Identifying Therapeutic Targets for Amyotrophic Lateral Sclerosis Through Modeling of Multi-Omics Data

 ,
,  ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
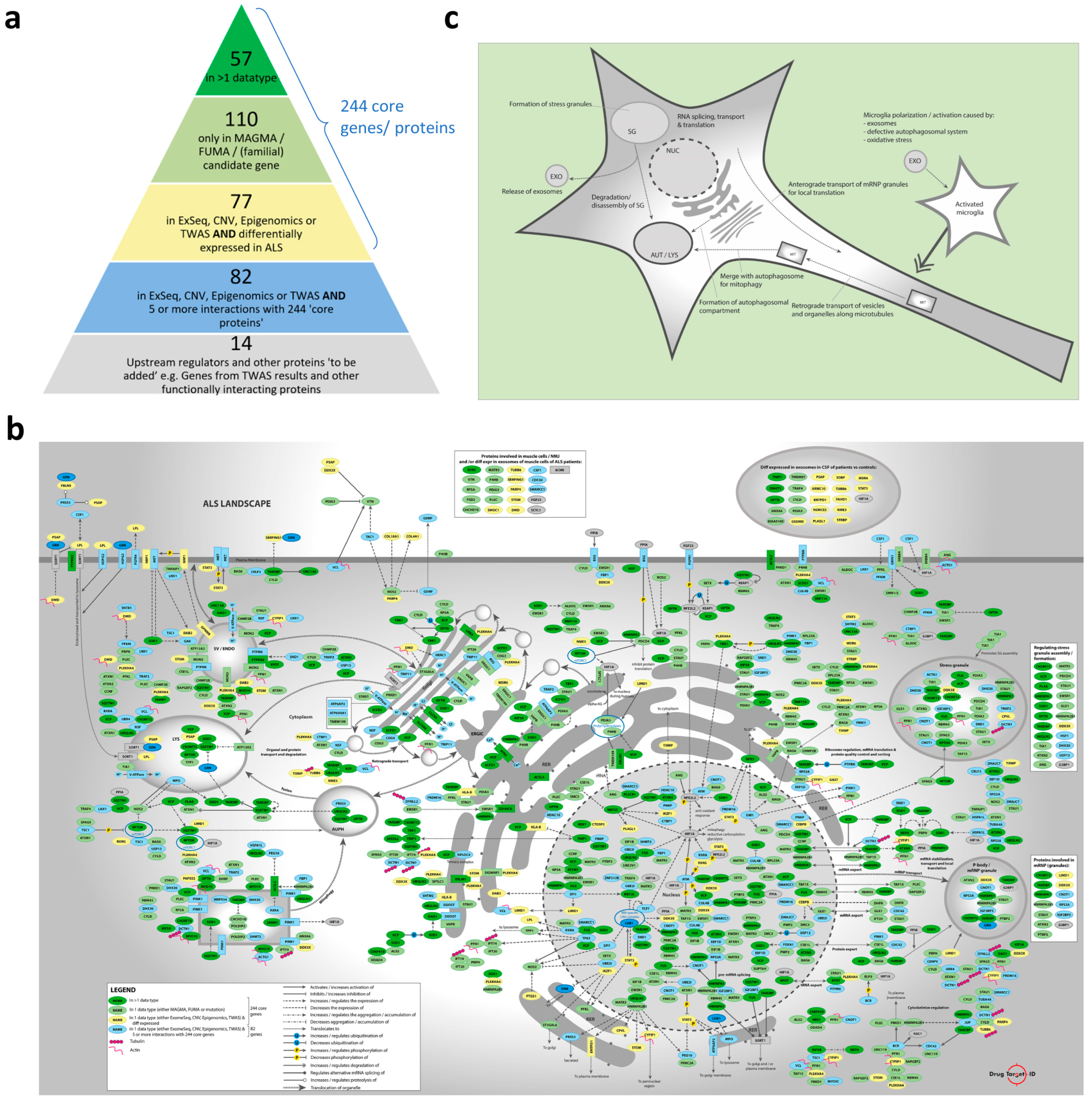
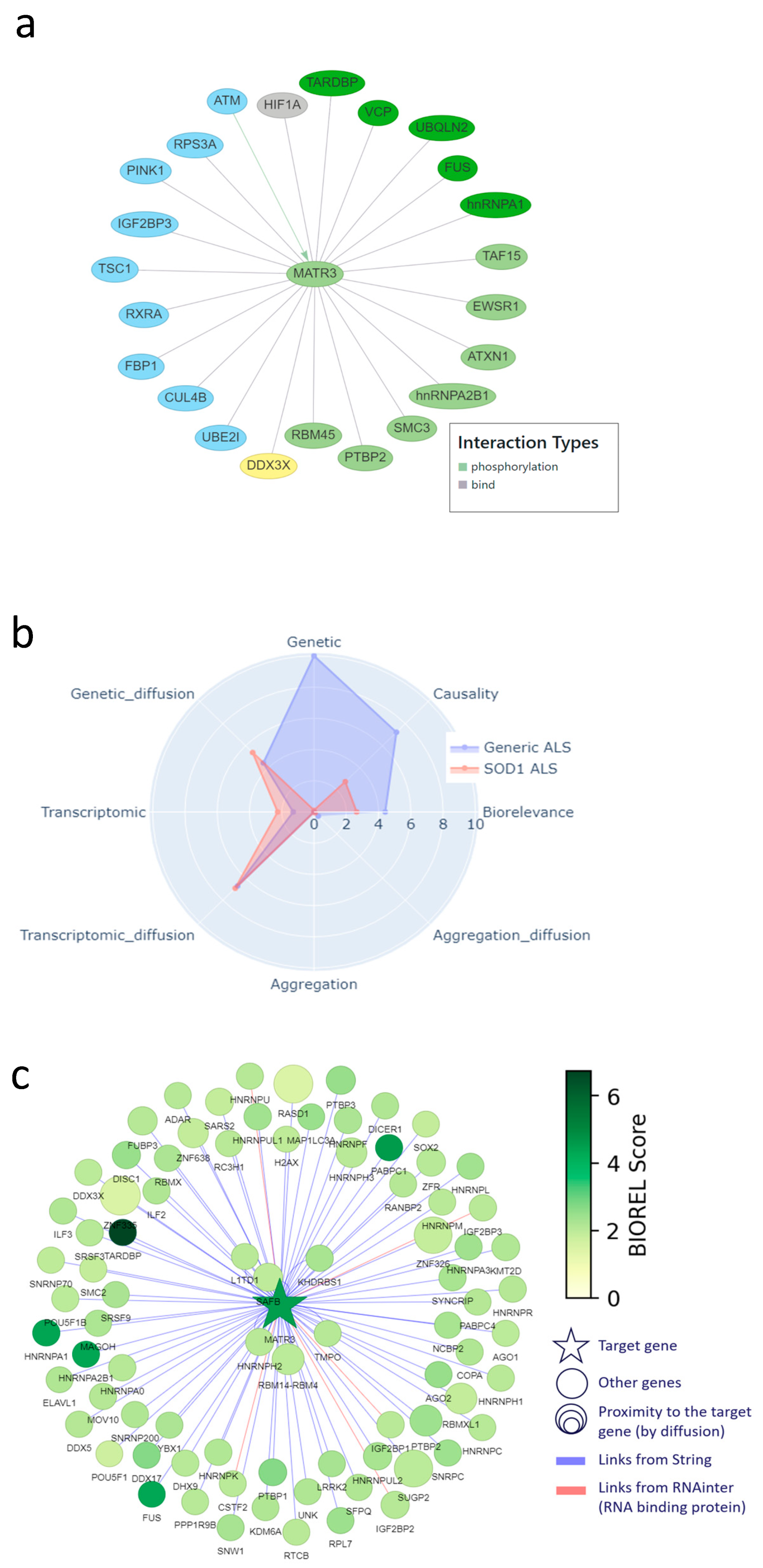
2.1. Candidate Genes/Proteins in the Molecular Landscape of ALS
2.2. Tissue Specificity Analyses
2.3. Upstream Regulator Analyses
2.4. Molecular Landscape and Identification/Selection of Therapeutic Targets
- Cytoskeleton-dependent transport of organelles and mRNP/stress granules.
- ER-ERGIC-Golgi regulation and function, including vesicle transport from and to the Golgi apparatus, Golgi acidification, and (indirect) involvement in Golgi fragmentation.
- Autophagosomal function, which includes autophagy, mitophagy, autophagosome-lysosome fusion, lysosomal function, and ‘autophagic flux’ in general.
- Hypoxia, which involves oxidative stress and processes that are protective in cerebral ischemia and ischemic stroke.
- mRNP/stress granules, which includes the expression or functional involvement in the regulation of mRNP and/or stress granules.
- Microglial regulation, which involves the polarization, activation/inhibition, pyroptosis, and (programmed) death of microglia.
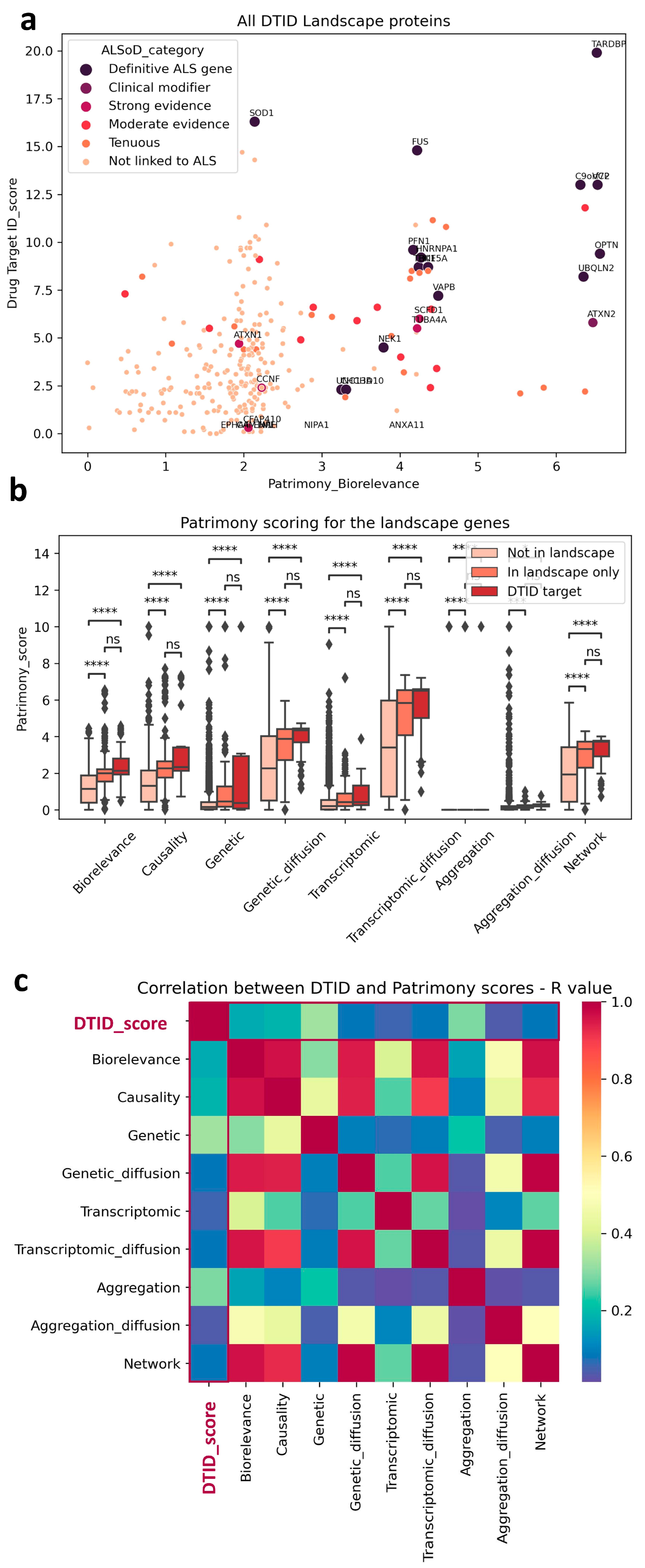
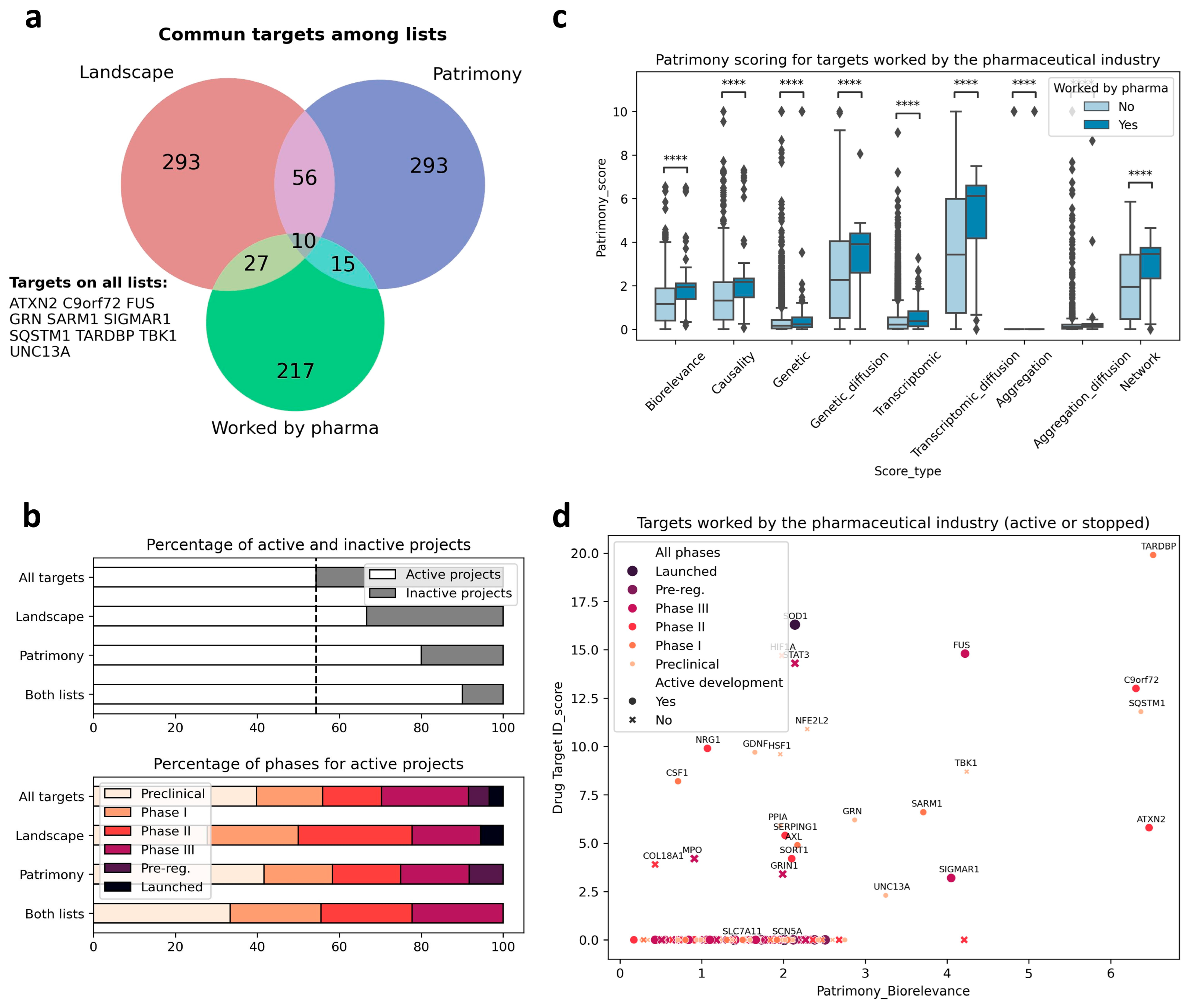
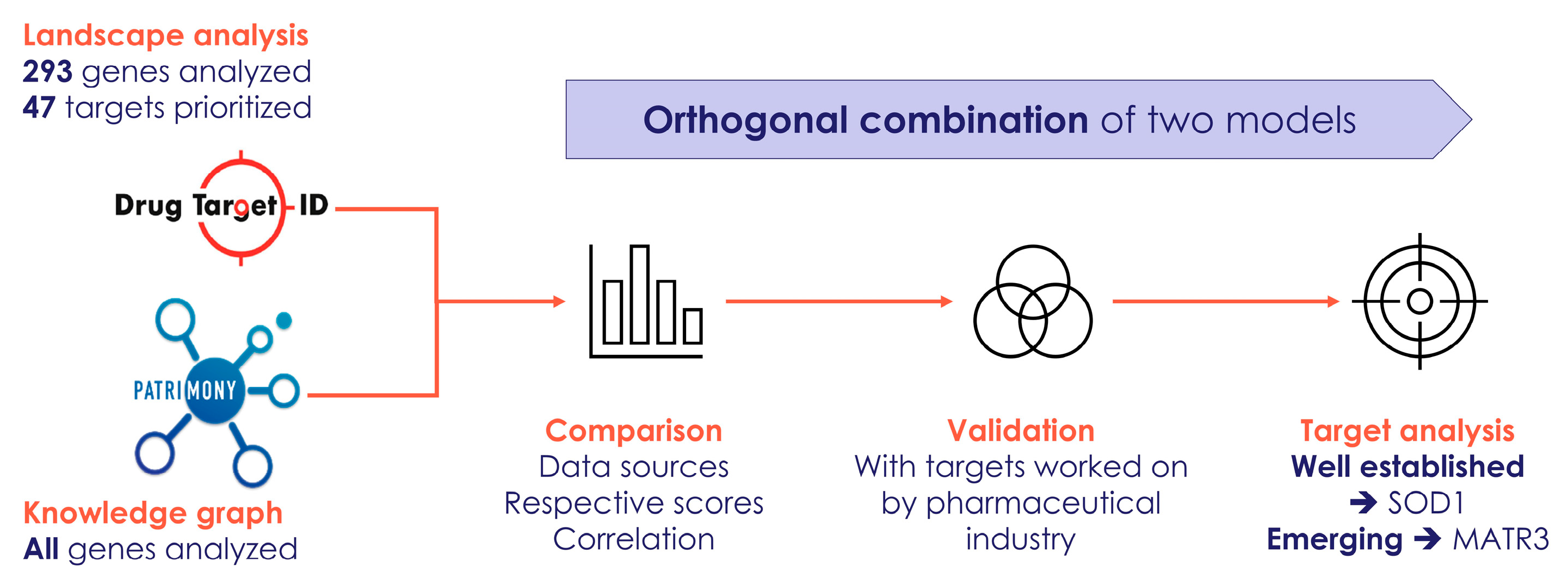
2.5. Cross Analysis of the ALS Landscape with Patrimony
2.6. Relevance of Targets Already Explored by the Pharmaceutical Industry
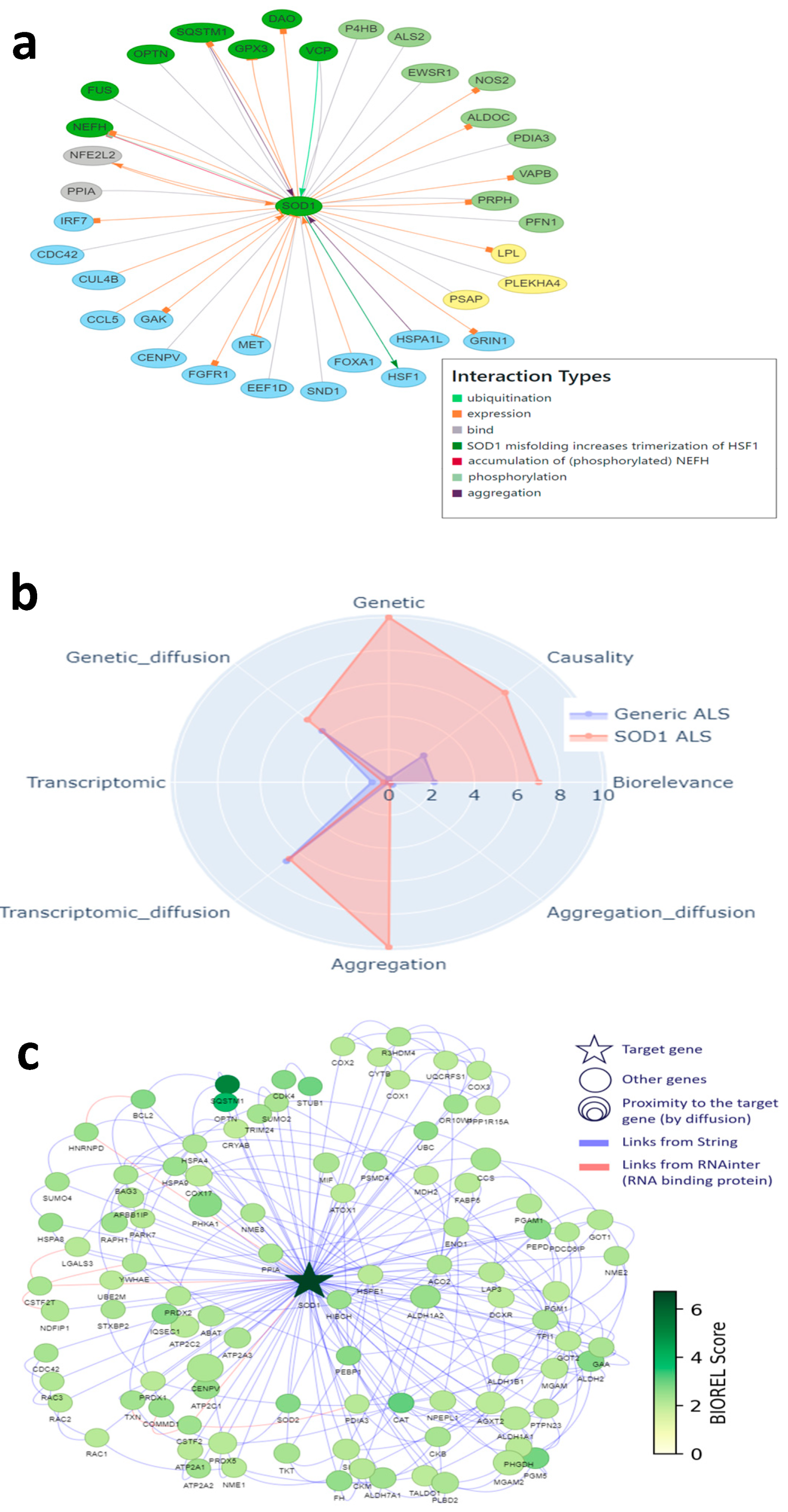
2.7. Corroboration of MATR3 as a New Target of Interest
2.8. Combining the Two Modeling Approaches
3. Discussion
4. Materials and Methods
4.1. Molecular Landscape for the Identification and Selection of Therapeutic Targets
4.1.1. Collecting/Compiling Omics Input Data for the ALS Landscape
4.1.2. MAGMA and FUMA Analyses of GWAS Data
4.1.3. Tissue Specificity Analyses of GWAS Data
4.1.4. From Input Genes to Candidate Genes
- (1)
- All genes associated through more than one type of genomics data were included (color code: dark green)
- (2)
- Genes associated only through MAGMA or FUMA analyses or identified through a (familial) mutation or mutations were included (color code: light green)
- (3)
- Genes associated through exome sequencing, CNVs, epigenomics, or TWAS studies were included, but only if they were also differentially expressed in ALS motor cortex, spinal cord, or CSF (color code: yellow). Of note, we would also have liked to include differential expression data from the cerebellum of ALS patients vs. controls, but these genome-wide data were not available
- (4)
- Remaining genes associated through exome sequencing, CNVs, epigenomics, or TWAS studies but that were not differentially expressed in ALS were only included if their encoded proteins had at least five interactions with the proteins/mRNAs encoded by the 244 genes from points 1–3 (color code: blue)
- (5)
- Lastly, genes/proteins not genetically associated with ALS were added. These include the two most significantly enriched upstream regulators (from the IPA analysis, see below)—i.e., GRN and LDB1—and genes coding for proteins that were strongly regulated/interacting in the landscape (color code: grey genes)
- (6)
- This prioritization process resulted in a list of 340 candidate genes for the ALS landscape, i.e., 326 candidate genes with a genetic link to ALS, and 14 ‘grey’ candidate genes without a known link to ALS (Table S3)
4.1.5. Upstream Regulator Analyses
4.1.6. From Candidate Genes to Landscape Genes/Proteins
4.1.7. Molecular Landscape of ALS
4.1.8. Scoring of ALS Landscape Proteins
4.1.9. Identifying Promising Novel Therapeutic Targets
4.2. Patrimony Platform for Target Discovery
4.3. Complementarity Between the Two Modeling Approaches
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| AI | artificial intelligence |
| ALS | amyotrophic lateral sclerosis |
| CNV | copy number variation |
| DTID | Drug Target ID |
| FUS | fused in sarcoma |
| GWAS | genome-wide association study |
| KG | knowledge graph |
| MATR3 | matrin-3 |
| PPIs | Protein–protein interactions |
| SOD1 | superoxide dismutase 1 |
| TDP-43 | TAR DNA-binding protein 43 |
| TWAS | transcriptome-wide association study |
References
- Feldman, E.L.; Goutman, S.A.; Petri, S.; Mazzini, L.; Savelieff, M.G.; Shaw, P.J.; Sobue, G. Amyotrophic lateral sclerosis. Lancet 2022, 400, 1363–1380. [Google Scholar] [CrossRef] [PubMed]
- Akçimen, F.; Lopez, E.R.; Landers, J.E.; Nath, A.; Chiò, A.; Chia, R.; Traynor, B.J. Amyotrophic lateral sclerosis: Translating genetic discoveries into therapies. Nat. Rev. Genet. 2023, 24, 642–658. [Google Scholar] [CrossRef] [PubMed]
- Klemann, C.J.H.M.; Visser, J.E.; van den Bosch, L.; Martens, G.J.M.; Poelmans, G. Integrated molecular landscape of amyotrophic lateral sclerosis provides insights into disease etiology. Brain Pathol. 2018, 28, 203–211. [Google Scholar] [CrossRef] [PubMed]
- Blair, H.A. Tofersen: First Approval. Drugs 2023, 83, 1039–1043. [Google Scholar] [CrossRef] [PubMed]
- Maragakis, N.J.; de Carvalho, M.; Weiss, M.D. Therapeutic targeting of ALS pathways: Refocusing an incomplete picture. Ann. Clin. Transl. Neurol. 2023, 10, 1948–1971. [Google Scholar] [CrossRef] [PubMed]
- Mead, R.J.; Shan, N.; Reiser, H.J.; Marshall, F.; Shaw, P.J. Amyotrophic lateral sclerosis: A neurodegenerative disorder poised for successful therapeutic translation. Nat. Rev. Drug Discov. 2023, 22, 185–212. [Google Scholar] [CrossRef] [PubMed]
- van Rheenen, W.; van der Spek, R.A.A.; Bakker, M.K.; van Vugt, J.J.F.A.; Hop, P.J.; Zwamborn, R.A.J.; de Klein, N.; Westra, H.-J.; Bakker, O.B.; Deelen, P.; et al. Common and rare variant association analyses in amyotrophic lateral sclerosis identify 15 risk loci with distinct genetic architectures and neuron-specific biology. Nat. Genet. 2021, 53, 1636–1648. [Google Scholar] [CrossRef] [PubMed]
- Nicolas, A.; Kenna, K.P.; Renton, A.E.; Ticozzi, N.; Faghri, F.; Chia, R.; Dominov, J.A.; Kenna, B.J.; Nalls, M.A.; Keagle, P.; et al. Genome-wide Analyses Identify KIF5A as a Novel ALS Gene. Neuron 2018, 97, 1267–1288. [Google Scholar] [CrossRef] [PubMed]
- Blokhuis, A.M.; Groen, E.J.N.; Koppers, M.; van den Berg, L.H.; Pasterkamp, R.J. Protein aggregation in amyotrophic lateral sclerosis. Acta Neuropathol. 2013, 125, 777–794. [Google Scholar] [CrossRef] [PubMed]
- Suk, T.R.; Rousseaux, M.W.C. The role of TDP-43 mislocalization in amyotrophic lateral sclerosis. Mol. Neurodegener. 2020, 15, 45. [Google Scholar] [CrossRef] [PubMed]
- Miller, R.G.; Mitchell, J.D.; Moore, D.H. Riluzole for amyotrophic lateral sclerosis (ALS)/motor neuron disease (MND). Cochrane Database Syst. Rev. 2012, 2012, CD001447. [Google Scholar] [CrossRef] [PubMed]
- Jaiswa, M.K. Riluzole and edaravone: A tale of two amyotrophic lateral sclerosis drugs. Med. Res. Rev. 2019, 39, 733–748. [Google Scholar] [CrossRef] [PubMed]
- Santos, J.R.; Park, J. MATR3’s Role beyond the Nuclear Matrix: From Gene Regulation to Its Implications in Amyotrophic Lateral Sclerosis and Other Diseases. Cells 2024, 13, 980. [Google Scholar] [CrossRef] [PubMed]
- Italsgen; Johnson, J.O.; Pioro, E.P.; Boehringer, A.; Chia, R.; Feit, H.; Renton, A.E.; Pliner, H.A.; Abramzon, Y.; Marangi, G.; et al. Mutations in the Matrin 3 gene cause familial amyotrophic lateral sclerosis. Nat. Neurosci. 2014, 17, 664–666. [Google Scholar] [CrossRef] [PubMed]
- Tada, M.; Doi, H.; Koyano, S.; Kubota, S.; Fukai, R.; Hashiguchi, S.; Hayashi, N.; Kawamoto, Y.; Kunii, M.; Tanaka, K.; et al. Matrin 3 is a Component of Neuronal Cytoplasmic Inclusions of Motor Neurons in Sporadic Amyotrophic Lateral Sclerosis. Am. J. Pathol. 2018, 188, 507–514. [Google Scholar] [CrossRef] [PubMed]
- Jones, A.R.; Troakes, C.; King, A.; Sahni, V.; De Jong, S.; Bossers, K.; Papouli, E.; Mirza, M.; Al-Sarraj, S.; Shaw, C.E.; et al. Stratified gene expression analysis identifies major amyotrophic lateral sclerosis genes. Neurobiol. Aging 2015, 36, 2006.e1–2006.e9. [Google Scholar] [CrossRef] [PubMed]
- Le Gall, L.; Duddy, W.J.; Martinat, C.; Mariot, V.; Connolly, O.; Milla, V.; Anakor, E.; Ouandaogo, Z.G.; Millecamps, S.; Lainé, J.; et al. Muscle cells of sporadic amyotrophic lateral sclerosis patients secrete neurotoxic vesicles. J. Cachexia Sarcopenia Muscle 2022, 13, 1385–1402. [Google Scholar] [CrossRef] [PubMed]
- Kao, C.S.; van Bruggen, R.; Kim, J.R.; Chen, X.X.L.; Chan, C.; Lee, J.; Cho, W.I.; Zhao, M.; Arndt, C.; Maksimovic, K.; et al. Selective neuronal degeneration in MATR3 S85C knock-in mouse model of early-stage ALS. Nat. Commun. 2020, 11, 5304. [Google Scholar] [CrossRef] [PubMed]
- You, J.; Maksimovic, K.; Lee, J.; Khan, M.; Masuda, R.; Park, J. Selective Loss of MATR3 in Spinal Interneurons, Upper Motor Neurons and Hippocampal CA1 Neurons in a MATR3 S85C Knock-In Mouse Model of Amyotrophic Lateral Sclerosis. Biology 2022, 11, 298. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Yamashita, S.; Hara, K.; Doki, T.; Tawara, N.; Ikeda, T.; Misumi, Y.; Zhang, Z.; Matsuo, Y.; Nagai, M.; et al. Mutant MATR3 mouse model to explain multisystem proteinopathy. J. Pathol. 2019, 249, 182–192. [Google Scholar] [CrossRef] [PubMed]
- Salem, A.; Wilson, C.J.; Rutledge, B.S.; Dilliott, A.; Farhan, S.; Choy, W.-Y.; Duennwald, M.L. Matrin3: Disorder and ALS Pathogenesis. Front. Mol. Biosci. 2022, 8, 794646. [Google Scholar] [CrossRef] [PubMed]
- Malik, A.M.; Wu, J.J.; Gillies, C.A.; Doctrove, Q.A.; Li, X.; Huang, H.; Tank, E.H.M.; Shakkottai, V.G.; Barmada, S. Neuronal activity regulates Matrin 3 abundance and function in a calcium-dependent manner through calpain-mediated cleavage and calmodulin binding. Proc. Natl. Acad. Sci. USA 2023, 120, e2206217120. [Google Scholar] [CrossRef] [PubMed]
- Weeks, H.M.; Therrien, A.S.; Bastian, A.J. The cerebellum contributes to proprioception during motion. J. Neurophysiol. 2017, 118, 693–702. [Google Scholar] [CrossRef] [PubMed]
- Akay, T.; Murray, A.J. Relative Contribution of Proprioceptive and Vestibular Sensory Systems to Locomotion: Opportunities for Discovery in the Age of Molecular Science. Int. J. Mol. Sci. 2021, 22, 1467. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Xing, H.; Ernst, A.F.; Liu, C.; Maugee, C.; Yokoi, F.; Lakshmana, M.; Li, Y. Hyperactivity of Purkinje cell and motor deficits in C9orf72 knockout mice. Mol. Cell. Neurosci. 2022, 121, 103756. [Google Scholar] [CrossRef] [PubMed]
- Chipika, R.H.; Mulkerrin, G.; Pradat, P.-F.; Murad, A.; Ango, F.; Raoul, C.; Bede, P. Cerebellar pathology in motor neuron disease: Neuroplasticity and neurodegeneration. Neural Regen. Res. 2022, 17, 2335–2341. [Google Scholar] [CrossRef] [PubMed]
- Bede, P.; Chipika, R.H.; Christidi, F.; Hengeveld, J.C.; Karavasilis, E.; Argyropoulos, G.D.; Lope, J.; Shing, S.L.H.; Velonakis, G.; Dupuis, L.; et al. Genotype-associated cerebellar profiles in ALS: Focal cerebellar pathology and cerebro-cerebellar connectivity alterations. J. Neurol. Neurosurg. Psychiatry 2021, 92, 1197–1205. [Google Scholar] [CrossRef] [PubMed]
- Tan, R.H.; Devenney, E.; Dobson-Stone, C.; Kwok, J.B.; Hodges, J.R.; Kiernan, M.C.; Halliday, G.M.; Hornberger, M.; Kassubek, J. Cerebellar Integrity in the Amyotrophic Lateral Sclerosis—Frontotemporal Dementia Continuum. PLoS ONE 2014, 8, e105632. [Google Scholar] [CrossRef] [PubMed]
- Herrera, M.; Pretelli, G.; Desai, J.; Garralda, E.; Siu, L.L.; Steiner, T.M.; Au, L. Bispecific antibodies: Advancing precision oncology. Trends Cancer 2024, 10, 893–919. [Google Scholar] [CrossRef] [PubMed]
- Fu, Z.; Li, S.; Han, S.; Shi, C.; Zhang, Y. Antibody drug conjugate: The “biological missile” for targeted cancer therapy. Signal Transduct. Target. Ther. 2022, 7, 93. [Google Scholar] [CrossRef] [PubMed]
- Klemann, C.J.H.M.; Martens, G.J.M.; Sharma, M.; Martens, M.B.; Isacson, O.; Gasser, T.; Visser, J.E.; Poelmans, G. Integrated molecular landscape of Parkinson’s disease. NPJ Parkinson Dis. 2017, 3, 14. [Google Scholar] [CrossRef] [PubMed]
- Widomska, J.; De Witte, W.; Buitelaar, J.K.; Glennon, J.C.; Poelmans, G. Molecular Landscape of Tourette’s Disorder. Int. J. Mol. Sci. 2023, 24, 1428. [Google Scholar] [CrossRef] [PubMed]
- Kluivers, K.B.; Lince, S.L.; Ruiz-Zapata, A.M.; Post, W.M.; Cartwright, R.; Kerkhof, M.H.; Widomska, J.; De Witte, W.; Pecanka, J.; Kiemeney, L.A.; et al. Molecular Landscape of Pelvic Organ Prolapse Provides Insights into Disease Etiology. Int. J. Mol. Sci. 2023, 24, 6087. [Google Scholar] [CrossRef] [PubMed]
- Ruisch, I.H.; Widomska, J.; De Witte, W.; Mota, N.R.; Fanelli, G.; Van Gils, V.; Jansen, W.J.; Vos, S.J.B.; Fóthi, A.; Barta, C.; et al. Molecular landscape of the overlap between Alzheimer’s disease and somatic insulin-related diseases. Alzheimer Res. Ther. 2024, 16, 239. [Google Scholar] [CrossRef] [PubMed]
- de Leeuw, C.A.; Mooij, J.M.; Heskes, T.; Posthuma, D. MAGMA: Generalized Gene-Set Analysis of GWAS Data. PLoS Comput. Biol. 2015, 11, e1004219. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, K.; Taskesen, E.; van Bochoven, A.; Posthuma, D. Functional mapping and annotation of genetic associations with FUMA. Nat. Commun. 2017, 8, 1826. [Google Scholar] [CrossRef] [PubMed]
- Wishart, D.S.; Feunang, Y.D.; Guo, A.C.; Lo, E.J.; Marcu, A.; Grant, J.R.; Sajed, T.; Johnson, D.; Li, C.; Sayeeda, Z.; et al. DrugBank 5.0: A major update to the DrugBank database for 2018. Nucleic Acids Res. 2018, 46, D1074–D1082. [Google Scholar] [CrossRef] [PubMed]
- Pinero, J.; Bravo, A.; Queralt-Rosinach, N.; Gutierrez-Sacristan, A. DisGeNET: A comprehensive platform integrating information on human disease-associated genes and variants. Nucleic Acids Res. 2017, 45, D833–D839. [Google Scholar] [CrossRef] [PubMed]
- Szklarczyk, D.; Gable, A.L.; Nastou, K.C.; Lyon, D.; Kirsch, R.; Pyysalo, S.; Doncheva, N.T.; Legeay, M.; Fang, T.; Bork, P.; et al. The STRING database in 2021: Customizable protein-protein networks, and functional characterization of user-uploaded gene/measurement sets. Nucleic Acids Res. 2021, 49, D605–D612. [Google Scholar] [CrossRef] [PubMed]
- Moingeon, P. Artificial intelligence-driven drug development against autoimmune diseases. Trends Pharmacol. Sci. 2023, 44, 411–424. [Google Scholar] [CrossRef] [PubMed]
- Bowden, J.; Holmes, M.V. Meta-analysis and Mendelian randomization: A review. Res. Synth. Methods 2019, 10, 486–496. [Google Scholar] [CrossRef] [PubMed]
- Kang, J.; Tang, Q.; He, J.; Li, L.; Yang, N.; Yu, S.; Wang, M.; Zhang, Y.; Lin, J.; Cui, T.; et al. RNAInter v4.0: RNA interactome repository with redefined confidence scoring system and improved accessibility. Nucleic Acids Res. 2022, 50, D326–D332. [Google Scholar] [CrossRef] [PubMed]
- Barabási, A.-L.; Gulbahce, N.; Loscalzo, J. Network medicine: A network-based approach to human disease. Nat. Rev. Genet. 2011, 12, 56–68. [Google Scholar] [CrossRef] [PubMed]
- Blaudin de Thé, F.-X.; Baudier, C.; Andrade Pereira, R.; Lefebvre, C.; Moingeon, P. Transforming drug discovery with a high-throughput AI-powered platform: A 5-year experience with Patrimony. Drug Discov. Today 2023, 28, 103772. [Google Scholar] [CrossRef] [PubMed]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Blaudin de Thé, F.X.; Klemann, C.J.H.M.; De Witte, W.; Widomska, J.; Delagrange, P.; Mannoury La Cour, C.; Fouesnard, M.; Elouej, S.; Mayl, K.; Lévy, N.; et al. Identifying Therapeutic Targets for Amyotrophic Lateral Sclerosis Through Modeling of Multi-Omics Data. Int. J. Mol. Sci. 2025, 26, 7087. https://doi.org/10.3390/ijms26157087
Blaudin de Thé FX, Klemann CJHM, De Witte W, Widomska J, Delagrange P, Mannoury La Cour C, Fouesnard M, Elouej S, Mayl K, Lévy N, et al. Identifying Therapeutic Targets for Amyotrophic Lateral Sclerosis Through Modeling of Multi-Omics Data. International Journal of Molecular Sciences. 2025; 26(15):7087. https://doi.org/10.3390/ijms26157087
Chicago/Turabian StyleBlaudin de Thé, François Xavier, Cornelius J. H. M. Klemann, Ward De Witte, Joanna Widomska, Philippe Delagrange, Clotilde Mannoury La Cour, Mélanie Fouesnard, Sahar Elouej, Keith Mayl, Nicolas Lévy, and et al. 2025. "Identifying Therapeutic Targets for Amyotrophic Lateral Sclerosis Through Modeling of Multi-Omics Data" International Journal of Molecular Sciences 26, no. 15: 7087. https://doi.org/10.3390/ijms26157087
APA StyleBlaudin de Thé, F. X., Klemann, C. J. H. M., De Witte, W., Widomska, J., Delagrange, P., Mannoury La Cour, C., Fouesnard, M., Elouej, S., Mayl, K., Lévy, N., Krupp, J., Jeggo, R., Moingeon, P., & Poelmans, G. (2025). Identifying Therapeutic Targets for Amyotrophic Lateral Sclerosis Through Modeling of Multi-Omics Data. International Journal of Molecular Sciences, 26(15), 7087. https://doi.org/10.3390/ijms26157087