Role of Kinins in Hypertension and Heart Failure

Abstract
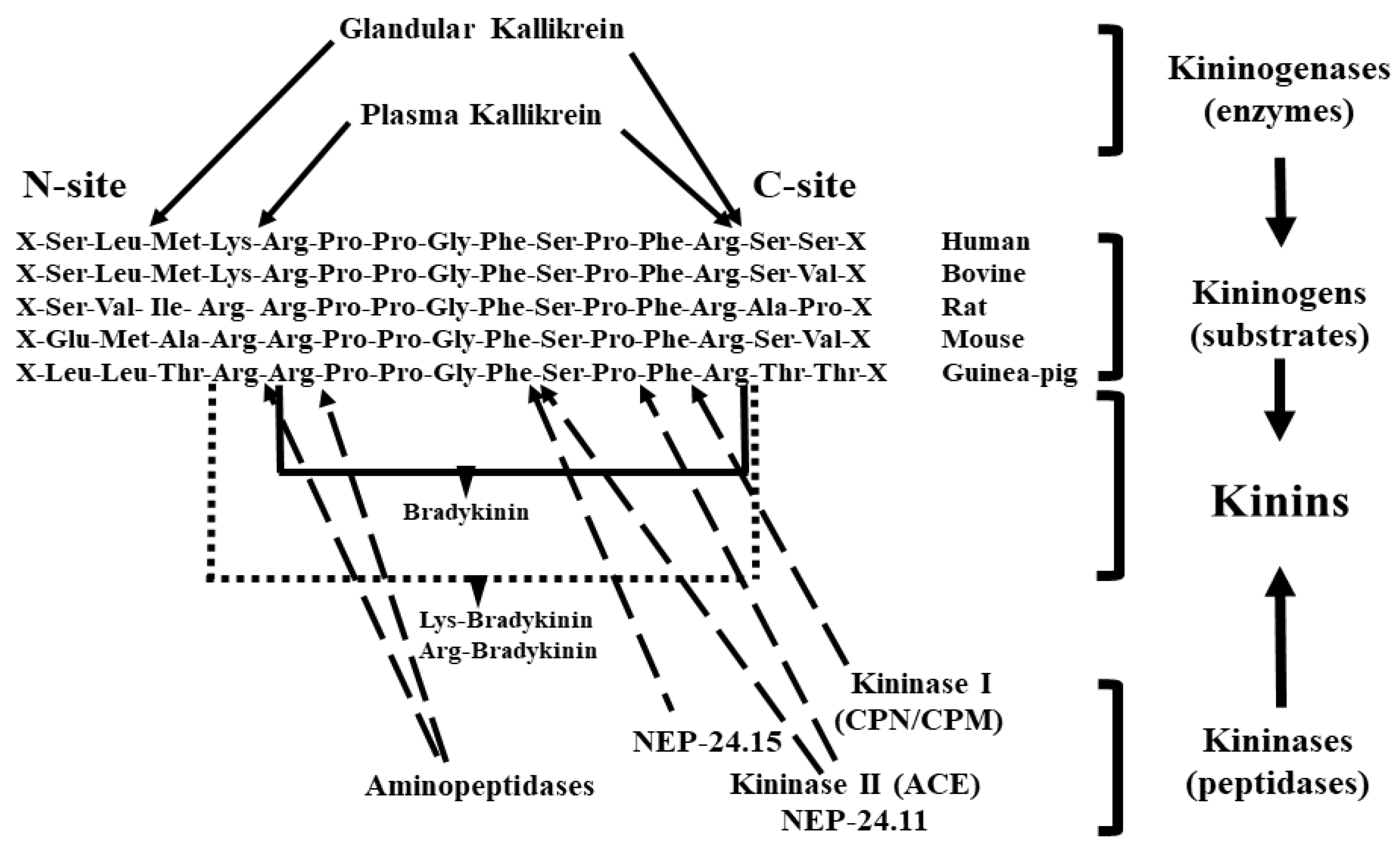
1. Kallikrein–Kinin System
1.1. Tissue (Glandular) Kallikrein
1.2. Kininogens or Kallikrein Substrates
1.3. Kinins
1.4. Kininases
1.5. Receptors
2. The KKS in the Vasculature and Regulation of Local Blood Flow
3. Kinins in Regulation of Cardiac and Renal Blood Flow
4. Kinins in Blood Pressure Regulation and the Pathogenesis of Hypertension
5. Role of Kinins in Thermoregulation
6. KKS Versus SARS-CoV2 in COVID-19 Patients
7. Role of Kinins in the Therapeutic Effect of ACE Inhibitors and Angiotensin Receptor Blockers (ARBs)
8. Role of Kinins in the Effects of ACE Inhibitors on Hypertensive Target Organ Damage and in Heart Failure Post-MI
9. Role of Kinins in the Cardioprotective Effect of ARBs
10. Material and Methods
10.1. For Renovascular Hypertension in B2-/- Mice
10.2. Induction of 2K-1C Hypertension
10.3. Systolic Blood Pressure (SBP)
10.4. Data Analysis
11. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Carretero, O.A. Kinins: Local hormones in regulation of blood pressure and renal function. Choices Cardiol. 1993, 7 (Suppl. 1), 10–14. [Google Scholar]
- Rhaleb, N.-E.; Yang, X.P.; Carretero, O.A. The kallikrein-kinin system as a regulator of cardiovascular and renal function. In Comprehensive Physiology; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2011; pp. 971–993. [Google Scholar]
- Carretero, O.A.; Scicli, A.G. Kinins paracrine hormone. Kidney Int. Suppl. 1988, 26, S52–S59. [Google Scholar] [PubMed]
- Yu, H.; Bowden, D.W.; Spray, B.J.; Rich, S.S.; Freedman, B.I. Identification of human plasma kallikrein gene polymorphisms and evaluation of their role in end-stage renal disease. Hypertension 1998, 31, 906–911. [Google Scholar] [PubMed]
- Yu, H.; Anderson, P.J.; Freedman, B.I.; Rich, S.S.; Bowden, D.W. Genomic structure of the human plasma prekallikrein gene, identification of allelic variants, and analysis in end-stage renal disease. Genomics 2000, 69, 225–234. [Google Scholar] [PubMed]
- Lu, X.; Zhao, W.; Huang, J.; Li, H.; Yang, W.; Wang, L.; Huang, W.; Chen, S.; Gu, D. Common variation in KLKB1 and essential hypertension risk: Tagging-SNP haplotype analysis in a case-control study. Hum. Genet. 2007, 121, 327–335. [Google Scholar] [PubMed]
- Scicli, A.G.; Mindroiu, T.; Scicli, G.; Carretero, O.A. Blood kinins, their concentration in normal subjects and in patients with congenital deficiency in plasma prekallikrein and kininogen. J. Lab. Clin. Med. 1982, 100, 81–93. [Google Scholar]
- Colman, R.W. Patho-physiology of kallikrein system. Ann. Clin. Lab. Sci. 1980, 10, 220–226. [Google Scholar]
- Kaplan, A.P.; Silverberg, M. The coagulation-kinin pathway of human plasma. Blood 1987, 70, 1–15. [Google Scholar]
- Sundsmo, J.S.; Fair, D.S. Relationships among the complement, kinin, coagulation and fibrinolytic systems in the inflammatory reaction. Clin. Physiol. Biochem. 1983, 1, 225–284. [Google Scholar]
- Couture, R.; Blaes, N.; Girolami, J.P. Kinin receptors in vascular biology and pathology. Curr. Vasc. Pharmacol. 2014, 12, 223–248. [Google Scholar]
- Clements, J.A. The glandular kallikrein family of enzymes: Tissue-specific expression and hormonal regulation. Endocr. Rev. 1989, 10, 393–419. [Google Scholar] [CrossRef]
- Boucher, R.; Demassieux, S.; Garcia, R.; Genest, J. Tonin, angiotensin II system. Circ. Res. 1977, 41, 26–29. [Google Scholar] [PubMed]
- Lilja, H. A kallikrein-like serine protease in prostatic fluid cleaves the predominant seminal vesicle protein. J. Clin. Investig. 1985, 76, 1899–1903. [Google Scholar] [CrossRef] [PubMed]
- Carbini, L.A.; Scicli, A.G.; Carretero, O.A. The molecular biology of the kallikrein-kinin system: III. The human kallikrein gene family and kallikrein substrate. J. Hypertens. 1993, 11, 893–898. [Google Scholar] [CrossRef] [PubMed]
- Scicli, A.G.; Carbini, L.A.; Carretero, O.A. The molecular biology of the kallikrein-kinin system: II. The rat gene family. J. Hypertens. 1993, 11, 775–780. [Google Scholar] [CrossRef] [PubMed]
- Saed, G.M.; Carretero, O.A.; MacDonald, R.J.; Scicli, A.G. Kallikrein messenger RNA in rat arteries and veins. Circ. Res. 1990, 67, 510–516. [Google Scholar] [PubMed]
- Nolly, H.; Saed, G.; Carretero, O.A.; Scicli, G.; Scicli, A.G. Adrenal kallikrein. Hypertension 1993, 21, 911–915. [Google Scholar]
- Nolly, H.; Scicli, A.G.; Scicli, G.; Carretero, O.A. Characterization of a kininogenase from rat vascular tissue resembling tissue kallikrein. Circ. Res. 1985, 56, 816–821. [Google Scholar]
- Nolly, H.; Carbini, L.A.; Scicli, G.; Carretero, O.A.; Scicli, A.G. A local kallikrein-kinin system is present in rat hearts. Hypertension 1994, 23, 919–923. [Google Scholar]
- Chao, J.; Chao, L.; Swain, C.C.; Tsai, J.; Margolius, H.S. Tissue kallikrein in rat brain and pituitary: Regional distribution and estrogen induction in the anterior pituitary. Endocrinology 1987, 120, 475–482. [Google Scholar]
- Chao, J.; Chao, L.; Margolius, H.S. Isolation of tissue kallikrein in rat spleen by monoclonal antibody-affinity chromatography. Biochim. Biophys. Acta 1984, 801, 244–249. [Google Scholar] [CrossRef]
- Scicli, G.; Nolly, H.; Carretero, O.A.; Scicli, A.G. Glandular kallikrein-like enzyme in adrenal glands. In Kinins V—Part B, 247B ed.; Plenum Press: New York, NY, USA, 1989; pp. 217–222. [Google Scholar]
- Clements, J.A.; Matheson, B.A.; MacDonald, R.J.; Funder, J.W. The expression of the kallikrein gene family in the rat pituitary: Oestrogen effects and the expression of an additional family member in the neurointermediate lobe. J. Neuroendocrinol. 1989, 1, 199–203. [Google Scholar] [CrossRef] [PubMed]
- Powers, C.A.; Nasjletti, A. A major sex difference in kallikrein-like activity in the rat anterior pituitary. Endocrinology 1984, 114, 1841–1844. [Google Scholar] [CrossRef] [PubMed]
- Frey, E.K.; Kraut, H.; Werle, E. Kallikrein Padutin; Vogel, R., Ed.; Ferdinand Enke Verlag: Stuttgart, Germany, 1950. [Google Scholar]
- Zimmermann, A.; Geiger, R.; Kortmann, H. Similarity between a kininogenase (kallikrein) from human large intestine and human urinary kallikrein. Hoppe-Seylers Z. Physiol. Chem. 1979, 360, 1767–1773. [Google Scholar] [CrossRef] [PubMed]
- Schachter, M.; Longridge, D.J.; Wheeler, G.D.; Mehta, J.G.; Uchida, Y. Immunocytochemical and enzyme histochemical localization of kallikrein-like enzymes in colon, intestine, and stomach of rat and cat. J. Histochem. Cytochem. 1986, 34, 927–934. [Google Scholar] [CrossRef]
- Hilton, S.M. The physiological role of glandular kallikreins. In Handbook of Experimental Pharmacology, 25th ed.; Vol 25: Bradykinin, Kallidin and Kallikrein; Springer: New York, NY, USA, 1970; Volume 25, pp. 389–399. [Google Scholar]
- Rabito, S.F.; Scicli, A.G.; Carretero, O.A. Immunoreactive glandular kallikrein in plasma. In Enzymatic Release of Vasoactive Peptides; Gross, F., Vogel, G., Eds.; Raven Press: New York, NY, USA, 1980; pp. 247–256. [Google Scholar]
- Rabito, S.F.; Scicli, A.G.; Kher, V.; Carretero, O.A. Immunoreactive glandular kallikrein in rat plasma: A radioimmunoassay for its determination. Am. J. Physiol. 1982, 242, H602–H610. [Google Scholar] [CrossRef] [PubMed]
- Geiger, R.; Stuckstedte, U.; Fritz, H. Isolation and characterization of human urinary kallikrein. Hoppe Seylers. Z. Physiol. Chem. 1980, 361, 1003–1016. [Google Scholar] [CrossRef]
- Scicli, A.G.; írstavik, T.B.; Rabito, S.F.; Murray, R.D.; Carretero, O.A. Blood kinins after sympathetic nerve stimulation of the rat submandibular gland. Hypertension 1983, 5 (Suppl. I), I-101. [Google Scholar] [CrossRef]
- Pisano, J.J.; Corthorn, J.; Yates, K.; Pierce, J.V. The kallikrein-kinin system in the kidney. Contrib. Nephrol. 1978, 12, 116–125. [Google Scholar]
- Omata, K.; Carretero, O.A.; Itoh, S.; Scicli, A.G. Active and inactive kallikrein in rabbit connecting tubules and urine during low and normal sodium intake. Kidney Int. 1983, 24, 714–718. [Google Scholar] [CrossRef]
- Noda, Y.; Yamada, K.; Igic, R.; Erdös, E.G. Regulation of rat urinary and renal kallikrein and prekallikrein by corticosteroids. Proc. Natl. Acad. Sci. USA 1983, 80, 3059–3063. [Google Scholar] [CrossRef] [PubMed]
- Alhenc-Gelas, F.; Marchetti, J.; Allegrini, J.; Corvol, P.; Menard, J. Measurement of urinary kallikrein activity. Species differences in kinin production. Biochim. Biophys. Acta 1981, 677, 477–488. [Google Scholar] [CrossRef]
- Mindroiu, T.; Scicli, G.; Perini, F.; Carretero, O.A.; Scicli, A.G. Identification of a new kinin in human urine. J. Biol. Chem. 1986, 261, 7407–7411. [Google Scholar] [PubMed]
- Jacobsen, S. Substrates for plasma kinin-forming enzymes in human, dog and rabbit plasmas. Br. J. Pharmacol. 1966, 26, 403–411. [Google Scholar] [CrossRef] [PubMed]
- Merkulov, S.; Zhang, W.M.; Komar, A.A.; Schmaier, A.H.; Barnes, E.; Zhou, Y.; Lu, X.; Iwaki, T.; Castellino, F.J.; Luo, G.; et al. Deletion of murine kininogen gene 1 (mKng1) causes loss of plasma kininogen and delays thrombosis. Blood 2008, 111, 1274–1281. [Google Scholar] [CrossRef]
- Adam, A.; Damas, J.; Calay, G.; Renard, C.; Remacle-Volon, G.; Bourdon, V. Quantification of rat T-kininogen using immunological methods Application to inflammatory processes. Biochem. Pharmacol. 1989, 38, 1569–1575. [Google Scholar] [CrossRef]
- Chen, H.-M.; Considine, K.B.; Liao, W.S.L. Interleukin-6 responsiveness and cell-specific expression of the rat kininogen gene. J. Biol. Chem. 1991, 266, 2946–2952. [Google Scholar]
- DeLa Cadena, R.A.; Laskin, K.J.; Pixley, R.A.; Sartor, R.B.; Schwab, J.H.; Back, N.; Bedi, G.S.; Fisher, R.S.; Colman, R.W. Role of kallikrein-kinin system in pathogenesis of bacterial cell wall-induced inflammation. Am. J. Physiol. 1991, 260, G213–G219. [Google Scholar] [CrossRef]
- Okamoto, H.; Hatta, A.; Itoh, N.; Ohashi, Y.; Arakawa, K.; Nakanishi, S. Acute phase responses of plasma angiotensinogen and T-kininogen in rats. Biochem. Pharmacol. 1987, 36, 3069–3073. [Google Scholar] [CrossRef]
- Kaplan, A.P.; Silverberg, M.; Ghebrehiwet, B.; Atkins, P.; Zweiman, B. The kallikrein-kinin system in inflammation. Adv. Exp. Med. Biol. 1989, 247, 125–136. [Google Scholar]
- Campbell, D.J.; Kladis, A.; Duncan, A.-M. Bradykinin peptides in kidney, blood, and other tissues of the rat. Hypertension 1993, 21, 155–165. [Google Scholar] [CrossRef]
- Cherry, P.D.; Furchgott, R.F.; Zawadzki, J.V.; Jothianandan, D. Role of endothelial cells in relaxation of isolated arteries by bradykinin. Proc. Natl. Acad. Sci. USA 1982, 79, 2106–2110. [Google Scholar] [CrossRef] [PubMed]
- Vane, J.R.; Änggård, E.E.; Botting, R.M. Regulatory functions of the vascular endothelium. N. Engl. J. Med. 1990, 323, 27–36. [Google Scholar] [PubMed]
- Tiffany, C.W.; Burch, R.M. Bradykinin stimulates tumor necrosis factor and interleukin-1 release from macrophages. FEBS Lett. 1989, 247, 189–192. [Google Scholar] [CrossRef]
- Smith, D.; Gilbert, M.; Owen, W.G. Tissue plasminogen activator release in vivo in response to vasoactive agents. Blood 1985, 66, 835–839. [Google Scholar] [CrossRef]
- Erdös, E. Kininases. In Handbook of Experimental Pharmacology; Erdös, E.G., Ed.; Vol. XXV Suppl: Bradykinin, Kallidin and Kallikrein; Springer: Berlin/Heidelberg, Germany, 1979; pp. 427–487. [Google Scholar]
- Erdös, E.G. Angiotensin I converting enzyme. Circ. Res. 1975, 36, 247–255. [Google Scholar] [CrossRef] [PubMed]
- Skidgel, R.A.; Schulz, W.W.; Tam, L.-T.; Erdös, E.G. Human renal angiotensin I converting enzyme and neutral endopeptidase. Kidney Int. 1987, 31 (Suppl. 20), S-45–S-48. [Google Scholar]
- Vijayaraghavan, J.; Scicli, A.G.; Carretero, O.A.; Slaughter, C.; Moomaw, C.; Hersh, L.B. The hydrolysis of endothelins by neutral endopeptidase 24.11 (enkephalinase). J. Biol. Chem. 1990, 265, 14150–14155. [Google Scholar]
- Ura, N.; Carretero, O.A.; Erdös, E.G. Role of renal endopeptidase 24.11 in kinin metabolism in vitro and in vivo. Kidney Int. 1987, 32, 507–513. [Google Scholar] [CrossRef]
- Ishida, H.; Scicli, A.G.; Carretero, O.A. Role of angiotensin converting enzyme and other peptidases in in vivo metabolism of kinins. Hypertension 1989, 14, 322–327. [Google Scholar] [CrossRef]
- Ndjekembo Shango, D.; Simonet, O.; Vallot, F.; Defendi, F.; De Kock, M. ACE-inhibition and bradykinin-mediated angioedema. Rev. Med. Liege 2020, 75, 151–153. [Google Scholar]
- Regoli, D.; Gobeil, F. Kallikrein-kinin system as the dominant mechanism to counteract hyperactive renin-angiotensin system. Can. J. Physiol. Pharmacol. 2017, 95, 1117–1124. [Google Scholar] [CrossRef] [PubMed]
- Regoli, D.; Barabe, J. Pharmacology of bradykinin and related kinins. Pharmacol. Rev. 1980, 32, 1–46. [Google Scholar] [PubMed]
- Regoli, D.; Drapeau, G.; Rovero, P.; Dion, S.; Rhaleb, N.-E.; Barabé, J.; D‘Orléans-Juste, P.; Ward, P. Conversion of kinins and their antagonists into B1 receptor activators and blockers in isolated vessels. Eur. J. Pharmacol. 1986, 127, 219–224. [Google Scholar] [CrossRef]
- Sörensen-Zender, I.; Chen, R.; Rong, S.; David, S.; Melk, A.; Haller, H.; Schmitt, R. Binding to carboxypeptidase M mediates protective effects of fibrinopeptide Bβ(15-42). Transl. Res. J. Lab. Clin. Med. 2019, 213, 124–135. [Google Scholar]
- Guimarães, P.B.; da Silva, R.F.; Hoff, C.C.; Fernandes, L.; Nakaie, C.R.; Chagas, J.R.; Carmona, A.K.; Bader, M.; Pesquero, J.B. Interactions between carboxypeptidase M and kinin B1 receptor in endothelial cells. Inflamm. Res. Off. J. Eur. Histamine Res. Soc. 2019, 68, 845–855. [Google Scholar] [CrossRef] [PubMed]
- Regoli, D. Pharmacology of bradykinin and related kinins. Adv. Exp. Med. Biol. 1983, 156, 569–584. [Google Scholar]
- Regoli, D.; Rhaleb, N.-E.; Drapeau, G.; Dion, S.; Tousignant, C.; D‘Orl:eans-Juste, P.; Devillier, P. Basic pharmacology of kinins: Pharmacologic receptors and other mechanisms. Adv. Exp. Med. Biol. 1989, 247, 399–407. [Google Scholar]
- McEachern, A.E.; Shelton, E.R.; Bhakta, S.; Obernolte, R.; Bach, C.; Zuppan, P.; Fujisaki, J.; Aldrich, R.W.; Jarnagin, K. Expression cloning of a rat B2 bradykinin receptor. Proc. Natl. Acad. Sci. USA 1991, 88, 7724–7728. [Google Scholar] [CrossRef]
- Regoli, D.; Jukic, D.; Gobell, F.; Rhaleb, N.-E. Receptor for bradykinin and related kinin: A critical analysis. Can. J. Physiol. Pharmacol. 1993, 71, 556–567. [Google Scholar] [CrossRef]
- Hecquet, C.; Tan, F.; Marcic, B.M.; Erdos, E.G. Human bradykinin B(2) receptor is activated by kallikrein and other serine proteases. Mol. Pharmacol. 2000, 58, 828–836. [Google Scholar] [CrossRef]
- Abadir, P.M.; Periasamy, A.; Carey, R.M.; Siragy, H.M. Angiotensin II type 2 receptor-bradykinin B2 receptor functional heterodimerization. Hypertension 2006, 48, 316–322. [Google Scholar] [CrossRef] [PubMed]
- Barki-Harrington, L.; Bookout, A.L.; Wang, G.; Lamb, M.E.; Leeb-Lundberg, L.M.; Daaka, Y. Requirement for direct cross-talk between B1 and B2 kinin receptors for the proliferation of androgen-insensitive prostate cancer PC3 cells. Biochem. J. 2003, 371, 581–587. [Google Scholar] [CrossRef] [PubMed]
- Sabatini, R.A.; Guimaraes, P.B.; Fernandes, L.; Reis, F.C.; Bersanetti, P.A.; Mori, M.A.; Navarro, A.; Hilzendeger, A.M.; Santos, E.L.; Andrade, M.C.; et al. ACE activity is modulated by kinin B2 receptor. Hypertension 2008, 51, 689–695. [Google Scholar] [CrossRef] [PubMed]
- AbdAlla, S.; Zaki, E.; Lother, H.; Quitterer, U. Involvement of the amino terminus of the B(2) receptor in agonist-induced receptor dimerization. J. Biol. Chem. 1999, 274, 26079–26084. [Google Scholar] [CrossRef] [PubMed]
- Oza, N.B.; Schwartz, J.H.; Goud, H.D.; Levinsky, N.G. Rat aortic smooth muscle cells in culture express kallikrein, kininogen, and bradykininase activity. J. Clin. Investig. 1990, 85, 597–600. [Google Scholar] [CrossRef]
- Bergaya, S.; Meneton, P.; Bloch-Faure, M.; Mathieu, E.; Alhenc-Gelas, F.; Levy, B.I.; Boulanger, C.M. Decreased flow-dependent dilation in carotid arteries of tissue kallikrein-knockout mice. Circ. Res. 2001, 88, 593–599. [Google Scholar] [CrossRef] [PubMed]
- Meneton, P.; Bloch-Faure, M.; Hagege, A.A.; Ruetten, H.; Huang, W.; Bergaya, S.; Ceiler, D.; Gehring, D.; Martins, I.; Salmon, G.; et al. Cardiovascular abnormalities with normal blood pressure in tissue kallikrein-deficient mice. Proc. Natl. Acad. Sci. USA 2001, 98, 2634–2639. [Google Scholar] [CrossRef]
- Azizi, M.; Boutouyrie, P.; Bissery, A.; Agharazii, M.; Verbeke, F.; Stern, N.; Bura-Riviere, A.; Laurent, S.; Ahenc-Gelas, F.; Jeunemaitre, X. Arterial and renal consequences of partial genetic deficiency in tissue kallikrein activity in humans. J. Clin. Investig. 2005, 115, 780–787. [Google Scholar] [CrossRef]
- Berg, T.; Carretero, O.A.; Scicli, A.G.; Tilley, B.; Stewart, J.M. Role of kinin in regulation of rat submandibular gland blood flow. Hypertension 1989, 14, 73–80. [Google Scholar] [CrossRef]
- Seino, M.; Carretero, O.A.; Albertini, R.; Scicli, A.G. Kinins in regulation of uteroplacental blood flow in the pregnant rabbit. Am. J. Physiol. 1982, 242, H142–H147. [Google Scholar] [CrossRef]
- Roman, R.J.; Kaldunski, M.L.; Scicli, A.G.; Carretero, O.A. Influence of kinins and angiotensin II on the regulation of papillary blood flow. Am. J. Physiol. 1988, 255, F690–F698. [Google Scholar] [CrossRef] [PubMed]
- Seino, M.; Abe, K.; Nushiro, N.; Omata, K.; Kasai, Y.; Yoshinaga, K. Effects of a competitive antagonist of bradykinin on blood pressure and renal blood flow in anesthetized rats. J. Hypertens. 1988, 6, 867–871. [Google Scholar] [CrossRef] [PubMed]
- Beierwaltes, W.H.; Carretero, O.A.; Scicli, A.G. Renal hemodynamics in response to a kinin analogue antagonist. Am. J. Physiol. 1988, 255, F408–F414. [Google Scholar] [CrossRef] [PubMed]
- Zimmerman, B.G.; Raich, P.C.; Vavrek, R.J.; Stewart, J.M. Bradykinin contribution to renal blood flow effect of angiotensin converting enzyme inhibitor in the conscious sodium-restricted dog. Circ. Res. 1990, 66, 234–240. [Google Scholar] [CrossRef]
- Nakagawa, M.; Nasjletti, A. Renal function as affected by inhibitors of kininase II and of neutral endopeptidase 24.11 in rats with and without desoxycorticosterone pretreatment. Adv. Exp. Med. Biol. 1989, 247, 495–499. [Google Scholar]
- Omoro, S.A.; Majid, D.S.A.; El-Dahr, S.S.; Navar, L.G. Kinin influences on renal regional blood flow responses to angiotensin-converting enzyme inhibition in dogs. Am. J. Physiol. 1999, 276, F271–F277. [Google Scholar] [CrossRef]
- Tomiyama, H.; Scicli, A.G.; Scicli, G.M.; Carretero, O.A. Renal effects of Fab fragments of kinin antibodies on deoxycorticosterone acetate-salt-treated rats. Hypertension 1990, 15, 761–766. [Google Scholar] [CrossRef]
- Ruocco, N.A., Jr.; Bergelson, B.A.; Yu, T.-K.; Gavras, I.; Gavras, H. Augmentation of coronary blood flow by ACE inhibition: Role of angiotensin and bradykinin. Clin. Exp. Hypertens. 1995, 17, 1059–1072. [Google Scholar] [CrossRef]
- Magrini, F.; Shimizu, M.; Roberts, N.; Fouad, F.M.; Tarazi, R.C.; Zanchetti, A. Converting-enzyme inhibition and coronary blood flow. Circulation 1987, 75, I168–I174. [Google Scholar]
- Westermann, D.; Schultheiss, H.P.; Tschope, C. New perspective on the tissue kallikrein-kinin system in myocardial infarction: Role of angiogenesis and cardiac regeneration. Int. Immunopharmacol. 2008, 8, 148–154. [Google Scholar] [CrossRef]
- Xu, J.; Carretero, O.A.; Zhu, L.; Shesely, E.G.; Rhaleb, N.E.; Dai, X.; Wang, L.; Yang, J.J.; Yang, X.P. Protective role of AT(2) and B(1) receptors in kinin B(2)-receptor-knockout mice with myocardial infarction. Clin. Sci. 2013, 124, 87–96. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.H.; Yang, X.P.; Shesely, E.G.; Sankey, S.S.; Carretero, O.A. Role of angiotensin II type 2 receptors and kinins in the cardioprotective effect of angiotensin II type 1 receptor antagonists in rats with heart failure. J. Am. Coll. Cardiol. 2004, 43, 1473–1480. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.H.; Yang, X.P.; Mehta, D.; Bulagannawar, M.; Scicli, G.M.; Carretero, O.A. Role of kinins in chronic heart failure and in the therapeutic effect of ACE inhibitors in kininogen-deficient rats. Am. J. Physiol. Heart Circ. Physiol. 2000, 278, H507–H514. [Google Scholar] [CrossRef]
- Liu, Y.H.; Yang, X.P.; Sharov, V.G.; Nass, O.; Sabbah, H.N.; Peterson, E.; Carretero, O.A. Effects of angiotensin-converting enzyme inhibitors and angiotensin II type 1 receptor antagonists in rats with heart failure. Role of kinins and angiotensin II type 2 receptors. J. Clin. Investig. 1997, 99, 1926–1935. [Google Scholar] [CrossRef] [PubMed]
- Carretero, O.A.; Roman, R.; Beierwaltes, W.H.; Madeddu, P.; Scicli, A.G. Kinins as Paracrine Renal Hormones. In Renal Function, Hypertension and Kallikrein-Kinin System, Proceedings of the Symposium on Renal Function, Hypertension and Kallikrein-Kinin System, Sapporo, Japan, 7–9 December 1987; Iimura, O., Margolius, H.S., Eds.; University of Tokyo Press: Tokyo, Japan, 1988; p. 25. [Google Scholar]
- Parratt, J.R.; Vegh, A.; Papp, J.G. Bradykinin as an endogenous myocardial protective substance with particular reference to ischemic preconditioning—A brief review of the evidence. Can. J. Physiol. Pharmacol. 1995, 73, 837–842. [Google Scholar] [CrossRef]
- Yang, X.P.; Liu, Y.H.; Scicli, G.M.; Webb, C.R.; Carretero, O.A. Role of kinins in the cardioprotective effect of preconditioning. Study of myocardial ischemia/reperfusion injury in B2 kinin receptor knockout mice and kininogen-deficient rats. Hypertension 1997, 30, 735–740. [Google Scholar] [CrossRef]
- Yang, X.P.; Liu, Y.H.; Mehta, D.; Cavasin, M.A.; Shesely, E.; Xu, J.; Liu, F.; Carretero, O.A. Diminished cardioprotective response to inhibition of angiotensin-converting enzyme and angiotensin II type 1 receptor in B(2) kinin receptor gene knockout mice. Circ. Res. 2001, 88, 1072–1079. [Google Scholar] [CrossRef]
- Sinaiko, A.R.; Glasser, R.J.; Gillum, R.F.; Prineas, R.J. Urinary kallikrein excretion in grade school children with high and low blood pressure. J. Pediatrics 1982, 100, 938–940. [Google Scholar] [CrossRef]
- Uchiyama, M.; Otsuka, T.; Sakai, K. Urinary kallikrein excretion in children of parents with essential hypertension. Arch. Dis. Child. 1985, 60, 974–975. [Google Scholar] [CrossRef]
- Wollheim, E.; Peterknecht, S.; Dees, C.; Wiener, A.; Wollheim, C.B. Defect in the excretion of a vasoactive polypeptide fraction A possible genetic marker of primary hypertension. Hypertension 1981, 3, 574–579. [Google Scholar] [CrossRef] [PubMed]
- Zinner, S.H.; Margolius, H.S.; Rosner, B.; Keiser, H.R.; Kass, E.H. Familial aggregation of urinary kallikrein concentration in childhood: Relation to blood pressure, race and urinary electrolytes. Am. J. Epidemiol. 1976, 104, 124–132. [Google Scholar] [CrossRef] [PubMed]
- Pravenec, M.; Kren, V.; Kunes, J.; Scicli, A.G.; Carretero, O.A.; Simonet, L.; Kurtz, T.W. Cosegregation of blood pressure with a kallikrein gene family polymorphism. Hypertension 1991, 17, 242–246. [Google Scholar] [CrossRef] [PubMed]
- Carretero, O.A.; Amin, V.M.; Ocholik, T.; Scicli, A.G.; Koch, J. Urinary kallikrein in rats bred for their susceptibility and resistance to the hypertensive effect of salt. A new radioimmunoassay for its direct determination. Circ. Res. 1978, 42, 727–731. [Google Scholar] [CrossRef]
- Carretero, O.A.; Polomski, C.; Hampton, A.; Scicli, A.G. Urinary kallikrein, plasma renin and aldosterone in New Zealand genetically hypertensive (GH) rats. Clin. Exp. Pharmacol. Physiol. 1976, 3, 55–59. [Google Scholar]
- Carretero, O.A.; Scicli, A.G.; Piwonska, A.; Koch, J. Urinary kallikrein in rats bred for susceptibility and resistance to the hypertensive effect of salt and in New Zealand genetically hypertensive rats. Mayo Clin. Proc. 1977, 52, 465–467. [Google Scholar]
- Keiser, H.R.; Geller, R.G.; Margolius, H.S.; Pisano, J.J. Urinary kallikrein in hypertensive animal models. Fed. Proc. 1976, 35, 199–202. [Google Scholar]
- Carretero, O.A.; Scicli, A.G. The renal kallikrein-kinin system in human and in experimental hypertension. Klin. Wochenschr. 1978, 56 (Suppl. I), 113–125. [Google Scholar] [CrossRef]
- Holland, O.B.; Chud, J.M.; Braunstein, H. Urinary kallikrein excretion in essential and mineralocorticoid hypertension. J. Clin. Investig. 1980, 65, 347–356. [Google Scholar] [CrossRef]
- Margolius, H.S.; Horwitz, D.; Pisano, J.J.; Keiser, H.R. Urinary kallikrein excretion in hypertensive man. Relationships to sodium intake and sodium-retaining steroids. Circ. Res. 1974, 35, 820–825. [Google Scholar] [CrossRef]
- Seino, M.; Abe, K.; Otsuka, Y.; Saito, T.; Irokawa, N.; Yasujima, M.; Ciba, S.; Yoshinaga, K. Urinary kallikrein excretion and sodium metabolism in hypertensive patients. Tohoku J. Exp. Med. 1975, 116, 359–367. [Google Scholar] [CrossRef]
- Sustarsic, D.L.; McPartland, R.P.; Rapp, J.P.; Schlager, G.; Tan, S.Y. Urinary kallikrein and urinary prostaglandin E2 in genetically hypertensive mice. Proc. Soc. Exp. Biol. Med. 1980, 163, 193–199. [Google Scholar] [CrossRef] [PubMed]
- Rhaleb, N.-E.; Yang, X.-P.; Peng, H.; Cavasin, M.A.; Liu, Y.H.; Yang, F.; Xu, J.; Carretero, O.A. Cardiovascular phenotype of male 129/SvEvTac, 129/SvJ and B2-KO mice [abstract]. FASEB J. 2001, 15, A101. [Google Scholar]
- Rhaleb, N.-E.; Yang, X.P.; Nanba, M.; Shesely, E.G.; Carretero, O.A. Effect of Chronic Blockade of the Kallikrein-Kinin System on the Development of Hypertension in Rats. Hypertension 2001, 37, 121–128. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Carretero, O.A.; Shesely, E.G.; Rhaleb, N.-E.; Yang, J.J.; Bader, M.; Yang, X.P. The kinin B1 receptor contributes to the cardioprotective effect of angiotensin-converting enzyme inhibitors and angiotensin receptor blockers in mice. Exp. Physiol. 2009, 94, 322–329. [Google Scholar] [CrossRef] [PubMed]
- Rhaleb, N.-E.; Peng, H.; Alfie, M.; Shesely, E.G.; Carretero, O.A. Effect of ACE inhibitor on DOCA-salt- and aortic coarctation-induced hypertension in mice. Do kinin B2 receptors play a role? Hypertension 1999, 33, 329–334. [Google Scholar] [CrossRef]
- Rhaleb, N.-E.; Yang, X.P.; Scicli, A.G.; Carretero, O.A. Role of kinins and nitric oxide in the antihypertrophic effect of ramipril. Hypertension 1994, 23, 865–868. [Google Scholar] [CrossRef]
- Majima, M.; Katori, M.; Hanazuka, M.; Mizogami, S.; Nakano, T.; Nakao, Y.; Mikami, R.; Uryu, H.; Okamura, R.; Mohsin, S.S.J.; et al. Suppression of rat deoxycorticosterone-salt hypertension by kallikrein-kinin system. Hypertension 1991, 17, 806–813. [Google Scholar] [CrossRef]
- Majima, M.; Yoshida, O.; Mihara, H.; Muto, T.; Mizogami, S.; Kuribayashi, Y.; Katori, M.; Oh-Ishi, S. High sensitivity to salt in kininogen-deficient Brown Norway Katholiek rats. Hypertension 1993, 22, 705–714. [Google Scholar] [CrossRef]
- Majima, M.; Mizogami, S.; Kuribayashi, Y.; Katori, M.; Oh-Ishi, S. Hypertension induced by a nonpressor dose of angiotensin II in kininogen-deficient rats. Hypertension 1994, 24, 111–119. [Google Scholar] [CrossRef]
- Madeddu, P.; Parpaglia, P.P.; Demontis, M.P.; Varoni, M.V.; Fattaccio, M.C.; Glorioso, N. Chronic inhibition of bradykinin B2-receptors enhances the slow vasopressor response to angiotensin II. Hypertension 1994, 23, 646–652. [Google Scholar] [CrossRef]
- Alfie, M.E.; Yang, X.P.; Hess, F.; Carretero, O.A. Salt-sensitive hypertension in bradykinin B2 receptor knockout mice. Biochem. Biophys. Res. Commun. 1996, 224, 625–630. [Google Scholar] [CrossRef]
- Cervenka, L.; Harrison-Bernard, L.M.; Dipp, S.; Primrose, G.; Imig, J.D.; El-Dahr, S.S. Early onset salt-sensitive hypertension in bradykinin B2 receptor null mice. Hypertension 1999, 34, 176–180. [Google Scholar] [CrossRef] [PubMed]
- Emanueli, C.; Maestri, R.; Corradi, D.; Marchione, R.; Minasi, A.; Tozzi, M.G.; Salis, M.B.; Straino, S.; Capogrossi, M.C.; Olivetti, G.; et al. Dilated and failing cardiomyopathy in bradykinin B2 receptor knockout mice. Circulation 1999, 100, 2359–2365. [Google Scholar] [CrossRef] [PubMed]
- Emanueli, C.; Fink, E.; Milia, A.F.; Salis, M.B.; Conti, M.; Demontis, M.P.; Madeddu, P. Enhanced blood pressure sensitivity to deoxycorticosterone in mice with disruption of bradykinin B2 receptor gene. Hypertension 1998, 31, 1278–1283. [Google Scholar] [CrossRef] [PubMed]
- Madeddu, P.; Milia, A.F.; Salis, M.B.; Gaspa, L.; Gross, W.; Lippoldt, A.; Emanueli, C. Renovascular hypertension in bradykinin B2-receptor knockout mice. Hypertension 1998, 32, 503–509. [Google Scholar] [CrossRef]
- Harrison-Bernard, L.M.; Dipp, S.; El-Dahr, S.S. Renal and blood pressure phenotype in 18 month-old bradykinin B2R-/-CRD mice. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2003, 285, 782–790. [Google Scholar] [CrossRef][Green Version]
- Milia, A.F.; Gross, V.; Plehm, R.; De, S.J., Jr.; Bader, M.; Luft, F.C. Normal blood pressure and renal function in mice lacking the bradykinin B(2) receptor. Hypertension 2001, 37, 1473–1479. [Google Scholar] [CrossRef]
- Trabold, F.; Pons, S.; Hagege, A.A.; Bloch-Faure, M.; Alhenc-Gelas, F.; Giudicelli, J.-F.; Richer-Giudicelli, C.; Meneton, P. Cardiovascular phenotypes of kinin B2 receptor- and tissue kallikrein-deficient mice. Hypertension 2002, 40, 90–95. [Google Scholar] [CrossRef]
- Kakoki, M.; Sullivan, K.A.; Backus, C.; Hayes, J.M.; Oh, S.S.; Hua, K.; Gasim, A.M.; Tomita, H.; Grant, R.; Nossov, S.B.; et al. Lack of both bradykinin B1 and B2 receptors enhances nephropathy, neuropathy, and bone mineral loss in Akita diabetic mice. Proc. Natl. Acad. Sci. USA 2010, 107, 10190–10195. [Google Scholar] [CrossRef]
- Regoli, D.; Plante, G.E.; Gobeil, F., Jr. Impact of kinins in the treatment of cardiovascular diseases. Pharmacol. Ther. 2012, 135, 94–111. [Google Scholar] [PubMed]
- Marceau, F.; Bachelard, H.; Bouthillier, J.; Fortin, J.P.; Morissette, G.; Bawolak, M.T.; Charest-Morin, X.; Gera, L. Bradykinin receptors: Agonists, antagonists, expression, signaling, and adaptation to sustained stimulation. Int. Immunopharmacol. 2020, 82, 106305. [Google Scholar] [PubMed]
- Feldberg, W.; Lewis, G.P. The action of peptides on the adrenal medulla. Release of adrenaline by bradykinin and angiotensin. J. Physiol. 1964, 171, 98–108. [Google Scholar] [PubMed]
- Starke, K.; Peskar, B.A.; Schumacher, K.A.; Taube, H.D. Bradykinin and postganglionic sympathetic transmission. Naunyn-Schmiedeberg’s Arch. Pharmacol. 1977, 299, 23–32. [Google Scholar]
- Rump, L.C.; Berlit, T.; Schwertfeger, E.; Beyersdorf, F.; Schollmeyer, P.; Bohmann, C. Angiotensin converting enzyme inhibition unmasks the sympathofacilitatory effect of bradykinin in human right atrium. J. Hypertens. 1997, 15, 1263–1270. [Google Scholar]
- Chulak, C.; Couture, R.; Foucart, S. Modulatory effect of bradykinin on noradrenaline release in isolated atria from normal and B2 knockout transgenic mice. Eur. J. Pharmacol. 1998, 346, 167–174. [Google Scholar]
- Kansui, Y.; Fujii, K.; Goto, K.; Abe, I. Bradykinin enhances sympathetic neurotransmission in rat blood vessels. Hypertension 2002, 39, 29–34. [Google Scholar]
- Farnell, G.S.; Pierce, K.E.; Collinsworth, T.A.; Murray, L.K.; Demes, R.N.; Juvancic-Heltzel, J.A.; Glickman, E.L. The influence of ethnicity on thermoregulation after acute cold exposure. Wilderness Environ. Med. 2008, 19, 238–244. [Google Scholar]
- Song, C.K.; Martinez, J.A.; Kailasam, M.T.; Dao, T.T.; Wong, C.M.; Parmer, R.J.; O’Connor, D.T. Renal kallikrein excretion: Role of ethnicity, gender, environment, and genetic risk of hypertension. J. Hum. Hypertens. 2000, 14, 461–468. [Google Scholar]
- Gainer, J.V.; Brown, N.J.; Bachvarova, M.; Bastien, L.; Maltais, I.; Marceau, F.; Bachvarov, D.R. Altered frequency of a promoter polymorphism of the kinin B2 receptor gene in hypertensive African-Americans. Am. J. Hypertens. 2000, 13, 1268–1273. [Google Scholar]
- Talbot, S.; De Brito Gariépy, H.; Saint-Denis, J.; Couture, R. Activation of kinin B1 receptor evokes hyperthermia through a vagal sensory mechanism in the rat. J. Neuroinflamm. 2012, 9, 214. [Google Scholar] [CrossRef]
- Manolis, A.S.; Manolis, A.A.; Manolis, T.A.; Apostolopoulos, E.J.; Papatheou, D.; Melita, H. COVID-19 infection and cardiac arrhythmias. Trends Cardiovasc. Med. 2020. [Google Scholar] [CrossRef] [PubMed]
- Bader, F.; Manla, Y.; Atallah, B.; Starling, R.C. Heart failure and COVID-19. Heart Fail. Rev. 2020, 27, 1–10. [Google Scholar]
- Andersen, K.G.; Rambault, A.; Lipkin, W.I.; Holmes, E.C.; Garry, R.F. The proximal origin of SARS-CoV-2. Nat. Med. 2020, 26, 450–452. [Google Scholar] [CrossRef] [PubMed]
- Aquila, I.; Sacco, M.A.; Abenavoli, L.; Malara, N.; Arena, V.; Grassi, S.; Ausania, F.; Boccuto, L.; Ricci, C.; Gratteri, S.; et al. SARS-CoV-2 Pandemic: Review of the Literature and Proposal for Safe Autopsy Practice. Arch. Pathol. Lab. Med. 2020, 144, 1048–1056. [Google Scholar] [PubMed]
- Anderson, D.E.; Tan, C.W.; Chia, W.N.; Young, B.E.; Linster, M.; Low, J.H.; Tan, Y.J.; Chen, M.I.; Smith, G.J.D.; Leo, Y.S.; et al. Lack of cross-neutralization by SARS patient sera towards SARS-CoV-2. Emerg. Microbes Infect. 2020, 9, 900–902. [Google Scholar]
- Amor, S.; Baker, D.; Khoury, S.J.; Schmierer, K.; Giovanonni, G. SARS-CoV-2 and Multiple Sclerosis: Not all immune depleting DMTs are equal or bad. Ann. Neurol. 2020, 87, 794–797. [Google Scholar] [CrossRef]
- Kuster, G.M.; Pfister, O.; Burkard, T.; Zhou, Q.; Twerenbold, R.; Haaf, P.; Widmer, A.F.; Osswald, S. SARS-CoV2: Should inhibitors of the renin-angiotensin system be withdrawn in patients with COVID-19? Eur. Heart J. 2020, 41, 1801–1803. [Google Scholar] [CrossRef]
- Zhang, H.; Penninger, J.M.; Li, Y.; Zhong, N.; Slutsky, A.S. Angiotensin-converting enzyme 2 (ACE2) as a SARS-CoV-2 receptor: Molecular mechanisms and potential therapeutic target. Intensive Care Med. 2020, 46, 586–590. [Google Scholar] [CrossRef]
- Hoffmann, M.; Kleine-Weber, H.; Schroeder, S.; Kruger, N.; Herrler, T.; Erichsen, S.; Schiergens, T.S.; Herrler, G.; Wu, N.H.; Nitsche, A.; et al. SARS-CoV-2 Cell Entry Depends on ACE2 and TMPRSS2 and Is Blocked by a Clinically Proven Protease Inhibitor. Cell 2020, 181, 271–280. [Google Scholar] [CrossRef]
- Danser, A.H.J.; Epstein, M.; Batlle, D. Renin-Angiotensin System Blockers and the COVID-19 Pandemic: At Present There Is No Evidence to Abandon Renin-Angiotensin System Blockers. Hypertension 2020, 75, 1382–1385. [Google Scholar] [CrossRef] [PubMed]
- Donoghue, M.; Wakimoto, H.; Maguire, C.T.; Acton, S.; Hales, P.; Stagliano, N.; Fairchild-Huntress, V.; Xu, J.; Lorenz, J.N.; Kadambi, V.; et al. Heart block, ventricular tachycardia, and sudden death in ACE2 transgenic mice with downregulated connexins. J. Mol. Cell. Cardiol. 2003, 35, 1043–1053. [Google Scholar] [CrossRef]
- Donoghue, M.; Hsieh, F.; Baronas, E.; Godbout, K.; Gosselin, M.; Stagliano, N.; Donovan, M.; Woolf, B.; Robison, K.; Jeyaseelan, R.; et al. A novel angiotensin-converting enzyme-related carboxypeptidase (ACE2) converts angiotensin I to angiotensin 1-9. Circ. Res. 2000, 87, E1–E9. [Google Scholar] [CrossRef]
- Meini, S.; Zanichelli, A.; Sbrojavacca, R.; Iuri, F.; Roberts, A.T.; Suffritti, C.; Tascini, C. Understanding the Pathophysiology of COVID-19: Could the Contact System Be the Key? Front. Immunol. 2020, 11, 2014. [Google Scholar] [CrossRef] [PubMed]
- Mahmudpour, M.; Roozbeh, J.; Keshavarz, M.; Farrokhi, S.; Nabipour, I. COVID-19 cytokine storm: The anger of inflammation. Cytokine 2020, 133, 155151. [Google Scholar] [CrossRef]
- Zhu, L.; Carretero, O.A.; Xu, J.; Wang, L.; Harding, P.; Rhaleb, N.E.; Yang, J.J.; Sumners, C.; Yang, X.P. Angiotensin II type 2 receptor-stimulated activation of plasma prekallikrein and bradykinin release: Role of SHP-1. Am. J. Physiol. Heart Circ. Physiol. 2012, 302, H2553–H2559. [Google Scholar] [CrossRef]
- Carretero, O.A.; Yang, X.P.; Rhaleb, N.-E. Kinins and Cardiovascular Disease. In Renin Angiotensin System and Cardiovascular Disease; Walmor, C.D., Edward, D.F., Eds.; Humana Press: Totowa, NJ, USA, 2010; pp. 151–185. [Google Scholar]
- Carretero, O.A.; Yang, X.-P.; Rhaleb, N.-E. The kallikrein-kinin system as a regulator of cardiovascular and renal function. In Hypertension: A Companion to Brenner and Rector’s the Kidney, 2nd ed.; Oparil, S., Weber, M.A., Eds.; Elsevier: Philadelphia, PA, USA, 2005; pp. 203–218. [Google Scholar]
- Van de Veerdonk, F.L.; Netea, M.G.; van Deuren, M.; van der Meer, J.W.; de Mast, Q.; Brüggemann, R.J.; van der Hoeven, H. Kallikrein-kinin blockade in patients with COVID-19 to prevent acute respiratory distress syndrome. ELife 2020, 9, e57555. [Google Scholar]
- Rhaleb, N.-E.; Gobeil, F.; Regoli, D. Non-selectivity of new bradykinin for bradykinin B1 for receptors. Pharmacol. Lett. 1992, 51, PL125–PL129. [Google Scholar]
- Carretero, O.A.; Kuk, P.; Piwonska, S.; Houle, J.A.; Marin-Grez, M. Role of the renin-angiotensin system in the pathogenesis of severe hypertension in rats. Circ. Res. 1971, 29, 654–663. [Google Scholar] [CrossRef]
- Marks, E.S.; Bing, R.F.; Thurston, H.; Swales, J.D. Vasodepressor property of the converting enzyme inhibitor captopril (SQ 14 225): The role of factors other than renin-angiotensin blockade in the rat. Clin. Sci. 1980, 58, 1–6. [Google Scholar] [CrossRef]
- Marcic, B.M.; Erdös, E.G. Protein kinase C and phosphatase inhibitors block the ability of angiotensin I-converting enzyme inhibitors to resensitize the receptor to bradykinin without altering the primary effects of bradykinin. J. Pharmacol. Exp. Ther. 2000, 294, 605–612. [Google Scholar]
- Cachofeiro, V.; Sakakibara, T.; Nasjletti, A. Kinins, nitric oxide, and the hypotensive effect of captopril and ramiprilat in hypertension. Hypertension 1992, 19, 138–145. [Google Scholar] [CrossRef] [PubMed]
- Carbonell, L.F.; Carretero, O.A.; Stewart, J.M.; Scicli, A.G. Effect of a kinin antagonist on the acute antihypertensive activity of enalaprilat in severe hypertension. Hypertension 1988, 11, 239–243. [Google Scholar] [CrossRef] [PubMed]
- Carretero, O.A.; Scicli, A.G. The kallikrein-kinin system as a regulator of cardiovascular and renal function. In Hypertension: Physiology, Diagnosis, and Management, 2nd ed.; Laragh, J.H., Brenner, B.M., Eds.; Raven Press: New York, NY, USA, 1995; pp. 983–999. [Google Scholar]
- Campbell, D.J. The kallikrein-kinin system in humans. Clin. Exp. Pharmacol. Physiol. 2001, 28, 1060–1065. [Google Scholar] [CrossRef] [PubMed]
- Clappison, B.H.; Anderson, W.P.; Johnston, C.I. Role of the kallikrein-kinin system in the renal effects of angiotensin-converting enzyme inhibition in anaesthetized dogs. Clin. Exp. Pharmacol. Physiol. 1981, 8, 509–513. [Google Scholar] [CrossRef]
- McCaa, R.E. Studies in vivo with angiotensin I converting enzyme (kininase II) inhibitors. Fed. Proc. 1979, 38, 2783–2787. [Google Scholar]
- Nasjletti, A.; Colina-Chourio, J.; McGiff, J.C. Disappearance of bradykinin in the renal circulation of dogs. Effects of kininase inhibition. Circ. Res. 1975, 37, 59–65. [Google Scholar] [CrossRef] [PubMed]
- Vinci, J.M.; Horwitz, D.; Zusman, R.M.; Pisano, J.J.; Catt, K.J.; Keiser, H.R. The effect of converting enzyme inhibition with SQ20,881 on plasma and urinary kinins, prostaglandin E and angiotensin II in hypertensive man. Hypertension 1979, 1, 416–426. [Google Scholar] [CrossRef]
- Carretero, O.A.; Miyazaki, S.; Scicli, A.G. Role of kinins in the acute antihypertensive effect of the converting enzyme inhibitor, captopril. Hypertension 1981, 3, 18–22. [Google Scholar] [CrossRef]
- Carretero, O.A.; Orstavik, T.B.; Rabito, S.F.; Scicli, A.G. Interference of converting enzyme inhibitors with the kallikrein-kinin system. In Clinical and Experimental Hypertension; Proceedings of the Symposium on Renin-Angiotension System: Biochemistry, Pharmacology, Clinical Aspects, 1983, 5th ed.; Ganten, D., Hackenthal, E., Hofbauer, K.G., Inagami, T., Eds.; Marcel Dekker, Inc.: New York, NY, USA, 1983; pp. 1277–1285. [Google Scholar]
- Carretero, O.A.; Scicli, A.G.; Maitra, S.R. Role of kinins in the pharmacological effects of converting enzyme inhibitors. In Angiotensin Converting Enzyme Inhibitors. Mechanisms of Action and Clinical Implications; Horovitz, Z.P., Ed.; Urban & Schwarzenberg: Baltimore, MD, USA, 1981; pp. 105–121. [Google Scholar]
- Danckwardt, L.; Shimizu, I.; Bönner, G.; Rettig, R.; Unger, T. Converting enzyme inhibition in kinin-deficient Brown Norway rats. Hypertension 1990, 16, 429–435. [Google Scholar] [CrossRef]
- Benetos, A.; Gavras, H.; Stewart, J.M.; Vavrek, R.J.; Hatinoglou, S.; Gavras, I. Vasodepressor role of endogenous bradykinin assessed by a bradykinin antagonist. Hypertension 1986, 8, 971–974. [Google Scholar] [CrossRef] [PubMed]
- Salgado, M.C.O.; Rabito, S.F.; Carretero, O.A. Blood kinin in one-kidney, one clip hypertensive rats. Hypertension 1986, 8 (Suppl. I), I-110. [Google Scholar]
- Pontieri, V.; Lopes, O.U.; Ferreira, S.H. Hypotensive effect of captopril. Role of bradykinin and prostaglandinlike substances. Hypertension 1990, 15 (Suppl. I), I-55–I-58. [Google Scholar] [CrossRef] [PubMed]
- Murphey, L.J.; Gainer, J.V.; Vaughan, D.E.; Brown, N.J. Angiotensin-converting enzyme insertion/deletion polymorphism modulates the human in vivo metabolism of bradykinin. Circulation 2000, 102, 829–832. [Google Scholar] [CrossRef]
- Overlack, A.; Stumpe, K.O.; Heck, I.; Ressel, C.; Kühnert, M.; Krück, F. Identification of angiotensin II- and kinin-dependent mechanisms in essential hypertension. In Hypertension: Mechanisms and Management; Philipp, T., Distler, A., Eds.; Springer: Berlin/Heidelberg, Germany, 1980; pp. 183–191. [Google Scholar]
- Gainer, J.V.; Morrow, J.D.; Loveland, A.; King, D.J.; Brown, N.J. Effect of bradykinin-receptor blockade on the response to angiotensin-converting-enzyme inhibitor in normotensive and hypertensive subjects. N. Engl. J. Med. 1998, 339, 1285–1292. [Google Scholar] [CrossRef]
- Bao, G.; Gohlke, P.; Qadri, F.; Unger, T. Chronic kinin receptor blockade attenuates the antihypertensive effect of ramipril. Hypertension 1992, 20, 74–79. [Google Scholar] [CrossRef]
- Nakagawa, M.; Nasjletti, A. Plasma kinin concentration in deoxycorticosterone-salt hypertension. Hypertension 1988, 11, 411–415. [Google Scholar] [CrossRef]
- Carretero, O.A. High-mineralocorticoid conditions: Kinins (paracrine hormones) in the regulation of renal function and blood pressure. In Progress in Endocrinology, Proceedings of the Ninth International Congress of Endocrinology, Nice 1992, Carnforth, Lancastershire, UK; Mornex, R., Jaffiol, C., Leclère, J., Eds.; Parthenon Publications Group: Lancashire, UK, 1993; pp. 536–540. [Google Scholar]
- Gohlke, P.; Linz, W.; Schölkens, B.A.; Kuwer, I.; Bartenbach, S.; Schnell, A.; Unger, T. Angiotensin-converting enzyme inhibition improves cardiac function. Role of bradykinin. Hypertension 1994, 23, 411–418. [Google Scholar] [CrossRef]
- Pradelles, P.; Frobert, Y.; Creminon, C.; Ivonine, H.; Frindel, E. Distribution of a negative regulator of haematopoietic stem cell proliferation (AcSDKP) and thymosin beta 4 in mouse tissues. FEBS Lett. 1991, 289, 171–175. [Google Scholar] [CrossRef]
- Pradelles, P.; Frobert, Y.; Creminon, C.; Liozon, E.; Masse, A.; Frindel, E. Negative regulator of pluripotent hematopoietic stem cell proliferation in human white blood cells and plasma as analysed by enzyme immunoassay. Biochem. Biophys. Res. Commun. 1990, 170, 986–993. [Google Scholar] [CrossRef]
- Cavasin, M.A.; Rhaleb, N.E.; Yang, X.P.; Carretero, O.A. Prolyl oligopeptidase is involved in release of the antifibrotic peptide Ac-SDKP. Hypertension 2004, 43, 1140–1145. [Google Scholar] [CrossRef] [PubMed]
- Cavasin, M.A.; Liao, T.D.; Yang, X.P.; Yang, J.J.; Carretero, O.A. Decreased endogenous levels of Ac-SDKP promote organ fibrosis. Hypertension 2007, 50, 130–136. [Google Scholar] [CrossRef]
- Myohanen, T.T.; Tenorio-Laranga, J.; Jokinen, B.; Vazquez-Sanchez, R.; Moreno-Baylach, M.J.; Garcia-Horsman, J.A.; Mannisto, P.T. Prolyl oligopeptidase induces angiogenesis both in vitro and in vivo in a novel regulatory manner. Br. J. Pharmacol. 2011, 163, 1666–1678. [Google Scholar] [CrossRef] [PubMed]
- Kumar, N.; Nakagawa, P.; Janic, B.; Romero, C.A.; Worou, M.E.; Monu, S.R.; Peterson, E.L.; Shaw, J.; Valeriote, F.; Ongeri, E.M.; et al. The anti-inflammatory peptide Ac-SDKP is released from thymosin-beta4 by renal meprin-alpha and prolyl oligopeptidase. Am. J. Physiol. Ren. Physiol. 2016, 310, F1026–F1034. [Google Scholar] [CrossRef] [PubMed]
- Azizi, M.; Rousseau, A.; Ezan, E.; Guyene, T.T.; Michelet, S.; Grognet, J.M.; Lenfant, M.; Corvol, P.; Menard, J. Acute angiotensin-converting enzyme inhibition increases the plasma level of the natural stem cell regulator N-acetyl-seryl-aspartyl-lysyl-proline. J. Clin. Investig. 1996, 97, 839–844. [Google Scholar] [CrossRef]
- Yang, F.; Yang, X.P.; Liu, Y.H.; Xu, J.; Cingolani, O.; Rhaleb, N.-E.; Carretero, O.A. Ac-SDKP reverses inflammation and fibrosis in rats with heart failure after myocardial infarction. Hypertension 2004, 43, 229–236. [Google Scholar] [CrossRef] [PubMed]
- Peng, H.; Carretero, O.A.; Liao, T.D.; Peterson, E.L.; Rhaleb, N.-E. Role of N-acetyl-seryl-aspartyl-lysyl-proline in the antifibrotic and anti-inflammatory effects of the angiotensin-converting enzyme inhibitor captopril in hypertension. Hypertension 2007, 49, 695–703. [Google Scholar] [CrossRef]
- Rhaleb, N.-E.; Peng, H.; Yang, X.P.; Liu, Y.H.; Mehta, D.; Ezan, E.; Carretero, O.A. Long-term effect of N-acetyl-seryl-aspartyl-lysyl-proline on left ventricular collagen deposition in rats with 2-kidney, 1-clip hypertension. Circulation 2001, 103, 3136–3141. [Google Scholar] [CrossRef]
- Linz, W.; Schölkens, B.A. A specific B2-bradykinin receptor antagonist HOE 140 abolishes the antihypertrophic effect of ramipril. Br. J. Pharmacol. 1992, 105, 771–772. [Google Scholar] [CrossRef]
- Pfeffer, M.A.; Braunwald, E.; Moyé, L.A.; Basta, L.; Brown, E.J., Jr.; Cuddy, T.E.; Davis, B.R.; Geltman, E.M.; Goldman, S.; Flaker, G.C.; et al. Effect of captopril on mortality and morbidity in patients with left ventricular dysfunction after myocardial infarction. Results of the Survival and Ventricular Enlargement trial. N. Engl. J. Med. 1992, 327, 669–677. [Google Scholar] [CrossRef]
- Gertz, S.D.; Kurgan, A. Tissue plasminogen activator and selective coronary vasodilation [letter]. Am. J. Cardiol. 1988, 62, 173. [Google Scholar] [PubMed]
- Loke, K.E.; Curran, C.M.L.; Messina, E.J.; Laycock, S.K.; Shesely, E.G.; Carretero, O.A.; Hintze, T.H. Role of nitric oxide in the control of cardiac oxygen consumption in B2-kinin receptor knockout mice. Hypertension 1999, 34, 563–567. [Google Scholar] [CrossRef]
- Liu, Y.H.; Xu, J.; Yang, X.P.; Yang, F.; Shesely, E.; Carretero, O.A. Effect of ACE inhibitors and angiotensin II type 1 receptor antagonists on endothelial NO synthase knockout mice with heart failure. Hypertension 2002, 39, 375–381. [Google Scholar] [CrossRef] [PubMed]
- Mombouli, J.-V.; Vanhoutte, P.M. Kinins and endothelial control of vascular smooth muscle. Annu. Rev. Pharmacol. Toxicol. 1995, 35, 679–705. [Google Scholar] [CrossRef] [PubMed]
- Mombouli, J.-V.; Vanhoutte, P.M. Endothelium-derived hyperpolarizing factor(s) and the potentiation of kinins by converting enzyme inhibitors. Am. J. Hypertens. 1995, 8, 19S–27S. [Google Scholar] [CrossRef]
- Baragatti, B.; Brizzi, F.; Barogi, S.; Laubach, V.E.; Sodini, D.; Shesely, E.G.; Regan, R.F.; Coceani, F. Interactions between NO, CO and an endothelium-derived hyperpolarizing factor (EDHF) in maintaining patency of the ductus arteriosus in the mouse. Br. J. Pharmacol. 2007, 151, 54–62. [Google Scholar] [CrossRef] [PubMed]
- Inokuchi, K.; Hirooka, Y.; Shimokawa, H.; Sakai, K.; Kishi, T.; Ito, K.; Kimura, Y.; Takeshita, A. Role of endothelium-derived hyperpolarizing factor in human forearm circulation. Hypertension 2003, 42, 919–924. [Google Scholar] [CrossRef][Green Version]
- Pomposiello, S.; Rhaleb, N.-E.; Alva, M.; Carretero, O.A. Reactive oxygen species: Role in the relaxation induced by bradykinin or arachidonic acid via EDHF in isolated porcine coronary arteries. J. Cardiovasc. Pharmacol. 1999, 34, 567–574. [Google Scholar] [CrossRef]
- Matsuda, H.; Hayashi, K.; Wakino, S.; Kubota, E.; Honda, M.; Tokuyama, H.; Takamatsu, I.; Tatematsu, S.; Saruta, T. Role of endothelium-derived hyperpolarizing factor in ACE inhibitor-induced renal vasodilation in vivo. Hypertension 2004, 43, 603–609. [Google Scholar] [CrossRef]
- Peng, H.; Carretero, O.A.; Vuljaj, N.; Liao, T.D.; Motivala, A.; Peterson, E.L.; Rhaleb, N.-E. Angiotensin-converting enzyme inhibitors: A new mechanism of action. Circulation 2005, 112, 2436–2445. [Google Scholar] [CrossRef]
- Azizi, M.; Ezan, E.; Nicolet, L.; Grognet, J.M.; Menard, J. High plasma level of N-acetyl-seryl-aspartyl-lysyl-proline: A new marker of chronic angiotensin-converting enzyme inhibition. Hypertension 1997, 30, 1015–1019. [Google Scholar] [CrossRef] [PubMed]
- Azizi, M.; Ezan, E.; Reny, J.-L.; Wdzieczak-Bakala, J.; Gerineau, V.; Ménard, J. Renal and metabolic clearance of N-acetyl-seryl-aspartyl-lysyl-proline (AcSDKP) during angiotensin-converting enzyme inhibition in humans. Hypertension 1999, 33, 879–886. [Google Scholar] [CrossRef] [PubMed]
- Srivastava, S.P.; Goodwin, J.E.; Kanasaki, K.; Koya, D. Inhibition of Angiotensin-Converting Enzyme Ameliorates Renal Fibrosis by Mitigating DPP-4 Level and Restoring Antifibrotic MicroRNAs. Genes 2020, 11, 211. [Google Scholar] [CrossRef] [PubMed]
- Duka, A.; Kintsurashvili, E.; Duka, I.; Ona, D.; Hopkins, T.A.; Bader, M.; Gavras, I.; Gavras, H. Angiotensin-converting enzyme inhibition after experimental myocardial infarct: Role of the kinin B1 and B2 receptors. Hypertension 2008, 51, 1352–1357. [Google Scholar] [CrossRef] [PubMed]
- Marketou, M.; Kintsurashvili, E.; Papanicolaou, K.N.; Lucero, H.A.; Gavras, I.; Gavras, H. Cardioprotective effects of a selective B(2) receptor agonist of bradykinin post-acute myocardial infarct. Am. J. Hypertens. 2010, 23, 562–568. [Google Scholar] [CrossRef] [PubMed]
- Yin, H.; Chao, J.; Bader, M.; Chao, L. Differential role of kinin B1 and B2 receptors in ischemia-induced apoptosis and ventricular remodeling. Peptides 2007, 28, 1383–1389. [Google Scholar] [CrossRef]
- Messadi, E.; Vincent, M.P.; Griol-Charhbili, V.; Mandet, C.; Colucci, J.; Krege, J.H.; Bruneval, P.; Bouby, N.; Smithies, O.; Alhenc-Gelas, F.; et al. Genetically determined angiotensin converting enzyme level and myocardial tolerance to ischemia. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2010, 24, 4691–4700. [Google Scholar]
- Messadi-Laribi, E.; Griol-Charhbili, V.; Gaies, E.; Vincent, M.P.; Heudes, D.; Meneton, P.; Alhenc-Gelas, F.; Richer, C. Cardioprotection and kallikrein-kinin system in acute myocardial ischaemia in mice. Clin. Exp. Pharmacol. Physiol. 2008, 35, 489–493. [Google Scholar] [CrossRef]
- Lagneux, C.; Adam, A.; Lamontagne, D. A study of the mediators involved in the protection induced by exogenous kinins in the isolated rat heart. Int. Immunopharmacol. 2003, 3, 1511–1518. [Google Scholar] [CrossRef]
- Yang, X.P.; Liu, Y.H.; Peterson, E.; Carretero, O.A. Effect of neutral endopeptidase 24.11 inhibition on myocardial ischemia/reperfusion injury: The role of kinins. J. Cardiovasc. Pharmacol. 1997, 29, 250–256. [Google Scholar] [CrossRef]
- Wollert, K.C.; Studer, R.; Doerfer, K.; Schieffer, E.; Holubarsch, C.; Just, H.; Drexler, H. Differential effects of kinins on cardiomyocyte hypertrophy and interstitial collagen matrix in the surviving myocardium after myocardial infarction in the rat. Circulation 1997, 95, 1910–1917. [Google Scholar] [CrossRef] [PubMed]
- Ito, H.; Hayashi, I.; Izumi, T.; Majima, M. Bradykinin inhibits development of myocardial infarction through B2 receptor signalling by increment of regional blood flow around the ischaemic lesions in rats. Br. J. Pharmacol. 2003, 138, 225–233. [Google Scholar] [CrossRef]
- Koch, M.; Bonaventura, K.; Spillmann, F.; Dendorfer, A.; Schultheiss, H.P.; Tschope, C. Attenuation of left ventricular dysfunction by an ACE inhibitor after myocardial infarction in a kininogen-deficient rat model. Biol. Chem. 2008, 389, 719–723. [Google Scholar] [CrossRef]
- Wollert, K.C.; Drexler, H. The kallikrein-kinin system in post-myocardial infarction cardiac remodeling. Am. J. Cardiol. 1997, 80, 158A–161A. [Google Scholar] [CrossRef]
- Chahine, R.; Adam, A.; Yamaguchi, N.; Gaspo, R.; Regoli, D.; Nadeau, R. Protective effects of bradykinin on the ischaemic heart: Implication of the B1 receptor. Br. J. Pharmacol. 1993, 108, 318–322. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Hartman, J.C.; Wall, T.M.; Hullinger, T.G.; Shebuski, R.J. Reduction of myocardial infarct size in rabbits by ramiprilat: Reversal by the bradykinin antagonist HOE 140. J. Cardiovasc. Pharmacol. 1993, 21, 996–1003. [Google Scholar] [CrossRef]
- Hoshida, S.; Yamashita, N.; Kuzuya, T.; Hori, M. Differential effects of long-term renin-angiotensin system blockade on limitation of infarct size in cholesterol-fed rabbits. Atherosclerosis 2000, 149, 287–294. [Google Scholar] [CrossRef]
- Lamontagne, D.; Nakhostine, N.; Couture, R.; Nadeau, R. Mechanisms of kinin B1-receptor-induced hypotension in the anesthetized dog. J. Cardiovasc. Pharmacol. 1996, 28, 645–650. [Google Scholar] [CrossRef]
- Belichard, P.; Loillier, B.; Paquet, J.L.; Luccarini, J.M.; Pruneau, D. Haemodynamic and cardiac effects of kinin B1 and B2 receptor stimulation in conscious instrumented dogs. Br. J. Pharmacol. 1996, 117, 1565–1571. [Google Scholar] [CrossRef][Green Version]
- Rastegar, M.A.; Marchini, F.; Morazzoni, G.; Vegh, A.; Papp, J.G.; Parratt, J.R. The effects of Z13752A, a combined ACE/NEP inhibitor, on responses to coronary artery occlusion; a primary protective role for bradykinin. Br. J. Pharmacol. 2000, 129, 671–680. [Google Scholar] [CrossRef]
- McMurray, J.J.; Packer, M.; Desai, A.S.; Gong, J.; Lefkowitz, M.P.; Rizkala, A.R.; Rouleau, J.L.; Shi, V.C.; Solomon, S.D.; Swedberg, K.; et al. Angiotensin-neprilysin inhibition versus enalapril in heart failure. N. Engl. J. Med. 2014, 371, 993–1004. [Google Scholar] [CrossRef] [PubMed]
- Solomon, S.D.; McMurray, J.J.V.; Anand, I.S.; Ge, J.; Lam, C.S.P.; Maggioni, A.P.; Martinez, F.; Packer, M.; Pfeffer, M.A.; Pieske, B.; et al. Angiotensin-Neprilysin Inhibition in Heart Failure with Preserved Ejection Fraction. N. Engl. J. Med. 2019, 381, 1609–1620. [Google Scholar] [CrossRef] [PubMed]
- Campbell, D.J. Neprilysin Inhibitors and Bradykinin. Front. Med. 2018, 5, 257. [Google Scholar] [CrossRef]
- Seyedi, N.; Xu, X.; Nasjletti, A.; Hintze, T.H. Coronary kinin generation mediates nitric oxide release after angiotensin receptor stimulation. Hypertension 1995, 26, 164–170. [Google Scholar] [CrossRef]
- Kurisu, S.; Ozono, R.; Oshima, T.; Kambe, M.; Ishida, T.; Sugino, H.; Matsuura, H.; Chayama, K.; Teranishi, Y.; Iba, O.; et al. Cardiac angiotensin II type 2 receptor activates the kinin/NO system and inhibits fibrosis. Hypertension 2003, 41, 99–107. [Google Scholar] [CrossRef] [PubMed]
- Gohlke, P.; Pees, C.; Unger, T. AT2 receptor stimulation increases aortic cyclic GMP in SHRSP by a kinin-dependent mechanism. Hypertension 1998, 31, 349–355. [Google Scholar] [CrossRef] [PubMed]
- Pees, C.; Unger, T.; Gohlke, P. Effect of angiotensin AT2 receptor stimulation on vascular cyclic GMP production in normotensive Wistar Kyoto rats. Int. J. Biochem. Cell Biol. 2003, 35, 963–972. [Google Scholar] [CrossRef]
- Abadir, P.M.; Carey, R.M.; Siragy, H.M. Angiotensin AT2 receptors directly stimulate renal nitric oxide in bradykinin B2-receptor-null mice. Hypertension 2003, 42, 600–604. [Google Scholar] [CrossRef]
- Xu, J.; Carretero, O.A.; Liu, Y.H.; Shesely, E.G.; Yang, F.; Kapke, A.; Yang, X.P. Role of AT2 receptors in the cardioprotective effect of AT1 antagonists in mice. Hypertension 2002, 40, 244–250. [Google Scholar] [CrossRef]
- Ignjatovic, T.; Tan, F.; Brovkovych, V.; Skidgel, R.A.; Erdos, E.G. Activation of bradykinin B1 receptor by ACE inhibitors. Int. Immunopharmacol. 2002, 2, 1787–1793. [Google Scholar] [CrossRef]
- Sun, Y.; Carretero, O.A.; Xu, J.; Rhaleb, N.-E.; Yang, J.J.; Pagano, P.J.; Yang, X.P. Deletion of inducible nitric oxide synthase provides cardioprotection in mice with 2-kidney, 1-clip hypertension. Hypertension 2009, 53, 49–56. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.P.; Liu, Y.H.; Rhaleb, N.-E.; Kurihara, N.; Kim, H.E.; Carretero, O.A. Echocardiographic assessment of cardiac function in conscious and anesthetized mice. Am. J. Physiol. Heart Circ. Physiol. 1999, 277, H1967–H1974. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Animal Specie | Receptors | Effects | Methods | Tools | References |
|---|---|---|---|---|---|
| Mice | B1 and B2 receptors | Both contribute in cardioprotective effect of ACE-I | MI by left anterior descending coronary artery (LAD) ligation | B1-R(-/-) B2-R(-/-) | [208] |
| Mice | B2 receptors | Improve cardiac function, tissue remodeling, and inflammation (structural and functional benefits) | MI by LAD ligation | B2 receptor selective agonist | [209] |
| Mice | B1 receptors | Cardioprotective effects (improve cardiac function and remodeling) | MI by LAD ligation | B1-R(-/-) | [112] |
| Mice | B2 receptors | Cardioprotective effect of ACE-I and ARB | MI by LAD ligation | B2-R(-/-) | [95] |
| Mice | B2 receptors | Cardiac remodeling, hypertrophy and dysfunction | MI by LAD ligation | B2-R(-/-) B1 or AT2 receptor antagonist | [88] |
| Mice | B1 receptors | Mediated part of the cardioprotective effects of ACE-I and ARB | MI by LAD ligation | B1-R(-/-) B1 or AT2 receptor antagonist | [112] |
| Mice | B2 receptors | Reduced infarct size reduced cardiomyocyte apoptosis | Ischemia reperfusion | B1 and B2 receptor agonists, B2-R(-/-), B2 receptor antagonist, preconditioning | [94,210,211] |
| Mice | B2 receptors | Reduced infarct size and cardio-protection | Ischemia reperfusion | Tissue-kallikrein deficient mice, AT1 and AT2 receptor antagonists | [212] |
| Rats | B2 receptors | Reduced infarct size | Isolated heart, ischemia reperfusion | Brown Norway Katholiek (BN-Ka) rats, B1 or B2 receptor antagonists or agonists, neutral endopeptidase (NEP) inhibitor, ACE-I | [94,213,214] |
| Rats | B2 receptors | Inhibits collagen deposition, reduce myocardial collagen accumulation by ACE-I and ARB | MI by LAD ligation | B2 receptor antagonist | [215] |
| Rats | B2 receptors | Mediated protective effects of ARB and ACE-I | MI by LAD ligation | ACE-I, ARBs, AT2 receptor antagonist | [91] |
| Rats | B2 receptors | Reduce infarct size | MI by LAD ligation | BN-Ka rats, B2 receptor antagonist, and a nonpeptide B2 receptor agonist | [216] |
| BN-Ka | kininogen | Kinin do not mediate the beneficial effects of ACE-I | MI by LAD ligation | BN-Ka versus BN Norway Hannover (wild-type rats) | [217] |
| Rat | B2 receptors | Inhibit the interstitial accumulation of collagen, no effects on cardio myocyte hypertrophy | Morphometric analysis, collagen deposition in left ventricular interstitial | B2 receptor antagonist | [218] |
| Rats | B1 receptors | Inhibited myocardial noradrenaline, reduced ventricular fibrillation | Ischemia reperfusion | B1 receptor agonist and antagonist | [219] |
| Rabbit | B2 receptors | Reduction in infarction size | Ischemia/reperfusion | B2 receptor antagonist | [220] |
| Rabbit | B2 receptors | Mediated the effect of ACE-I on infarct size | MI by LAD ligation plus high cholesterol diet | B2 receptor antagonist | [221] |
| Dog | B1-receptor | Hypotensive effect, peripheral vasodilation | Intra-arterial and intravenous injection | B1 agonist | [222] |
| Dogs | B1 and B2 receptors | Decreases mean arterial pressure (MAP) and coronary vascular resistance (CVR) | i.v. infusion | B1 receptor agonist and antagonist | [223] |
| Dogs | B2 receptors | Reduced infarct size | Isolated heart, ischemia reperfusion | Combined NEP/ACE inhibitor, B2 receptor antagonist | [224] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hamid, S.; Rhaleb, I.A.; Kassem, K.M.; Rhaleb, N.-E. Role of Kinins in Hypertension and Heart Failure. Pharmaceuticals 2020, 13, 347. https://doi.org/10.3390/ph13110347
Hamid S, Rhaleb IA, Kassem KM, Rhaleb N-E. Role of Kinins in Hypertension and Heart Failure. Pharmaceuticals. 2020; 13(11):347. https://doi.org/10.3390/ph13110347
Chicago/Turabian StyleHamid, Suhail, Imane A. Rhaleb, Kamal M. Kassem, and Nour-Eddine Rhaleb. 2020. "Role of Kinins in Hypertension and Heart Failure" Pharmaceuticals 13, no. 11: 347. https://doi.org/10.3390/ph13110347
APA StyleHamid, S., Rhaleb, I. A., Kassem, K. M., & Rhaleb, N.-E. (2020). Role of Kinins in Hypertension and Heart Failure. Pharmaceuticals, 13(11), 347. https://doi.org/10.3390/ph13110347