1. Introduction
The
Parechovirus genus includes six species (Parechovirus A-F), with genotypes that infect humans belonging to the Parechovirus-A (PeV-A) species. To date, 19 PeV-A types have been identified [
1]. PeV-A viruses have been detected in stool and respiratory specimens of healthy and ill (gastroenteritis and respiratory infections) patients, mostly children. PeV-A viruses have also been detected in blood and cerebrospinal fluid (CSF) specimens from patients, mostly in children and particularly infants less than 6 months of age, with severe illnesses (e.g., CNS infections or sepsis-like illness) [
2,
3]. The most frequently detected type is PeV-A1 [
4], found most often in stool samples of infected patients. However, PeV-A3 is the type most frequently linked to severe illness and potential neurological symptoms or sequelae [
5]. PeV-A3 was first reported in 2004 in Japan [
6] and thereafter reported elsewhere globally [
2].
PeV-A contains a single-stranded positive-sense RNA genome that is approximately 7300 nucleotides long and has a single open reading frame (ORF). The genome encodes a single large polyprotein, which is cleaved into three structural proteins (VP0, VP3, and VP1) and seven non-structural proteins (2A–C and 3A–D) [
7]. Over time, drastic changes can occur across the PeV-A genome driven by the lack of proofreading activity of the PeV-A RNA polymerase, leading to a high mutation rate of approximately 2.5 × 10
−3 substitutions per site per year [
8]. Recombination has also been shown to occur between PeV-A3 and other PeV-A types resulting in changes across large stretches of the genome [
9,
10,
11]. From an epidemiological standpoint, emergence of recombinant and/or mutated strains can lead to an increase in hospitalizations and clinical severity in infants [
11,
12]. Sanger-based sequencing of the VP1 capsid encoding gene and primers targeting the 5′untranslated region (UTR) have been the standard method for routine surveillance and genotyping of PeV-A [
13,
14]. However, this approach captures changes occurring only across a small region in the genome and hinders the understanding of changes across the entire viral genome. Analyzing whole genome sequences of PeV-A3 isolates will improve understanding of how PeV-A3 strains evolve and will be an important tool to predict changes in preparation for potential PeV-A3 outbreaks.
2. Results/Genome Announcement
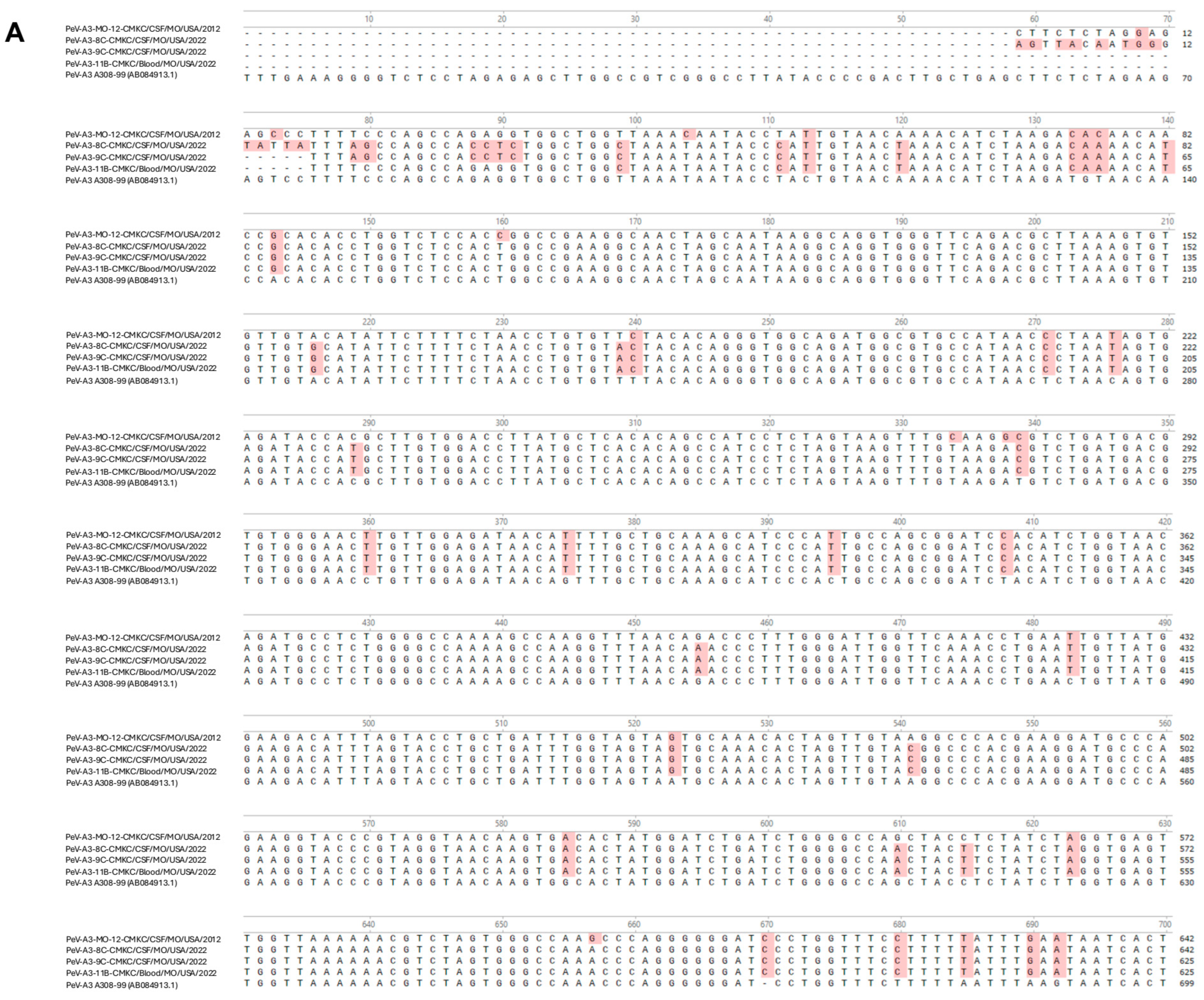
Names of the sequenced viral isolates as well as their GenBank accession numbers, sequence length and closest identity to a reported sequence are provided in
Table 1. A BLASTn analysis provided the highest alignment score for the PeV-A3-MO-12-CMKC/CSF/MO/USA/2012 isolate to the PeV-A3 isolate from Yamagata, Japan (LC864495.1). The PeV-A3-8C-CMKC/CSF/MO/USA/2022, PeV-A3-9C-CMKC/CSF/MO/USA/2022 and PeV-A3-11B-CMKC/Blood/MO/USA/2022 isolates aligned to another PeV-A3 isolate reported from Yamagata, Japan (LC864514.1). When compared with the prototypic sequence of PeV-A3-308/99 (AB084913), we identified multiple nucleotide changes spanning across the 5′ untranslated region (UTR) (
Figure 1A) and 3′ UTR (
Figure 1B) of the PeV-A3 isolates. Amino acid substitutions were also detected throughout the viral polyprotein (
Figure 1C). Unique amino acid substitutions were observed in the PeV-A3 isolates from 2022 (PeV-A3-8C-CMKC/CSF/MO/USA/2022, PeV-A3-9C-CMKC/CSF/MO/USA/2022 and PeV-A3-11B-CMKC/Blood/MO/USA/2022) that were absent in the 2012 isolate (PeV-A3-MO-12-CMKC/CSF/MO/USA/2012). These substitutions were seen in non-structural proteins [2A (L776P, K781N, V832I, N877S, V885I, V915I); 2C (S1129A, I1178V, N1206D, A1311S, T1322I, K1344Q, Q1348K, Q1355E); 3A (S1418N, R1426K, E1440D, A1463V, V1466A, K1482R); 3B (A1498V, S1507T); 3C (I1577V, S1620N); 3D (I1714V, A1766S, S1811T, A1858T, Y1943F, S1958A, E1966D, D1976N, G2059N, I2060V)] and structural protein VP1 (R764G, A765V, A767V) (
Figure 1C).
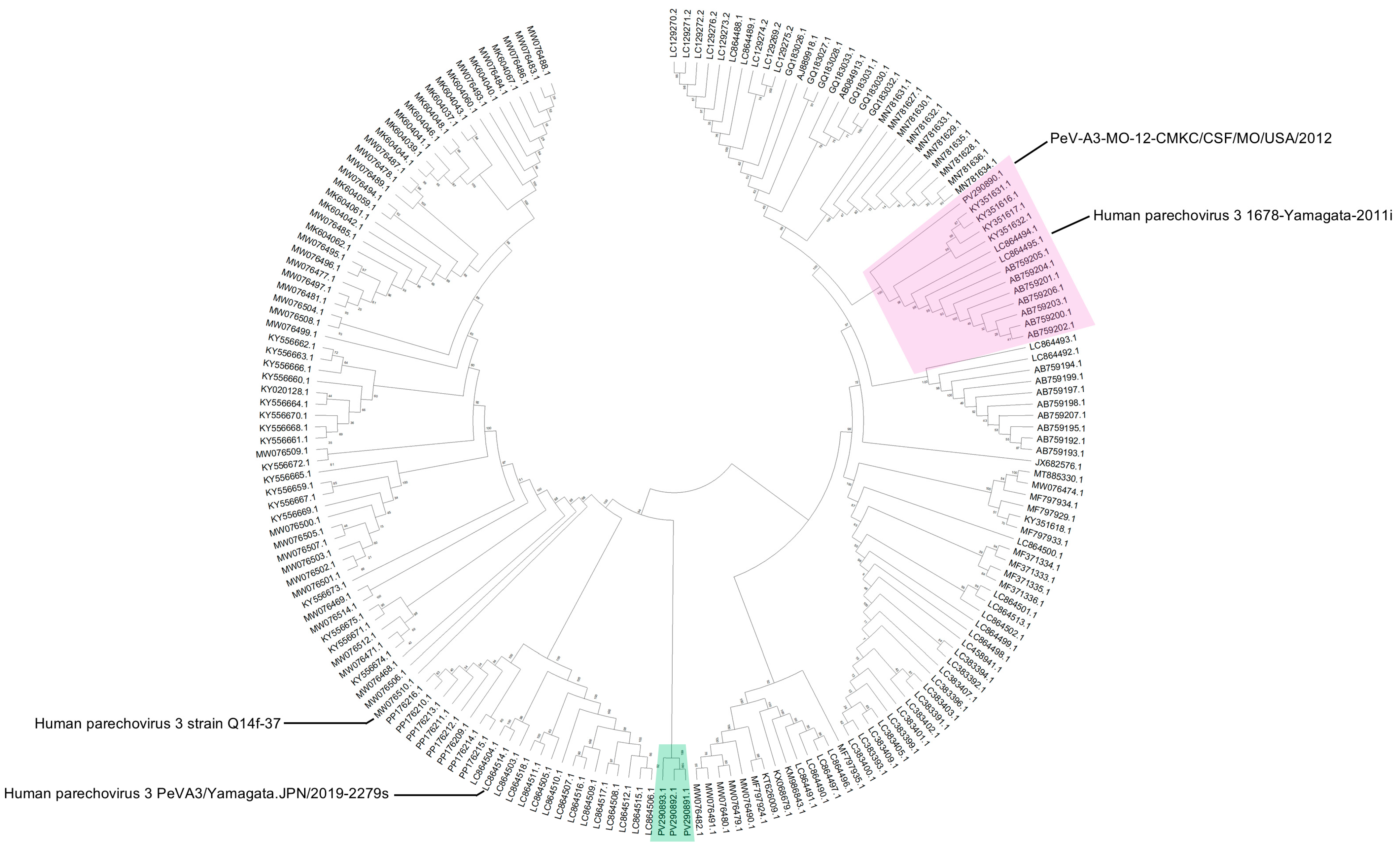
Phylogenetic analysis of whole genome sequences revealed that PeV-A3 isolate from 2012 (PeV-A3-MO-12-CMKC/CSF/MO/USA/2012) clustered together with isolates from Germany and Yamagata, Japan, that were collected in 2011 and 2012 (
Figure 2, highlighted in red). The three isolates from 2022 (PeV-A3-8C-CMKC/CSF/MO/USA/2022, PeV-A3-9C-CMKC/CSF/MO/USA/2022 and PeV-A3-11B-CMKC/Blood/MO/USA/2022) are highly divergent and cluster as a distinct clade in the phylogenetic tree when compared with the closest available PeV-A3 genomes (
Figure 2, highlighted in green). The closest genetic relatives of the 2022 isolates is to a PeV-A3 strain reported from Australia in 2019 (strain Q14f-37, MW076510.1) (
Figure 2).
3. Discussion
PeV-A3 circulation in the United States follows a biennial pattern with peaks observed every alternate year [
15]. Whole genome sequences of PeV-A3 isolates from outbreaks can provide genomic data on how PeV-A3 viral strains change over time. In this study, we sequenced the whole genome of four PeV-A3 clinical isolates from the United States, one from the 2012 outbreak (PeV-A3-MO-12-CMKC/CSF/MO/USA/2012) and three from 2022 (PeV-A3-8C-CMKC/CSF/MO/USA/2022, PeV-A3-9C-CMKC/CSF/MO/USA/2022 and PeV-A3-11B-CMKC/Blood/MO/USA/2022).
All the isolates have nucleotide changes in their 5′UTR and 3′UTR, plus amino acid substitutions in the viral polypeptide when compared to the genome of the prototypic PeV-A3-A308/99 strain (
Figure 1A–C). The isolates from 2022 (PeV-A3-8C-CMKC/CSF/MO/USA/2022, PeV-A3-9C-CMKC/CSF/MO/USA/2022, PeV-A3-11B-CMKC/Blood/MO/USA/2022) have unique amino acid substitutions that were absent in the PeV-A3-MO-12-CMKC/CSF/MO/USA/2012 isolate and PeV-A3-A308/99 (
Figure 1C). A higher propensity of C to U transitions relative to other mutations has been previously reported in picornaviruses [
16]. Certainly, within our dataset of 188 PeV-A3 sequences used for multiple sequence alignments, we observed 378 sites of transitions and 119 sites of transversions. Within the transition sites, there were 120 sites with C to U transition and 105 sites that had a U to C transition. The implication of this transition is yet to be explored for PeV-A3.
While it is not surprising to detect PeV-A3-MO-12-CMKC/CSF/MO/USA/2012 as a close relative of other circulating PeV-As, the sequence divergence of 2022 PeV-A3 isolates (PeV-A3-8C-CMKC/CSF/MO/USA/2022, PeV-A3-9C-CMKC/CSF/MO/USA/2022, PeV-A3-11B-CMKC/Blood/MO/USA/2022) represented a new strain (<95% whole genome identity) that has not been characterized before and that is concurrent with their distinct phylogenetic position (
Figure 2). Our method of phylogenetic analysis used a classical approach, assuming the generation of recombinant viruses from a recent common ancestor. Recent studies have provided validation of a phylogeny-free approach to better predict viral evolution [
17,
18], but the classical approach is still robust and computationally practical for a dataset like ours. Furthermore, we are aware that for many non-polio enteroviruses, routine Sanger sequencing is performed across the capsid region, particularly the VP1 region. Our phylogenetic tree has been constructed using a dataset of whole genome sequences and sequences >80% coverage to our input sequences, reported on GenBank. Therefore, these sequences used for analysis should have most of the 5′UTR and VP1 sequences included and so we do not envision any significant differences with a tree, if it would have been made exclusively with these specific regions.
Overall, our results highlight the importance of continuous molecular surveillance of emerging PeV-A strains. Ongoing research will assess the phenotypic impact of mutations on viral replication and pathogenesis.
4. Materials and Methods
Cells and Viruses: The Vero-Polio (Vero-P) cell line, as previously described [
19], and four PeV-A3 isolates were provided by the Centers for Disease Control and Prevention, Atlanta, USA. Upon receipt, these viral isolates were passaged twice in Vero-P cells prior to nucleic acid extraction. Vero-P cells were maintained in Eagle’s Minimum Essential Medium (EMEM) (ATCC, Catalog #30-2003) supplemented with 10% Fetal Bovine Serum (Catalog #16140071, Thermo Fisher Scientific) and 1% Penicillin–Streptomycin (Catalog #15070063, Gibco).
Reverse transcription and random amplification of PeV-A3 nucleic acid isolates: Viral nucleic acids were extracted from PeV-A3 isolates using a QIAamp viral RNA minikit (Qiagen, Catalog # 52904) followed by DNase I treatment (rDNase I; Ambion of Thermo Fisher Scientific, Catalog# AM2222) following the manufacturer’s instructions. The extracted nucleic acids were amplified using a sequence-independent, single-primer amplification (SISPA) protocol that has been described previously [
20]. Briefly, reverse transcription of viral RNA was performed using SuperScript III reverse transcriptase (Invitrogen, Thermo Fisher ScientificCatalog# 18080093) with a 28-base-pair reverse primer consisting of a 3′ end with eight random nucleotides (N1_8N, CCTTGAAGGCGGACTGTGAGNNNNNNNN). A complementary DNA (cDNA) strand was synthesized using the Klenow fragment of DNA polymerase I [(3′ to 5′exonuclease) (New England BioLabs, Catalog# M0209)]. Polymerase chain reaction (PCR) amplification was performed with AmpliTaq Gold polymerase (Thermo Fisher Scientific, Catalog# 4311806) and 100 mM of the forward primer in a 25 mL reaction volume using the following conditions: 1 cycle of 95 °C for 5 min, 5 cycles of 95 °C for 1 min, 59 °C for 1 min, and 72 °C for 90 s, followed by 25 cycles of 95 °C for 30 s, 59 °C for 30 s, and 72 °C for 90 s.
Library construction using NexteraXT and sequencing: Paired-end libraries from PeV-A3 isolates were generated using a NexteraXT DNA library preparation kit (Illumina, Catalog#FC-131-1024). Briefly, genomic DNA samples amplified from reverse transcription and random amplification were quantified on a Qubit High Sensitivity DNA kit (ThermoFisher Scientific, Catalog# Q32854) and visualized on a TapeStation (Agilent Technologies, Santa Clara, CA, USA). Thereafter, amplicons were tagged with adapter sequences using an Illumina DNA/RNA UD Indexes Set B, Tagmentation kit (Illumina, Catalog# 20091656) and then amplified to generate a library. The amplified libraries were cleaned using Illumina Purification beads (Illumina, Catalog# 20060057) and library quality was checked using an Agilent 2200 Tapestation system (Agilent Technologies, Santa Clara, CA, USA) using a High Sensitivity D1000 Screen Tape kit (Agilent Technologies, Catalog# 5067-5584, 5067-5585). Individual libraries were normalized and pooled to a final concentration of 1 nM. The pooled library was denatured and diluted, and 1.4 pM of denatured library was loaded for a 2 X 149 bp paired-end sequencing run on the MiniSeq platform (Illumina, Catalog# SY-420-1001) following the manufacturer’s protocol.
Data analysis: We analyzed the MiniSeq sequences by read mapping and gene annotation using Geneious 11.1.2 (Biomatters,
https://www.geneious.com accessed on 2 December 2024), as described previously [
20], using PeV-A3 genome references from GenBank. Identification of mutations across the genome was performed using SnapGene
® software, version 8.0.2 (from Dotmatics; available at
snapgene.com). To obtain related genomes for subsequent phylogenetic analysis, standard nucleotide basic local alignment search using BLASTn [
21] was performed on all four sequences against the NCBI non-redundant (nr) nucleotide database with default parameters. The output generated was 100 top hits per sequence. Redundant sequences were removed to eventually reduce the collection to a final of 188 PeV-A3 sequences. Sequences were aligned using MUSCLE [
22] in MEGA 12 (Molecular Evolutionary Genetics Analysis) [
23] with default parameters. The phylogenetic analysis was performed using the Neighbor-Joining (NJ) algorithm [
24] to generate a phylogenetic tree with 1000 bootstrap replicates on MEGA12 (
Figure 2).
5. Conclusions
In this manuscript, we report sequences of four PeV-A3 clinical isolates from the USA (PV290890-PV290893). Our data indicates that the 2022 clinical isolates of PeV-A3 (PeV-A3-8C-CMKC/CSF/MO/USA/2022, PeV-A3-9C-CMKC/CSF/MO/USA/2022, PeV-A3-11B-CMKC/Blood/MO/USA/2022) cluster as a separate clade and have unique mutations [2A (L776P, K781N, V832I, N877S, V885I, V915I); 2C (S1129A, I1178V, N1206D, A1311S, T1322I, K1344Q, Q1348K, Q1355E); 3A (S1418N, R1426K, E1440D, A1463V, V1466A, K1482R); 3B (A1498V, S1507T); 3C (I1577V, S1620N); 3D (I1714V, A1766S, S1811T, A1858T, Y1943F, S1958A, E1966D, D1976N, G2059N, I2060V); VP1 (R764G, A765V, A767V)] that were absent in the PeV-A3-MO-12-CMKC/CSF/MO/USA/2012 isolate.
Author Contributions
Conceptualization, D.D., T.F.F.N., C.J.H., E.R. and R.S.; methodology, T.F.F.N., C.J.H. and E.R.; software, T.F.F.N.; validation, T.F.F.N., C.J.H. and R.S.; formal analysis, D.D., C.J.H. and V.C.B.; investigation, D.D., T.F.F.N. and E.R.; resources, R.S.; data curation, D.D., T.F.F.N. and C.J.H.; writing—original draft preparation, D.D.; writing—review and editing, D.D., T.F.F.N., C.J.H., E.R., B.A.M., A.S. and K.E.V.; supervision, B.A.M.; project administration, R.S.; funding acquisition, R.S. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable
Informed Consent Statement
Not applicable
Data Availability Statement
The genome sequences of PeV-A3 isolates were deposited in GenBank under accession numbers PV290890-PV290893.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Zell, R.; Delwart, E.; Gorbalenya, A.E.; Hovi, T.; King, A.M.Q.; Knowles, N.J.; Lindberg, A.M.; Pallansch, M.A.; Palmenberg, A.C.; Reuter, G.; et al. ICTV Virus Taxonomy Profile: Picornaviridae. J. Gen. Virol. 2017, 98, 2421–2422. [Google Scholar] [CrossRef] [PubMed]
- Sridhar, A.; Karelehto, E.; Brouwer, L.; Pajkrt, D.; Wolthers, K.C. Parechovirus A Pathogenesis and the Enigma of Genotype A-3. Viruses 2019, 11, 1062. [Google Scholar] [CrossRef] [PubMed]
- Esposito, S.; Rahamat-Langendoen, J.; Ascolese, B.; Senatore, L.; Castellazzi, L.; Niesters, H.G.M. Pediatric parechovirus infections. J. Clin. Virol. 2014, 60, 84–89. [Google Scholar] [CrossRef]
- Olijve, L.; Jennings, L.; Walls, T. Human Parechovirus: An Increasingly Recognized Cause of Sepsis-Like Illness in Young Infants. Clin. Microbiol. Rev. 2018, 31, e00047-17. [Google Scholar] [CrossRef]
- Boivin, G.; Abed, Y.; Boucher, F.D. Human parechovirus 3 and neonatal infections. Emerg. Infect. Dis. 2005, 11, 103–105. [Google Scholar] [CrossRef]
- Ito, M.; Yamashita, T.; Tsuzuki, H.; Takeda, N.; Sakae, K. Isolation and identification of a novel human parechovirus. J. Gen. Virol. 2004, 85 Pt 2, 391–398. [Google Scholar] [CrossRef]
- Stanway, G.; Kalkkinen, N.; Roivainen, M.; Ghazi, F.; Khan, M.; Smyth, M.; Meurman, O.; Hyypiä, T. Molecular and biological characteristics of echovirus 22, a representative of a new picornavirus group. J. Virol. 1994, 68, 8232–8238. [Google Scholar] [CrossRef]
- Faria, N.R.; de Vries, M.; van Hemert, F.J.; Benschop, K.; van der Hoek, L. Rooting human parechovirus evolution in time. BMC Evol. Biol. 2009, 9, 164. [Google Scholar] [CrossRef]
- Mizuta, K.; Itagaki, T.; Chikaoka, S.; Wada, M.; Ikegami, T.; Sendo, D.; Iseki, C.; Shimizu, Y.; Abe, S.; Komabayashi, K.; et al. Recombinant parechovirus A3 possibly causes various clinical manifestations, including myalgia; findings in Yamagata, Japan in 2019. Infect. Dis. 2022, 54, 632–650. [Google Scholar] [CrossRef]
- Truong, T.C.; Park, H.; Kim, J.H.; Tran, V.T.; Kim, W. The evolutionary phylodynamics of human parechovirus A type 3 reveal multiple recombination events in South Korea. J. Med. Virol. 2024, 96, e29477. [Google Scholar] [CrossRef]
- Bialasiewicz, S.; May, M.; Tozer, S.; Day, R.; Bernard, A.; Zaugg, J.; Gartrell, K.; Alexandersen, S.; Chamings, A.; Wang, C.Y.T.; et al. Novel Human Parechovirus 3 Diversity, Recombination, and Clinical Impact Across 7 Years: An Australian Story. J. Infect. Dis. 2023, 227, 278–287. [Google Scholar] [CrossRef] [PubMed]
- Joseph, L.; May, M.; Thomas, M.; Smerdon, C.; Tozer, S.; Bialasiewicz, S.; McKenna, R.; Sargent, P.; Kynaston, A.; Heney, C.; et al. Human Parechovirus 3 in Infants: Expanding Our Knowledge of Adverse Outcomes. Pediatr. Infect. Dis. J. 2019, 38, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Nix, W.A.; Maher, K.; Pallansch, M.A.; Oberste, M.S. Parechovirus typing in clinical specimens by nested or semi-nested PCR coupled with sequencing. J. Clin. Virol. 2010, 48, 202–207. [Google Scholar] [CrossRef] [PubMed]
- Benschop, K.; Minnaar, R.; Koen, G.; van Eijk, H.; Dijkman, K.; Westerhuis, B.; Molenkamp, R.; Wolthers, K. Detection of human enterovirus and human parechovirus (HPeV) genotypes from clinical stool samples: Polymerase chain reaction and direct molecular typing, culture characteristics, and serotyping. Diagn. Microbiol. Infect. Dis. 2010, 68, 166–173. [Google Scholar] [CrossRef]
- Sasidharan, A.; Banerjee, D.; Harrison, C.J.; Selvarangan, R. Emergence of Parechovirus A3 as the Leading Cause of Central Nervous System Infection, Surpassing Any Single Enterovirus Type, in Children in Kansas City, Missouri, USA, from 2007 to 2016. J. Clin. Microbiol. 2021, 59, e02935-20. [Google Scholar] [CrossRef]
- Simmonds, P.; Ansari, M.A. Extensive C->U transition biases in the genomes of a wide range of mammalian RNA viruses; potential associations with transcriptional mutations, damage- or host-mediated editing of viral RNA. PLoS Pathog. 2021, 17, e1009596. [Google Scholar] [CrossRef]
- Rochman, N.D. It takes a village to build a virus. Proc. Natl. Acad. Sci. USA 2023, 120, e2219052120. [Google Scholar] [CrossRef]
- Preska Steinberg, A.; Silander, O.K.; Kussell, E. Correlated substitutions reveal SARS-like coronaviruses recombine frequently with a diverse set of structured gene pools. Proc. Natl. Acad. Sci. USA 2023, 120, e2206945119. [Google Scholar] [CrossRef]
- Rhoden, E.; Ng, T.F.F.; Campagnoli, R.; Nix, W.A.; Konopka-Anstadt, J.; Selvarangan, R.; Briesach, L.; Oberste, M.S.; Weldon, W.C. Antifungal Triazole Posaconazole Targets an Early Stage of the Parechovirus A3 Life Cycle. Antimicrob. Agents Chemother. 2020, 64, e02372-19. [Google Scholar] [CrossRef]
- Montmayeur, A.M.; Ng, T.F.; Schmidt, A.; Zhao, K.; Magaña, L.; Iber, J.; Castro, C.J.; Chen, Q.; Henderson, E.; Ramos, E.; et al. High-Throughput Next-Generation Sequencing of Polioviruses. J. Clin. Microbiol. 2017, 55, 606–615. [Google Scholar] [CrossRef]
- Altschul, S.F.; Madden, T.L.; Schäffer, A.A.; Zhang, J.; Zhang, Z.; Miller, W.; Lipman, D.J. Gapped BLAST and PSI-BLAST: A new generation of protein database search programs. Nucleic Acids Res. 1997, 25, 3389–3402. [Google Scholar] [CrossRef] [PubMed]
- Edgar, R.C. MUSCLE: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004, 32, 1792–1797. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Stecher, G.; Suleski, M.; Sanderford, M.; Sharma, S.; Tamura, K. MEGA12: Molecular Evolutionary Genetic Analysis Version 12 for Adaptive and Green Computing. Mol. Biol. Evol. 2024, 41, msae263. [Google Scholar] [CrossRef] [PubMed]
- Saitou, N.; Nei, M. The neighbor-joining method: A new method for reconstructing phylogenetic trees. Mol. Biol. Evol. 1987, 4, 406–425. [Google Scholar]
| Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).


 ,
, 




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}