The Role of Ubiquitination on Macrophages in Cardiovascular Diseases and Targeted Treatment

Abstract
1. Introduction
2. Ubiquitination and Macrophages
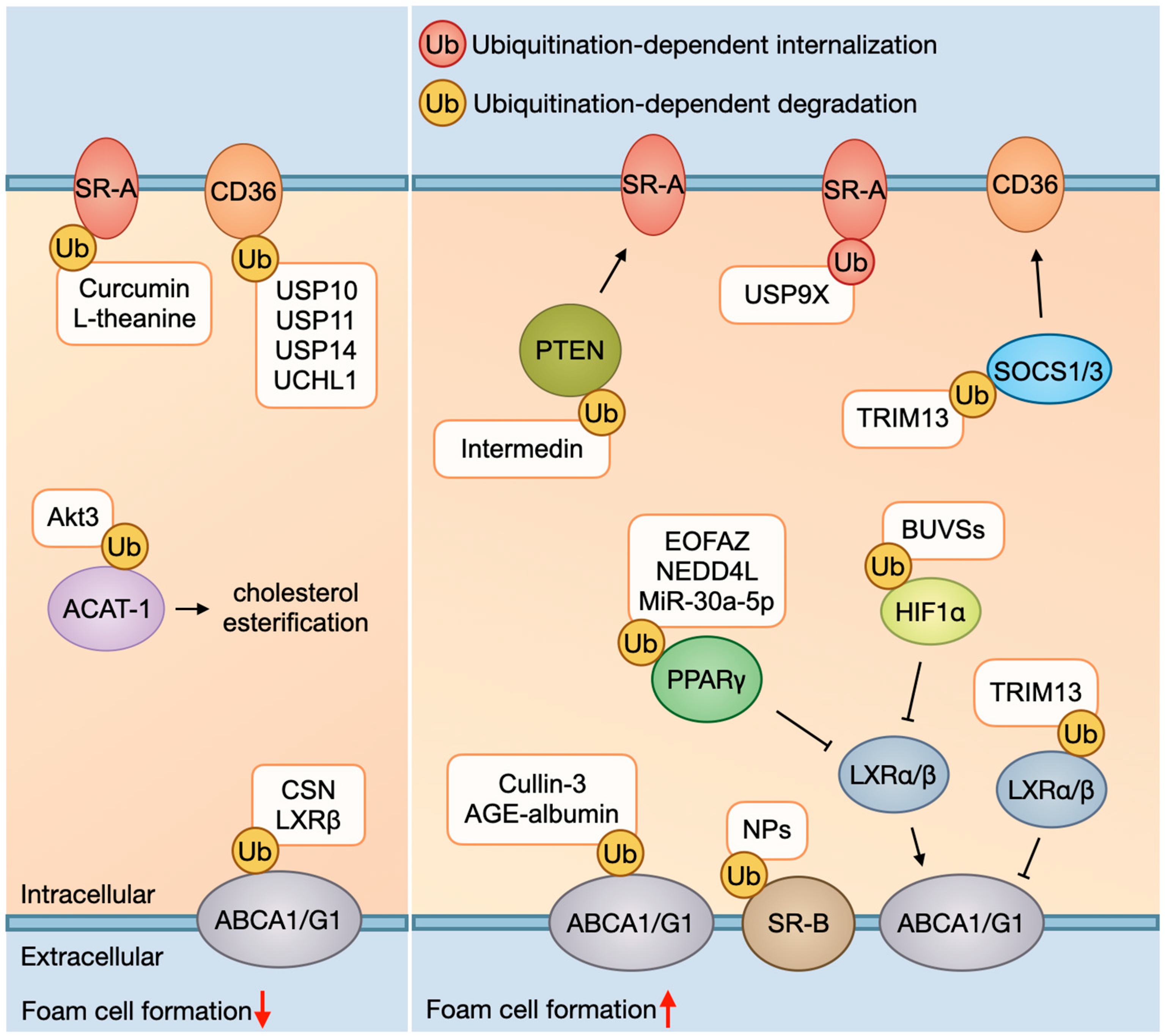
2.1. Foam Cell Formation and Ubiquitination
2.1.1. Ox-LDL Uptake and Ubiquitination
2.1.2. Cholesterol Esterification and Ubiquitination
2.1.3. Cholesterol Efflux and Ubiquitination
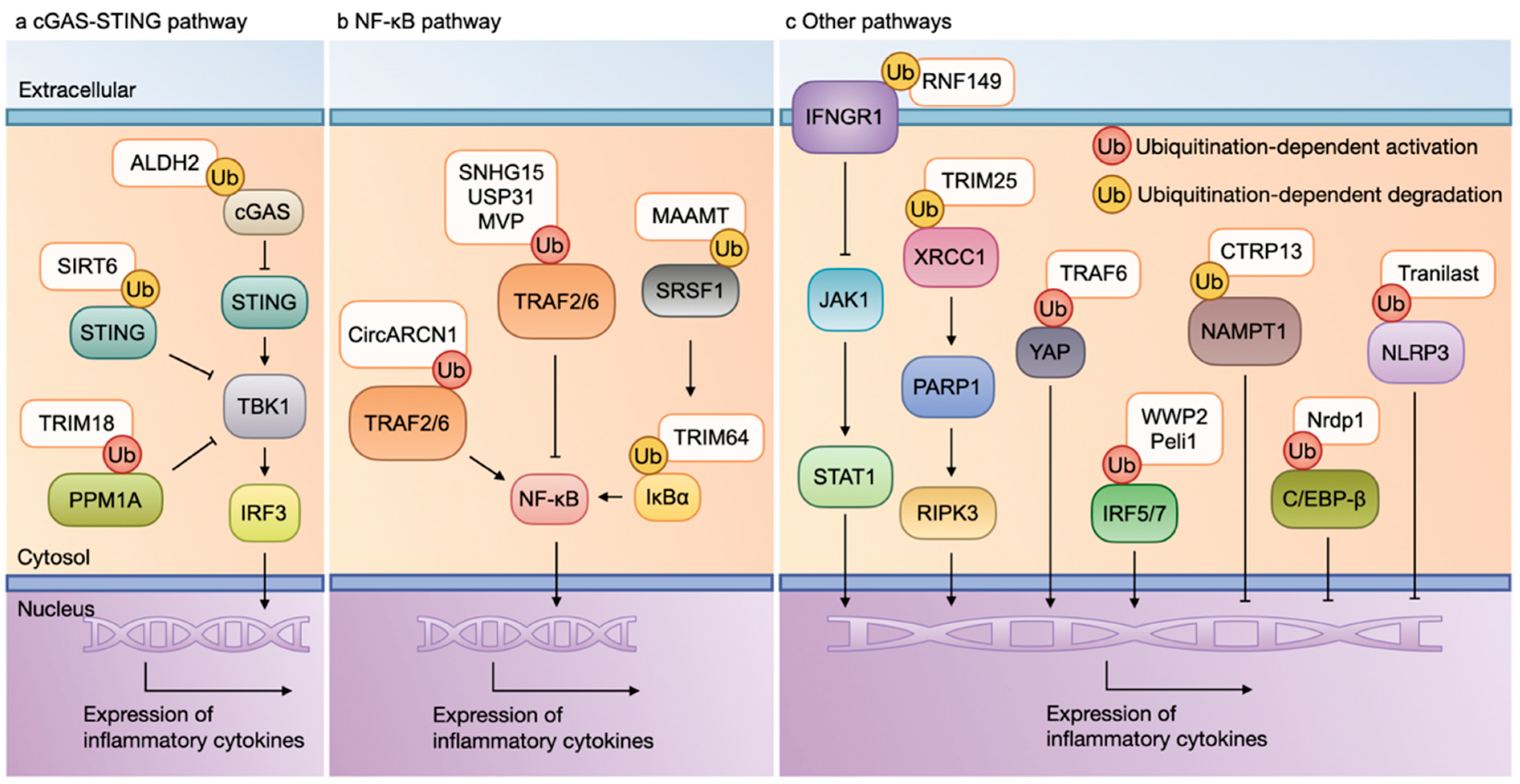
2.2. Inflammation and Ubiquitination
2.2.1. cGAS-STING Pathway and Ubiquitination
2.2.2. NF-κB Pathway and Ubiquitination
2.2.3. Other Pathways and Ubiquitination
2.3. Oxidative Stress and Ubiquitination
2.4. Programmed Cell Death and Ubiquitination
2.4.1. Apoptosis and Ubiquitination
2.4.2. Necroptosis and Ubiquitination
2.4.3. Ferroptosis and Ubiquitination
2.4.4. Pyroptosis and Ubiquitination
2.5. Autophagy and Ubiquitination
2.6. Mitophagy and Ubiquitination
2.7. Efferocytosis and Ubiquitination
3. Conclusions and Perspectives

{kind=link}
{kind=link}
| Substrate |
Ubiquitin
Chain Type | Detection Methods | Effects of Ubiquitination on Substrate | Effects of Ubiquitination on Macrophage | Models for Detecting Ubiquitination | Effects of Ubiquitination on Disease | Treatment Targeting Ubiquitination | Ref. |
|---|---|---|---|---|---|---|---|---|
| SR-A | not applicable | co-IP | promote degradation | inhibit SR-A-mediated ox-LDL uptake | J774.A1 cells | mitigate AS | curcumin | [18] |
| SR-A | not applicable | co-IP | promote degradation | inhibit SR-A-mediated ox-LDL uptake | RAW264.7 cells | mitigate AS | L-theanine | [19] |
| SR-A1 | Lys63, polyUb | co-IP | promote internalization | promote SR-A1-mediated ox-LDL uptake | HEK293 cells; HeLa cell; RAW264.7 cells | aggravate AS | USP9X | [24] |
| CD36 | polyUb | co-IP | promote degradation | inhibit CD36-mediated ox-LDL uptake | primary human macrophages | mitigate AS | CTRP9, deficiency of USP11 | [20] |
| CD36 | Lys48, polyUb | co-IP | promote degradation | inhibit CD36-mediated ox-LDL uptake | RAW264.7 cells; mouse peritoneal macrophages; THP1 macrophages; HEK293T cells | mitigate AS | UCHL1 deficiency | [21] |
| CD36 | polyUb | co-IP | promote degradation | inhibit CD36-mediated ox-LDL uptake | RAW264.7 cells; THP1 macrophages | mitigate AS | USP14 deficiency | [22] |
| CD36 | Lys48, polyUb | co-IP | promote degradation | inhibit CD36-mediated ox-LDL uptake | AW264.7 cells; THP1 macrophages | mitigate AS | USP10 deficiency | [23] |
| SOCS1/3 | not applicable | co-IP | promote degradation | promote CD36 expression via STAT1 activation | mouse peritoneal macrophages | aggravate AS | TRIM13 deficiency | [25] |
| PTEN | polyUb | co-IP | promote degradation | promote SR-A expression and ac-LDL uptake | RAW264.7 cells; mouse peritoneal macrophages | aggravate AS | Intermedin | [26] |
| ACAT-1 | polyUb | co-IP | promote degradation | inhibit cholesterol esterification and storage | murine peritoneal macrophages cultured with or without ac-LDL for 18 h | mitigate AS | Akt3 | [28] |
| ABCA1/G1 | not applicable | co-IP | promote degradation | inhibit ABCA1/G1-mediated cholesterol efflux | CHO cells incubated with cholesterol/cyclodextrin for 8 h | aggravate AS | not applicable | [32] |
| ABCA1 | not applicable | co-IP | promote degradation | inhibit ABCA1-mediated cholesterol efflux | HEK293 cells | aggravate AS | CSN | [35] |
| ABCA1 | not applicable | co-IP | promote degradation | inhibit ABCA1-mediated cholesterol efflux | COS1 cells; liver plasma membrane fractions from mice | aggravate AS | LXRβ | [33] |
| ABCA1 | not applicable | co-IP | promote degradation | inhibit ABCA1-mediated cholesterol efflux | J774 cells | aggravate AS | not applicable | [36] |
| ABCA1 | not applicable | co-IP | promote degradation | inhibit ABCA1-mediated cholesterol efflux | mouse peritoneal macrophages | aggravate AS | not applicable | [34] |
| SR-B1 | not applicable | co-IP | promote degradation | inhibit SR-B1-mediated cholesterol efflux | RAW264.7 cells; THP-1 macrophages treated with ox-LDL | aggravate AS | avoid magnetite NPs exposure | [37] |
| HIF1α | not applicable | not applicable | promote degradation | promote foam cell formation | RAW264.7 cells | aggravate AS | avoid BUVSs exposure | [38] |
| LXRα/β | not applicable | co-IP | promote degradation | inhibit ABCA1/G1-mediated cholesterol efflux | mouse peritoneal macrophages | aggravate AS | TRIM13 deficiency | [25] |
| PPARγ | not applicable | co-IP | promote degradation | inhibit ABCA1/G1-mediated cholesterol efflux | THP1 macrophages cultured with ox-LDL | aggravate AS | EOFAZ | [39] |
| PPARγ | Lys48, Lys63, polyUb | co-IP | not applicable | inhibit cholesterol efflux via downregulating ABCA1, ABCG1, LDLR, and PCSK9 | RAW264.7 cells cultured with ox-LDL | aggravate AS | miR-30a-5p | [40] |
| cGAS | Lys48, polyUb | co-IP | promote degradation | inhibit inflammatory responses via inactivating TBK1-IRF3 pathway | RAW264.7 cells stimulated by ox-LDL | mitigate AS | ALDH2 | [43] |
| STING | not applicable | co-IP | promote degradation | inhibit pro-inflammatory status | RAW264.7 cells | mitigate MI | SIRT6 deficiency | [44] |
| PPM1A | Lys63, polyUb | co-IP | promote stability | inhibit type-I IFN-mediated antiviral immunity via dephosphorylating TBK1-IRF3 pathway | THP-1 macrophages | aggravate viral myocarditis | TRIM18 deficiency | [45] |
| IRF7 | monoUb | co-IP | promote activation | promote infiltration, inflammatory responses, and profibrotic potential via upregulating CCL5 and IFN signaling | BMDMs from mice with LPS stimulation | aggravate NICM | WWP2 deficiency | [46] |
| IRF5 | Lys63 | co-IP | promote activation | promote M1 polarization and migratory ability | BMDMs from mice stimulated with conditioned medium for 24 h | aggravate MIRI | Peli1 deficiency | [47] |
| TRAF2 | Lys63 | co-IP | promote activation | promote inflammatory responses via activation of MAPK and NF-κB pathways | HEK293T cells | mitigate acute ischemic stroke | SNHG15 deficiency | [58] |
| TRAF2/6 | Lys63, polyUb | co-IP | promote activation | promote macrophage inflammation via NF-κB activation | THP-1 macrophages treated with LPS | aggravate AS | circARCN1 deficiency | [59] |
| TRAF6 | polyUb | co-IP | promote activation | promote inflammatory responses via NF-κB activation | BMDMs from mice stimulated with LPS; HEK293T cells | exacerbate AS | MVP | [60] |
| IκBα | not applicable | co-IP | promote degradation | activate NF-κB signaling, promoting pyroptosis, inflammation, and foam cell formation | THP-1 macrophages treated with ox-LDL | aggravate AS | TRIM64 deficiency | [56] |
| SRSF1 | not applicable | co-IP | promote degradation | promote inflammatory responses via NF-κB activation | BMDMs | aggravate DCM/EAM | MAAMT deficiency | [57] |
| IFNGR1 | Lys48, polyUb | co-IP | promote degradation | inhibit pro-inflammatory state | HEK293T cells | mitigate MI | RNF149 | [65] |
| XRCC1 | not applicable | Western blotting | promote degradation | promote necroptosis and pro-inflammatory status via PARP1 activation | BMDMs from mice stimulated with ox-LDL | aggravate AS | TRIM25 deficiency | [66] |
| C/EBP-β | not applicable | not applicable | not applicable | promote M2 polarization | not applicable | mitigate ICH | Nrdp1 | [67] |
| NAMPT1 | not applicable | co-IP | promote degradation | promote macrophage infiltration and pro-inflammatory status | HEK293T cells | aggravate AAA | CTRP13 stimulation | [69] |
| YAP | Lys63, polyUb | co-IP | increase nuclear translocation and stability | promote chemokine production and migration | mouse peritoneal macrophages; HEK293 cells | aggravate AS | YAP deficiency | [61] |
| NLRP3 | Lys63, polyUb | co-IP | inhibit activation | inhibit inflammatory responses | 293T cells; J774A.1 cells | mitigate AS | tranilast | [71] |
| Nrf2 | not applicable | co-IP | promote degradation | increase oxidative stress | RAW264.7 cells | aggravate AS | oridonin | [75] |
| p53 | polyUb | co-IP | promote degradation | suppress lipid-bearing macrophages apoptosis | mouse peripheral blood monocytes cultured with and ag-LDL | aggravate AS | LIG deficiency | [77] |
| LXRα | polyUb | co-IP | promote degradation | promote ER stress-dependent apoptosis | mouse peritoneal macrophages | aggravate neointimal hyperplasia | IFN-γ deficiency | [79] |
| Keap1 | not applicable | co-IP | promote degradation | inhibit ferroptosis and ferroptosis-mediated foam cell formation and inflammation | mouse peritoneal macrophages | mitigate AS | PNS | [82] |
| Sirt1 | ubiquitination | co-IP | promote degradation | inhibit autophagy | primary human macrophages incubated with ox-LDL | aggravate AS | USP22 upregulation via CTRP9 | [85] |
| MFN1/2 | polyUb | co-IP | not applicable | promote mitophagy | BMDMs incubated with ox-LDL | mitigate AS | AIBP | [87] |
| LRP-1 | not applicable | co-IP | promote degradation | decrease efferocytosis | RAW264.7 cells incubated with ox-LDL | aggravate AS | Epsins deficiency | [89] |
| PPARγ | not applicable | not applicable | promote degradation | promote pro-inflammatory status and impair efferocytosis | not applicable | aggravate AS | SHP2 deficiency | [90] |
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| CVD | Cardiovascular disease |
| Ub | Ubiquitin |
| DUB | Deubiquitinating enzyme |
| USP | Ub-specific protease |
| OTU | Ovarian tumor protease |
| UCH | Ub C-terminal hydrolase |
| MJD | Machado–Joseph domain-containing protease |
| JAMM | JAMM/MPN domain-associated Zn-dependent metalloprotease |
| MINDY | Motif interacting with Ub-containing novel DUB family |
| MCPIP | Monocyte chemotactic protein-induced protein |
| PPPDE | Permuted papain fold peptidases of dsRNA virus and eukaryote |
| ZUP1 | Zinc finger-containing Ub peptidase 1 |
| AS | Atherosclerosis |
| M-CSF | Macrophage colony-stimulating factor |
| LXRα/β | Liver X receptor α/β |
| Ox-LDL | Oxidize low-density lipoprotein |
| SR | Scavenger receptor |
| Lox-1 | Lectin-like ox-LDL receptor 1 |
| CD36 | Cluster of differentiation 36 |
| SOCS1/3 | Suppressor of cytokine signaling 1/3 |
| TRIM | Tripartite motif-containing protein |
| PTEN | Phosphatase and tensin homolog |
| Ac-LDL | Acetylated LDL |
| ACAT-1 | Acyl coenzyme A: cholesterol acyltransferase-1 |
| ABCA1/G1 | ATP-binding cassette transporters A1/G1 |
| CSN | COP9 signalosome |
| AGE | Advanced glycation end product |
| Par1 | Protease-activated receptor 1 |
| NP | Nanoparticle |
| HIF1α | Hypoxia-inducible factor 1α |
| BUVS | Benzotriazole ultraviolet stabilizer |
| EOFAZ | Essential oil from Fructus Alpinia zerumbet |
| cGAS | Cyclic GMP-AMP synthase |
| STING | Stimulator of interferon genes |
| TBK1 | TANK-binding kinase 1 |
| IRF3 | Interferon regulatory factor 3 |
| MI | Myocardial infarction |
| PPM1A | Protein phosphatase 1A |
| MAVS | Mitochondrial antiviral signaling |
| IFN | Interferon |
| IRF | Interferon regulatory factor |
| NICM | Non-ischemic cardiomyopathy |
| PAD | Peripheral artery disease |
| IL-1R | Interlukein-1 receptor |
| TRAF | Tumor necrosis factor receptor-associated factor |
| MAPK | Mitogen-activated protein kinase |
| NF-κB | Nuclear factor-κB |
| IκB | Inhibitor of κB |
| SNHG15 | Small nucleolar RNA host gene 15 |
| MVP | Major vault protein |
| SRSF1 | Serine/arginine-rich splicing factor 1 |
| EAM | Experimental autoimmune myocarditis |
| IFNGR1 | Interferon gamma receptor 1 |
| JAK1 | Janus kinase 1 |
| STAT1 | Signal transducer and activator of transcription 1 |
| RNF149 | Ring finger protein 149 |
| Nrdp1 | Neuregulin receptor degradation protein 1 |
| C/EBP-β | CCAAT enhancer-binding protein-β |
| ICH | Intracerebral hemorrhage |
| NAMPT1 | Nicotinamide phosphoribosyl-transferase 1 |
| AAA | Abdominal aortic aneurysm |
| YAP | Yes-associated protein |
| NLRP3 | NOD-like receptor thermal protein domain-associated protein 3 |
| Nrf2 | Nuclear factor erythroid 2-related factor 2 |
| Keap1 | Kelch-like ECH-associated protein 1 |
| PARP1 | Poly (ADP-ribose) polymerase 1 |
| RIPK3 | Receptor-interacting protein kinase 3 |
| Ag-LDL | Aggregated LDL |
| LIG | LDL-inducible gene |
| PCI | Percutaneous coronary intervention |
| PNS | Panax notoginseng saponins |
| Sirt1 | Sirtuin 1 |
| CTRP | C1q/TNF-related protein |
| AIBP | Apolipoprotein A-I binding protein |
| PARK2 | Parkin 2 |
| MFN1/2 | Mitofusin 1/2 |
| ROS | Reactive oxygen species |
| LRP-1 | LDLR (low-density lipoprotein receptor)-related protein 1 |
| PPARγ | peroxisome proliferator-activated receptor γ |
| SHP2 | Src homology 2-containing protein tyrosine phosphatase 2 |
| MIRI | Myocardial ischemic and reperfusion injury |
| DCM | Dilated cardiomyopathy |
| EAM | Experimental autoimmune myocarditis |
References
- Tsao, C.W.; Aday, A.W.; Almarzooq, Z.I.; Anderson, C.A.M.; Arora, P.; Avery, C.L.; Baker-Smith, C.M.; Beaton, A.Z.; Boehme, A.K.; Buxton, A.E.; et al. Heart Disease and Stroke Statistics—2023 Update: A Report from the American Heart Association. Circulation 2023, 147, E93–E621. [Google Scholar] [CrossRef] [PubMed]
- Koelwyn, G.J.; Corr, E.M.; Erbay, E.; Moore, K.J. Regulation of Macrophage Immunometabolism in Atherosclerosis. Nat. Immunol. 2018, 19, 526–537. [Google Scholar] [CrossRef] [PubMed]
- Hershko, A.; Ciechanover, A. The Ubiquitin System. Annu. Rev. Biochem. 1998, 67, 425–479. [Google Scholar] [CrossRef] [PubMed]
- Popovic, D.; Vucic, D.; Dikic, I. Ubiquitination in Disease Pathogenesis and Treatment. Nat. Med. 2014, 20, 1242–1253. [Google Scholar] [CrossRef]
- Grice, G.L.; Nathan, J.A. The Recognition of Ubiquitinated Proteins by the Proteasome. Cell. Mol. Life Sci. 2016, 73, 3497–3506. [Google Scholar] [CrossRef]
- Zhao, Y.; Mudge, M.C.; Soll, J.M.; Rodrigues, R.B.; Byrum, A.K.; Schwarzkopf, E.A.; Bradstreet, T.R.; Gygi, S.P.; Edelson, B.T.; Mosammaparast, N. OTUD4 Is a Phospho-Activated K63 Deubiquitinase That Regulates MyD88-Dependent Signaling. Mol. Cell 2018, 69, 505–516.e5. [Google Scholar] [CrossRef]
- Cockram, P.E.; Kist, M.; Prakash, S.; Chen, S.H.; Wertz, I.E.; Vucic, D. Ubiquitination in the Regulation of Inflammatory Cell Death and Cancer. Cell Death Differ. 2021, 28, 591–605. [Google Scholar] [CrossRef]
- Wang, B.; Cai, W.; Ai, D.; Zhang, X.; Yao, L. The Role of Deubiquitinases in Vascular Diseases. J. Cardiovasc. Transl. Res. 2020, 13, 131–141. [Google Scholar] [CrossRef]
- Ren, J.; Yu, P.; Liu, S.; Li, R.; Niu, X.; Chen, Y.; Zhang, Z.; Zhou, F.; Zhang, L. Deubiquitylating Enzymes in Cancer and Immunity. Adv. Sci. 2023, 10, e2303807. [Google Scholar] [CrossRef]
- Björkegren, J.L.M.; Lusis, A.J. Atherosclerosis: Recent Developments. Cell 2022, 185, 1630–1645. [Google Scholar] [CrossRef]
- Kobiyama, K.; Ley, K. Atherosclerosis a Chronic Inflammatory Disease with an Autoimmune Component. Circ. Res. 2018, 123, 1118–1120. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.; Shim, D.; Lee, J.S.; Zaitsev, K.; Williams, J.W.; Kim, K.W.; Jang, M.Y.; Jang, H.S.; Yun, T.J.; Lee, S.H.; et al. Transcriptome Analysis Reveals Nonfoamy Rather than Foamy Plaque Macrophages Are Proinflammatory in Atherosclerotic Murine Models. Circ. Res. 2018, 123, 1127–1142. [Google Scholar] [CrossRef] [PubMed]
- De Villiers, W.J.S.; Smart, E.J. Macrophage Scavenger Receptors and Foam Cell Formation. J. Leukoc. Biol. 1999, 66, 740–746. [Google Scholar] [CrossRef]
- Orekhov, A.N. LDL and Foam Cell Formation as the Basis of Atherogenesis. Curr. Opin. Lipidol. 2018, 29, 279–284. [Google Scholar] [CrossRef]
- Ouimet, M.; Franklin, V.; Mak, E.; Liao, X.; Tabas, I.; Marcel, Y.L. Autophagy Regulates Cholesterol Efflux from Macrophage Foam Cells via Lysosomal Acid Lipase. Cell Metab. 2011, 13, 655–667. [Google Scholar] [CrossRef]
- Robichaud, S.; Fairman, G.; Vijithakumar, V.; Mak, E.; Cook, D.P.; Pelletier, A.R.; Huard, S.; Vanderhyden, B.C.; Figeys, D.; Lavallée-Adam, M.; et al. Identification of Novel Lipid Droplet Factors That Regulate Lipophagy and Cholesterol Efflux in Macrophage Foam Cells. Autophagy 2021, 17, 3671–3689. [Google Scholar] [CrossRef]
- Kunjathoor, V.V.; Febbraio, M.; Podrez, E.A.; Moore, K.J.; Andersson, L.; Koehn, S.; Rhee, J.S.; Silverstein, R.; Hoff, H.F.; Freeman, M.W. Scavenger Receptors Class A-I/II and CD36 Are the Principal Receptors Responsible for the Uptake of Modified Low Density Lipoprotein Leading to Lipid Loading in Macrophages. J. Biol. Chem. 2002, 277, 49982–49988. [Google Scholar] [CrossRef]
- Zhao, J.F.; Ching, L.C.; Huang, Y.C.; Chen, C.Y.; Chiang, A.N.; Kou, Y.R.; Shyue, S.K.; Lee, T.S. Molecular Mechanism of Curcumin on the Suppression of Cholesterol Accumulation in Macrophage Foam Cells and Atherosclerosis. Mol. Nutr. Food Res. 2012, 56, 691–701. [Google Scholar] [CrossRef]
- Lei, J.; Ye, J.; She, R.; Zhang, R.; Wang, Y.; Yang, G.; Yang, J.; Luo, L. L-Theanine Inhibits Foam Cell Formation via Promoting the Scavenger Receptor A Degradation. Eur. J. Pharmacol. 2021, 904, 174181. [Google Scholar] [CrossRef]
- Zeng, M.; Wei, X.; He, Y.; Yang, Y. Ubiquitin-Specific Protease 11-Mediated CD36 Deubiquitination Acts on C1q/TNF-Related Protein 9 against Atherosclerosis. ESC Heart Fail. 2023, 10, 2499–2509. [Google Scholar] [CrossRef]
- Xia, X.; Xu, Q.; Liu, M.; Chen, X.; Liu, X.; He, J.; Hu, T.; Yu, C.; Huang, H.; Liu, S.; et al. Deubiquitination of CD36 by UCHL1 Promotes Foam Cell Formation. Cell Death Dis. 2020, 11, 636. [Google Scholar] [CrossRef] [PubMed]
- Zhang, F.; Xia, X.; Chai, R.; Xu, R.; Xu, Q.; Liu, M.; Chen, X.; Liu, B.; Liu, S.; Liu, N. Inhibition of USP14 Suppresses the Formation of Foam Cell by Promoting CD36 Degradation. J. Cell Mol. Med. 2020, 24, 3292–3302. [Google Scholar] [CrossRef] [PubMed]
- Xia, X.; Xu, Q.; Liu, X.; Liu, N.; Hu, T.; He, J.; Yu, C.; Shao, Z.; Liao, Y.; Huang, H. USP10 Deletion Inhibits Macrophage-Derived Foam Cell Formation and Cellular-Oxidized Low Density Lipoprotein Uptake by Promoting the Degradation of CD36. Aging 2020, 12, 22892–22905. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Tang, X.; Yao, L.; Wang, Y.; Chen, Z.; Li, M.; Wu, N.; Wu, D.; Dai, X.; Jiang, H.; et al. Disruption of USP9X in Macrophages Promotes Foam Cell Formation and Atherosclerosis. J. Clin. Investig. 2022, 132, e154217. [Google Scholar] [CrossRef]
- Govatati, S.; Kumar, R.; Boro, M.; Traylor, J.G.; Orr, A.W.; Lusis, A.J.; Rao, G.N. TRIM13 Reduces Cholesterol Efflux and Increases Oxidized LDL Uptake Leading to Foam Cell Formation and Atherosclerosis. J. Biol. Chem. 2024, 300, 107224. [Google Scholar] [CrossRef]
- Dai, X.Y.; Cai, Y.; Mao, D.D.; Qi, Y.F.; Tang, C.; Xu, Q.; Zhu, Y.; Xu, M.J.; Wang, X. Increased Stability of Phosphatase and Tensin Homolog by Intermedin Leading to Scavenger Receptor A Inhibition of Macrophages Reduces Atherosclerosis in Apolipoprotein E-Deficient Mice. J. Mol. Cell Cardiol. 2012, 53, 509–520. [Google Scholar] [CrossRef]
- Miyazaki, A.; Sakashita, N.; Lee, O.; Takahashi, K.; Horiuchi, S.; Hakamata, H.; Morganelli, P.M.; Chang, C.C.Y.; Chang, T.Y. Expression of ACAT-1 Protein in Human Atherosclerotic Lesions and Cultured Human Monocytes-Macrophages. Arterioscler. Thromb. Vasc. Biol. 1998, 18, 1568–1574. [Google Scholar] [CrossRef]
- Ding, L.; Biswas, S.; Morton, R.E.; Smith, J.D.; Hay, N.; Byzova, T.V.; Febbraio, M.; Podrez, E.A. Akt3 Deficiency in Macrophages Promotes Foam Cell Formation and Atherosclerosis in Mice. Cell Metab. 2012, 15, 861–872. [Google Scholar] [CrossRef]
- Chistiakov, D.A.; Melnichenko, A.A.; Myasoedova, V.A.; Grechko, A.V.; Orekhov, A.N. Mechanisms of Foam Cell Formation in Atherosclerosis. J. Mol. Med. 2017, 95, 1153–1165. [Google Scholar] [CrossRef]
- Loix, M.; Zelcer, N.; Bogie, J.F.J.; Hendriks, J.J.A. The Ubiquitous Role of Ubiquitination in Lipid Metabolism. Trends Cell Biol. 2024, 34, 416–429. [Google Scholar] [CrossRef]
- Marfella, R.; D’Amico, M.; Di Filippo, C.; Baldi, A.; Siniscalchi, M.; Sasso, F.C.; Portoghese, M.; Carbonara, O.; Crescenzi, B.; Sangiuolo, P.; et al. Increased Activity of the Ubiquitin-Proteasome System in Patients With Symptomatic Carotid Disease Is Associated With Enhanced Inflammation and May Destabilize the Atherosclerotic Plaque: Effects of Rosiglitazone Treatment. J. Am. Coll. Cardiol. 2006, 47, 2444–2455. [Google Scholar] [CrossRef] [PubMed]
- Hsieh, V.; Kim, M.J.; Gelissen, I.C.; Brown, A.J.; Sandoval, C.; Hallab, J.C.; Kockx, M.; Traini, M.; Jessup, W.; Kritharides, L. Cellular Cholesterol Regulates Ubiquitination and Degradation of the Cholesterol Export Proteins ABCA1 and ABCG1. J. Biol. Chem. 2014, 289, 7524–7536. [Google Scholar] [CrossRef] [PubMed]
- Mizuno, T.; Hayashi, H.; Kusuhara, H. Cellular Cholesterol Accumulation Facilitates Ubiquitination and Lysosomal Degradation of Cell Surface-Resident ABCA1. Arterioscler. Thromb. Vasc. Biol. 2015, 35, 1347–1356. [Google Scholar] [CrossRef] [PubMed]
- Raghavan, S.; Singh, N.K.; Mani, A.M.; Rao, G.N. Protease-Activated Receptor 1 Inhibits Cholesterol Efflux and Promotes Atherogenesis via Cullin 3-Mediated Degradation of the ABCA1 Transporter. J. Biol. Chem. 2018, 293, 10574–10589. [Google Scholar] [CrossRef]
- Azuma, Y.; Takada, M.; Maeda, M.; Kioka, N.; Ueda, K. The COP9 Signalosome Controls Ubiquitinylation of ABCA1. Biochem. Biophys. Res. Commun. 2009, 382, 145–148. [Google Scholar] [CrossRef]
- Iborra, R.T.; Machado-Lima, A.; Okuda, L.S.; Pinto, P.R.; Nakandakare, E.R.; Machado, U.F.; Correa-Giannella, M.L.; Pickford, R.; Woods, T.; Brimble, M.A.; et al. AGE-Albumin Enhances ABCA1 Degradation by Ubiquitin-Proteasome and Lysosomal Pathways in Macrophages. J. Diabetes Complicat. 2018, 32, 1–10. [Google Scholar] [CrossRef]
- Yu, H.; Xu, L.; Cui, T.; Wang, Y.; Wang, B.; Zhang, Z.; Su, R.; Zhang, J.; Zhang, R.; Wei, Y.; et al. The Foam Cell Formation Associated with Imbalanced Cholesterol Homeostasis Due to Airborne Magnetite Nanoparticles Exposure. Toxicol. Sci. 2022, 189, 287–300. [Google Scholar] [CrossRef]
- Shen, X.; Hu, W.; Xu, C.; Xu, C.; Wan, Y.; Hu, J. Benzotriazole Ultraviolet Stabilizer UV-234 Promotes Foam Cell Formation in RAW264.7 Macrophages. Environ. Pollut. 2023, 316, 120560. [Google Scholar] [CrossRef]
- Wang, S.; Xiang, J.; Zhang, G.; Fu, L.; Xu, Y.; Chen, Y.; Tao, L.; Hu, X.; Shen, X. Essential Oil from Fructus Alpinia Zerumbet Ameliorates Atherosclerosis by Activating PPARγ-LXRα-ABCA1/G1 Signaling Pathway. Phytomedicine 2024, 123, 155227. [Google Scholar] [CrossRef]
- Song, F.; Li, J.Z.; Wu, Y.; Wu, W.Y.; Wang, Y.; Li, G. Ubiquitinated Ligation Protein NEDD4L Participates in MiR-30a-5p Attenuated Atherosclerosis by Regulating Macrophage Polarization and Lipid Metabolism. Mol. Ther. Nucleic Acids 2021, 26, 1303–1317. [Google Scholar] [CrossRef]
- Moore, K.J.; Sheedy, F.J.; Fisher, E.A. Macrophages in Atherosclerosis: A Dynamic Balance. Nat. Rev. Immunol. 2013, 13, 709–721. [Google Scholar] [CrossRef] [PubMed]
- Bi, X.; Du, C.; Wang, X.; Wang, X.Y.; Han, W.; Wang, Y.; Qiao, Y.; Zhu, Y.; Ran, L.; Liu, Y.; et al. Mitochondrial Damage-Induced Innate Immune Activation in Vascular Smooth Muscle Cells Promotes Chronic Kidney Disease-Associated Plaque Vulnerability. Adv. Sci. 2021, 8, 2002738. [Google Scholar] [CrossRef] [PubMed]
- Rui, H.; Yu, H.; Chi, K.; Han, Z.; Zhu, W.; Zhang, J.; Guo, H.; Zou, W.; Wang, F.; Xu, P.; et al. ALDH2 Deficiency Augments Atherosclerosis through the USP14-CGAS-Dependent Polarization of Proinflammatory Macrophages. Redox Biol. 2024, 76, 103318. [Google Scholar] [CrossRef]
- Kong, W.; Chen, J.; Ruan, X.; Xu, X.; Li, X.; Bao, M.; Shao, Y.; Bian, X.; Li, R.; Jiang, Q.; et al. Cardiac Injury Activates STING Signaling via Upregulating SIRT6 in Macrophages after Myocardial Infarction. Life Sci. 2024, 341, 122474. [Google Scholar] [CrossRef]
- Fang, M.; Zhang, A.; Du, Y.; Lu, W.; Wang, J.; Minze, L.J.; Cox, T.C.; Li, X.C.; Xing, J.; Zhang, Z. TRIM18 Is a Critical Regulator of Viral Myocarditis and Organ Inflammation. J. Biomed. Sci. 2022, 29, 55. [Google Scholar] [CrossRef]
- Chen, H.; Chew, G.; Devapragash, N.; Loh, J.Z.; Huang, K.Y.; Guo, J.; Liu, S.; Tan, E.L.S.; Chen, S.; Tee, N.G.Z.; et al. The E3 Ubiquitin Ligase WWP2 Regulates Pro-Fibrogenic Monocyte Infiltration and Activity in Heart Fibrosis. Nat. Commun. 2022, 13, 7375. [Google Scholar] [CrossRef]
- Chen, H.; Hou, Y.; Zhai, Y.; Yang, J.; Que, L.; Liu, J.; Lu, L.; Ha, T.; Li, C.; Xu, Y.; et al. Peli1 Deletion in Macrophages Attenuates Myocardial Ischemia/Reperfusion Injury by Suppressing M1 Polarization. J. Leukoc. Biol. 2023, 113, 95–108. [Google Scholar] [CrossRef]
- Seneviratne, A.N.; Edsfeldt, A.; Cole, J.E.; Kassiteridi, C.; Swart, M.; Park, I.; Green, P.; Khoyratty, T.; Saliba, D.; Goddard, M.E.; et al. Interferon Regulatory Factor 5 Controls Necrotic Core Formation in Atherosclerotic Lesions by Impairing Efferocytosis. Circulation 2017, 136, 1140–1154. [Google Scholar] [CrossRef]
- Edsfeldt, A.; Swart, M.; Singh, P.; Dib, L.; Sun, J.; Cole, J.E.; Park, I.; Al-Sharify, D.; Persson, A.; Nitulescu, M.; et al. Interferon Regulatory Factor-5-Dependent CD11c+ Macrophages Contribute to the Formation of Rupture-Prone Atherosclerotic Plaques. Eur. Heart J. 2022, 43, 1864–1877. [Google Scholar] [CrossRef]
- Liu, H.; Cheng, W.L.; Jiang, X.; Wang, P.X.; Fang, C.; Zhu, X.Y.; Huang, Z.; She, Z.G.; Li, H. Ablation of Interferon Regulatory Factor 3 Protects Against Atherosclerosis in Apolipoprotein E-Deficient Mice. Hypertension 2017, 69, 510–520. [Google Scholar] [CrossRef]
- Zafar, A.; Pong Ng, H.; Diamond-Zaluski, R.; Kim, G.D.; Ricky Chan, E.; Dunwoodie, S.L.; Smith, J.D.; Mahabeleshwar, G.H. CITED2 Inhibits STAT1-IRF1 Signaling and Atherogenesis. FASEB J. 2021, 35, e21833. [Google Scholar] [CrossRef] [PubMed]
- Ganta, V.C.; Choi, M.H.; Kutateladze, A.; Fox, T.E.; Farber, C.R.; Annex, B.H. A MicroRNA93-Interferon Regulatory Factor-9-Immunoresponsive Gene-1-Itaconic Acid Pathway Modulates M2-Like Macrophage Polarization to Revascularize Ischemic Muscle. Circulation 2017, 135, 2403–2425. [Google Scholar] [CrossRef] [PubMed]
- Al Mamun, A.; Chauhan, A.; Qi, S.; Ngwa, C.; Xu, Y.; Sharmeen, R.; Hazen, A.L.; Li, J.; Aronowski, J.A.; McCullough, L.D.; et al. Microglial IRF5-IRF4 Regulatory Axis Regulates Neuroinflammation after Cerebral Ischemia and Impacts Stroke Outcomes. Proc. Natl. Acad. Sci. USA 2020, 117, 1742–1752. [Google Scholar] [CrossRef] [PubMed]
- Ye, S.; Lawlor, M.A.; Rivera-Reyes, A.; Egolf, S.; Chor, S.; Pak, K.; Ciotti, G.E.; Lee, A.C.; Marino, G.E.; Shah, J.; et al. YAP1-Mediated Suppression of USP31 Enhances NFκB Activity to Promote Sarcomagenesis. Cancer Res. 2018, 78, 2705–2720. [Google Scholar] [CrossRef]
- Zhu, W.; Chu, H.; Zhang, Y.; Luo, T.; Yu, H.; Zhu, H.; Liu, Y.; Gao, H.; Zhao, Y.; Li, Q.; et al. Fructose-1,6-Bisphosphatase 1 Dephosphorylates IκBα and Suppresses Colorectal Tumorigenesis. Cell Res. 2023, 33, 245–257. [Google Scholar] [CrossRef]
- Zhu, C.; Chen, W.; Cui, H.; Huang, Z.; Ding, R.; Li, N.; Wang, Q.; Wu, F.; Zhao, Y.; Cong, X. TRIM64 Promotes Ox-LDL-Induced Foam Cell Formation, Pyroptosis, and Inflammation in THP-1-Derived Macrophages by Activating a Feedback Loop with NF-ΚB via IκBα Ubiquitination. Cell Biol. Toxicol. 2023, 39, 607–620. [Google Scholar] [CrossRef]
- Gan, T.; Liu, W.; Wang, Y.; Huang, D.; Hu, J.; Wang, Y.; Xiong, J.; Wang, X.; Xu, Q.; Xiong, N.; et al. LncRNA MAAMT Facilitates Macrophage Recruitment and Proinflammatory Activation and Exacerbates Autoimmune Myocarditis through the SRSF1/NF-ΚB Axis. Int. J. Biol. Macromol. 2024, 278, 134193. [Google Scholar] [CrossRef]
- Sun, H.; Li, S.; Xu, Z.; Liu, C.; Gong, P.; Deng, Q.; Yan, F. SNHG15 Is a Negative Regulator of Inflammation by Mediating TRAF2 Ubiquitination in Stroke-Induced Immunosuppression. J. Neuroinflamm. 2022, 19, 1. [Google Scholar] [CrossRef]
- Pan, Z.; Lv, J.; Zhao, L.; Xing, K.; Ye, R.; Zhang, Y.; Chen, S.; Yang, P.; Yu, H.; Lin, Y.; et al. CircARCN1 Aggravates Atherosclerosis by Regulating HuR-Mediated USP31 MRNA in Macrophages. Cardiovasc. Res. 2024, 120, 1531–1549. [Google Scholar] [CrossRef]
- Ben, J.; Jiang, B.; Wang, D.; Liu, Q.; Zhang, Y.; Qi, Y.; Tong, X.; Chen, L.; Liu, X.; Zhang, Y.; et al. Major Vault Protein Suppresses Obesity and Atherosclerosis through Inhibiting IKK-NF-ΚB Signaling Mediated Inflammation. Nat. Commun. 2019, 10, 1801. [Google Scholar] [CrossRef]
- Liu, M.; Yan, M.; Lv, H.; Wang, B.; Lv, X.; Zhang, H.; Xiang, S.; Du, J.; Liu, T.; Tian, Y.; et al. Macrophage K63-Linked Ubiquitination of YAP Promotes Its Nuclear Localization and Exacerbates Atherosclerosis. Cell Rep. 2020, 32, 107990. [Google Scholar] [CrossRef] [PubMed]
- Mia, M.M.; Cibi, D.M.; Ghani, S.A.B.A.; Song, W.; Tee, N.; Ghosh, S.; Mao, J.; Olson, E.N.; Singh, M.K. YAP/TAZ Deficiency Reprograms Macrophage Phenotype and Improves Infarct Healing and Cardiac Function after Myocardial Infarction. PLoS Biol. 2020, 18, e3000941. [Google Scholar] [CrossRef] [PubMed]
- Francisco, J.; Guan, J.; Zhang, Y.; Nakada, Y.; Mareedu, S.; Sung, E.; Hu, C.M.; Oka, S.; Zhai, P.; Sadoshima, J.; et al. Suppression of Myeloid YAP Antagonizes Adverse Cardiac Remodeling during Pressure Overload Stress. J. Mol. Cell Cardiol. 2023, 181, 1–14. [Google Scholar] [CrossRef]
- Zhang, Y.; Wang, Y.; Li, J.; Li, C.; Liu, W.; Long, X.; Wang, Z.; Zhao, R.; Ge, J.; Shi, B. Annexin A2 Facilitates Neovascularization to Protect against Myocardial Infarction Injury via Interacting with Macrophage YAP and Endothelial Integrin Β3. Shock 2023, 60, 573–584. [Google Scholar] [CrossRef]
- Huang, C.K.; Chen, Z.; Zhou, Z.; Chen, S.; Chen, L.; Li, L.; Li, T.; Yan, X.; Chai, D. RNF149 Destabilizes IFNGR1 in Macrophages to Favor Postinfarction Cardiac Repair. Circ. Res. 2024, 135, 518–536. [Google Scholar] [CrossRef]
- Wu, H.; Gao, W.; Ma, Y.; Zhong, X.; Qian, J.; Huang, D.; Ge, J. TRIM25-Mediated XRCC1 Ubiquitination Accelerates Atherosclerosis by Inducing Macrophage M1 Polarization and Programmed Death. Inflamm. Res. 2024, 73, 1445–1458. [Google Scholar] [CrossRef]
- Wu, X.; Chen, Z.; Chen, Q.; Lin, C.; Zheng, X.; Yuan, B. Nrdp1-Mediated Macrophage Phenotypic Regulation Promotes Functional Recovery in Mice with Mild Neurological Impairment after Intracerebral Hemorrhage. Neuroscience 2024, 545, 16–30. [Google Scholar] [CrossRef]
- Imai, S. Nicotinamide Phosphoribosyltransferase (Nampt): A Link between NAD Biology, Metabolism, and Diseases. Curr. Pharm. Des. 2009, 15, 20–28. [Google Scholar] [CrossRef]
- Xu, W.; Chao, Y.; Liang, M.; Huang, K.; Wang, C. CTRP13 Mitigates Abdominal Aortic Aneurysm Formation via NAMPT1. Mol. Ther. 2021, 29, 324–337. [Google Scholar] [CrossRef]
- Broz, P.; Dixit, V.M. Inflammasomes: Mechanism of Assembly, Regulation and Signalling. Nat. Rev. Immunol. 2016, 16, 407–420. [Google Scholar] [CrossRef]
- Chen, S.; Wang, Y.; Pan, Y.; Liu, Y.; Zheng, S.; Ding, K.; Mu, K.; Yuan, Y.; Li, Z.; Song, H.; et al. Novel Role for Tranilast in Regulating NLRP3 Ubiquitination, Vascular Inflammation, and Atherosclerosis. J. Am. Heart Assoc. 2020, 9, e015513. [Google Scholar] [CrossRef] [PubMed]
- Senoner, T.; Dichtl, W. Oxidative Stress in Cardiovascular Diseases: Still a Therapeutic Target? Nutrients 2019, 11, 2090. [Google Scholar] [CrossRef] [PubMed]
- Hybertson, B.M.; Gao, B.; Bose, S.K.; McCord, J.M. Oxidative Stress in Health and Disease: The Therapeutic Potential of Nrf2 Activation. Mol. Asp. Med. 2011, 32, 234–246. [Google Scholar] [CrossRef] [PubMed]
- Piotrowska, M.; Swierczynski, M.; Fichna, J.; Piechota-Polanczyk, A. The Nrf2 in the Pathophysiology of the Intestine: Molecular Mechanisms and Therapeutic Implications for Inflammatory Bowel Diseases. Pharmacol. Res. 2021, 163, 105243. [Google Scholar] [CrossRef]
- Wang, L.; Zhao, X.; Ding, J.; Liu, Y.; Liu, H.; Zheng, L.; Zhao, H.; Sun, Z.; Li, K.; Cai, J.; et al. Oridonin Attenuates the Progression of Atherosclerosis by Inhibiting NLRP3 and Activating Nrf2 in Apolipoprotein E-Deficient Mice. Inflammopharmacology 2023, 31, 1993–2005. [Google Scholar] [CrossRef]
- Goldstein, J.L.; Brown, M.S. The Low-Density Lipoprotein Pathway and Its Relation to Atherosclerosis. Annu. Rev. Biochem. 1977, 46, 897–930. [Google Scholar] [CrossRef]
- Kikuchi, J.; Furukawa, Y.; Kubo, N.; Tokura, A.; Hayashi, N.; Nakamura, M.; Matsuda, M.; Sakurabayashi, I. Induction of Ubiquitin-Conjugating Enzyme by Aggregated Low Density Lipoprotein in Human Macrophages and Its Implications for Atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 2000, 20, 128–134. [Google Scholar] [CrossRef]
- Satish, M.; Gunasekar, P.; Asensio, J.A.; Agrawal, D.K. Vitamin D Attenuates HMGB1-Mediated Neointimal Hyperplasia after Percutaneous Coronary Intervention in Swine. Mol. Cell Biochem. 2020, 474, 219–228. [Google Scholar] [CrossRef]
- Zhao, Q.; Zhou, D.; You, H.; Lou, B.; Zhang, Y.; Tian, Y.; Guo, N.; Chen, X.; Liu, Y.; Wu, Y.; et al. IFN-γ Aggravates Neointimal Hyperplasia by Inducing Endoplasmic Reticulum Stress and Apoptosis in Macrophages by Promoting Ubiquitin-Dependent Liver X Receptor-α Degradation. FASEB J. 2017, 31, 5321–5331. [Google Scholar] [CrossRef]
- Cruz-Gregorio, A.; Amezcua-Guerra, L.M.; Fisher-Bautista, B.; Romero-Beltrán, A.; Fonseca-Camarillo, G. The Protective Role of Interleukin-37 in Cardiovascular Diseases through Ferroptosis Modulation. Int. J. Mol. Sci. 2024, 25, 9758. [Google Scholar] [CrossRef]
- Anandhan, A.; Dodson, M.; Shakya, A.; Chen, J.; Liu, P.; Wei, Y.; Tan, H.; Wang, Q.; Jiang, Z.; Yang, K.; et al. NRF2 Controls Iron Homeostasis and Ferroptosis through HERC2 and VAMP8. Sci. Adv. 2023, 9, eade9585. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Zheng, G.; Yang, S.; Liu, S.; Wu, Y.; Miao, Y.; Liang, Z.; Hua, Y.; Zhang, J.; Shi, J.; et al. The Plant Extract PNS Mitigates Atherosclerosis via Promoting Nrf2-Mediated Inhibition of Ferroptosis through Reducing USP2-Mediated Keap1 Deubiquitination. Br. J. Pharmacol. 2024, 181, 4822–4844. [Google Scholar] [CrossRef] [PubMed]
- Wei, Y.; Lan, B.; Zheng, T.; Yang, L.; Zhang, X.; Cheng, L.; Tuerhongjiang, G.; Yuan, Z.; Wu, Y. GSDME-Mediated Pyroptosis Promotes the Progression and Associated Inflammation of Atherosclerosis. Nat. Commun. 2023, 14, 929. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Sultana, N.; Siraj, N.; Ward, L.J.; Pawlik, M.; Levy, E.; Jovinge, S.; Bengtsson, E.; Yuan, X.M. Autophagy Dysfunction and Regulatory Cystatin C in Macrophage Death of Atherosclerosis. J. Cell Mol. Med. 2016, 20, 1664–1672. [Google Scholar] [CrossRef]
- Zeng, M.; Yang, Y.; Wang, Z.; Zhao, X.; Zhu, D.; Wang, M.; Chen, Y.; Wei, X. CTRP9 Prevents Atherosclerosis Progression through Changing Autophagic Status of Macrophages by Activating USP22 Mediated-de-Ubiquitination on Sirt1 in Vitro. Mol. Cell Endocrinol. 2024, 584, 112161. [Google Scholar] [CrossRef]
- Rodger, C.E.; McWilliams, T.G.; Ganley, I.G. Mammalian Mitophagy—From in Vitro Molecules to in Vivo Models. FEBS J. 2018, 285, 1185–1202. [Google Scholar] [CrossRef]
- Choi, S.H.; Agatisa-Boyle, C.; Gonen, A.; Kim, A.; Kim, J.; Alekseeva, E.; Tsimikas, S.; Miller, Y.I. Intracellular AIBP (Apolipoprotein A-I Binding Protein) Regulates Oxidized LDL (Low-Density Lipoprotein)-Induced Mitophagy in Macrophages. Arterioscler. Thromb. Vasc. Biol. 2021, 41, E82–E96. [Google Scholar] [CrossRef]
- Doddapattar, P.; Dev, R.; Ghatge, M.; Patel, R.B.; Jain, M.; Dhanesha, N.; Lentz, S.R.; Chauhan, A.K. Myeloid Cell PKM2 Deletion Enhances Efferocytosis and Reduces Atherosclerosis. Circ. Res. 2022, 130, 1289–1305. [Google Scholar] [CrossRef]
- Brophy, M.L.; Dong, Y.; Tao, H.; Yancey, P.G.; Song, K.; Zhang, K.; Wen, A.; Wu, H.; Lee, Y.; Malovichko, M.V.; et al. Myeloid-Specific Deletion of Epsins 1 and 2 Reduces Atherosclerosis by Preventing LRP-1 Downregulation. Circ. Res. 2019, 124, E6–E19. [Google Scholar] [CrossRef]
- Wu, C.; Zheng, P.; Ma, L.; Xu, C.; Hu, L.; Yang, Z.; Fei, F.; Shen, Z.; Zhang, X.; Wu, Z.; et al. Protein Tyrosine Phosphatase SHP2 in Macrophages Acts as an Antiatherosclerotic Regulator in Mice. Arterioscler. Thromb. Vasc. Biol. 2024, 44, 202–217. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, L.; Zhang, Y.; Yue, J.; Zhou, R. The Role of Ubiquitination on Macrophages in Cardiovascular Diseases and Targeted Treatment. Int. J. Mol. Sci. 2025, 26, 4260. https://doi.org/10.3390/ijms26094260
Wang L, Zhang Y, Yue J, Zhou R. The Role of Ubiquitination on Macrophages in Cardiovascular Diseases and Targeted Treatment. International Journal of Molecular Sciences. 2025; 26(9):4260. https://doi.org/10.3390/ijms26094260
Chicago/Turabian StyleWang, Li, Yan Zhang, Jianming Yue, and Ronghua Zhou. 2025. "The Role of Ubiquitination on Macrophages in Cardiovascular Diseases and Targeted Treatment" International Journal of Molecular Sciences 26, no. 9: 4260. https://doi.org/10.3390/ijms26094260
APA StyleWang, L., Zhang, Y., Yue, J., & Zhou, R. (2025). The Role of Ubiquitination on Macrophages in Cardiovascular Diseases and Targeted Treatment. International Journal of Molecular Sciences, 26(9), 4260. https://doi.org/10.3390/ijms26094260