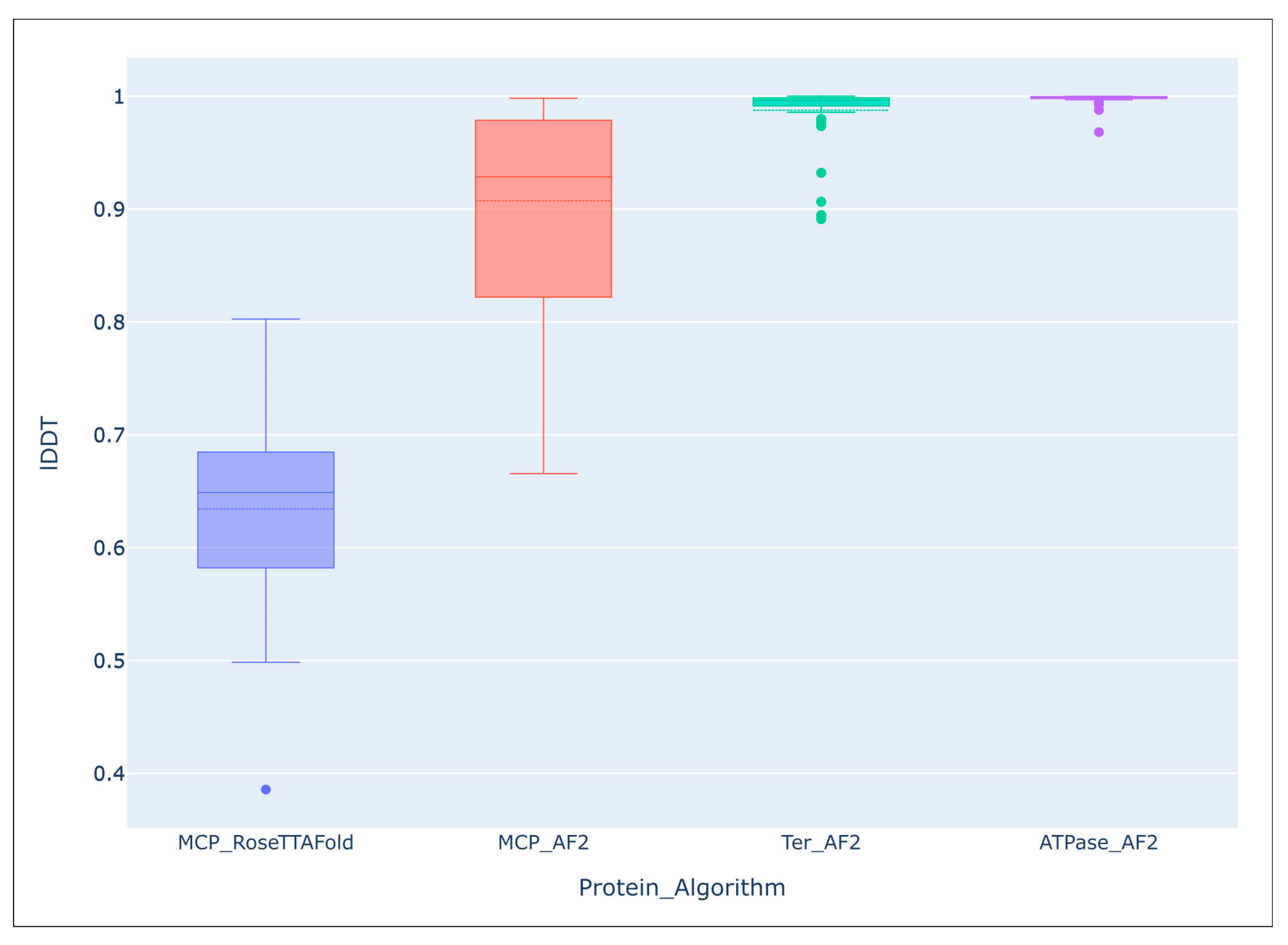
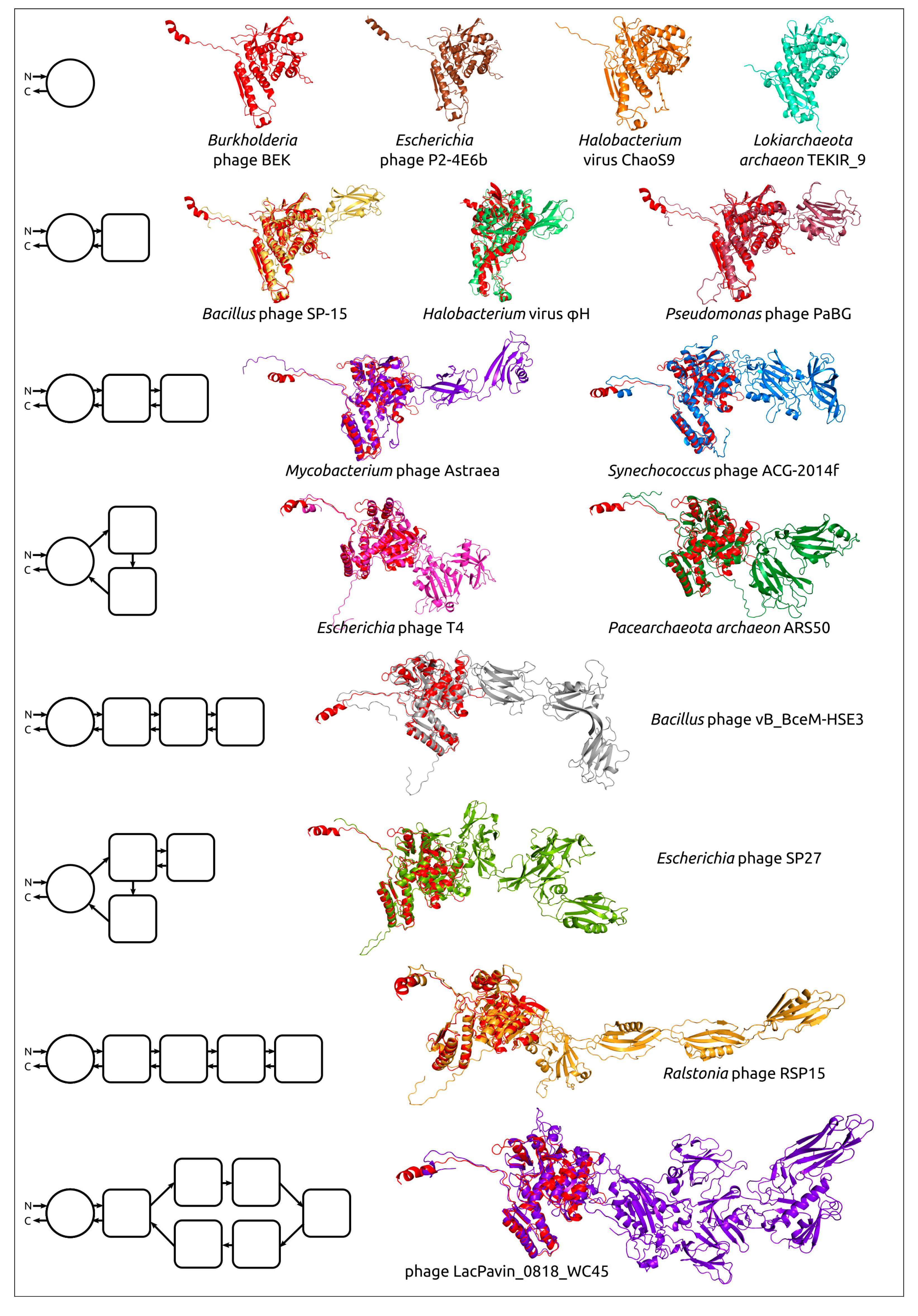
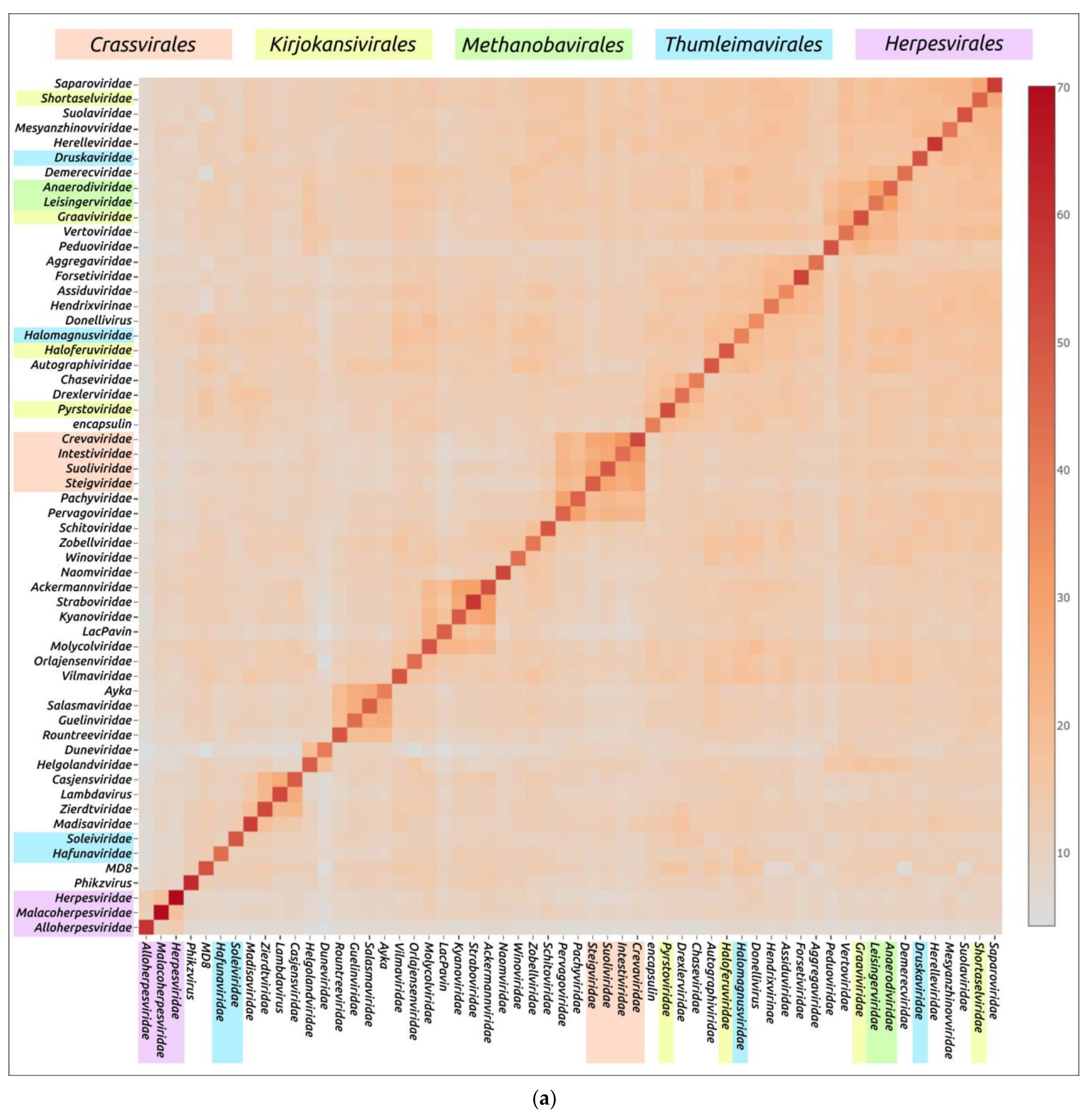
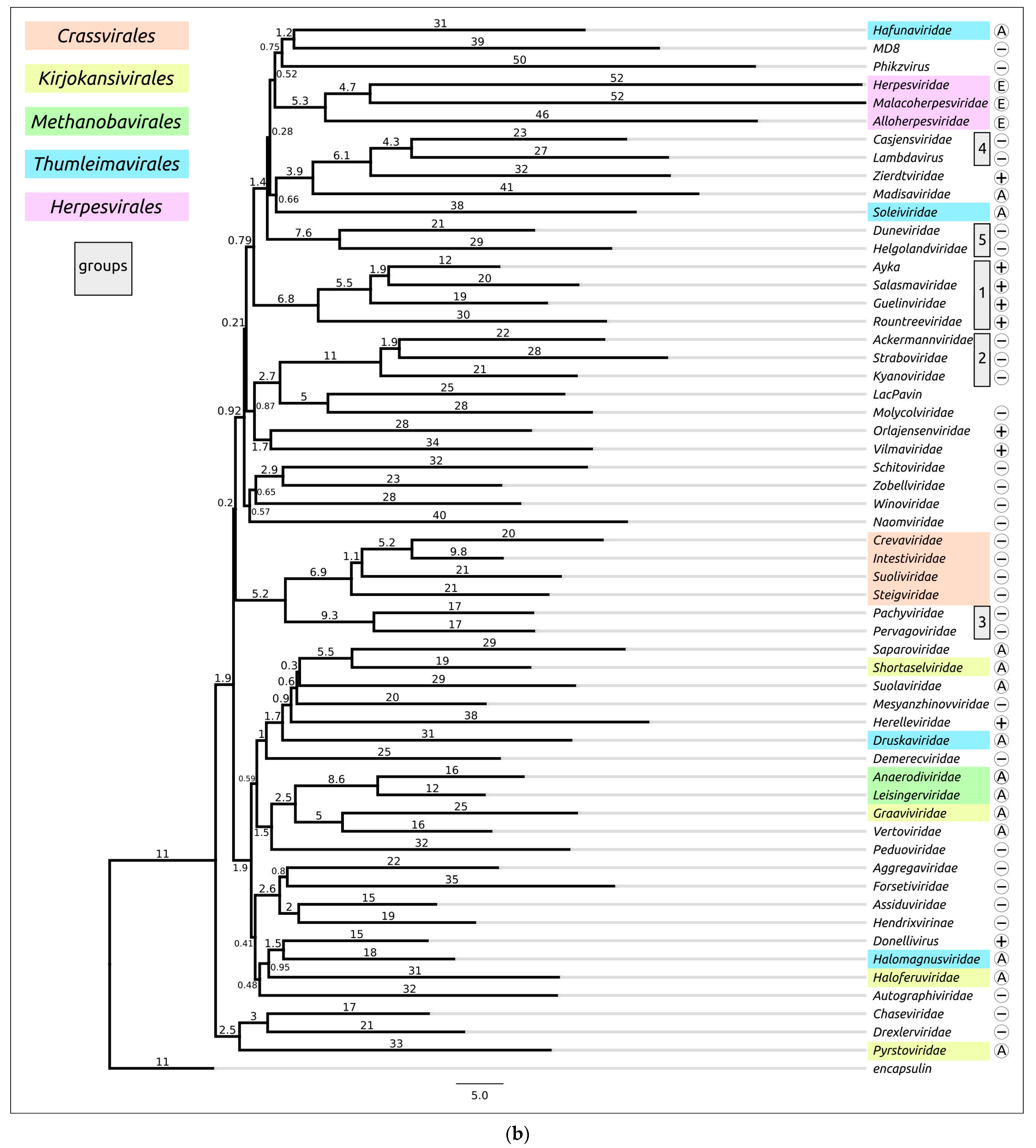
4. Application of AlphaFold for Evolutionary and Taxonomic Studies
5. Further Development of AlphaFold and Machine Learning Techniques
5.1. AlphaFold-Multimer and Prediction of Multi-Chain Protein Complexes
5.2. AlphaFill
6. Critique of AlphaFold
6.1. Intrinsically Disordered Proteins and Intrinsically Disordered Protein Regions
6.2. Protein Interactions with Metal Ions, DNA, RNA, Cofactors, Ligands and Post-Translational Modifications
6.3. Protein Conformations
6.4. Mutations
6.5. Database Loopholes
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Creighton, T.E. Protein Folding. Biochem. J. 1990, 270, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Marcu, Ş.-B.; Tăbîrcă, S.; Tangney, M. An Overview of Alphafold’s Breakthrough. Front. Artif. Intell. 2022, 5, 875587. [Google Scholar] [CrossRef] [PubMed]
- Anfinsen, C.B. The Formation and Stabilization of Protein Structure. Biochem. J. 1972, 128, 737–749. [Google Scholar] [CrossRef] [PubMed]
- Richardson, J.S. The Anatomy and Taxonomy of Protein Structure. In Advances in Protein Chemistry; Anfinsen, C.B., Edsall, J.T., Richards, F.M., Eds.; Academic Press: Cambridge, USA, 1981; Volume 34, pp. 167–339. [Google Scholar]
- Rose, G.D.; Fleming, P.J.; Banavar, J.R.; Maritan, A. A Backbone-Based Theory of Protein Folding. Proc. Natl. Acad. Sci. USA 2006, 103, 16623–16633. [Google Scholar] [CrossRef]
- Janin, J.; Bahadur, R.P.; Chakrabarti, P. Protein–Protein Interaction and Quaternary Structure. Q. Rev. Biophys. 2008, 41, 133–180. [Google Scholar] [CrossRef]
- Xu, C.; Wang, Y.; Liu, C.; Zhang, C.; Han, W.; Hong, X.; Wang, Y.; Hong, Q.; Wang, S.; Zhao, Q.; et al. Conformational Dynamics of SARS-CoV-2 Trimeric Spike Glycoprotein in Complex with Receptor ACE2 Revealed by Cryo-EM. Sci. Adv. 2021, 7, eabe5575. [Google Scholar] [CrossRef]
- Śledź, P.; Caflisch, A. Protein Structure-Based Drug Design: From Docking to Molecular Dynamics. Curr. Opin. Struct. Biol. 2018, 48, 93–102. [Google Scholar] [CrossRef]
- Smyth, M.S.; Martin, J.H.J. X Ray Crystallography. Mol. Pathol. 2000, 53, 8. [Google Scholar] [CrossRef]
- Klukowski, P.; Riek, R.; Güntert, P. Rapid Protein Assignments and Structures from Raw NMR Spectra with the Deep Learning Technique ARTINA. Nat. Commun. 2022, 13, 6151. [Google Scholar] [CrossRef]
- Burley, S.K.; Berman, H.M.; Chiu, W.; Dai, W.; Flatt, J.W.; Hudson, B.P.; Kaelber, J.T.; Khare, S.D.; Kulczyk, A.W.; Lawson, C.L.; et al. Electron Microscopy Holdings of the Protein Data Bank: The Impact of the Resolution Revolution, New Validation Tools, and Implications for the Future. Biophys. Rev. 2022, 14, 1281–1301. [Google Scholar] [CrossRef]
- Agnihotry, S.; Pathak, R.K.; Singh, D.B.; Tiwari, A.; Hussain, I. Chapter 11—Protein Structure Prediction. In Bioinformatics; Singh, D.B., Pathak, R.K., Eds.; Academic Press: Cambridge, USA, 2022; pp. 177–188. ISBN 978-0-323-89775-4. [Google Scholar]
- Kuhlman, B.; Bradley, P. Advances in Protein Structure Prediction and Design. Nat. Rev. Mol. Cell. Biol. 2019, 20, 681–697. [Google Scholar] [CrossRef] [PubMed]
- Dhingra, S.; Sowdhamini, R.; Cadet, F.; Offmann, B. A Glance into the Evolution of Template-Free Protein Structure Prediction Methodologies. Biochimie 2020, 175, 85–92. [Google Scholar] [CrossRef] [PubMed]
- Bouatta, N.; AlQuraishi, M. Structural Biology at the Scale of Proteomes. Nat. Struct. Mol. Biol. 2023, 30, 129–130. [Google Scholar] [CrossRef] [PubMed]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Žídek, A.; Potapenko, A.; et al. Highly Accurate Protein Structure Prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef]
- AlQuraishi, M. AlphaFold at CASP13. Bioinformatics 2019, 35, 4862–4865. [Google Scholar] [CrossRef]
- Callaway, E. What’s next for AlphaFold and the AI Protein-Folding Revolution. Nature 2022, 604, 234–238. [Google Scholar] [CrossRef]
- Evans, R.; O’Neill, M.; Pritzel, A.; Antropova, N.; Senior, A.; Green, T.; Žídek, A.; Bates, R.; Blackwell, S.; Yim, J.; et al. Protein Complex Prediction with AlphaFold-Multimer. bioRxiv 2021. [Google Scholar] [CrossRef]
- Baek, M.; DiMaio, F.; Anishchenko, I.; Dauparas, J.; Ovchinnikov, S.; Lee, G.R.; Wang, J.; Cong, Q.; Kinch, L.N.; Schaeffer, R.D.; et al. Accurate Prediction of Protein Structures and Interactions Using a Three-Track Neural Network. Science 2021, 373, 871–876. [Google Scholar] [CrossRef]
- Antonelli, G.; Pistello, M. Virology: A Scientific Discipline Facing New Challenges. Clin. Microbiol. Infect. 2019, 25, 133–135. [Google Scholar] [CrossRef]
- Summers, W.C. The Strange History of Phage Therapy. Bacteriophage 2012, 2, 130–133. [Google Scholar] [CrossRef]
- Miroshnikov, K.A.; Evseev, P.V.; Lukianova, A.A.; Ignatov, A.N. Tailed Lytic Bacteriophages of Soft Rot Pectobacteriaceae. Microorganisms 2021, 9, 1819. [Google Scholar] [CrossRef]
- Brives, C.; Pourraz, J. Phage Therapy as a Potential Solution in the Fight against AMR: Obstacles and Possible Futures. Palgrave Commun. 2020, 6, 100. [Google Scholar] [CrossRef]
- Abdelkader, A.; Elzemrany, A.A.; El-Nadi, M.; Elsabbagh, S.A.; Shehata, M.A.; Eldehna, W.M.; El-Hadidi, M.; Ibrahim, T.M. In-Silico Targeting of SARS-CoV-2 NSP6 for Drug and Natural Products Repurposing. Virology 2022, 573, 96–110. [Google Scholar] [CrossRef] [PubMed]
- Flower, T.G.; Hurley, J.H. Crystallographic Molecular Replacement Using an in Silico-Generated Search Model of SARS-CoV-2 ORF8. Protein Sci. 2021, 30, 728–734. [Google Scholar] [CrossRef]
- Jansen van Vuren, P.; McAuley, A.J.; Kuiper, M.J.; Singanallur, N.B.; Bruce, M.P.; Riddell, S.; Goldie, S.; Mangalaganesh, S.; Chahal, S.; Drew, T.W.; et al. Highly Thermotolerant SARS-CoV-2 Vaccine Elicits Neutralising Antibodies against Delta and Omicron in Mice. Viruses 2022, 14, 800. [Google Scholar] [CrossRef]
- Singanallur, N.B.; van Vuren, P.J.; McAuley, A.J.; Bruce, M.P.; Kuiper, M.J.; Gwini, S.M.; Riddell, S.; Goldie, S.; Drew, T.W.; Blasdell, K.R.; et al. At Least Three Doses of Leading Vaccines Essential for Neutralisation of SARS-CoV-2 Omicron Variant. Front. Immunol. 2022, 13, 883612. [Google Scholar] [CrossRef] [PubMed]
- Bhowmick, S.; Jing, T.; Wang, W.; Zhang, E.Y.; Zhang, F.; Yang, Y. In Silico Protein Folding Prediction of COVID-19 Mutations and Variants. Biomolecules 2022, 12, 1665. [Google Scholar] [CrossRef] [PubMed]
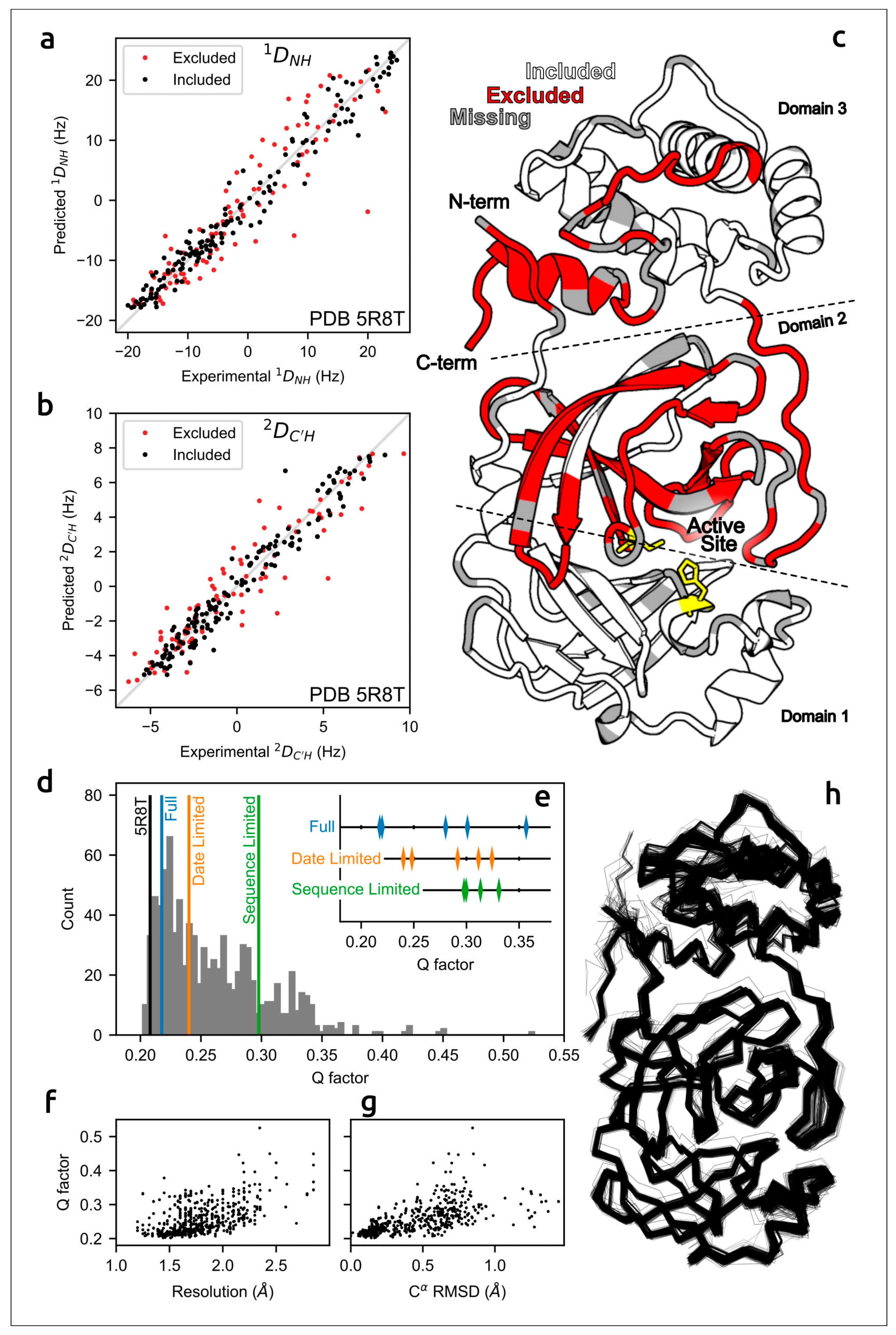
- Robertson, A.J.; Courtney, J.M.; Shen, Y.; Ying, J.; Bax, A. Concordance of X-Ray and AlphaFold2 Models of SARS-CoV-2 Main Protease with Residual Dipolar Couplings Measured in Solution. J. Am. Chem. Soc. 2021, 143, 19306–19310. [Google Scholar] [CrossRef] [PubMed]
- Kumari, S.; Chakraborty, S.; Ahmad, M.; Kumar, V.; Tailor, P.B.; Biswal, B.K. Identification of Probable Inhibitors for the DNA Polymerase of the Monkeypox Virus through the Virtual Screening Approach. Int. J. Biol. Macromol. 2023, 229, 515–528. [Google Scholar] [CrossRef] [PubMed]
- Kannan, S.R.; Sachdev, S.; Reddy, A.S.; Kandasamy, S.L.; Byrareddy, S.N.; Lorson, C.L.; Singh, K. Mutations in the Monkeypox Virus Replication Complex: Potential Contributing Factors to the 2022 Outbreak. J. Autoimmun. 2022, 133, 102928. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Liu, Y.; Li, K.; Zhang, L. Targeting F13 from Monkeypox Virus and Variola Virus by Tecovirimat: Molecular Simulation Analysis. J. Infect. 2022, 85, e99–e101. [Google Scholar] [CrossRef]
- Yefet, R.; Friedel, N.; Tamir, H.; Polonsky, K.; Mor, M.; Cherry-Mimran, L.; Taleb, E.; Hagin, D.; Sprecher, E.; Israely, T.; et al. Monkeypox Infection Elicits Strong Antibody and B Cell Response against A35R and H3L Antigens. iScience 2023, 26, 105957. [Google Scholar] [CrossRef] [PubMed]
- Benedyk, T.H.; Connor, V.; Caroe, E.R.; Shamin, M.; Svergun, D.I.; Deane, J.E.; Jeffries, C.M.; Crump, C.M.; Graham, S.C. Herpes Simplex Virus 1 Protein PUL21 Alters Ceramide Metabolism by Activating the Interorganelle Transport Protein CERT. J. Biol. Chem. 2022, 298, 102589. [Google Scholar] [CrossRef] [PubMed]
- Collantes, T.M.A.; Clark, C.M.; Musarrat, F.; Jambunathan, N.; Jois, S.; Kousoulas, K.G. Predicted Structure and Functions of the Prototypic Alphaherpesvirus Herpes Simplex Virus Type-1 UL37 Tegument Protein. Viruses 2022, 14, 2189. [Google Scholar] [CrossRef] [PubMed]
- Fieulaine, S.; Tubiana, T.; Bressanelli, S. De Novo Modelling of HEV Replication Polyprotein: Five-Domain Breakdown and Involvement of Flexibility in Functional Regulation. Virology 2023, 578, 128–140. [Google Scholar] [CrossRef]
- Liu, H.; Peck, X.Y.; Choong, Y.K.; Ng, W.S.; Engl, W.; Raghuvamsi, P.V.; Zhao, Z.W.; Anand, G.S.; Zhou, Y.; Sivaraman, J.; et al. Identification of Putative Binding Interface of PI(3,5)P2 Lipid on Rice Black-Streaked Dwarf Virus (RBSDV) P10 Protein. Virology 2022, 570, 81–95. [Google Scholar] [CrossRef]
- Chen, L.; Chen, L.; Chen, H.; Zhang, H.; Dong, P.; Sun, L.; Huang, X.; Lin, P.; Wu, L.; Jing, D.; et al. Structural Insights into the CP312R Protein of the African Swine Fever Virus. Biochem. Biophys. Res. Commun. 2022, 624, 68–74. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.Y.; Kwak, J.S.; Jung, W.; Kim, M.S.; Kim, K.H. Compensatory Mutations in the Matrix Protein of Viral Hemorrhagic Septicemia Virus (VHSV) Genotype IVa in Response to Artificial Mutation of Two Amino Acids (D62A E181A). Virus Res. 2023, 326, 199067. [Google Scholar] [CrossRef] [PubMed]
- Veit, M.; Gadalla, M.R.; Zhang, M. Using Alphafold2 to Predict the Structure of the Gp5/M Dimer of Porcine Respiratory and Reproductive Syndrome Virus. Int. J. Mol. Sci. 2022, 23, 13209. [Google Scholar] [CrossRef]
- Hötzel, I. Domain Organization of Lentiviral and Betaretroviral Surface Envelope Glycoproteins Modeled with AlphaFold. J. Virol. 2022, 96, e01348-21. [Google Scholar] [CrossRef]
- Weaver, G.C.; Arya, R.; Schneider, C.L.; Hudson, A.W.; Stern, L.J. Structural Models for Roseolovirus U20 And U21: Non-Classical MHC-I Like Proteins From HHV-6A, HHV-6B, and HHV-7. Front. Immunol. 2022, 13, 864898. [Google Scholar] [CrossRef]
- Al-Shayeb, B.; Skopintsev, P.; Soczek, K.M.; Stahl, E.C.; Li, Z.; Groover, E.; Smock, D.; Eggers, A.R.; Pausch, P.; Cress, B.F.; et al. Diverse Virus-Encoded CRISPR-Cas Systems Include Streamlined Genome Editors. Cell 2022, 185, 4574–4586.e16. [Google Scholar] [CrossRef] [PubMed]
- Klumpp, J.; Dunne, M.; Loessner, M.J. A Perfect Fit: Bacteriophage Receptor-Binding Proteins for Diagnostic and Therapeutic Applications. Curr. Opin. Microbiol. 2023, 71, 102240. [Google Scholar] [CrossRef] [PubMed]
- Goulet, A.; Cambillau, C. Structure and Topology Prediction of Phage Adhesion Devices Using AlphaFold2: The Case of Two Oenococcus Oeni Phages. Microorganisms 2021, 9, 2151. [Google Scholar] [CrossRef]
- Evseev, P.; Lukianova, A.; Tarakanov, R.; Tokmakova, A.; Popova, A.; Kulikov, E.; Shneider, M.; Ignatov, A.; Miroshnikov, K. Prophage-Derived Regions in Curtobacterium Genomes: Good Things, Small Packages. Int. J. Mol. Sci. 2023, 24, 1586. [Google Scholar] [CrossRef]
- Hawkins, N.C.; Kizziah, J.L.; Hatoum-Aslan, A.; Dokland, T. Structure and Host Specificity of Staphylococcus Epidermidis Bacteriophage Andhra. Sci. Adv. 2022, 8, eade0459. [Google Scholar] [CrossRef]
- Nieweglowska, E.S.; Brilot, A.F.; Méndez-Moran, M.; Kokontis, C.; Baek, M.; Li, J.; Cheng, Y.; Baker, D.; Bondy-Denomy, J.; Agard, D.A. The ΦPA3 Phage Nucleus Is Enclosed by a Self-Assembling 2D Crystalline Lattice. Nat. Commun. 2023, 14, 927. [Google Scholar] [CrossRef] [PubMed]
- Šiborová, M.; Füzik, T.; Procházková, M.; Nováček, J.; Benešík, M.; Nilsson, A.S.; Plevka, P. Tail Proteins of Phage SU10 Reorganize into the Nozzle for Genome Delivery. Nat. Commun. 2022, 13, 5622. [Google Scholar] [CrossRef]
- Conners, R.; McLaren, M.; Łapińska, U.; Sanders, K.; Stone, M.R.L.; Blaskovich, M.A.T.; Pagliara, S.; Daum, B.; Rakonjac, J.; Gold, V.A.M. CryoEM Structure of the Outer Membrane Secretin Channel PIV from the F1 Filamentous Bacteriophage. Nat. Commun. 2021, 12, 6316. [Google Scholar] [CrossRef]
- Eskenazi, A.; Lood, C.; Wubbolts, J.; Hites, M.; Balarjishvili, N.; Leshkasheli, L.; Askilashvili, L.; Kvachadze, L.; van Noort, V.; Wagemans, J.; et al. Combination of Pre-Adapted Bacteriophage Therapy and Antibiotics for Treatment of Fracture-Related Infection Due to Pandrug-Resistant Klebsiella Pneumoniae. Nat. Commun. 2022, 13, 302. [Google Scholar] [CrossRef]
- McGinnis, R.J.; Brambley, C.A.; Stamey, B.; Green, W.C.; Gragg, K.N.; Cafferty, E.R.; Terwilliger, T.C.; Hammel, M.; Hollis, T.J.; Miller, J.M.; et al. A Monomeric Mycobacteriophage Immunity Repressor Utilizes Two Domains to Recognize an Asymmetric DNA Sequence. Nat. Commun. 2022, 13, 4105. [Google Scholar] [CrossRef]
- Zhang, T.; Tamman, H.; Coppieters ’t Wallant, K.; Kurata, T.; LeRoux, M.; Srikant, S.; Brodiazhenko, T.; Cepauskas, A.; Talavera, A.; Martens, C.; et al. Direct Activation of a Bacterial Innate Immune System by a Viral Capsid Protein. Nature 2022, 612, 132–140. [Google Scholar] [CrossRef] [PubMed]
- Evseev, P.; Gutnik, D.; Shneider, M.; Miroshnikov, K. Use of an Integrated Approach Involving AlphaFold Predictions for the Evolutionary Taxonomy of Duplodnaviria Viruses. Biomolecules 2023, 13, 110. [Google Scholar] [CrossRef]
- Liu, Y.; Demina, T.A.; Roux, S.; Aiewsakun, P.; Kazlauskas, D.; Simmonds, P.; Prangishvili, D.; Oksanen, H.M.; Krupovic, M. Diversity, Taxonomy, and Evolution of Archaeal Viruses of the Class Caudoviricetes. PLOS Biol. 2021, 19, e3001442. [Google Scholar] [CrossRef] [PubMed]
- Podgorski, J.M.; Freeman, K.; Gosselin, S.; Huet, A.; Conway, J.F.; Bird, M.; Grecco, J.; Patel, S.; Jacobs-Sera, D.; Hatfull, G.; et al. A Structural Dendrogram of the Actinobacteriophage Major Capsid Proteins Provides Important Structural Insights into the Evolution of Capsid Stability. Structure 2023, 31, 282–294.e5. [Google Scholar] [CrossRef]
- Evseev, P.; Shneider, M.; Miroshnikov, K. Evolution of Phage Tail Sheath Protein. Viruses 2022, 14, 1148. [Google Scholar] [CrossRef]
- Hötzel, I. Deep-Time Structural Evolution of Retroviral and Filoviral Surface Envelope Proteins. J. Virol. 2022, 96, e00063-22. [Google Scholar] [CrossRef]
- Callaway, E. “The Entire Protein Universe”: AI Predicts Shape of Nearly Every Known Protein. Nature 2022, 608, 15–16. [Google Scholar] [CrossRef] [PubMed]
- Perrakis, A.; Sixma, T.K. AI Revolutions in Biology. EMBO Rep. 2021, 22, e54046. [Google Scholar] [CrossRef]
- Akdel, M.; Pires, D.E.V.; Pardo, E.P.; Jänes, J.; Zalevsky, A.O.; Mészáros, B.; Bryant, P.; Good, L.L.; Laskowski, R.A.; Pozzati, G.; et al. A Structural Biology Community Assessment of AlphaFold2 Applications. Nat. Struct. Mol. Biol. 2022, 29, 1056–1067. [Google Scholar] [CrossRef]
- Mirdita, M.; Schütze, K.; Moriwaki, Y.; Heo, L.; Ovchinnikov, S.; Steinegger, M. ColabFold: Making Protein Folding Accessible to All. Nat. Methods 2022, 19, 679–682. [Google Scholar] [CrossRef] [PubMed]
- Humphreys, I.; Pei, J.; Baek, M.; Krishnakumar, A.; Anishchenko, I.; Ovchinnikov, S.; Zhang, J.; Ness, T.J.; Banjade, S.; Bagde, S.R.; et al. Computed Structures of Core Eukaryotic Protein Complexes. Science 2021, 374, eabm4805. [Google Scholar] [CrossRef]
- Gomes, P.S.F.C.; Gomes, D.E.B.; Bernardi, R.C. Protein Structure Prediction in the Era of AI: Challenges and Limitations When Applying to in Silico Force Spectroscopy. Front. Bioinform. 2022, 2. [Google Scholar] [CrossRef] [PubMed]
- Subramaniam, S.; Kleywegt, G. A Paradigm Shift in Structural Biology. Nat. Methods 2022, 19, 20–23. [Google Scholar] [CrossRef]
- Drake, Z.C.; Seffernick, J.T.; Lindert, S. Protein Complex Prediction Using Rosetta, AlphaFold, and Mass Spectrometry Covalent Labeling. Nat. Commun. 2022, 13, 7846. [Google Scholar] [CrossRef] [PubMed]
- He, J.; Lin, P.; Chen, J.; Cao, H.; Huang, S.Y. Model Building of Protein Complexes from Intermediate-Resolution Cryo-EM Maps with Deep Learning-Guided Automatic Assembly. Nat. Commun. 2022, 13, 4066. [Google Scholar] [CrossRef]
- Bryant, P.; Pozzati, G.; Elofsson, A. Improved Prediction of Protein-Protein Interactions Using AlphaFold2. Nat. Commun. 2022, 13, 1265. [Google Scholar] [CrossRef] [PubMed]
- Bryant, P.; Pozzati, G.; Zhu, W.; Shenoy, A.; Kundrotas, P.; Elofsson, A. Predicting the Structure of Large Protein Complexes Using AlphaFold and Monte Carlo Tree Search. Nat. Commun. 2022, 13, 6028. [Google Scholar] [CrossRef]
- Ruff, K.M.; Pappu, R.V. AlphaFold and Implications for Intrinsically Disordered Proteins. J. Mol. Biol. 2021, 433, 167208. [Google Scholar] [CrossRef]
- Laurents, D.V. AlphaFold 2 and NMR Spectroscopy: Partners to Understand Protein Structure, Dynamics and Function. Front. Mol. Biosci. 2022, 9, 906437. [Google Scholar] [CrossRef] [PubMed]
- Edich, M.; Briggs, D.C.; Kippes, O.; Gao, Y.; Thorn, A. The Impact of AlphaFold on Experimental Structure Solution. Faraday Discuss. 2022, 240, 184–195. [Google Scholar] [CrossRef]
- Wong, F.; Krishnan, A.; Zheng, E.J.; Stärk, H.; Manson, A.L.; Earl, A.M.; Jaakkola, T.; Collins, J.J. Benchmarking AlphaFold -enabled Molecular Docking Predictions for Antibiotic Discovery. Mol. Syst. Biol. 2022, 18, e11081. [Google Scholar] [CrossRef]
- Hekkelman, M.L.; de Vries, I.; Joosten, R.P.; Perrakis, A. AlphaFill: Enriching AlphaFold Models with Ligands and Cofactors. Nat. Methods 2022, 20, 205–213. [Google Scholar] [CrossRef]
- Bagdonas, H.; Fogarty, C.; Fadda, E.; Agirre, J. The Case for Post-Predictional Modifications in the AlphaFold Protein Structure Database. Nat. Struct. Mol. Biol. 2021, 28, 869–870. [Google Scholar] [CrossRef] [PubMed]
- Van Breugel, M.; Rosa e Silva, I.; Andreeva, A. Structural Validation and Assessment of AlphaFold2 Predictions for Centrosomal and Centriolar Proteins and Their Complexes. Commun. Biol. 2022, 5, 312. [Google Scholar] [CrossRef]
- Lane, T.J. Protein Structure Prediction Has Reached the Single-Structure Frontier. Nat. Methods 2023, 20, 170–173. [Google Scholar] [CrossRef]
- Bertoline, L.M.F.; Lima, A.N.; Krieger, J.E.; Teixeira, S.K. Before and after AlphaFold2: An Overview of Protein Structure Prediction. Front. Bioinform. 2023, 3, 1120370. [Google Scholar] [CrossRef] [PubMed]
- Buel, G.; Walters, K. Can AlphaFold2 Predict the Impact of Missense Mutations on Structure? Nat. Struct. Mol. Biol. 2022, 29, 1–2. [Google Scholar] [CrossRef]
- Pak, M.A.; Markhieva, K.A.; Novikova, M.S.; Petrov, D.S.; Vorobyev, I.S.; Maksimova, E.S.; Kondrashov, F.A.; Ivankov, D.N. Using AlphaFold to Predict the Impact of Single Mutations on Protein Stability and Function. PLOS ONE 2023, 18, e0282689. [Google Scholar] [CrossRef]
- Walls, A.C.; Park, Y.-J.; Tortorici, M.A.; Wall, A.; McGuire, A.T.; Veesler, D. Structure, Function, and Antigenicity of the SARS-CoV-2 Spike Glycoprotein. Cell 2020, 181, 281–292.e6. [Google Scholar] [CrossRef] [PubMed]
- Han, Y.; Král, P. Computational Design of ACE2-Based Peptide Inhibitors of SARS-CoV-2. ACS Nano 2020, 14, 5143–5147. [Google Scholar] [CrossRef]
- Zhang, L.; Lin, D.; Sun, X.; Curth, U.; Drosten, C.; Sauerhering, L.; Becker, S.; Rox, K.; Hilgenfeld, R. Crystal Structure of SARS-CoV-2 Main Protease Provides a Basis for Design of Improved α-Ketoamide Inhibitors. Science 2020, 368, 409–412. [Google Scholar] [CrossRef] [PubMed]
- Cao, Y.; Yang, R.; Wang, W.; Jiang, S.; Yang, C.; Liu, N.; Dai, H.; Lee, I.; Meng, X.; Yuan, Z. Probing the Formation, Structure and Free Energy Relationships of M Protein Dimers of SARS-CoV-2. Comput. Struct. Biotechnol. J. 2022, 20, 573–582. [Google Scholar] [CrossRef]
- Heo, L.; Feig, M. High-accuracy protein structures by combining machine-learning with physics-based refinement. Proteins 2020, 88, 637–642. [Google Scholar] [CrossRef] [PubMed]
- Hiranuma, N.; Park, H.; Baek, M.; Anishchenko, I.; Dauparas, J.; Baker, D. Improved Protein Structure Refinement Guided by Deep Learning Based Accuracy Estimation. Nat. Commun. 2021, 12, 1340. [Google Scholar] [CrossRef]
- Li, Z.; Hirst, J.D. Computed Optical Spectra of SARS-CoV-2 Proteins. Chem. Phys. Lett. 2020, 758, 137935. [Google Scholar] [CrossRef]
- Du, Z.; Su, H.; Wang, W.; Ye, L.; Wei, H.; Peng, Z.; Anishchenko, I.; Baker, D.; Yang, J. The TrRosetta Server for Fast and Accurate Protein Structure Prediction. Nat. Protoc. 2021, 16, 5634–5651. [Google Scholar] [CrossRef]
- Rizk, J.G.; Lippi, G.; Henry, B.M.; Forthal, D.N.; Rizk, Y. Prevention and Treatment of Monkeypox. Drugs 2022, 82, 957–963. [Google Scholar] [CrossRef] [PubMed]
- Delaune, D.; Iseni, F. Drug Development against Smallpox: Present and Future. Antimicrob. Agents Chemother. 2020, 64, e01683-19. [Google Scholar] [CrossRef]
- Peng, Q.; Xie, Y.; Kuai, L.; Wang, H.; Qi, J.; Gao, G.F.; Shi, Y. Structure of Monkeypox Virus DNA Polymerase Holoenzyme. Science 2023, 379, 100–105. [Google Scholar] [CrossRef]
- Sehrawat, S.; Kumar, D.; Rouse, B.T. Herpesviruses: Harmonious Pathogens but Relevant Cofactors in Other Diseases? Front. Cell. Infect. Microbiol. 2018, 8, 177. [Google Scholar] [CrossRef] [PubMed]
- Current ICTV Taxonomy Release | ICTV. Available online: https://ictv.global/taxonomy (accessed on 9 November 2022).
- Nahas, K.L.; Connor, V.; Scherer, K.M.; Kaminski, C.F.; Harkiolaki, M.; Crump, C.M.; Graham, S.C. Near-Native State Imaging by Cryo-Soft-X-Ray Tomography Reveals Remodelling of Multiple Cellular Organelles during HSV-1 Infection. PLOS Pathog. 2022, 18, e1010629. [Google Scholar] [CrossRef]
- Bigalke, J.M.; Heldwein, E.E. Nuclear Exodus: Herpesviruses Lead the Way. Annu. Rev. Virol. 2016, 3, 387–409. [Google Scholar] [CrossRef]
- Wommack, K.E.; Colwell, R.R. Virioplankton: Viruses in Aquatic Ecosystems. Microbiol. Mol. Biol. Rev. 2000, 64, 69–114. [Google Scholar] [CrossRef] [PubMed]
- Simmonds, P.; Adams, M.J.; Benkő, M.; Breitbart, M.; Brister, J.R.; Carstens, E.B.; Davison, A.J.; Delwart, E.; Gorbalenya, A.E.; Harrach, B.; et al. Consensus Statement: Virus Taxonomy in the Age of Metagenomics. Nat. Rev. Microbiol. 2017, 15, 161–168. [Google Scholar] [CrossRef] [PubMed]
- Hendrix, R.W.; Smith, M.C.M.; Burns, R.N.; Ford, M.E.; Hatfull, G.F. Evolutionary Relationships among Diverse Bacteriophages and Prophages: All the World’s a Phage. Proc. Natl. Acad. Sci. USA 1999, 96, 2192–2197. [Google Scholar] [CrossRef]
- Shkoporov, A.N.; Hill, C. Bacteriophages of the Human Gut: The “Known Unknown” of the Microbiome. Cell Host Microbe 2019, 25, 195–209. [Google Scholar] [CrossRef]
- Loc-Carrillo, C.; Abedon, S.T. Pros and Cons of Phage Therapy. Bacteriophage 2011, 1, 111–114. [Google Scholar] [CrossRef]
- Fischetti, V.A. Bacteriophage Endolysins: A Novel Anti-Infective to Control Gram-Positive Pathogens. Int J. Med. Microbiol. 2010, 300, 357–362. [Google Scholar] [CrossRef]
- Ouyang, R.; Costa, A.R.; Cassidy, C.K.; Otwinowska, A.; Williams, V.C.J.; Latka, A.; Stansfeld, P.J.; Drulis-Kawa, Z.; Briers, Y.; Pelt, D.M.; et al. High-Resolution Reconstruction of a Jumbo-Bacteriophage Infecting Capsulated Bacteria Using Hyperbranched Tail Fibers. Nat. Commun. 2022, 13, 7241. [Google Scholar] [CrossRef]
- Krupovic, M.; Koonin, E.V. Multiple Origins of Viral Capsid Proteins from Cellular Ancestors. Proc. Natl. Acad. Sci. USA 2017, 114, E2401–E2410. [Google Scholar] [CrossRef]
- Salemme, F.R.; Miller, M.D.; Jordan, S.R. Structural Convergence during Protein Evolution. Proc. Natl. Acad. Sci. USA 1977, 74, 2820–2824. [Google Scholar] [CrossRef] [PubMed]
- Holm, L. Using Dali for Protein Structure Comparison. Methods Mol. Biol. 2020, 2112, 29–42. [Google Scholar] [CrossRef]
- Bisio, H.; Legendre, M.; Giry, C.; Philippe, N.; Alempic, J.-M.; Jeudy, S.; Abergel, C. Evolution of Giant Pandoravirus Revealed by CRISPR/Cas9. Nat. Commun. 2023, 14, 428. [Google Scholar] [CrossRef] [PubMed]
- Fokine, A.; Leiman, P.G.; Shneider, M.M.; Ahvazi, B.; Boeshans, K.M.; Steven, A.C.; Black, L.W.; Mesyanzhinov, V.V.; Rossmann, M.G. Structural and Functional Similarities between the Capsid Proteins of Bacteriophages T4 and HK97 Point to a Common Ancestry. Proc. Natl. Acad. Sci. USA 2005, 102, 7163–7168. [Google Scholar] [CrossRef]
- Fang, Q.; Tang, W.-C.; Fokine, A.; Mahalingam, M.; Shao, Q.; Rossmann, M.G.; Rao, V.B. Structures of a Large Prolate Virus Capsid in Unexpanded and Expanded States Generate Insights into the Icosahedral Virus Assembly. Proc. Natl. Acad. Sci. USA 2022, 119, e2203272119. [Google Scholar] [CrossRef]
- Steven, A.C.; Greenstone, H.L.; Booy, F.P.; Black, L.W.; Ross, P.D. Conformational Changes of a Viral Capsid Protein. Thermodynamic Rationale for Proteolytic Regulation of Bacteriophage T4 Capsid Expansion, Co-Operativity, and Super-Stabilization by Soc Binding. J. Mol. Biol. 1992, 228, 870–884. [Google Scholar] [CrossRef] [PubMed]
- Bowman, B.R.; Baker, M.L.; Rixon, F.J.; Chiu, W.; Quiocho, F.A. Structure of the Herpesvirus Major Capsid Protein. EMBO J. 2003, 22, 757–765. [Google Scholar] [CrossRef]
- Hark Gan, H.; Perlow, R.A.; Roy, S.; Ko, J.; Wu, M.; Huang, J.; Yan, S.; Nicoletta, A.; Vafai, J.; Sun, D.; et al. Analysis of Protein Sequence/Structure Similarity Relationships. Biophys. J. 2002, 83, 2781–2791. [Google Scholar] [CrossRef]
- An, H.; Froehlich, J.; Lebrilla, C. Determination of Glycosylation Sites and Site-Specific Heterogeneity in Glycoproteins. Curr. Opin. Chem. Biol. 2009, 13, 421–426. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Authors, Year | Virus or Viral Group | Study Aim (s) | Results and AF2 Usage |
|---|---|---|---|
| Callaway 2022 [18] | to explore how AF2 changes biology | AF2 affects many studies and provides a quality of prediction not previously achievable by computational tools. At the same time, it has limitations and it is important to consider them when conducting research. | |
| Evans et al., 2021 [19] | to present the extension of AlphaFold for protein complexes—AlphaFold-Multimer | AlphaFold-Multimer significantly improves the quality of predicted multimeric interfaces, compared with basic AlphaFold adapted to input data, while maintaining a high level of accuracy within the chain. | |
| Abdelkader et al., 2022 [25] | SARS-CoV-2 | to find inhibitors of non-structural protein 6 (NSP6) | Using the AF2 predictions, candidate inhibitors were suggested and recommended for biological testing. |
| Flower et al., 2021 [26] | SARS-CoV-2 | to test the in silico prediction of β-rich ORF8 protein for finding an MR solution to the crystallographic phase problem | It was shown that the ORF8 protein model, predicted by AF2, is sufficiently accurate to provide a phase solution by MR. |
| Vuren et al., 2022 [27] | SARS-CoV-2 | to test highly thermo-tolerant monomeric receptor-binding domain derivatives on mice for the development of new vaccines | The monomeric formulation of the vaccine was observed to produce a slightly superior immune response, possibly because it presents more antigenic epitopes, as shown using AF2 predictions. |
| Singanallur et al., 2022 [28] | SARS-CoV-2 | to assess leading vaccines in virus neutralisation assays against Delta and Omicron variants of concern (VOC) and a reference isolate | At least a third dose of these vaccines is necessary to generate sufficient neutralising antibodies against emerging VOC. AF2 was used to find an explanation for the observed reduction in neutralisation of Omicron compared with other variants. |
| Bhowmick et al., 2022 [29] | SARS-CoV-2 | to study the effects of various mutations in the RBD of the SARS-CoV-2 spike and its key interactions with the ACE-2 receptor, using protein structure prediction algorithms along with molecular docking | AF2-generated and trRosetta-generated models of RBD were compared. trRosetta predictions appeared to be more accurate and have been used for docking with the ACE-2 receptor of other mutated RBD variants. |
| Robertson et al., 2021 [30] | SARS-CoV-2 | to evaluate the concordance of AF2 models with residual dipolar couplings data | Close agreement between all sets of AlphaFold models and experimental residual dipolar couplings data was found for most of the protein. |
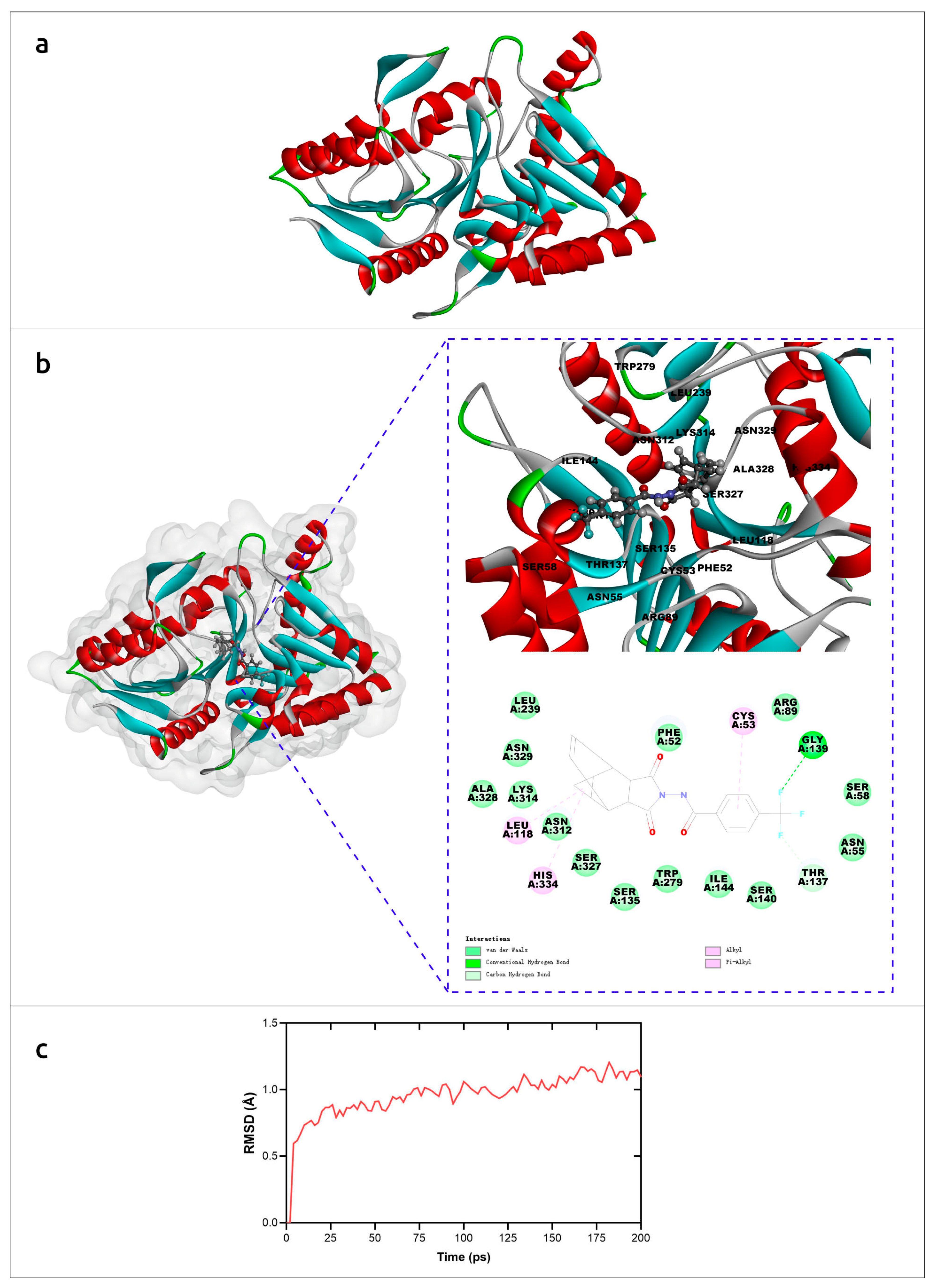
| Kumari et al., 2023 [31] | Monkeypox virus (MPXV) | to search for inhibitors of MPXV DNA polymerase (DNAP) for antiviral therapy | DNAP inhibitors were found using an AF2-generated model and virtual screening of ZINC and antiviral libraries. |
| Kannan et al., 2022 [32] | MPXV | to study the effects of mutations in DNA replication complex (RC) | Mutations in RC that are likely to contribute to the 2022 monkeypox outbreak were identified. AF2 predictions were used to model an RC component. |
| Li et al., 2022 [33] | MPXV | to study the mechanisms of inhibition of poxvirus phospholipase D (F13) by tecovirimat, which have been demonstrated to be effective against monkeypox in vitro and in anima | The potential binding pocket and the possible binding mode for tecovirimat with F13 were revealed using AF2 structure predictions and molecular docking. |
| Yefet et al., 2023 [34] | MPXV | to characterise the main serological and B cell markers accompanying MPXV infection in humans | The reactivity of three MPXV antigens to MPXV-11convalescent sera and responses caused by vaccinia virus-based vaccine (VACV) were tested. AF2 modelling indicated similar conformations of MPXV and VACV antigens. |
| Benedyk et al., 2022 [35] | Herpes Simplex Virus Type-1 (HSV-1) | to study the mechanisms of influence of HSV-1 on sphingolipid metabolism | Using AF2 predictions, the residues essential for the binding of involved proteins were identified and experiments demonstrating that HSV-1 modifies the sphingolipid metabolism via specific protein–protein interactions were conducted. |
| Collantes et al., 2022 [36] | HSV-1 | to study details of the transport of the viral particle towards the nucleus | Structural features of the UL37 tegument protein, which is important for retrograde transport and viral replication, were revealed. AF2 and other computational techniques were used for prediction of structures of UL37 and binding surface. |
| Fieulaine et al., 2023 [37] | Hepatitis E virus (HEV) | to study HEV replication polyprotein (pORF1) | The structure of HEV pORF1 was obtained with AF2 and then analysed. The protocol to express and purify the full-length HEV pORF1 was developed. |
| Liu et al., 2022 [38] | Rice black-streaked dwarf virus (RBSDV) | to reveal lipid-binding sites of major outer capsid protein (also known as P10) | The use of AF2 predictions and the results of experimental studies enabled the suggestion of putative binding sites of lipids on RBSDV P10 protein. |
| Chen et al., 2022 [39] | African swine fever virus (ASFV) | to study the mechanism of interactions of ssDNA and ssDNA-binding protein CP312R | With the assistance of AF2 predictions, the crystal structure of ASFV CP312R was determined, and the putative ssDNA binding core domain was suggested. |
| Kim et al., 2023 [40] | Viral hemorrhagic septicemia virus (VHSV) | to study the genesis of secondary mutations in the matrix (M) protein | VHSV was found to respond to the artificial mutation of M protein through secondary mutations. These secondary mutations occurred when the artificial mutations were harmful for the virus. AlphaFold was used to predict the structure of the M protein. |
| Veit et al., 2022 [41] | Porcine reproductive and respiratory syndrome virus (PRRSV) | to study the Gp5/M protein dimer, the major component of the viral envelope required for virus budding | Detailed bioinformatic analysis of Gp5/M was conducted using various bioinformatic tools. AlphaFold was used to obtain a model of the Gp5/M dimer. |
| Hötzel 2022 [42] | Several lentiviruses and betaretroviruses | to study the surface envelope glycoproteins of nonprimate lentiviruses and betaretroviruses | The consistence of AF2 models of small ruminant lentiviruses and betaretroviruses and experimental data was shown. Structural features of gp135 of small ruminant lentiviruses were discussed. |
| Weaver et al., 2022 [43] | Human roseolovirus | to clarify structural features of membrane glycoproteins U20 and U21 | AlphaFold and RoseTTAfold were used to predict the structures of U20 and U21. Structural features of these proteins were discussed. |
| Al-Shayeb et al., 2022 [44] | Bacteriophage metagenomic sequences | to study CRISPR systems encoded in phage genomes | Bacteriophage-encoded CRISPR systems were found and classified using genome-resolved metagenomics. The Casλ-RNA-DNA structure was determined using Cryo-EM. AF2 was used to obtain the initial model of the Casλ protein. |
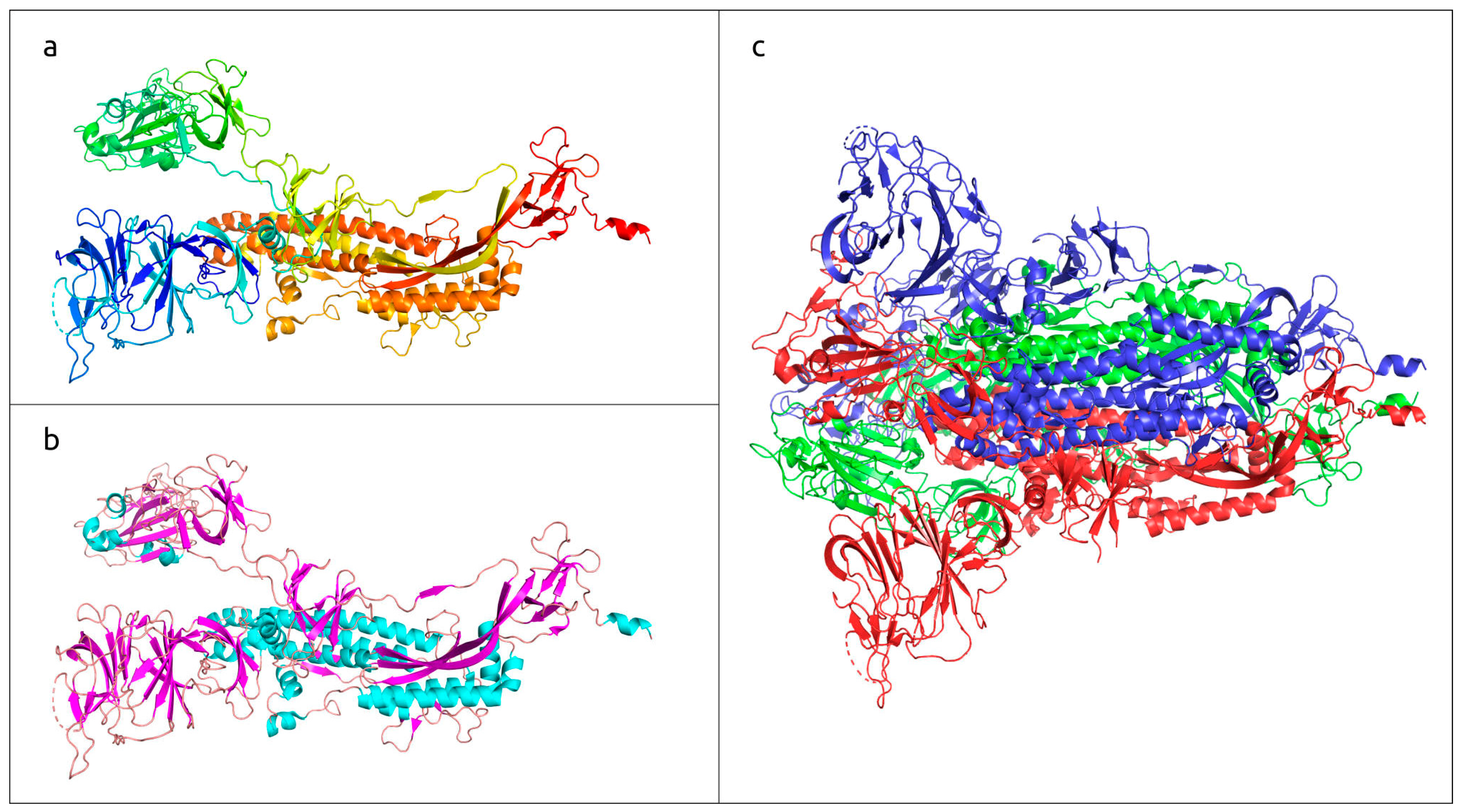
| Klumpp et al., 2023 [45] | Various bacteriophages | to review the features and use of phage receptor-binding proteins (RBPs) | Distinctive features of phage RBPs, the use of RBPs as antibacterial agents and the application of AlphaFold for the prediction of RBPs’ structure were described. |
| Goulet et al., 2021 [46] | Oenococcus oeni phages OE33PA and Vinitor162 | to reveal the structural features of different phage adhesion devices | The topology and structure of phage adhesion proteins was studied using AF2 modelling. Based on known models, a topological model of the OE33PA adhesion device was proposed. |
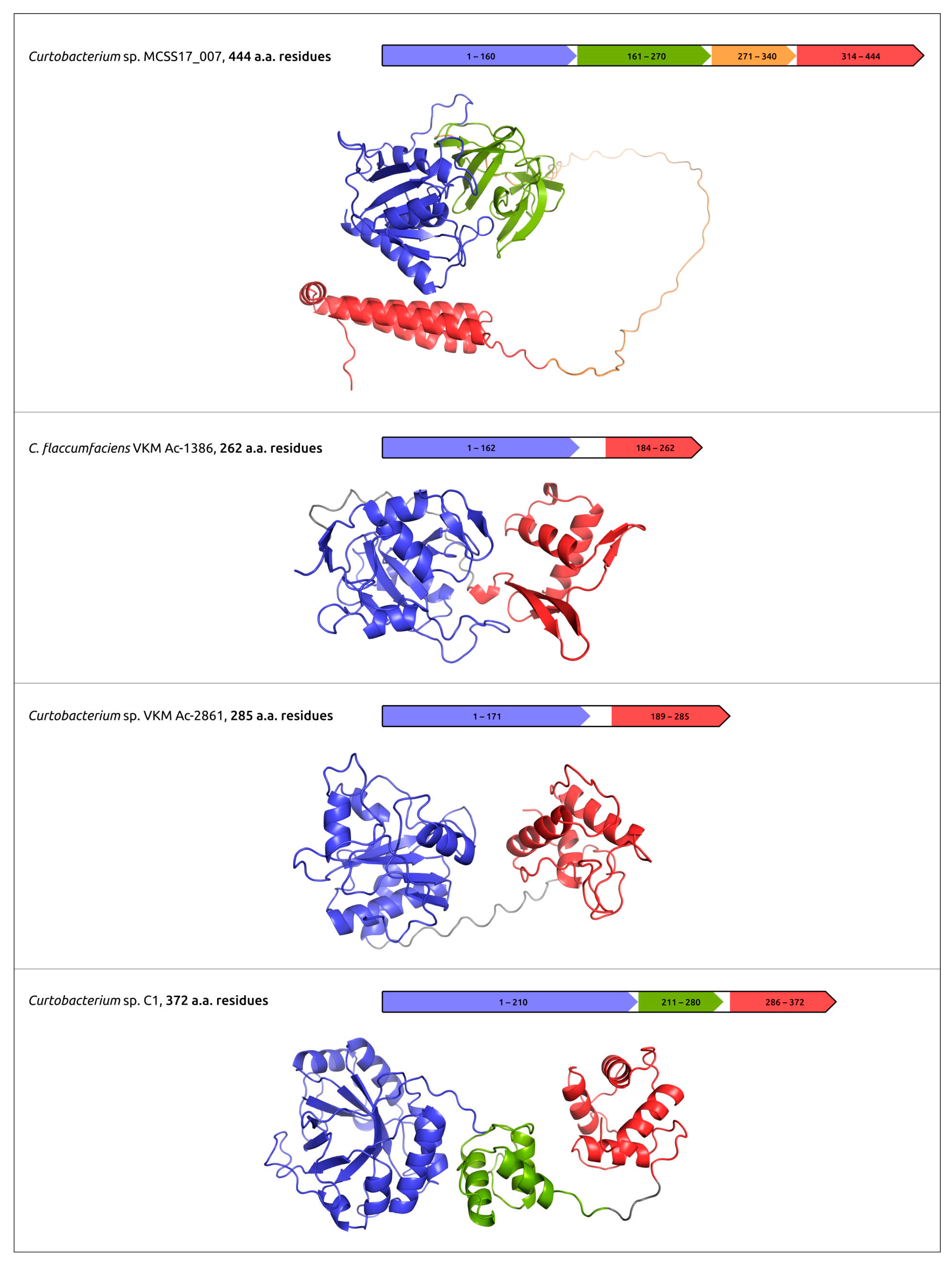
| Evseev et al., 2023 [47] | Curtobacterium prophages | to reveal and characterise Curtobacterium prophage-derived regions and glycopolymer-degrading enzymes of prophage origin | Prophage-derived regions were found and annotated. Glycopolymer-degrading enzymes of prophage origin were modelled using AF2, characterised and clustered. |
| Hawkins et al., 2022 [48] | Staphylococcus phage Andhra | to study the phage’s structural features | The Cryo-EM structure was reported. Using AlphaFold predictions, the distal tail model was built. |
| Nieweglowska et al., 2023 [49] | Pseudomonas phage ϕPA3 | to explore the mechanism of formation of the phage nucleus | The ability of Phage Nuclear Enclosure (PhuN) protein to spontaneously assemble into 2D sheets with p2 and p4 symmetry was shown. The p2 symmetric state was resolved by Cryo-EM. AF2 was used to build a model of the 2D array. |
| Šiborová et al., 2022 [50] | Escherichia phage SU10 | to study the mechanism of phage genome delivery | Cryo-EM and Cryo-ET characterisation of the attachment of the phage to the host cell was presented. The formation of a tail nozzle after rearrangement was shown. AF2 was used to build tail models. |
| Conners et al., 2021 [51] | Klebsiella phage f1 | to study the structural bases of the mechanism of phage egress and its practical application | Cryo-EM structure phage-encoded pIV secretin was determined, and the mechanism for phage egress was proposed. AF2 was used to predict the structure of the N0 domain of pIV. |
| Eskenazi et al., 2022 [52] | Klebsiella phage M1 | to investigate the effectiveness of combined pre-adapted bacteriophage therapy and antibiotics for the treatment of fracture-related infection | The therapy resulted in an objective improvement in the patient’s wounds and overall condition. The combination of phage and antibiotic therapy was demonstrated to be highly effective against the patient’s K. pneumoniae strain. AlphaFold was used for the modelling of original and mutated phage proteins. |
| McGinnis et al., 2022 [53] | Mycobacterium phage TipsytheTRex | to study the mechanism of the interaction of the immunity repressor and DNA | A Dual DNA binding domains model of the repressor was proposed. An AlphaFold model of the repressor protein was used to significantly improve the structure obtained using single-wavelength anomalous diffraction phasing. |
| Zhang et al., 2022 [54] | to investigate the functioning of the toxin–antitoxin system CapRelSJ46 that protects E. coli against phages | It was shown that the C-terminal domain of CapRelSJ46 controls the toxic N-terminal region. Major capsid proteins of some phages bind to the C-terminal domain to relieve autoinhibition, enabling the toxin domain. AF2 was used for predicting different conformations of CapRelSJ46. | |
| Evseev et al., 2023 [55] | Various archaeal and bacterial Duplodnaviria viruses | to clarify the classification of high-ranked taxa | Using the results of AlphaFold predictions, combined with the results of sequence-based phylogeny, suggestions for possible upgrades to taxonomic classifications of Duplodnaviria viruses were made. |
| Liu et al., 2021 [56] | Archaeal tailed viruses | to study and classify archaeal tailed viruses, including newly sequenced ones | A total of 37 newly sequenced genomes and published sequences were classified using genomic similarity and network-based analysis. AF2 was used for modelling major capsid proteins and further structural comparisons. |
| Podgorski et al., 2023 [57] | Actinobacteriophages | to classify actinobacteriophage major capsid proteins | AlphaFold predictions, together with experimentally obtained structures, were used to construct a detailed structural dendrogram describing the evolution of capsid structural stability within actinobacteriophages. |
| Evseev et al., 2022 [58] | Various myoviruses | to reveal patterns of structural evolution of tail sheath protein | Based on AF2 predictions and laboratory-derived structures, patterns of evolution of phage sheath protein were revealed. |
| Hötzel et al., 2022 [59] | Various retroviruses | to clarify common structural features of the retroviral surface envelope protein subunit (SU) | Analysis of structures predicted with AF2 revealed the common conserved core of Sus and enabled the identification of a homologue structure in the SU equivalent GP1 of filoviruses, demonstrating their common origin. |
| Callaway 2022 [60] | to present the results of the first year of AF2 | The AlphaFold tool predicted about 200 million protein structures. About 35% of these structures were highly accurate and 45% could be used for specific purposes. | |
| Perrakis et al., 2021 [61] | to consider the scope and implications of AF2 applications in structural biology | Despite a number of limitations, the analysis of models obtained with AlphaFold can generate new and testable hypotheses about protein function, which is necessary for structural biology. | |
| Akdel et al., 2022 [62] | to evaluate the use of AF2 predictions for different structural biology challenges, such as variant effect prediction, pocket detection, and model | AF2 predictions, given their limitations, can be applied to existing structural biology problems, and their accuracy is close to that of experimental models. | |
| Mirdita et al., 2022 [63] | to present ColabFold and its comparison with other tools | ColabFold goes beyond the original AF2 functions by improving sequence searches, providing tools for modelling protein complexes, extending databases and determining protein structures, with about 90 times the speed of AF2. | |
| Humphreys et al., 2021 [64] | to obtain models for 106 previously unidentified protein complexes and 806 proteins, for which detailed structural information was lacking | The combination of AlphaFold and Ro-seTTAfold expanded the scope of deep-learning-based tools for modelling protein complexes. | |
| Gomes et al., 2022 [65] | to assess the reliability of AlphaFold predictions of Staphylococcus bacteria adhesins proteins, using single-molecule force spectroscopy | AlphaFold generates extremely robust protein structures, but in some cases cannot accurately predict protein multimers. Even AlphaFold Multimer failed to predict important structural features for some of the investigated complexes, such as the locking strand of adhesin. | |
| Subramaniam et al., 2022 [66] | to study a combination of computational and experimental tools for protein structure prediction | It was concluded that the development of structural biology in the future will be closely related to the synergy between deep-machine-learning-based predictions, as in AF2, and cryo-EM technology. | |
| Drake et al., 2022 [67] | to propose a new hybrid method of Alphafold, Rosetta and mass spectrometry covalent labelling for predicting protein complexes | Combining AF2 models of protein complexes with differential covalent labelling mass spectrometry data via the application of RosettaDock demonstrated a lower root-mean-square deviation than complexes predicted without covalent labelling data. | |
| He et al., 2022 [68] | to present EMBuild, an automatic model-building tool for protein complexes | EMBuild automatically builds models from intermediate-resolution cryo-EM maps integrating AlphaFold structure prediction. It provides quality and reliable models that are comparable to manually built structures. | |
| Bryant et al., 2022 [69] | to offer a new protocol for AF2 prediction of protein complexes | The use of optimised multiple sequence alignment together with AF2 showed acceptable quality for 63% of the dimers. | |
| Bryant et al., 2022 [70] | to propose the use of Monte Carlo tree search for predicting protein complexes with AF2 | The application of a Monte Carlo tree search for the predicted AF2 subcomponents yielded 91 of 175 complexes, with a median TM-score of 0.51, and 30 of them demonstrated high accuracy. | |
| Ruff et al., 2021 [71] | to study the implications of AlphaFold for intrinsically disordered proteins | Predicted structures obtained with AlphaFold emphasised the importance of intrinsically disordered proteins/regions. A huge number of protein regions that AlphaFold predicted with low accuracy overlapped with regions predicted as IDRs. | |
| Laurents et al., 2022 [72] | to provide information on the prediction of protein folding using a combination of NMR and AF2 spectroscopy | In the future, NMR spectroscopy may strengthen Alphafold predictions in areas where it has limitations: conformations, ligand and cofactor interactions, post-translational modifications and intrinsically disordered proteins. | |
| Edich et al., 2022 [73] | to study the impact of AF2 on experimental structure solution | Although AF2 has some drawbacks, it can help in the design of the experiment and determine which part of the protein sequence may be intrinsically disordered. It also encourages the conducting of more experimental studies, as data from them can improve deep-machine-learning’s ability to predict. | |
| Wong et al., 2022 [74] | to assess AlphaFold-enabled molecular docking predictions for drug discovery | The use of AF2 together with molecular docking simulations to predict protein-ligand bindings demonstrated poor performance. The prediction accuracy might be improved by the integration of machine-learning-based approaches. | |
| Hekkelman et al., 2022 [75] | to present AlphaFill, a tool for improving AlphaFold predictions with ligands and cofactors | The developed algorithm, employing sequence and structure similarity analysis, received a good validation performed against experimental structures. | |
| Bagdonas et al., 2021 [76] | to propose an approach that addresses the absence of cofactors and co- or post-translational modifications in AF2 models | This approach combines sequence and structure data to transfer protein glycosylation from a library of structurally balanced glycan blocks to the AlphaFold model. The algorithm was integrated into the Privateer software. | |
| Van Breugel et al., 2022 [77] | to assess the quality of AF2 models in the study of centrosome and centriole biogenesis | AF2 models can reveal important insights into the structural features of two key proteins in centrosome and centriole biogenesis, CEP192 and CEP44. The AF2 algorithm was used to predict, with subsequent experimental validation, previously unknown primary features in the structure of TTBK2 associated with CEP164, as well as the Chibby1-FAM92A complex. | |
| Lane 2023 [78] | to discuss AF2 restrictions concerning structural distribution and other issues | As deep-machine-learning algorithms develop, they require more and more experimental data. In the author’s opinion, experimental methods such as time-resolved crystallography, cryo-EM data and others can provide information that enables researchers to penetrate the essence of protein functioning. | |
| Bertoline et al., 2023 [79] | to provide an overview of changes in protein structure prediction before and after the advent of AF2 | The advent of AF2 has taken the protein folding prediction problem to the next step; however, it has several limitations. AF2 instigated the emergence of new tools, such as ESMfold, which, although inferior in accuracy, use different approaches, which enable very fast predictions. | |
| Buel et al., 2022 [80] | to study the ability of AF2 to predict the effect of missense mutations on structure | AF2 seems not to be able to predict the effect of missense mutations on the 3D structure of proteins. Differences between mutated and wild-type structures predicted by AlphaFold were extremely small. | |
| Pak et al., 2021 [81] | to evaluate the ability of AlphaFold to predict the impact of single mutations on protein stability | It seems impossible to obtain a reliable evaluation of the impact of mutation on protein stability with the direct application of AI predictions. |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gutnik, D.; Evseev, P.; Miroshnikov, K.; Shneider, M. Using AlphaFold Predictions in Viral Research. Curr. Issues Mol. Biol. 2023, 45, 3705-3732. https://doi.org/10.3390/cimb45040240
Gutnik D, Evseev P, Miroshnikov K, Shneider M. Using AlphaFold Predictions in Viral Research. Current Issues in Molecular Biology. 2023; 45(4):3705-3732. https://doi.org/10.3390/cimb45040240
Chicago/Turabian StyleGutnik, Daria, Peter Evseev, Konstantin Miroshnikov, and Mikhail Shneider. 2023. "Using AlphaFold Predictions in Viral Research" Current Issues in Molecular Biology 45, no. 4: 3705-3732. https://doi.org/10.3390/cimb45040240
APA StyleGutnik, D., Evseev, P., Miroshnikov, K., & Shneider, M. (2023). Using AlphaFold Predictions in Viral Research. Current Issues in Molecular Biology, 45(4), 3705-3732. https://doi.org/10.3390/cimb45040240