
TFProtBert: Detection of Transcription Factors Binding to Methylated DNA Using ProtBert Latent Space Representation



Abstract
1. Introduction
2. Results
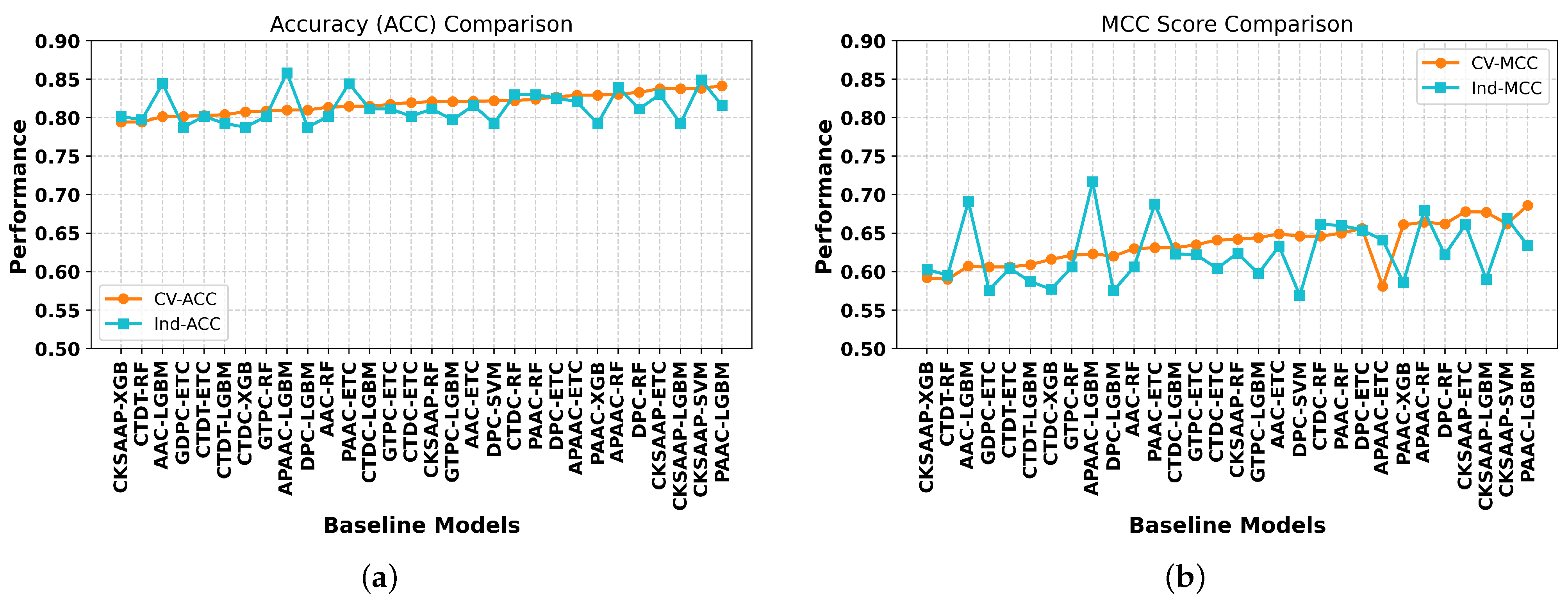
2.1. Performance Evaluation of the Baseline Models on Different Feature Encodings
2.2. Performance Evaluation of the Meta-Models Using Probabilistic Feature Vectors
2.3. Performance Evaluation of the ML Classifiers with TFProtBert Using 1024-D Vector
2.4. Performance Evaluation Between TFProtBert and the Existing Methods
2.5. Construction of the Second-Layer Model to Predict TFPMs and Its Comparison with the Existing Predictors
2.6. Performance of TFProtBert on Imbalanced Dataset
2.7. Two-Dimensional Feature Set Representation
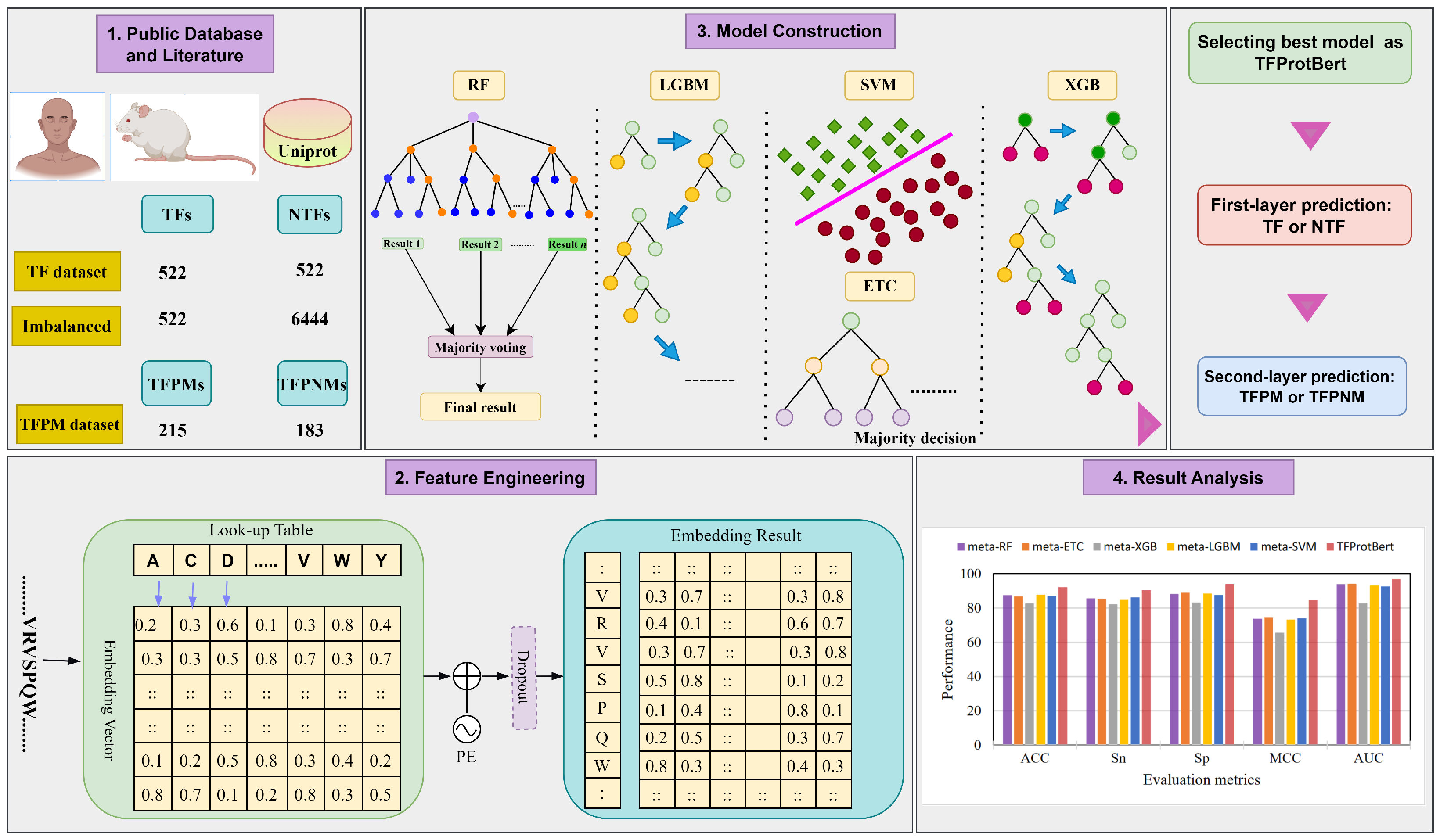
3. Materials and Methods
3.1. Data Collection
3.2. Feature Extraction
- (i)
- Amino Acid Composition (AAC): AAC [33] is a feature vector of length 20, representing the frequency of each amino acid’s occurrence within a given peptide sequence. The mathematical expression for AAC is as follows:Here, L represents the total length of the sequence, while denotes frequency of the occurrence of the amino acid type m.
- (ii)
- Pseudo-amino acid composition (PAAC): PAAC [34] translates protein or peptide sequences into numerical characteristics, capturing both the intrinsic attributes of each amino acid and its significant position within the sequence. The PAAC can be described as:withwhere is the number of times the amino acid z occurs, w is the weight, set at 0.5, L is the sequence length, and represents sequence-correlated factors. implies a correlation function, and the value of is set at 3, making the PAAC feature a 23-D-long vector.
- (iii)
- Amphiphilic pseudo-amino acid composition (APAAC): APAAC [35] takes into consideration the amphiphilic properties of amino acids, allowing for the depiction of protein sequences in terms of their hydrophobic and hydrophilic features. The computation process for APAAC is outlined as follows:within this context, w denotes the weight, which is assigned a value of 0.5, is the normalized occurrence of the amino acid z, and implies the sequence-order factor. These factors related to sequence order can be denoted as:the value is established as 3, resulting in an APAAC encoding of a 26-dimensional vector, while L is the peptide sequence.
- (iv)
- Composition of k-spaced amino acid pairs (CKSAAP): The method described in [36] for CKSAAP feature encoding is utilized to analyze protein or peptide sequences. To find the frequency of k-spaced amino acid pairs in a protein or peptide sequence, this method involves a series of computations. Since the parameter k, in this case, fluctuates from 0 to 5, we concentrated on descriptors, especially for k = 5, which produced a 2400-dimensional feature vector for CKSAAP.
- (v)
- Composition, transition, distribution, and triplet (CTDT): CTDT [37] is a feature representation approach employed in bioinformatics to analyze protein or peptide sequences. Its objective is to encompass diverse elements of the sequence, such as amino acid composition, shifts between distinct amino acid types, their arrangement patterns, and triplet arrangements.
- (vi)
- Composition, transition, distribution, and composition (CTDC): CTDC [37] encoding aims to capture various facets of the sequence, including the frequency of amino acids, transitions between different types of amino acids, their spatial distribution, and other aspects related to amino acid composition.
- (vii)
- Di-peptide composition (DPC): DPC [33] gives 400 descriptors based on the frequency of the two amino acids in a given sequence. It can be defined as:where is the number of the di-peptides of type m and n, and Z denotes the sequence length.
- (viii)
- Grouped di-peptide composition (GDPC): GDPC [38] is a special variation of the TDPC descriptor of 125 descriptors.where represents a di-peptide of amino acid types m and n, and Z represents the total length of the sequence. represents the five different classes.
- (ix)
- Grouped tri-peptide composition (GTPC): GTPC [38] is a special variation of the TPC descriptor of 125 descriptors.where represents a tri-peptide of amino acid types m, n, and o, and Z represents the total length of the sequence. represents the five different classes.
3.3. Conventional Machine Learning-Based Classifiers
3.4. Construction of Meta-Models
3.5. Construction of TFProtBert
3.6. Performance Evaluation
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Kummerfeld, S.K.; Teichmann, S.A. DBD: A transcription factor prediction database. Nucleic Acids Res. 2006, 34, D74–D81. [Google Scholar] [CrossRef] [PubMed]
- Bushweller, J.H. Targeting transcription factors in cancer—From undruggable to reality. Nat. Rev. Cancer 2019, 19, 611–624. [Google Scholar] [CrossRef] [PubMed]
- Yin, Y.; Morgunova, E.; Jolma, A.; Kaasinen, E.; Sahu, B.; Khund-Sayeed, S.; Das, P.K.; Kivioja, T.; Dave, K.; Zhong, F.; et al. Impact of cytosine methylation on DNA binding specificities of human transcription factors. Science 2017, 356, eaaj2239. [Google Scholar] [CrossRef]
- Shen, Z.; Zou, Q. Basic polar and hydrophobic properties are the main characteristics that affect the binding of transcription factors to methylation sites. Bioinformatics 2020, 36, 4263–4268. [Google Scholar] [CrossRef]
- Weirauch, M.T.; Yang, A.; Albu, M.; Cote, A.G.; Montenegro-Montero, A.; Drewe, P.; Najafabadi, H.S.; Lambert, S.A.; Mann, I.; Cook, K.; et al. Determination and inference of eukaryotic transcription factor sequence specificity. Cell 2014, 158, 1431–1443. [Google Scholar] [CrossRef]
- Wang, G.; Luo, X.; Wang, J.; Wan, J.; Xia, S.; Zhu, H.; Qian, J.; Wang, Y. MeDReaders: A database for transcription factors that bind to methylated DNA. Nucleic Acids Res. 2018, 46, D146–D151. [Google Scholar] [CrossRef]
- Gaston, K.; Fried, M. CpG methylation has differential effects on the binding of YY1 and ETS proteins to the bi-directional promoter of the Surf-1 and Surf-2 genes. Nucleic Acids Res. 1995, 23, 901–909. [Google Scholar] [CrossRef]
- Mann, I.K.; Chatterjee, R.; Zhao, J.; He, X.; Weirauch, M.T.; Hughes, T.R.; Vinson, C. CG methylated microarrays identify a novel methylated sequence bound by the CEBPB| ATF4 heterodimer that is active in vivo. Genome Res. 2013, 23, 988–997. [Google Scholar] [CrossRef]
- Liu, Q.; Fang, L.; Yu, G.; Wang, D.; Xiao, C.L.; Wang, K. Detection of DNA base modifications by deep recurrent neural network on Oxford Nanopore sequencing data. Nat. Commun. 2019, 10, 2449. [Google Scholar] [CrossRef]
- Hu, S.; Wan, J.; Su, Y.; Song, Q.; Zeng, Y.; Nguyen, H.N.; Shin, J.; Cox, E.; Rho, H.S.; Woodard, C.; et al. DNA methylation presents distinct binding sites for human transcription factors. eLife 2013, 2, e00726. [Google Scholar] [CrossRef]
- Gkountela, S.; Castro-Giner, F.; Szczerba, B.M.; Vetter, M.; Landin, J.; Scherrer, R.; Krol, I.; Scheidmann, M.C.; Beisel, C.; Stirnimann, C.U.; et al. Circulating tumor cell clustering shapes DNA methylation to enable metastasis seeding. Cell 2019, 176, 98–112. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Gao, Q.; Tan, S.; You, J.; Lyu, C.; Zhang, Y.; Han, M.; Chen, Z.; Li, J.; Wang, H.; et al. SET8 prevents excessive DNA methylation by methylation-mediated degradation of UHRF1 and DNMT1. Nucleic Acids Res. 2019, 47, 9053–9068. [Google Scholar] [CrossRef] [PubMed]
- Yin, S.; Liu, L.; Brobbey, C.; Palanisamy, V.; Ball, L.E.; Olsen, S.K.; Ostrowski, M.C.; Gan, W. PRMT5-mediated arginine methylation activates AKT kinase to govern tumorigenesis. Nat. Commun. 2021, 12, 3444. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Hu, X.; Huang, M.; Liu, J.; Gu, Y.; Ma, L.; Zhou, Q.; Cao, X. Mettl3-mediated mRNA m6A methylation promotes dendritic cell activation. Nat. Commun. 2019, 10, 1898. [Google Scholar] [CrossRef]
- Roulet, E.; Busso, S.; Camargo, A.A.; Simpson, A.J.; Mermod, N.; Bucher, P. High-throughput SELEX–SAGE method for quantitative modeling of transcription-factor binding sites. Nat. Biotechnol. 2002, 20, 831–835. [Google Scholar] [CrossRef]
- Rockel, S.; Geertz, M.; Maerkl, S.J. MITOMI: A microfluidic platform for in vitro characterization of transcription factor–DNA interaction. In Gene Regulatory Networks: Methods and Protocols; Humana Press: Totowa, NJ, USA, 2012; pp. 97–114. [Google Scholar]
- Yashiro, T.; Hara, M.; Ogawa, H.; Okumura, K.; Nishiyama, C. Critical role of transcription factor PU. 1 in the function of the OX40L/TNFSF4 promoter in dendritic cells. Sci. Rep. 2016, 6, 34825. [Google Scholar] [CrossRef]
- Wingender, E.; Dietze, P.; Karas, H.; Knüppel, R. TRANSFAC: A database on transcription factors and their DNA binding sites. Nucleic Acids Res. 1996, 24, 238–241. [Google Scholar] [CrossRef]
- Riaño-Pachón, D.M.; Ruzicic, S.; Dreyer, I.; Mueller-Roeber, B. PlnTFDB: An integrative plant transcription factor database. BMC Bioinform. 2007, 8, 1–10. [Google Scholar] [CrossRef]
- Zhu, Q.H.; Guo, A.Y.; Gao, G.; Zhong, Y.F.; Xu, M.; Huang, M.; Luo, J. DPTF: A database of poplar transcription factors. Bioinformatics 2007, 23, 1307–1308. [Google Scholar] [CrossRef]
- Zhang, Q.; Liu, W.; Zhang, H.M.; Xie, G.Y.; Miao, Y.R.; Xia, M.; Guo, A.Y. hTFtarget: A comprehensive database for regulations of human transcription factors and their targets. Genom. Proteom. Bioinform. 2020, 18, 120–128. [Google Scholar] [CrossRef]
- Liu, M.L.; Su, W.; Wang, J.S.; Yang, Y.H.; Yang, H.; Lin, H. Predicting preference of transcription factors for methylated DNA using sequence information. Mol. Ther.-Nucleic Acids 2020, 22, 1043–1050. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, Q.H.; Tran, H.V.; Nguyen, B.P.; Do, T.T. Identifying transcription factors that prefer binding to methylated DNA using reduced G-gap dipeptide composition. ACS Omega 2022, 7, 32322–32330. [Google Scholar] [CrossRef]
- Zheng, P.; Qi, Y.; Li, X.; Liu, Y.; Yao, Y.; Huang, G. A capsule network-based method for identifying transcription factors. Front. Microbiol. 2022, 13, 1048478. [Google Scholar] [CrossRef]
- Sabour, S.; Frosst, N.; Hinton, G.E. Dynamic routing between capsules. Adv. Neural Inf. Process. Syst. 2017, 30. [Google Scholar]
- Li, H.; Gong, Y.; Liu, Y.; Lin, H.; Wang, G. Detection of transcription factors binding to methylated DNA by deep recurrent neural network. Briefings Bioinform. 2022, 23, bbab533. [Google Scholar] [CrossRef]
- Van der Maaten, L.; Hinton, G. Visualizing data using t-SNE. J. Mach. Learn. Res. 2008, 9, 2579–2605. [Google Scholar]
- Hassan, M.T.; Tayara, H.; Chong, K.T. An integrative machine learning model for the identification of tumor T-cell antigens. BioSystems 2024, 237, 105177. [Google Scholar] [CrossRef]
- Kobak, D.; Berens, P. The art of using t-SNE for single-cell transcriptomics. Nat. Commun. 2019, 10, 5416. [Google Scholar] [CrossRef]
- Graves, A.; Schmidhuber, J. Framewise phoneme classification with bidirectional LSTM and other neural network architectures. Neural Netw. 2005, 18, 602–610. [Google Scholar] [CrossRef]
- Huang, Y.; Niu, B.; Gao, Y.; Fu, L.; Li, W. CD-HIT Suite: A web server for clustering and comparing biological sequences. Bioinformatics 2010, 26, 680–682. [Google Scholar] [CrossRef]
- Zou, Q.; Lin, G.; Jiang, X.; Liu, X.; Zeng, X. Sequence clustering in bioinformatics: An empirical study. Briefings Bioinform. 2020, 21, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Bhasin, M.; Raghava, G.P. Classification of nuclear receptors based on amino acid composition and dipeptide composition. J. Biol. Chem. 2004, 279, 23262–23266. [Google Scholar] [CrossRef]
- Chou, K.C. Pseudo amino acid composition and its applications in bioinformatics, proteomics and system biology. Curr. Proteom. 2009, 6, 262–274. [Google Scholar] [CrossRef]
- Chou, K.C. Using amphiphilic pseudo amino acid composition to predict enzyme subfamily classes. Bioinformatics 2005, 21, 10–19. [Google Scholar] [CrossRef]
- Chen, Z.; Zhao, P.; Li, F.; Marquez-Lago, T.T.; Leier, A.; Revote, J.; Zhu, Y.; Powell, D.R.; Akutsu, T.; Webb, G.I.; et al. iLearn: An integrated platform and meta-learner for feature engineering, machine-learning analysis and modeling of DNA, RNA and protein sequence data. Briefings Bioinform. 2020, 21, 1047–1057. [Google Scholar] [CrossRef]
- Dubchak, I.; Muchnik, I.; Holbrook, S.R.; Kim, S.H. Prediction of protein folding class using global description of amino acid sequence. Proc. Natl. Acad. Sci. USA 1995, 92, 8700–8704. [Google Scholar] [CrossRef]
- Chen, Z.; Zhao, P.; Li, F.; Leier, A.; Marquez-Lago, T.T.; Wang, Y.; Webb, G.I.; Smith, A.I.; Daly, R.J.; Chou, K.C.; et al. iFeature: A python package and web server for features extraction and selection from protein and peptide sequences. Bioinformatics 2018, 34, 2499–2502. [Google Scholar] [CrossRef]
- Noor, A.; Gaffar, S.; Hassan, M.; Junaid, M.; Mir, A.; Kaur, A. Hybrid image fusion method based on discrete wavelet transform (DWT), principal component analysis (PCA) and guided filter. In Proceedings of the 2020 First International Conference of Smart Systems and Emerging Technologies (SMARTTECH), Riyadh, Saudi Arabia, 3–5 November 2020; pp. 138–143. [Google Scholar]
- Eesaar, H.; Joe, S.; Rehman, M.U.; Jang, Y.; Chong, K.T. SEiPV-Net: An Efficient Deep Learning Framework for Autonomous Multi-Defect Segmentation in Electroluminescence Images of Solar Photovoltaic Modules. Energies 2023, 16, 7726. [Google Scholar] [CrossRef]
- Luo, J.Y.; Irisson, J.O.; Graham, B.; Guigand, C.; Sarafraz, A.; Mader, C.; Cowen, R.K. Automated plankton image analysis using convolutional neural networks. Limnol. Oceanogr. Methods 2018, 16, 814–827. [Google Scholar] [CrossRef]
- Solopov, M.; Chechekhina, E.; Kavelina, A.; Akopian, G.; Turchin, V.; Popandopulo, A.; Filimonov, D.; Ishchenko, R. Comparative Study of Deep Transfer Learning Models for Semantic Segmentation of Human Mesenchymal Stem Cell Micrographs. Int. J. Mol. Sci. 2025, 26, 2338. [Google Scholar] [CrossRef]
- Swanson, K.; Walther, P.; Leitz, J.; Mukherjee, S.; Wu, J.C.; Shivnaraine, R.V.; Zou, J. ADMET-AI: A machine learning ADMET platform for evaluation of large-scale chemical libraries. Bioinformatics 2024, 40, btae416. [Google Scholar] [CrossRef]
- Mir, B.A.; Tayara, H.; Chong, K.T. SB-Net: Synergizing CNN and LSTM Networks for Uncovering Retrosynthetic Pathways in Organic Synthesis. Comput. Biol. Chem. 2024, 112, 108130. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Pei, J.; Lai, L. Deep learning based regression and multiclass models for acute oral toxicity prediction with automatic chemical feature extraction. J. Chem. Inf. Model. 2017, 57, 2672–2685. [Google Scholar] [CrossRef] [PubMed]
- Setiya, A.; Jani, V.; Sonavane, U.; Joshi, R. MolToxPred: Small molecule toxicity prediction using machine learning approach. RSC Adv. 2024, 14, 4201–4220. [Google Scholar] [CrossRef] [PubMed]
- Gaffar, S.; Tayara, H.; Chong, K.T. Stack-AAgP: Computational prediction and interpretation of anti-angiogenic peptides using a meta-learning framework. Comput. Biol. Med. 2024, 174, 108438. [Google Scholar] [CrossRef]
- Zahid, H.; Tayara, H.; Chong, K.T. Harnessing machine learning to predict cytochrome P450 inhibition through molecular properties. Arch. Toxicol. 2024, 98, 2647–2658. [Google Scholar] [CrossRef]
- Arnal Segura, M.; Bini, G.; Krithara, A.; Paliouras, G.; Tartaglia, G.G. Machine Learning Methods for Classifying Multiple Sclerosis and Alzheimer’s Disease Using Genomic Data. Int. J. Mol. Sci. 2025, 26, 2085. [Google Scholar] [CrossRef]
- Wang, M.; Wang, J.; Rong, Z.; Wang, L.; Xu, Z.; Zhang, L.; He, J.; Li, S.; Cao, L.; Hou, Y.; et al. A bidirectional interpretable compound-protein interaction prediction framework based on cross attention. Comput. Biol. Med. 2024, 172, 108239. [Google Scholar] [CrossRef]
- Sharma, N.; Naorem, L.D.; Jain, S.; Raghava, G.P. ToxinPred2: An improved method for predicting toxicity of proteins. Briefings Bioinform. 2022, 23, bbac174. [Google Scholar] [CrossRef]
- Charoenkwan, P.; Chiangjong, W.; Nantasenamat, C.; Hasan, M.M.; Manavalan, B.; Shoombuatong, W. StackIL6: A stacking ensemble model for improving the prediction of IL-6 inducing peptides. Briefings Bioinform. 2021, 22, bbab172. [Google Scholar] [CrossRef]
- Hassan, M.T.; Tayara, H.; Chong, K.T. NaII-Pred: An ensemble-learning framework for the identification and interpretation of sodium ion inhibitors by fusing multiple feature representation. Comput. Biol. Med. 2024, 178, 108737. [Google Scholar] [CrossRef] [PubMed]
- Akiba, T.; Sano, S.; Yanase, T.; Ohta, T.; Koyama, M. Optuna: A next-generation hyperparameter optimization framework. In Proceedings of the 25th ACM SIGKDD International Conference on Knowledge Discovery & Data Mining, Anchorage, AK, USA, 4–8 August 2019; pp. 2623–2631. [Google Scholar]
- Hassan, M.T.; Tayara, H.; Chong, K.T. Meta-IL4: An ensemble learning approach for IL-4-inducing peptide prediction. Methods 2023, 217, 49–56. [Google Scholar] [CrossRef]
- Gaffar, S.; Hassan, M.T.; Tayara, H.; Chong, K.T. IF-AIP: A machine learning method for the identification of anti-inflammatory peptides using multi-feature fusion strategy. Comput. Biol. Med. 2023, 168, 107724. [Google Scholar] [CrossRef] [PubMed]
- Akbar, B.; Tayara, H.; Chong, K.T. Unveiling dominant recombination loss in perovskite solar cells with a XGBoost-based machine learning approach. Iscience 2024, 27, 109200. [Google Scholar] [CrossRef] [PubMed]
- Hassan, M.T.; Tayara, H.; Chong, K.T. Possum: Identification and interpretation of potassium ion inhibitors using probabilistic feature vectors. Arch. Toxicol. 2024, 99, 225–235. [Google Scholar] [CrossRef]
- Hassan, M.T.; Tayara, H.; Chong, K.T. iAnOxPep: A machine learning model for the identification of anti-oxidative peptides using ensemble learning. IEEE/ACM Trans. Comput. Biol. Bioinform. 2024, 22, 85–96. [Google Scholar] [CrossRef]
- Ming, Y.; Liu, H.; Cui, Y.; Guo, S.; Ding, Y.; Liu, R. Identification of DNA-binding proteins by Kernel Sparse Representation via L2, 1-matrix norm. Comput. Biol. Med. 2023, 159, 106849. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Models | Benchmark Dataset | Independent Dataset | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| ACC | Sn | Sp | MCC | AUC | ACC | Sn | Sp | MCC | AUC | |
| RF | 90.94 | 89.58 | 92.06 | 0.818 | 96.13 | 91.50 | 90.56 | 92.45 | 0.830 | 95.28 |
| ETC | 90.58 | 88.61 | 91.82 | 0.807 | 96.21 | 91.98 | 91.50 | 92.45 | 0.839 | 95.86 |
| XGB | 88.05 | 87.16 | 88.94 | 0.763 | 94.88 | 87.73 | 90.56 | 84.90 | 0.755 | 94.97 |
| LGBM | 91.43 | 90.07 | 90.08 | 0.810 | 96.17 | 93.86 | 95.28 | 92.45 | 0.877 | 97.18 |
| TFProtBert | 92.15 | 90.31 | 93.99 | 0.845 | 96.93 | 96.22 | 97.16 | 95.28 | 0.906 | 97.59 |
| Method | ACC | Sn | Sp | MCC | AUC |
|---|---|---|---|---|---|
| TFPred | 83.02 | 80.19 | 85.85 | 0.661 | 91.16 |
| Li_RNN | 86.63 | 88.68 | 83.96 | 0.727 | 91.30 |
| PSSM+CNN | 87.26 | 90.56 | 83.96 | 0.746 | 95.96 |
| Capsule_TF | 88.20 | 91.51 | 84.96 | 0.765 | 92.54 |
| TFProtBert | 96.22 | 97.16 | 95.28 | 0.906 | 97.59 |
| Method | ACC | Sn | Sp | MCC | AUC |
|---|---|---|---|---|---|
| Li_RNN | 26.42 | 30.43 | 18.93 | −0.483 | — |
| TFProtBert | 55.66 | 59.42 | 48.64 | 0.077 | 53.72 |
| Models | Imbalanced Benchmark Dataset | Independent Dataset | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ACC | Sn | Sp | MCC | AUC | Pr | F1 | ACC | Sn | Sp | MCC | AUC | Pr | F1 | |
| RF | 97.09 | 66.58 | 99.53 | 0.768 | 97.35 | 92.98 | 72.61 | 97.27 | 70.75 | 99.45 | 0.790 | 96.79 | 91.49 | 79.70 |
| ETC | 97.21 | 74.09 | 99.06 | 0.786 | 97.43 | 92.08 | 70.31 | 96.55 | 58.49 | 99.68 | 0.726 | 97.51 | 93.91 | 72.04 |
| XGB | 97.21 | 74.09 | 99.06 | 0.786 | 97.43 | 86.55 | 79.77 | 96.70 | 68.86 | 98.99 | 0.747 | 99.44 | 84.87 | 76.07 |
| LGBM | 97.09 | 75.54 | 98.81 | 0.780 | 96.50 | 83.90 | 79.31 | 96.91 | 79.24 | 98.37 | 0.779 | 96.84 | 80.00 | 79.64 |
| TFProtBert | 98.41 | 84.50 | 99.53 | 0.881 | 97.92 | 91.61 | 78.40 | 98.70 | 91.50 | 99.30 | 0.908 | 97.94 | 86.47 | 82.18 |
| Dataset | Training Set | Independent Set | ||
|---|---|---|---|---|
| Pos | Neg | Pos | Neg | |
| TF dataset | 416 | 416 | 106 | 106 |
| TFPM dataset | 146 | 146 | 69 | 37 |
| Imbalanced dataset | 416 | 5155 | 106 | 1289 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gaffar, S.; Chong, K.T.; Tayara, H. TFProtBert: Detection of Transcription Factors Binding to Methylated DNA Using ProtBert Latent Space Representation. Int. J. Mol. Sci. 2025, 26, 4234. https://doi.org/10.3390/ijms26094234
Gaffar S, Chong KT, Tayara H. TFProtBert: Detection of Transcription Factors Binding to Methylated DNA Using ProtBert Latent Space Representation. International Journal of Molecular Sciences. 2025; 26(9):4234. https://doi.org/10.3390/ijms26094234
Chicago/Turabian StyleGaffar, Saima, Kil To Chong, and Hilal Tayara. 2025. "TFProtBert: Detection of Transcription Factors Binding to Methylated DNA Using ProtBert Latent Space Representation" International Journal of Molecular Sciences 26, no. 9: 4234. https://doi.org/10.3390/ijms26094234
APA StyleGaffar, S., Chong, K. T., & Tayara, H. (2025). TFProtBert: Detection of Transcription Factors Binding to Methylated DNA Using ProtBert Latent Space Representation. International Journal of Molecular Sciences, 26(9), 4234. https://doi.org/10.3390/ijms26094234