
Duchenne Muscular Dystrophy: Integrating Current Clinical Practice with Future Therapeutic and Diagnostic Horizons

Abstract
1. Duchenne Muscular Dystrophy: Epidemiology and Pathophysiology
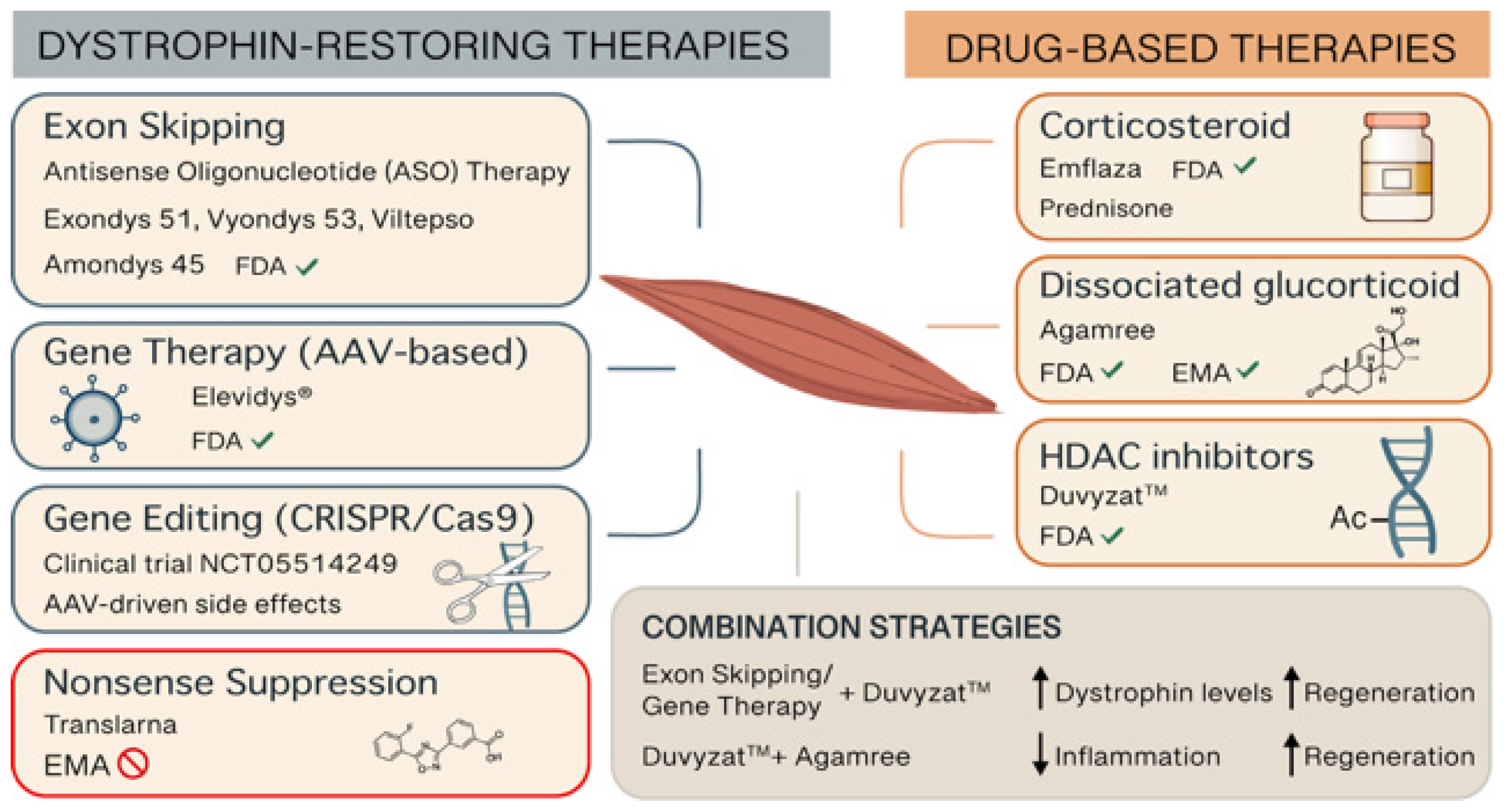
2. Current and Emerging Treatments
2.1. Corticosteroid Therapy
2.2. Nonsense Suppression Therapy
2.3. Antisense Oligonucleotide Therapy
2.4. Gene Therapy
2.5. Gene Editing Therapy
2.6. HDAC Inhibitor Therapy

{kind=link}
{kind=link}
| Brand Name | Active Ingredient | Manufacturer | Therapy Type | Target Patients | Approval Year | FDA/EMA | Administration | Side Effects | Additional Notes | References |
|---|---|---|---|---|---|---|---|---|---|---|
| Emflaza® | Deflazacort | PTC Therapeutics | Glucocorticoid (Steroid) | ≥2 years old patients | 2016 | Yes/no | Oral | Weight gain, Cushingoid appearance, behavioral changes, hypertension, bone fragility, growth delay | The first FDA-approved corticosteroid treatment for DMD | [17,18,19] |
| AGAMREE® | Vamorolone | Santhera Pharmaceuticals | Dissociative steroid | ≥2 years old patients | 2023 | Yes/yes | Oral | Mild GI symptoms, increased appetite, mild weight gain; fewer steroid-like side effects | The only approved medication for DMD in the European Union and the first DMD treatment approved in both the U.S. and EU | [20,21,22] |
| Translarna™ | Ataluren | PTC Therapeutics | Protein restoration therapy | ≥2 years old ambulatory patients | - | No/non-renewal | Oral | Headache, vomiting, diarrhea, flatulence, increased creatinine phosphokinase | Applies to DMD caused by nonsense mutations by inducing ribosomal readthrough | [23,24,25,26,27] |
| Exondys 51™ | Eteplirsen | Sarepta Therapeutics | Exon-skipping (exon 51) | Patients with mutations amenable to exon 51 skipping | 2016 | Yes/no | Weekly IV infusion | Balance disorder, vomiting, possible renal toxicity (kidney monitoring recommended) | First exon-skipping therapy approved for DMD; applies to 14% of DMD patients | [31,32,33] |
| Vyondys 53™ | Golodirsen | Sarepta Therapeutics | Exon-skipping (exon 53) | Patients with mutations amenable to exon 53 skipping | 2019 | Yes/no | Weekly IV infusion | Headache, fever, cough, vomiting, risk of kidney injury (kidney monitoring recommended) | Applies to 8–10% of DMD patients | [34,35,36,37] |
| Viltepso™ | Viltolarsen | NS Pharma | Exon-skipping (exon 53) | Patients with mutations amenable to exon 53 skipping | 2020 | Yes/no | Weekly IV infusion | Upper respiratory infections, injection site reactions, proteinuria (kidney monitoring recommended) | Applies to 8–10% of DMD patients | [38,39,40,41] |
| Amondys 45™ | Casimersen | Sarepta Therapeutics | Exon-skipping (exon 45) | Patients with mutations amenable to exon 45 skipping | 2021 | Yes/no | Weekly IV infusion | Headache, fever, increased liver enzymes, possible renal toxicity (kidney monitoring recommended) | Applies to 8–9% of DMD patients | [42,43] |
| Elevidys® | Delandistrogene moxeparvovec | Sarepta Therapeutics | Gene therapy (micro-dystrophin) | ≥4 years old 4 ambulatory and non-ambulatory patients | 2023 | Yes/no | Single IV infusion | Vomiting, fever, liver enzyme elevation, immune reaction (requires steroid prophylaxis) | One-time gene therapy | [48,49,50,51,52] |
| Duvyzat™ | Givinostat | Italfarmaco S.p.A. | HDAC inhibitor (epigenetic) | ≥6 years old with any dystrophin mutation | 2024 | Yes/no | Oral | GI disturbances, thrombocytopenia, elevated creatine kinase, fatigue | First nonsteroidal treatment for DMD approved for broad use; may be used alongside other therapies | [74,75,76,77,78,79] |
2.7. Future Directions for Combination Therapies
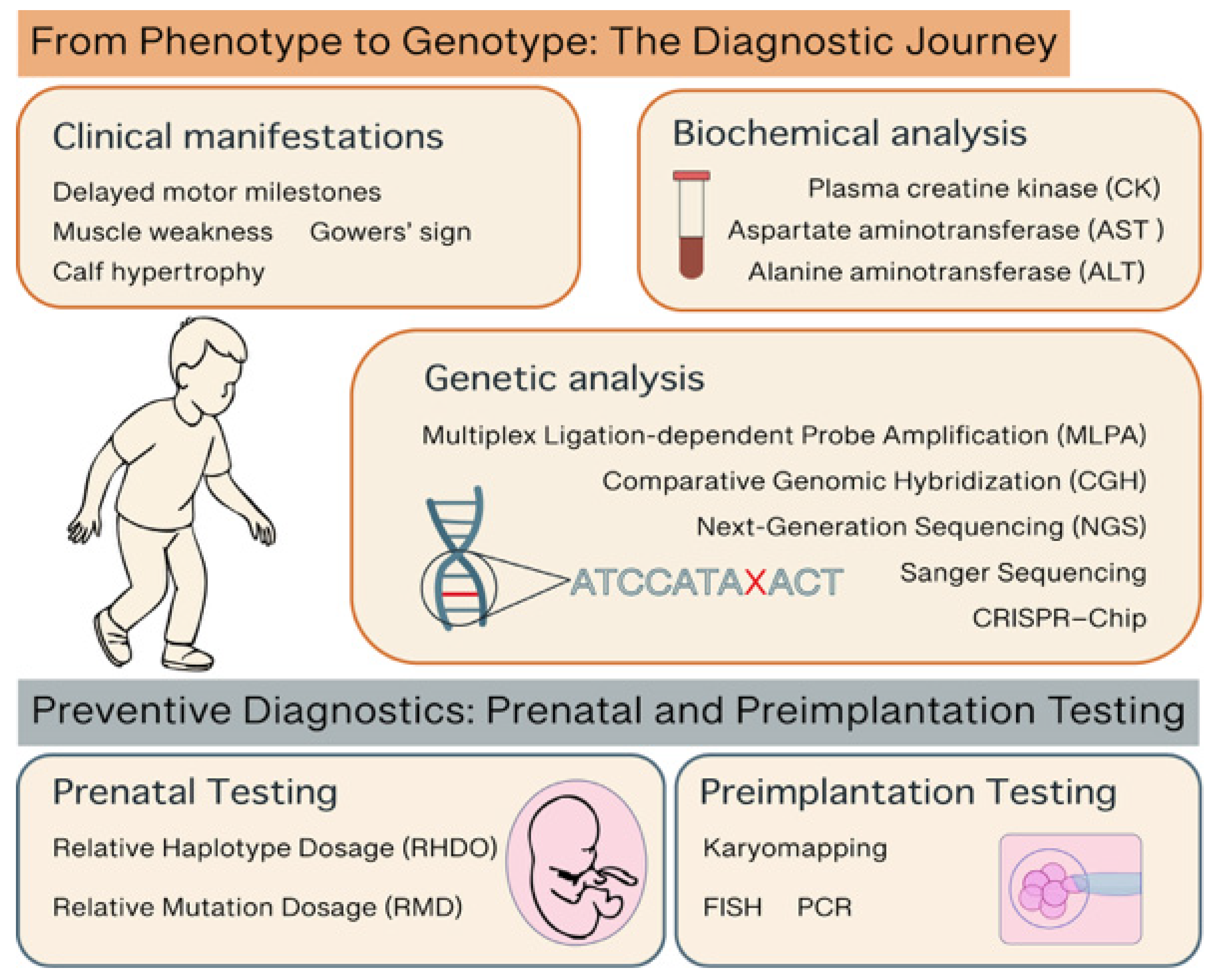
3. Diagnostic Approaches in DMD
3.1. From Phenotype to Genotype: The Diagnostic Journey
3.2. Preventive Diagnostics: Prenatal and Preimplantation Testing
| Diagnostic Method | Invasiveness | Purpose | What It Detects | When It Is Used | Notes | References |
|---|---|---|---|---|---|---|
| Creatine Kinase (CK) Test | Non-invasive | Initial screening | Elevated CK (>10× normal) suggests muscle damage | First step in suspected DMD | High CK is common but not specific to DMD | [81,83,84] |
| Multiplex Ligation-dependent Probe Amplification (MLPA) | Non-invasive | Definitive diagnosis | Detects large deletions/duplications in the DMD gene | Initial genetic test for diagnosis of common DMD mutations | Cannot detect small mutations | [2,83] |
| Next-Generation Sequences (NGS) | Non-invasive | Definitive diagnosis | Point mutations, deletions, duplications in the DMD gene | Gold standard for diagnosis of all DMD mutations | Most advanced and widely used today. | [2,81,83] |
| CRISPR–Chip | Non-invasive | Definitive diagnosis | Common mutations | Not yet available in clinical practice | Rapid (within 15 min), and bypass sequence amplification. | [85,86,87] |
| Muscle Biopsy | Invasive | Definitive diagnosis | Dystrophin expression via immunostaining | Rarely used today; reserved for unclear cases | Confirms lack or absence of dystrophin protein | [2,81] |
| Chorionic villus sampling (CVS)/Amniocentesis | Invasive | Prenatal Genetic Testing | In families with known DMD mutation | For at-risk families | Requires family history or prior diagnosis | [88,89] |
| Relative Haplotype Dosage (RHDO) | Non-invasive | Prenatal Genetic Testing | Mutation in cell-free fetal DNA | For at-risk families | Not suitable for detecting de novo mutations or maternal germline mosaicism. | [93] |
| Relative Mutation Dosage (RMD) | Non-invasive | Prenatal Genetic Testing | Mutation in cell-free fetal DNA | For at-risk families | Not suitable for detecting large deletions or duplications | [93] |
| FISH | Non-invasive | Preimplantation Testing | Mutation in embryonic cell | For at-risk families using IVF | Requires known familial mutation | [95,96,97] |
| PCR | Non-invasive | Preimplantation Testing | Mutation in embryonic cell | For at-risk families using IVF | Requires known familial mutation | [95,96,97] |
| Karyomapping | Non-invasive | Preimplantation Testing | Mutation in embryonic cell | For at-risk families using IVF | Faster and broader genetic analysis, including both mutation detection and chromosome balance | [98] |
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| DMD | Duchenne muscular dystrophy |
| BMD | Becker muscular dystrophy |
| HDAC | histone deacetylase |
| DAPC | dystrophin-associated protein complex |
| CK | creatine kinase |
| nNOS | neuronal nitric oxide synthase |
| FDA | Food and Drug Administration |
| EMA | European Medicines Agency |
| CHMP | Committee for Medicinal Products for Human Use |
| ASO | antisense oligonucleotides |
| PMO | phosphorodiamidate morpholino oligomer |
| AAV | adeno-associated virus |
| CRISPR | Clustered Regularly Interspaced Short Palindromic Repeats |
| gRNAs | guide RNAs |
| DSB | double-stranded breaks |
| NHEJ | non-homologous end joining |
| iPSC | induced pluripotent stem cells |
| FAPs | fibro-adipogenic progenitors |
| AST | aspartate aminotransferase |
| ALT | alanine aminotransferase |
| MLPA | multiplex ligation-dependent probe amplification |
| CGH | comparative genomic hybridization |
| NGS | next-generation sequencing |
| cffDNA | cell-free fetal DNA |
| NIPT | non-invasive prenatal testing |
| SGD | single-gene disorders |
| RHDO | relative haplotype dosage |
| RMD | relative mutation dosage |
| IVF | in vitro fertilization |
| PGD | preimplantation genetic diagnosis |
| FISH | fluorescence in situ hybridization |
References
- Mercuri, E.; Bonnemann, C.G.; Muntoni, F. Muscular Dystrophies. Lancet 2019, 394, 2025–2038. [Google Scholar] [CrossRef] [PubMed]
- Duan, D.; Goemans, N.; Takeda, S.; Mercuri, E.; Aartsma-Rus, A. Duchenne Muscular Dystrophy. Nat. Rev. Dis. Primers 2021, 7, 13. [Google Scholar] [CrossRef] [PubMed]
- Landfeldt, E.; Thompson, R.; Sejersen, T.; McMillan, H.J.; Kirschner, J.; Lochmüller, H. Life expectancy at birth in Duchenne muscular dystrophy: A systematic review and meta-analysis. Eur. J. Epidemiol. 2020, 35, 643–653. [Google Scholar] [CrossRef]
- Mah, J.K.; Korngut, L.; Dykeman, J.; Day, L.; Pringsheim, T.; Jette, N. A Systematic Review and Meta-Analysis on the Epidemiology of Duchenne and Becker Muscular Dystrophy. Neuromuscul. Disord. 2014, 24, 482–491. [Google Scholar] [CrossRef]
- Gao, Q.Q.; McNally, E.M. The Dystrophin Complex: Structure, Function, and Implications for Therapy. Compr. Physiol. 2015, 5, 1223–1239. [Google Scholar] [CrossRef]
- Ervasti, J.M.; Sonnemann, K.J. Biology of the Striated Muscle Dystrophin-Glycoprotein Complex. Int. Rev. Cytol. 2008, 265, 191–225. [Google Scholar] [CrossRef]
- Constantin, B. Dystrophin Complex Functions as a Scaffold for Signalling Proteins. Biochim. Biophys. Acta 2014, 1838, 635–642. [Google Scholar] [CrossRef]
- Allen, D.G.; Whitehead, N.P.; Froehner, S.C. Absence of Dystrophin Disrupts Skeletal Muscle Signaling: Roles of Ca2+, Reactive Oxygen Species, and Nitric Oxide in the Development of Muscular Dystrophy. Physiol. Rev. 2016, 96, 253–305. [Google Scholar] [CrossRef]
- Mokri, B.; Engel, A.G. Duchenne Dystrophy: Electron Microscopic Findings Pointing to a Basic or Early Abnormality in the Plasma Membrane of the Muscle Fiber. Neurology 1975, 25, 1111–1120. [Google Scholar] [CrossRef]
- Dubinin, M.V.; Belosludtsev, K.N. Ion Channels of the Sarcolemma and Intracellular Organelles in Duchenne Muscular Dystrophy: A Role in the Dysregulation of Ion Homeostasis and a Possible Target for Therapy. Int. J. Mol. Sci. 2023, 24, 2229. [Google Scholar] [CrossRef]
- Sander, M.; Chavoshan, B.; Harris, S.A.; Iannaccone, S.T.; Stull, J.T.; Thomas, G.D.; Victor, R.G. Functional muscle ischemia in neuronal nitric oxide synthase-deficient skeletal muscle of children with Duchenne muscular dystrophy. Proc. Natl. Acad. Sci. USA 2000, 97, 13818–13823. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.H.; Kwak, H.B.; Thompson, L.V.; Lawler, J.M. Contribution of Oxidative Stress to Pathology in Diaphragm and Limb Muscles with Duchenne Muscular Dystrophy. J. Muscle Res. Cell Motil. 2013, 34, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Sandri, M.; Coletto, L.; Grumati, P.; Bonaldo, P. Misregulation of Autophagy and Protein Degradation Systems in Myopathies and Muscular Dystrophies. J. Cell Sci. 2013, 126, 5325–5333. [Google Scholar] [CrossRef]
- Rosenberg, A.S.; Puig, M.; Nagaraju, K.; Hoffman, E.P.; Villalta, S.A.; Rao, V.A.; Wakefield, L.M.; Woodcock, J. Immune-Mediated Pathology in Duchenne Muscular Dystrophy. Sci. Transl. Med. 2015, 7, 299rv4. [Google Scholar] [CrossRef]
- Dumont, N.A.; Wang, Y.X.; von Maltzahn, J.; Pasut, A.; Bentzinger, C.F.; Brun, C.E.; Rudnicki, M.A. Dystrophin Expression in Muscle Stem Cells Regulates Their Polarity and Asymmetric Division. Nat. Med. 2015, 21, 1455–1463. [Google Scholar] [CrossRef]
- Eagle, M.; Bourke, J.; Bullock, R.; Gibson, M.; Mehta, J.; Giddings, D.; Straub, V.; Bushby, K. Managing Duchenne Muscular Dystrophy—The Additive Effect of Spinal Surgery and Home Nocturnal Ventilation in Improving Survival. Neuromuscul. Disord. 2007, 17, 470–475. [Google Scholar] [CrossRef]
- Czifrus, E.; Berlau, D.J. Corticosteroids for the Treatment of Duchenne Muscular Dystrophy: A Safety Review. Expert Opin. Drug Saf. 2024, 23, 1237–1247. [Google Scholar] [CrossRef]
- McDonald, C.M.; Henricson, E.K.; Abresch, R.T.; Duong, T.; Joyce, N.C.; Hu, F.; Clemens, P.R.; Hoffman, E.P.; Cnaan, A.; Gordish-Dressman, H.; et al. Long-Term Effects of Glucocorticoids on Function, Quality of Life, and Survival in Patients with Duchenne Muscular Dystrophy: A Prospective Cohort Study. Lancet 2018, 391, 451–461. [Google Scholar] [CrossRef]
- McDonald, C.M.; Sajeev, G.; Yao, Z.; McDonnell, E.; Elfring, G.; Souza, M.; Peltz, S.W.; Darras, B.T.; Shieh, P.B.; Cox, D.A.; et al. Deflazacort vs Prednisone Treatment for Duchenne Muscular Dystrophy: A Meta-Analysis of Disease Progression Rates in Recent Multicenter Clinical Trials. Muscle Nerve 2020, 61, 26–35. [Google Scholar] [CrossRef]
- Guglieri, M.; Clemens, P.R.; Perlman, S.J.; Smith, E.C.; Horrocks, I.; Finkel, R.S.; Mah, J.K.; Deconinck, N.; Goemans, N.; Haberlova, J.; et al. Efficacy and Safety of Vamorolone vs Placebo and Prednisone Among Boys With Duchenne Muscular Dystrophy: A Randomized Clinical Trial. JAMA Neurol. 2022, 79, 1005–1014. [Google Scholar] [CrossRef]
- Dang, U.J.; Damsker, J.M.; Guglieri, M.; Clemens, P.R.; Perlman, S.J.; Smith, E.C.; Horrocks, I.; Finkel, R.S.; Mah, J.K.; Deconinck, N.; et al. Efficacy and Safety of Vamorolone over 48 Weeks in Boys with Duchenne Muscular Dystrophy: A Randomized Controlled Trial. Neurology 2024, 102, e208112. [Google Scholar] [CrossRef] [PubMed]
- Keam, S.J. Vamorolone: First Approval. Drugs 2024, 84, 111–117. [Google Scholar] [CrossRef]
- Bushby, K.; Finkel, R.; Wong, B.; Barohn, R.; Campbell, C.; Comi, G.P.; Connolly, A.M.; Day, J.W.; Flanigan, K.M.; Goemans, N.; et al. Ataluren Treatment of Patients with Nonsense Mutation Dystrophinopathy. Muscle Nerve 2014, 50, 477–487. [Google Scholar] [CrossRef] [PubMed]
- McDonald, C.M.; Campbell, C.; Torricelli, R.E.; Finkel, R.S.; Flanigan, K.M.; Goemans, N.; Heydemann, P.; Kaminska, A.; Kirschner, J.; Muntoni, F.; et al. Ataluren in Patients with Nonsense Mutation Duchenne Muscular Dystrophy (ACT DMD): A Multicentre, Randomised, Double-Blind, Placebo-Controlled, Phase 3 Trial. Lancet 2017, 390, 1489–1498. [Google Scholar] [CrossRef]
- Haas, M.; Vlcek, V.; Balabanov, P.; Salmonson, T.; Bakchine, S.; Markey, G.; Weise, M.; Schlosser-Weber, G.; Brohmann, H.; Yerro, C.P.; et al. European Medicines Agency review of ataluren for the treatment of ambulant patients aged 5 years and older with Duchenne muscular dystrophy resulting from a nonsense mutation in the dystrophin gene. Neuromuscul. Disord. 2015, 25, 5–13. [Google Scholar] [CrossRef] [PubMed]
- Mercuri, E.; Osorio, A.N.; Muntoni, F.; Buccella, F.; Desguerre, I.; Kirschner, J.; Tulinius, M.; de Resende, M.B.D.; Morgenroth, L.P.; Gordish-Dressman, H.; et al. Safety and Effectiveness of Ataluren in Patients with Nonsense Mutation DMD in the STRIDE Registry Compared with the CINRG Duchenne Natural History Study (2015–2022): 2022 Interim Analysis. J. Neurol. 2023, 270, 3896–3913. [Google Scholar] [CrossRef]
- European Medicine Agency. EMA Confirms Recommendation for Non-Renewal of Authorisation of Duchenne Muscular Dystrophy Medicine Translarna. Available online: https://www.ema.europa.eu/en/news/ema-confirms-recommendation-non-renewal-authorisation-duchenne-muscular-dystrophy-medicine-translarna (accessed on 18 June 2025).
- Torres-Masjoan, L.; Aguti, S.; Zhou, H.; Muntoni, F. Clinical applications of exon-skipping antisense oligonucleotides in neuromuscular diseases. Mol Ther. 2025, 33, 2689–2704. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Sang, A.; Zhuo, S.; Bochanis, A.; Manautou, J.E.; Bahal, R.; Zhong, X.B.; Rasmussen, T.P. Mechanisms of Action of the US Food and Drug Administration-Approved Antisense Oligonucleotide Drugs. BioDrugs 2024, 38, 511–526. [Google Scholar] [CrossRef]
- Aartsma-Rus, A.; Goemans, N. A Sequel to the Eteplirsen Saga: Eteplirsen is Approved in the United States but Was Not Approved in Europe. Nucleic Acid Ther. 2019, 29, 13–15. [Google Scholar] [CrossRef]
- Syed, Y.Y. Eteplirsen: First Global Approval. Drugs 2016, 76, 1699–1704. [Google Scholar] [CrossRef]
- Lim, K.R.; Maruyama, R.; Yokota, T. Eteplirsen in the Treatment of Duchenne Muscular Dystrophy. Drug Des. Devel. Ther. 2017, 11, 533–545. [Google Scholar] [CrossRef] [PubMed]
- Aartsma-Rus, A.; Krieg, A.M. FDA Approves Eteplirsen for Duchenne Muscular Dystrophy: The Next Chapter in the Eteplirsen Saga. Nucleic Acid Ther. 2017, 27, 1–3. [Google Scholar] [CrossRef] [PubMed]
- Heo, Y.A. Golodirsen: First Approval. Drugs 2020, 80, 329–333. [Google Scholar] [CrossRef]
- Anwar, S.; Yokota, T. Golodirsen for Duchenne Muscular Dystrophy. Drugs Today 2020, 56, 491–504. [Google Scholar] [CrossRef]
- Aartsma-Rus, A.; Corey, D.R. The 10th Oligonucleotide Therapy Approved: Golodirsen for Duchenne Muscular Dystrophy. Nucleic Acid Ther. 2020, 30, 67–70. [Google Scholar] [CrossRef]
- Servais, L.; Mercuri, E.; Straub, V.; Guglieri, M.; Seferian, A.M.; Scoto, M.; Leone, D.; Koenig, E.; Khan, N.; Dugar, A.; et al. Long-Term Safety and Efficacy Data of Golodirsen in Ambulatory Patients with Duchenne Muscular Dystrophy Amenable to Exon 53 Skipping: A First-in-Human, Multicenter, Two-Part, Open-Label, Phase 1/2 Trial. Nucleic Acid Ther. 2022, 32, 29–39. [Google Scholar] [CrossRef]
- Roshmi, R.R.; Yokota, T. Pharmacological Profile of Viltolarsen for the Treatment of Duchenne Muscular Dystrophy: A Japanese Experience. Clin. Pharmacol. 2021, 13, 235–242. [Google Scholar] [CrossRef]
- Roshmi, R.R.; Yokota, T. Viltolarsen for the Treatment of Duchenne Muscular Dystrophy. Drugs Today 2019, 55, 627–639. [Google Scholar] [CrossRef]
- Komaki, H.; Nagata, T.; Saito, T.; Masuda, S.; Takeshita, E.; Sasaki, M.; Tachimori, H.; Nakamura, H.; Aoki, Y.; Takeda, S. Systemic Administration of the Antisense Oligonucleotide NS-065/NCNP-01 for Skipping of Exon 53 in Patients with Duchenne Muscular Dystrophy. Sci. Transl. Med. 2018, 10, eaan0713. [Google Scholar] [CrossRef]
- Komaki, H.; Takeshima, Y.; Matsumura, T.; Ozasa, S.; Funato, M.; Takeshita, E.; Iwata, Y.; Yajima, H.; Egawa, Y.; Toramoto, T.; et al. Viltolarsen in Japanese Duchenne Muscular Dystrophy Patients: A Phase 1/2 Study. Ann. Clin. Transl. Neurol. 2020, 7, 2393–2408. [Google Scholar] [CrossRef]
- Shirley, M. Casimersen: First Approval. Drugs 2021, 81, 875–879. [Google Scholar] [CrossRef] [PubMed]
- Assefa, M.; Gepfert, A.; Zaheer, M.; Hum, J.M.; Skinner, B.W. Casimersen (AMONDYS 45™): An Antisense Oligonucleotide for Duchenne Muscular Dystrophy. Biomedicines 2024, 12, 912. [Google Scholar] [CrossRef] [PubMed]
- Chwalenia, K.; Wood, M.J.A.; Roberts, T.C. Progress and prospects in antisense oligonucleotide-mediated exon skipping therapies for Duchenne muscular dystrophy. J. Muscle Res. Cell Motil. 2025. online ahead of print. [Google Scholar] [CrossRef] [PubMed]
- Arechavala-Gomeza, V.; López-Martínez, A.; Aartsma-Rus, A. Antisense RNA therapies for muscular dystrophies. J. Neuromuscul. Dis. 2025. online ahead of print. [Google Scholar] [CrossRef]
- Duan, D. Systemic AAV Micro-Dystrophin Gene Therapy for Duchenne Muscular Dystrophy. Mol. Ther. 2018, 26, 2337–2356. [Google Scholar] [CrossRef]
- Happi Mbakam, C.; Tremblay, J.P. Gene Therapy for Duchenne Muscular Dystrophy: An Update on the Latest Clinical Developments. Expert Rev. Neurother. 2023, 23, 905–920. [Google Scholar] [CrossRef]
- Baranello, G.; Muntoni, F. AAV Gene Therapy for Duchenne Muscular Dystrophy: Lessons Learned from a Phase 3 Trial. Hum. Gene Ther. 2024, 31, 541–543. [Google Scholar] [CrossRef]
- Hoy, S.M. Delandistrogene Moxeparvovec: First Approval. Drugs 2023, 83, 1323–1329. [Google Scholar] [CrossRef]
- Zaidman, C.M.; Proud, C.M.; McDonald, C.M.; Lehman, K.J.; Goedeker, N.L.; Mason, S.; Murphy, A.P.; Guridi, M.; Wang, S.; Reid, C.; et al. Delandistrogene Moxeparvovec Gene Therapy in Ambulatory Patients (Aged ≥4 to <8 Years) with Duchenne Muscular Dystrophy: 1-Year Interim Results from Study SRP-9001-103 (ENDEAVOR). Ann. Neurol. 2023, 94, 955–968. [Google Scholar] [CrossRef]
- US Food and Drug Administration. FDA Expands Approval of Gene Therapy for Patients with Duchenne Muscular Dystrophy. Available online: https://www.fda.gov/news-events/press-announcements/fda-expands-approval-gene-therapy-patients-duchenne-muscular-dystrophy (accessed on 18 June 2025).
- Bhattacharyya, M.; Miller, L.E.; Miller, A.L.; Bhattacharyya, R. The FDA Approval of Delandistrogene Moxeparvovec-Rokl for Duchenne Muscular Dystrophy: A Critical Examination of the Evidence and Regulatory Process. Expert Opin. Biol. Ther. 2024, 24, 869–871. [Google Scholar] [CrossRef]
- Wang, D.; Zhang, F.; Gao, G. CRISPR-Based Therapeutic Genome Editing: Strategies and In Vivo Delivery by AAV Vectors. Cell 2020, 181, 136–150. [Google Scholar] [CrossRef] [PubMed]
- Chemello, F.; Bassel-Duby, R.; Olson, E.N. Correction of Muscular Dystrophies by CRISPR Gene Editing. J. Clin. Investig. 2020, 130, 2766–2776. [Google Scholar] [CrossRef] [PubMed]
- Amoasii, L.; Hildyard, J.C.W.; Li, H.; Sanchez-Ortiz, E.; Mireault, A.; Caballero, D.; Harron, R.; Stathopoulou, T.R.; Massey, C.; Shelton, J.M.; et al. Gene editing restores dystrophin expression in a canine model of Duchenne muscular dystrophy. Science 2018, 362, 86–91. [Google Scholar] [CrossRef] [PubMed]
- Moretti, A.; Fonteyne, L.; Giesert, F.; Hoppmann, P.; Meier, A.B.; Bozoglu, T.; Baehr, A.; Schneider, C.M.; Sinnecker, D.; Klett, K.; et al. Somatic gene editing ameliorates skeletal and cardiac muscle failure in pig and human models of Duchenne muscular dystrophy. Nat. Med. 2020, 26, 207–214. [Google Scholar] [CrossRef]
- Nance, M.E.; Shi, R.; Hakim, C.H.; Wasala, N.B.; Yue, Y.; Pan, X.; Zhang, T.; Robinson, C.A.; Duan, S.X.; Yao, G.; et al. AAV9 Edits Muscle Stem Cells in Normal and Dystrophic Adult Mice. Mol. Ther. 2019, 27, 1568–1585. [Google Scholar] [CrossRef]
- Kwon, J.B.; Ettyreddy, A.R.; Vankara, A.; Bohning, J.D.; Devlin, G.; Hauschka, S.D.; Asokan, A.; Gersbach, C.A. In Vivo Gene Editing of Muscle Stem Cells with Adeno-Associated Viral Vectors in a Mouse Model of Duchenne Muscular Dystrophy. Mol. Ther. Methods Clin. Dev. 2020, 19, 320–329. [Google Scholar] [CrossRef]
- Haque, U.S.; Toshifumi Yokota, T. Gene Editing for Duchenne Muscular Dystrophy: From Experimental Models to Emerging Therapies. Degener. Neurol. Neuromuscul. Dis. 2025, 15, 17–40. [Google Scholar] [CrossRef]
- Ali, A.; Rahman, M.Y.; Sheikh, D. The Role of CRISPR/Cas9 in Revolutionizing Duchenne’s Muscular Dystrophy Treatment: Opportunities and Obstacles. Glob. Med. Genet. 2024, 11, 349–357. [Google Scholar] [CrossRef]
- Dhoke, N.R.; Kim, H.; Azzag, K.; Crist, S.B.; Kiley, J.; Perlingeiro, R.C.R. A Novel CRISPR-Cas9 Strategy to Target DYSTROPHIN Mutations Downstream of Exon 44 in Patient-Specific DMD iPSCs. Cells 2024, 13, 972. [Google Scholar] [CrossRef]
- Singh, A.; Irfan, H.; Fatima, E.; Nazir, Z.; Verma, A.; Akilimali, A. Revolutionary Breakthrough: FDA Approves CASGEVY, the First CRISPR/Cas9 Gene Therapy for Sickle Cell Disease. Ann. Med. Surg. 2024, 86, 4555–4559. [Google Scholar] [CrossRef]
- Lek, A.; Wong, B.; Keeler, A.; Blackwood, M.; Ma, K.; Huang, S.; Sylvia, K.; Batista, A.R.; Artinian, R.; Kokoski, D.; et al. Death after High-Dose rAAV9 Gene Therapy in a Patient with Duchenne’s Muscular Dystrophy. N. Engl. J. Med. 2023, 389, 1203–1210. [Google Scholar] [CrossRef] [PubMed]
- Cannan, W.J.; Pederson, D.S. Mechanisms and Consequences of Double-Strand DNA Break Formation in Chromatin. J. Cell Physiol. 2016, 231, 3–14. [Google Scholar] [CrossRef] [PubMed]
- Kleinstiver, B.P.; Pattanayak, V.; Prew, M.S.; Tsai, S.Q.; Nguyen, N.T.; Zheng, Z.; Joung, J.K. High-fidelity CRISPR-Cas9 nucleases with no detectable genome-wide off-target effects. Nature 2016, 529, 490–495. [Google Scholar] [CrossRef]
- Doench, J.G.; Fusi, N.; Sullender, M.; Hegde, M.; Vaimberg, E.W.; Donovan, K.F.; Smith, I.; Tothova, Z.; Wilen, C.; Orchard, R.; et al. Optimized sgRNA design to maximize activity and minimize off-target effects of CRISPR-Cas9. Nat. Biotechnol. 2016, 34, 184–191. [Google Scholar] [CrossRef]
- Sandonà, M.; Cavioli, G.; Renzini, A.; Cedola, A.; Gigli, G.; Coletti, D.; McKinsey, T.A.; Moresi, V.; Saccone, V. Histone Deacetylases: Molecular Mechanisms and Therapeutic Implications for Muscular Dystrophies. Int. J. Mol. Sci. 2023, 24, 4306. [Google Scholar] [CrossRef]
- Mozzetta, C.; Sartorelli, V.; Puri, P.L. HDAC Inhibitors as Pharmacological Treatment for Duchenne Muscular Dystrophy: A Discovery Journey From Bench to Patients. Trends Mol. Med. 2024, 30, 278–294. [Google Scholar] [CrossRef]
- Aartsma-Rus, A. Histone Deacetylase Inhibition with Givinostat: A Multi-Targeted Mode of Action with the Potential to Halt the Pathological Cascade of Duchenne Muscular Dystrophy. Front. Cell Dev. Biol. 2025, 12, 1514898. [Google Scholar] [CrossRef]
- Consalvi, S.; Saccone, V.; Mozzetta, C. Histone Deacetylase Inhibitors: A Potential Epigenetic Treatment for Duchenne Muscular Dystrophy. Epigenomics 2014, 6, 547–560. [Google Scholar] [CrossRef]
- Consalvi, S.; Saccone, V.; Giordani, L.; Minetti, G.; Mozzetta, C.; Puri, P.L. Histone Deacetylase Inhibitors in the Treatment of Muscular Dystrophies: Epigenetic Drugs for Genetic Diseases. Mol. Med. 2011, 17, 457–465. [Google Scholar] [CrossRef]
- Mozzetta, C.; Consalvi, S.; Saccone, V.; Tierney, M.; Diamantini, A.; Mitchell, K.J.; Marazzi, G.; Borsellino, G.; Battistini, L.; Sassoon, D.; et al. Fibroadipogenic Progenitors Mediate the Ability of HDAC Inhibitors to Promote Regeneration in Dystrophic Muscles of Young, But Not Old Mdx Mice. EMBO Mol. Med. 2013, 5, 626–639. [Google Scholar] [CrossRef]
- Saccone, V.; Consalvi, S.; Giordani, L.; Mozzetta, C.; Barozzi, I.; Sandonà, M.; Ryan, T.; Rojas-Muñoz, A.; Madaro, L.; Fasanaro, P.; et al. HDAC-Regulated MyomiRs Control BAF60 Variant Exchange and Direct the Functional Phenotype of Fibro-Adipogenic Progenitors in Dystrophic Muscles. Genes Dev. 2014, 28, 841–857. [Google Scholar] [CrossRef] [PubMed]
- Sandonà, M.; Consalvi, S.; Tucciarone, L.; De Bardi, M.; Scimeca, M.; Angelini, D.F.; Buffa, V.; D’Amico, A.; Bertini, E.S.; Cazzaniga, S.; et al. HDAC Inhibitors Tune miRNAs in Extracellular Vesicles of Dystrophic Muscle-Resident Mesenchymal Cells. EMBO Rep. 2020, 21, e50863. [Google Scholar] [CrossRef]
- Consalvi, S.; Tucciarone, L.; Macrì, E.; De Bardi, M.; Picozza, M.; Salvatori, I.; Renzini, A.; Valente, S.; Mai, A.; Moresi, V.; et al. Determinants of Epigenetic Resistance to HDAC Inhibitors in Dystrophic Fibro-Adipogenic Progenitors. EMBO Rep. 2022, 23, e54721. [Google Scholar] [CrossRef] [PubMed]
- Consalvi, S.; Mozzetta, C.; Bettica, P.; Germani, M.; Fiorentini, F.; Del Bene, F.; Rocchetti, M.; Leoni, F.; Monzani, V.; Mascagni, P.; et al. Preclinical Studies in the mdx Mouse Model of Duchenne Muscular Dystrophy with the Histone Deacetylase Inhibitor Givinostat. Mol. Med. 2013, 19, 79–87. [Google Scholar] [CrossRef] [PubMed]
- Bettica, P.; Petrini, S.; D’Oria, V.; D’Amico, A.; Catteruccia, M.; Pane, M.; Sivo, S.; Magri, F.; Brajkovic, S.; Messina, S.; et al. Histological Effects of Givinostat in Boys with Duchenne Muscular Dystrophy. Neuromuscul. Disord. 2016, 26, 643–649. [Google Scholar] [CrossRef]
- Mercuri, E.; Vilchez, J.J.; Boespflug-Tanguy, O.; Zaidman, C.M.; Mah, J.K.; Goemans, N.; Müller-Felber, W.; Niks, E.H.; Schara-Schmidt, U.; Bertini, E.; et al. Safety and Efficacy of Givinostat in Boys with Duchenne Muscular Dystrophy (EPIDYS): A Multicentre, Randomised, Double-Blind, Placebo-Controlled, Phase 3 Trial. Lancet Neurol. 2024, 23, 393–403. [Google Scholar] [CrossRef]
- Lamb, Y.N. Givinostat: First Approval. Drugs 2024, 84, 849–856. [Google Scholar] [CrossRef]
- García-Rodríguez, R.; Hiller, M.; Jiménez-Gracia, L.; van der Pal, Z.; Balog, J.; Adamzek, K.; Aartsma-Rus, A.; Spitali, P. Premature Termination Codons in the DMD Gene Cause Reduced Local mRNA Synthesis. Proc. Natl. Acad. Sci. USA 2020, 117, 16456–16464. [Google Scholar] [CrossRef]
- Mercuri, E.; Pane, M.; Cicala, G.; Brogna, C.; Ciafaloni, E. Detecting Early Signs in Duchenne Muscular Dystrophy: Comprehensive Review and Diagnostic Implications. Front. Pediatr. 2023, 11, 1276144. [Google Scholar] [CrossRef]
- Darmahkasih, A.J.; Rybalsky, I.; Tian, C.; Shellenbarger, K.C.; Horn, P.S.; Lambert, J.T.; Wong, B.L. Neurodevelopmental, behavioral, and emotional symptoms common in Duchenne muscular dystrophy. Muscle Nerve 2020, 61, 466–474. [Google Scholar] [CrossRef]
- Aartsma-Rus, A.; Ginjaar, I.B.; Bushby, K. The Importance of Genetic Diagnosis for Duchenne Muscular Dystrophy. J. Med. Genet. 2016, 53, 145–151. [Google Scholar] [CrossRef] [PubMed]
- Nakata, K.C.F.; Pereira, P.P.S.; Riveros, B.S. Creatine Kinase Test Diagnostic Accuracy in Neonatal Screening for Duchenne Muscular Dystrophy: A Systematic Review. Clin. Biochem. 2021, 98, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Gootenberg, J.S.; Abudayyeh, O.O.; Lee, J.W.; Essletzbichler, P.; Dy, A.J.; Joung, J.; Verdine, V.; Donghia, N.; Daringer, N.M.; Freije, C.A.; et al. Nucleic acid detection with CRISPR-Cas13a/C2c2. Science 2017, 356, 438–442. [Google Scholar] [CrossRef]
- Pardee, K.; Green, A.A.; Takahashi, M.K.; Braff, D.; Lambert, G.; Lee, J.W.; Ferrante, T.; Ma, D.; Donghia, N.; Fan, M.; et al. Rapid, Low-Cost Detection of Zika Virus Using Programmable Biomolecular Components. Cell 2016, 165, 1255–1266. [Google Scholar] [CrossRef]
- Hajian, R.; Balderston, S.; Tran, T.; deBoer, T.; Etienne, J.; Sandhu, M.; Wauford, N.A.; Chung, J.Y.; Nokes, J.; Athaiya, M.; et al. Detection of Unamplified Target Genes via CRISPR-Cas9 Immobilized on a Graphene Field-Effect Transistor. Nat. Biomed. Eng. 2019, 3, 427–437. [Google Scholar] [CrossRef]
- Abbs, S.; Tuffery-Giraud, S.; Bakker, E.; Ferlini, A.; Sejersen, T.; Mueller, C.R. Best practice guidelines on molecular diagnostics in Duchenne/Becker muscular dystrophies. Neuromuscul. Disord. 2010, 20, 422–427. [Google Scholar] [CrossRef]
- Bakker, M.; Birnie, E.; Robles de Medina, P.; Sollie, K.M.; Pajkrt, E.; Bilardo, C.M. Total pregnancy loss after chorionic villus sampling and amniocentesis: A cohort study. Ultrasound Obstet. Gynecol. 2017, 49, 599–606. [Google Scholar] [CrossRef]
- Allen, S.; Young, E.; Bowns, B. Noninvasive Prenatal Diagnosis for Single Gene Disorders. Curr. Opin. Obstet. Gynecol. 2017, 29, 73–79. [Google Scholar] [CrossRef]
- Chiu, R.W.; Chan, K.C.; Gao, Y.; Lau, V.Y.; Zheng, W.; Leung, T.Y.; Foo, C.H.; Xie, B.; Tsui, N.B.; Lun, F.M.; et al. Noninvasive prenatal diagnosis of fetal chromosomal aneuploidy by massively parallel genomic. Proc. Natl. Acad. Sci. USA 2008, 105, 20458–20463. [Google Scholar] [CrossRef]
- Parks, M.; Court, S.; Cleary, S.; Clokie, S.; Hewitt, J.; Williams, D.; Cole, T.; MacDonald, F.; Griffiths, M.; Allen, S. Non-Invasive Prenatal Diagnosis of Duchenne and Becker Muscular Dystrophies by Relative Haplotype Dosage. Prenat. Diagn. 2016, 36, 312–320. [Google Scholar] [CrossRef]
- Zaninović, L.; Bašković, M.; Ježek, D.; Bojanac, A.K. Accuracy of Non-Invasive Prenatal Testing for Duchenne Muscular Dystrophy in Families at Risk: A Systematic Review. Diagnostics 2023, 13, 183. [Google Scholar] [CrossRef] [PubMed]
- Parikh, F.R.; Athalye, A.S.; Kulkarni, D.K.; Sanap, R.R.; Dhumal, S.B.; Warang, D.J.; Naik, D.J.; Madon, P.F. Evolution and Utility of Preimplantation Genetic Testing for Monogenic Disorders in Assisted Reproduction—A Narrative Review. J. Hum. Reprod. Sci. 2021, 14, 329–339. [Google Scholar] [CrossRef] [PubMed]
- Malcov, M.; Ben-Yosef, D.; Schwartz, T.; Mey-Raz, N.; Azem, F.; Lessing, J.B.; Amit, A.; Yaron, Y. Preimplantation genetic diagnosis (PGD) for Duchenne muscular dystrophy (DMD) by triplex-nested PCR. Prenat. Diagn. 2005, 25, 1200–1205. [Google Scholar] [CrossRef] [PubMed]
- Malmgren, H.; White, I.; Johansson, S.; Levkov, L.; Iwarsson, E.; Fridström, M.; Blennow, E. PGD for dystrophin gene deletions using fluorescence in situ hybridization. Mol. Hum. Reprod. 2006, 12, 353–356. [Google Scholar] [CrossRef]
- Ren, Z.; Zeng, H.T.; Xu, Y.W.; Zhuang, G.L.; Deng, J.; Zhang, C.; Zhou, C.Q. Preimplantation genetic diagnosis for Duchenne muscular dystrophy by multiple displacement amplification. Fertil. Steril. 2009, 91, 359–364. [Google Scholar] [CrossRef]
- Mongkolchaipak, S.; Piyamongkol, W.; Piyamongkolx, W. Successful strategy of comprehensive pre-implantation genetic testing for Duchenne muscular dystrophy and chromosome balance using karyomapping and fluorescent PCR. Reprod. Biomed. Online 2019, 39, e64–e65. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Montagna, C.; Maiani, E.; Pieroni, L.; Consalvi, S. Duchenne Muscular Dystrophy: Integrating Current Clinical Practice with Future Therapeutic and Diagnostic Horizons. Int. J. Mol. Sci. 2025, 26, 6742. https://doi.org/10.3390/ijms26146742
Montagna C, Maiani E, Pieroni L, Consalvi S. Duchenne Muscular Dystrophy: Integrating Current Clinical Practice with Future Therapeutic and Diagnostic Horizons. International Journal of Molecular Sciences. 2025; 26(14):6742. https://doi.org/10.3390/ijms26146742
Chicago/Turabian StyleMontagna, Costanza, Emiliano Maiani, Luisa Pieroni, and Silvia Consalvi. 2025. "Duchenne Muscular Dystrophy: Integrating Current Clinical Practice with Future Therapeutic and Diagnostic Horizons" International Journal of Molecular Sciences 26, no. 14: 6742. https://doi.org/10.3390/ijms26146742
APA StyleMontagna, C., Maiani, E., Pieroni, L., & Consalvi, S. (2025). Duchenne Muscular Dystrophy: Integrating Current Clinical Practice with Future Therapeutic and Diagnostic Horizons. International Journal of Molecular Sciences, 26(14), 6742. https://doi.org/10.3390/ijms26146742