

Chromosome-Contiguous Reference Genome for Spirometra to Underpin Future Discovery Research

 , , , , and
, , , , and
Abstract
1. Introduction
2. Results
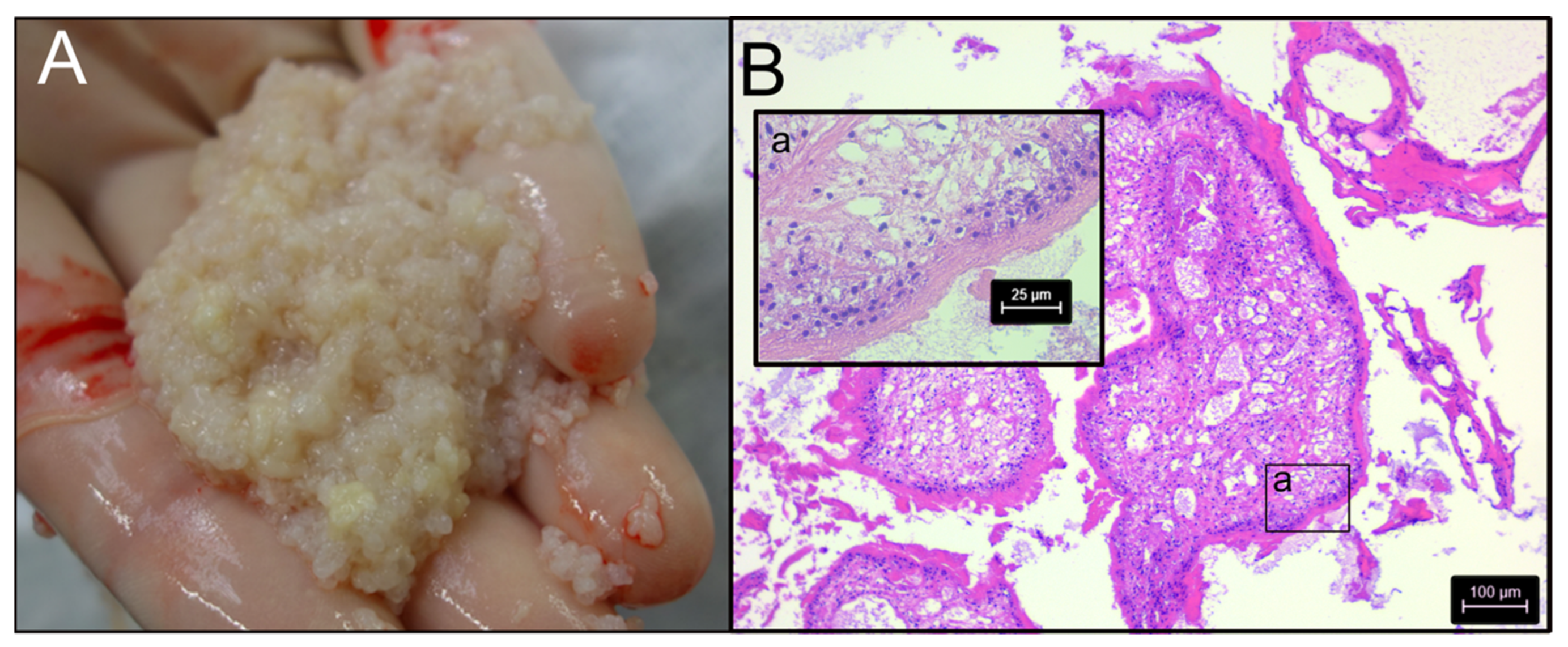
2.1. Initial Genetic Characterization
2.2. Inference of Ploidy and Nuclear Genome Assembly
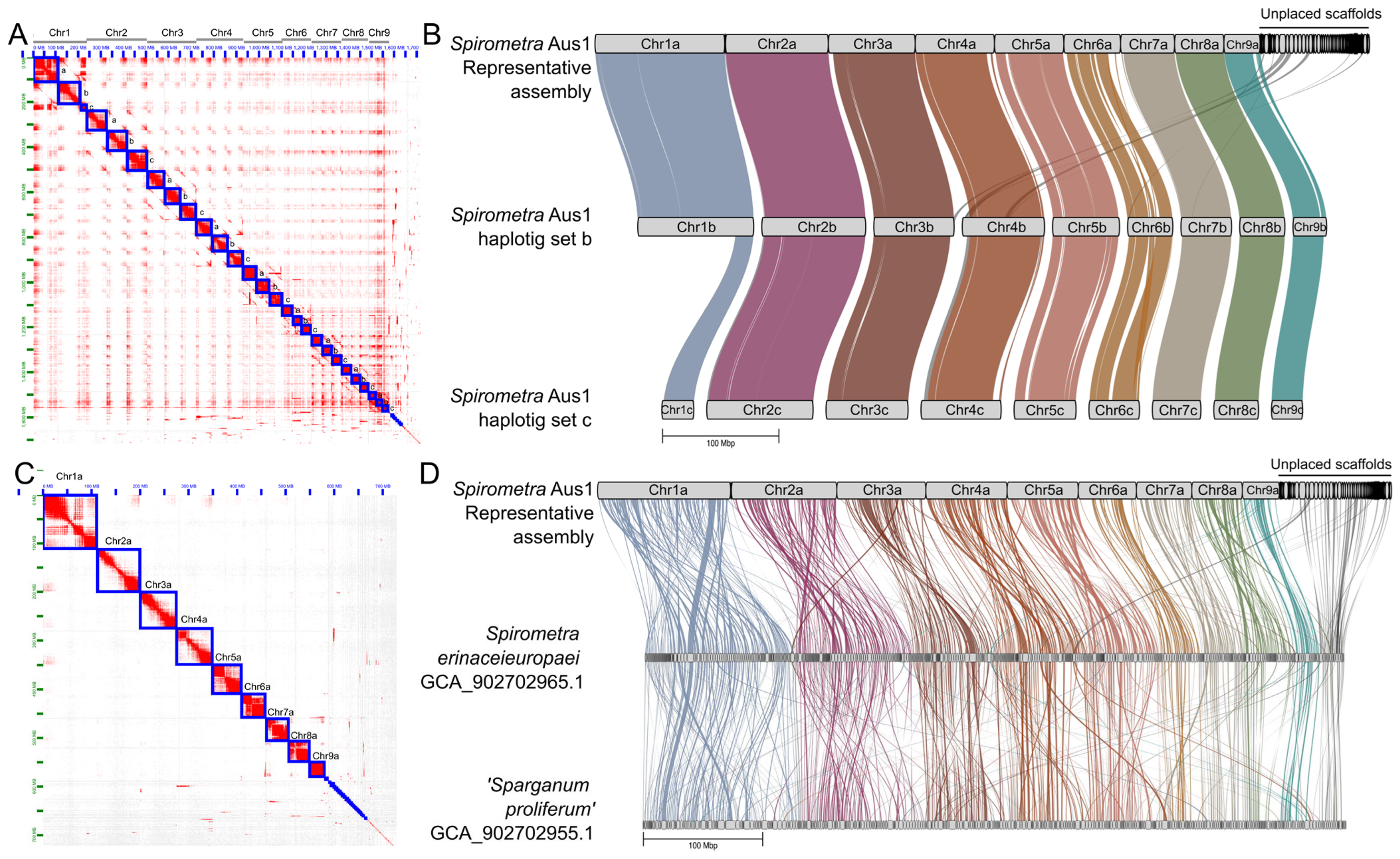
2.3. Inference of Nine Chromosomes
2.4. Nuclear Genome Annotation
2.5. Synteny
3. Discussion
4. Materials and Methods
4.1. Parasite Material
4.2. Isolation of Genomic DNA, and Construction and Sequencing of DNA Long Read, Short-Read and In Situ Hi-C Libraries
4.3. Isolation of Total RNA and Construction and Sequencing of Long-Read and Short-Read Libraries
4.4. Assessing Genome Size, Heterozygosity and Ploidy
4.5. Assembly and Scaffolding of Genomic Contigs and Removal of Potential Contaminants
4.6. Gene Models and Annotation
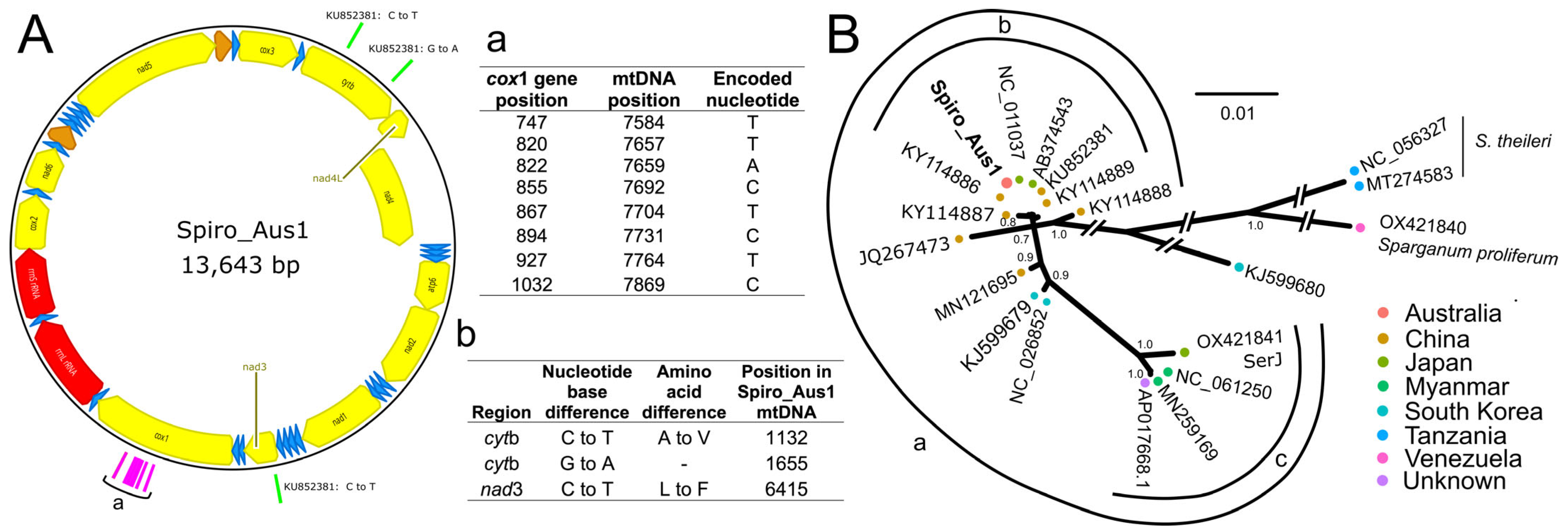
4.7. Comparative Mitochondrial Genomic Analyses
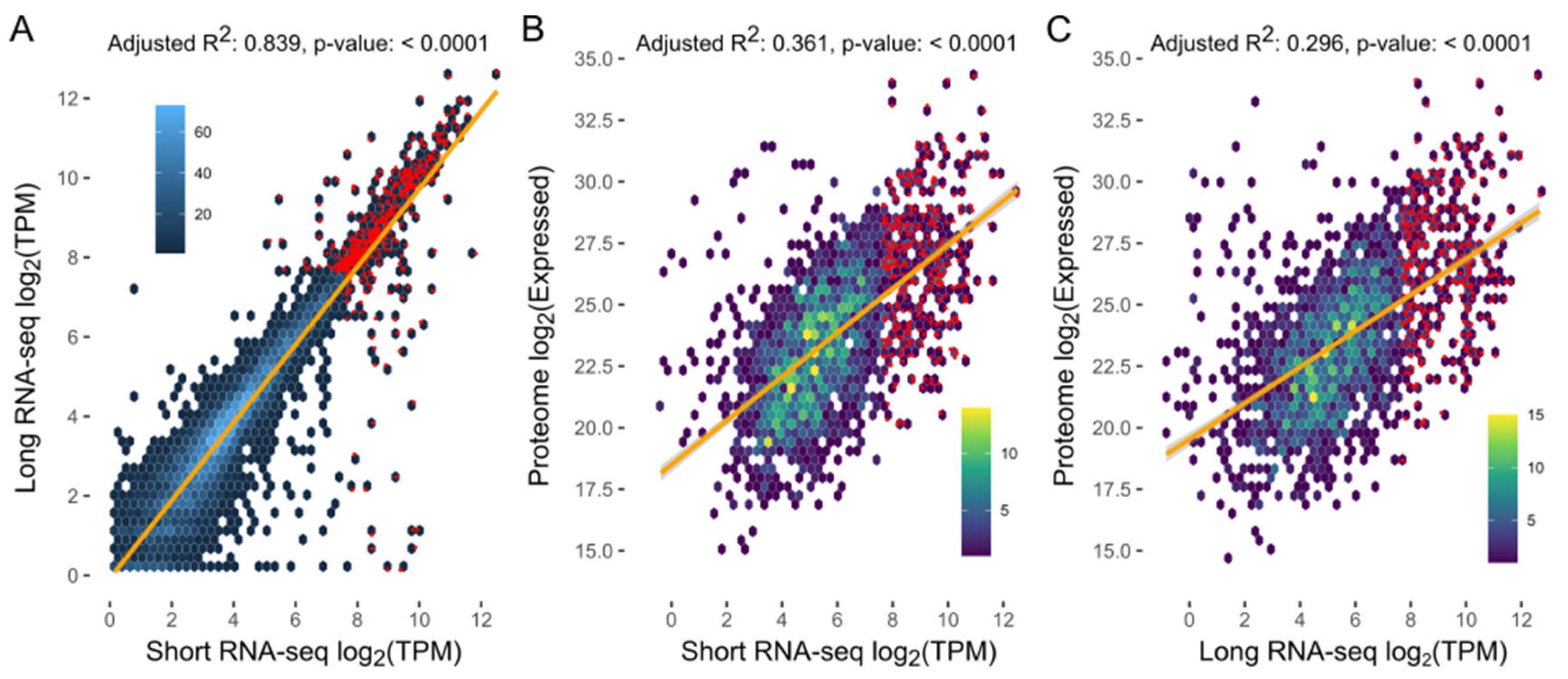
4.8. Proteomic Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Liu, Q.; Li, M.W.; Wang, Z.D.; Zhao, G.H.; Zhu, X.Q. Human sparganosis, a neglected food-borne zoonosis. Lancet Infect. Dis. 2015, 15, 1226–1235. [Google Scholar] [CrossRef] [PubMed]
- Kikuchi, T.; Maruyama, H. Human proliferative sparganosis update. Parasitol. Int. 2020, 75, 102036. [Google Scholar] [CrossRef]
- Tran, Q.R.; Tran, M.C.; Mehanna, D. Sparganosis: An under-recognised zoonosis in Australia? BMJ Case Rep. 2019, 12, e228396. [Google Scholar] [CrossRef]
- Kim, J.G.; Ahn, C.S.; Sohn, W.M.; Nawa, Y.; Kong, Y. Human sparganosis in Korea. J. Korean Med. Sci. 2018, 33, e273. [Google Scholar] [CrossRef]
- Kuchta, R.; Scholz, T.; Brabec, J.; Narduzzi-Wicht, B. Diphyllobothrium, Diplogonoporus and Spirometra. In Biology of Foodborne Parasites; Xiao, L., Ryan, U., Feng, F., Eds.; CRC Press: Boca Raton, FL, USA, 2015; pp. 299–326. [Google Scholar]
- Kuchta, R.; Kołodziej-Sobocińska, M.; Brabec, J.; Młocicki, D.; Sałamatin, R.; Scholz, T. Sparganosis (Spirometra) in Europe in the molecular era. Clin. Infect. Dis. 2021, 72, 882–890. [Google Scholar] [CrossRef] [PubMed]
- Bennett, H.M.; Mok, H.P.; Gkrania-Klotsas, E.; Tsai, I.J.; Stanley, E.J.; Antoun, N.M.; Coghlan, A.; Harsha, B.; Traini, A.; Ribeiro, D.M.; et al. The genome of the sparganosis tapeworm Spirometra erinaceieuropaei isolated from the biopsy of a migrating brain lesion. Genome Biol. 2014, 15, 510. [Google Scholar] [CrossRef]
- Kikuchi, T.; Dayi, M.; Hunt, V.L.; Ishiwata, K.; Toyoda, A.; Kounosu, A.; Sun, S.; Maeda, Y.; Kondo, Y.; de Noya, B.A.; et al. Genome of the fatal tapeworm Sparganum proliferum uncovers mechanisms for cryptic life cycle and aberrant larval proliferation. Commun. Biol. 2021, 4, 649. [Google Scholar] [CrossRef] [PubMed]
- Korhonen, P.K.; Young, N.D.; Gasser, R.B. Making sense of genomes of parasitic worms: Tackling bioinformatic challenges. Biotechnol. Adv. 2016, 34, 663–686. [Google Scholar] [CrossRef]
- Lightowlers, M.W.; Gasser, R.B.; Hemphill, A.; Romig, T.; Tamarozzi, F.; Deplazes, P.; Torgerson, P.; Garcia, H.H.; Kern, P. Advances in the treatment, diagnosis, control and scientific understanding of taeniid cestode parasite infections over the past 50 years. Int. J. Parasitol. 2021, 51, 1167–1192. [Google Scholar] [CrossRef]
- Kamenetzky, L.; Maldonado, L.L.; Cucher, M.A. Cestodes in the genomic era. Parasitol. Res. 2022, 121, 1077–1089. [Google Scholar] [CrossRef]
- Oey, H.; Zakrzewski, M.; Gravermann, K.; Young, N.D.; Korhonen, P.K.; Gobert, G.N.; Hasan, S.; Martine, D.M.; You, H.; Lavin, M.; et al. Whole-genome sequence of the bovine blood fluke Schistosoma bovis reveals evidence for introgressive hybridization with S. haematobium. PLoS Pathog. 2019, 15, e1007513. [Google Scholar] [CrossRef]
- Kinkar, L.; Korhonen, P.K.; Cai, H.; Gauci, C.G.; Lightowlers, M.W.; Saarma, U.; Jenkins, D.J.; Li, J.; Young, N.D.; Gasser, R.B. Long-read sequencing reveals a 4.4 kb tandem repeat region in the mitogenome of Echinococcus granulosus (sensu stricto) genotype G1. Parasites Vectors 2019, 12, 238. [Google Scholar] [CrossRef] [PubMed]
- Stroehlein, A.J.; Korhonen, P.K.; Chong, T.M.; Lim, Y.L.; Chan, K.G.; Webster, B.; Rollinson, D.; Brindley, P.J.; Gasser, R.B.; Young, N.D. High-quality Schistosoma haematobium genome achieved by single-molecule and long-range sequencing. GigaScience 2019, 8, giz108. [Google Scholar] [CrossRef]
- Kinkar, L.; Young, N.D.; Sohn, W.-M.; Stroehlein, A.J.; Korhonen, P.K.; Gasser, R.B. First record of a tandem-repeat region within the mitochondrial genome of Clonorchis sinensis using a long-read sequencing approach. PLoS Negl. Trop. Dis. 2020, 14, e0008552. [Google Scholar] [CrossRef] [PubMed]
- Young, N.D.; Stroehlein, A.J.; Kinkar, L.; Wang, W.; Sohn, W.-M.; Kaur, P.; Weisz, D.; Dudchenko, O.; Aiden, E.L.; Korhonen, P.K.; et al. High-quality reference genome for Clonorchis sinensis. Genomics 2021, 113, 1605–1615. [Google Scholar] [CrossRef] [PubMed]
- Stroehlein, A.J.; Korhonen, P.K.; Lee, V.V.; Ralph, S.A.; Mentink-Kane, M.; You, H.; McManus, D.P.; Tchuem Tchuenté, L.-A.; Stothard, J.R.; Kaur, P.; et al. Chromosome-level genome of Schistosoma haematobium underpins genome-wide explorations of molecular variation. PLoS Pathog. 2022, 18, e1010288. [Google Scholar] [CrossRef]
- Rao, S.S.; Huntley, M.H.; Durand, N.C.; Stamenova, E.K.; Bochkov, I.D.; Robinson, J.T.; Sanborn, A.L.; Machol, I.; Omer, A.D.; Lander, E.S.; et al. A 3D map of the human genome at kilobase resolution reveals principles of chromatin looping. Cell 2014, 159, 1665–1680. [Google Scholar] [CrossRef]
- Amarasinghe, S.L.; Su, S.; Dong, X.; Zappia, L.; Ritchie, M.E.; Gouil, Q. Opportunities and challenges in long-read sequencing data analysis. Genome Biol. 2020, 21, 30. [Google Scholar] [CrossRef]
- Wang, Y.; Yu, J.; Jiang, M.; Lei, W.; Zhang, X.; Tang, H. Sequencing and assembly of polyploid genomes. Methods Mol. Biol. 2023, 2545, 429–458. [Google Scholar] [CrossRef]
- Bowman, D.D. Georgis’ Parasitology for Veterinarians, 11th ed.; Elsevier: St. Louis, MO, USA, 2020; ISBN 9780323543965. [Google Scholar]
- Zhu, X.Q.; Beveridge, I.; Berger, L.; Barton, D.; Gasser, R.B. Single-strand conformation polymorphism-based analysis reveals genetic variation within Spirometra erinacei (Cestoda: Pseudophyllidea) from Australia. Mol. Cell. Probes 2002, 16, 159–165. [Google Scholar] [CrossRef]
- Okino, T.; Ushirogawa, H.; Matoba, K.; Nishimatsu, S.I.; Saito, M. Establishment of the complete life cycle of Spirometra (Cestoda: Diphyllobothriidae) in the laboratory using a newly isolated triploid clone. Parasitol. Int. 2017, 66, 116–118. [Google Scholar] [CrossRef] [PubMed]
- Lightowlers, M.W.; Rickard, M.D. Excretory-secretory products of helminth parasites: Effects on host immune responses. Parasitology 1988, 96, S123–S166. [Google Scholar] [CrossRef] [PubMed]
- Harnett, W. Secretory products of helminth parasites as immunomodulators. Mol. Biochem. Parasitol. 2014, 195, 130–136. [Google Scholar] [CrossRef]
- Doyle, S.R. Improving helminth genome resources in the post-genomic era. Trends Parasitol. 2022, 38, 831–840. [Google Scholar] [CrossRef]
- Hong, X.; Liu, S.N.; Xu, F.F.; Han, L.L.; Jiang, P.; Wang, Z.Q.; Cui, J.; Zhang, X. Global genetic diversity of Spirometra tapeworms. Trop. Biomed. 2020, 37, 237–250. [Google Scholar] [PubMed]
- Liu, W.; Gong, T.; Chen, S.; Liu, Q.; Zhou, H.; He, J.; Wu, Y.; Li, F.; Liu, Y. Epidemiology, diagnosis, and prevention of sparganosis in Asia. Animals 2022, 12, 1578. [Google Scholar] [CrossRef]
- Rubinoff, D.; Cameron, S.; Will, K. A genomic perspective on the shortcomings of mitochondrial DNA for “barcoding” identification. J. Hered. 2006, 97, 581–594. [Google Scholar] [CrossRef]
- Kress, W.J.; Erickson, D.L. DNA barcodes: Methods and protocols. Methods Mol. Biol. 2012, 858, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Adiconis, X.; Haber, A.L.; Simmons, S.K.; Moonshine, A.L.; Ji, Z.; Busby, M.A.; Shi, X.; Jacques, J.; Lancaster, M.A.; Pan, J.Q.; et al. Comprehensive comparative analysis of 5′-end RNA-sequencing methods. Nat. Methods 2018, 15, 505–511. [Google Scholar] [CrossRef]
- Huerta-Cepas, J.; Forslund, K.; Coelho, L.P.; Szklarczyk, D.; Jensen, L.J.; von Mering, C.; Bork, P. Fast genome-wide functional annotation through orthology assignment by eggNOG-Mapper. Mol. Biol. Evol. 2017, 34, 2115–2122. [Google Scholar] [CrossRef]
- Huerta-Cepas, J.; Szklarczyk, D.; Heller, D.; Hernández-Plaza, A.; Forslund, S.K.; Cook, H.; Mende, D.R.; Letunic, I.; Rattei, T.; Jensen, L.J.; et al. eggNOG 5.0: A hierarchical, functionally and phylogenetically annotated orthology resource based on 5090 organisms and 2502 viruses. Nucleic Acids Res. 2019, 47, D309–D314. [Google Scholar] [CrossRef] [PubMed]
- Mueller, J.F. The biology of Spirometra. J. Parasitol. 1974, 60, 3–14. [Google Scholar] [CrossRef] [PubMed]
- Mueller, J.F.; Acholonu, A.D. Sparganum growth factor in New World Spirometra spp. J. Parasitol. 1974, 60, 728–729. [Google Scholar] [CrossRef]
- Brehm, K. Echinococcus multilocularis as an experimental model in stem cell research and molecular host-parasite interaction. Parasitology 2010, 137, 537–555. [Google Scholar] [CrossRef]
- Pierson, L.; Mousley, A.; Devine, L.; Marks, N.J.; Day, T.A.; Maule, A.G. RNA interference in a cestode reveals specific silencing of selected highly expressed gene transcripts. Int. J. Parasitol. 2010, 40, 605–615. [Google Scholar] [CrossRef]
- Pouchkina-Stantcheva, N.N.; Cunningham, L.J.; Hrčkova, G.; Olson, P.D. RNA-mediated gene suppression and in vitro culture in Hymenolepis microstoma. Int. J. Parasitol. 2013, 43, 641–646. [Google Scholar] [CrossRef]
- Arunsan, P.; Ittiprasert, W.; Smout, M.J.; Cochran, C.J.; Mann, V.H.; Chaiyadet, S.; Karinshak, S.E.; Sripa, B.; Young, N.D.; Sotillo, J.; et al. Programmed knockout mutation of liver fluke granulin attenuates virulence of infection-induced hepatobiliary morbidity. eLife 2019, 8, e41463. [Google Scholar] [CrossRef]
- You, H.; Mayer, J.U.; Johnston, R.L.; Sivakumaran, H.; Ranasinghe, S.; Rivera, V.; Kondrashova, O.; Koufariotis, L.T.; Du, X.; Driguez, P.; et al. CRISPR/Cas9-mediated genome editing of Schistosoma mansoni acetylcholinesterase. FASEB J. 2021, 35, e21205. [Google Scholar] [CrossRef]
- Zhang, L.; Wang, L.; Xiang, S.; Hu, Y.; Zhao, S.; Liao, Y.; Zhu, Z.; Wu, X. CRISPR/Cas9-mediated gene knockout of Sj16 in Schistosoma japonicum eggs upregulates the host-to-egg immune response. FASEB J. 2022, 36, e22615. [Google Scholar] [CrossRef]
- Campos, T.L.; Korhonen, P.K.; Hofmann, A.; Gasser, R.B.; Young, N.D. Machine learning for the prediction and prioritisation of essential genes in eukaryotes—Challenges and prospects. Biotechnol. Adv. 2022, 54, 107822. [Google Scholar] [CrossRef]
- Hong, D.; Xie, H.; Wan, H.; An, N.; Xu, C.; Zhang, J. Efficacy comparison between long-term high-dose praziquantel and surgical therapy for cerebral sparganosis: A multicenter retrospective cohort study. PLoS Negl. Trop. Dis. 2018, 12, e0006918. [Google Scholar] [CrossRef]
- Chen, S.; Zhou, Y.; Chen, Y.; Gu, J. fastp: An ultra-fast all-in-one FASTQ pre-processor. Bioinformatics 2018, 34, i884–i890. [Google Scholar] [CrossRef]
- Ranallo-Benavidez, T.R.; Jaron, K.S.; Schatz, M.C. GenomeScope 2.0 and Smudgeplot for reference-free profiling of polyploid genomes. Nat. Commun. 2020, 11, 1432. [Google Scholar] [CrossRef] [PubMed]
- Kokot, M.; Dlugosz, M.; Deorowicz, S. KMC 3: Counting and manipulating k-mer statistics. Bioinformatics 2017, 33, 2759–2761. [Google Scholar] [CrossRef] [PubMed]
- Zhou, C.; McCarthy, S.A.; Durbin, R. YaHS: Yet another Hi-C scaffolding tool. Bioinformatics 2023, 39, btac808. [Google Scholar] [CrossRef]
- Durand, N.C.; Shamim, M.S.; Machol, I.; Rao, S.S.; Huntley, M.H.; Lander, E.S.; Aiden, E.L. Juicer provides a one-click system for analyzing loop-resolution Hi-C experiments. Cell Syst. 2016, 3, 95–98. [Google Scholar] [CrossRef] [PubMed]
- Simão, F.A.; Waterhouse, R.M.; Ioannidis, P.; Kriventseva, E.V.; Zdobnov, E.M. BUSCO: Assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 2015, 31, 3210–3212. [Google Scholar] [CrossRef]
- Flynn, J.M.; Hubley, R.; Goubert, C.; Rosen, J.; Clark, A.G.; Feschotte, C.; Smit, A.F. RepeatModeler2 for automated genomic discovery of transposable element families. Proc. Natl. Acad. Sci. USA 2020, 117, 9451–9457. [Google Scholar] [CrossRef]
- Cantalapiedra, C.P.; Hernández-Plaza, A.; Letunic, I.; Bork, P.; Huerta-Cepas, J. eggNOG-mapper v2: Functional annotation, orthology assignments, and domain prediction at the metagenomic scale. Mol. Biol. Evol. 2021, 38, 5825–5829. [Google Scholar] [CrossRef]
- Tarailo-Graovac, M.; Chen, N. Using RepeatMasker to identify repetitive elements in genomic sequences. Curr. Protoc. Bioinform. 2009, 4, 4.10.1–4.10.14. [Google Scholar] [CrossRef]
- Song, L.; Florea, L. Rcorrector: Efficient and accurate error correction for Illumina RNA-seq reads. GigaScience 2015, 4, 48. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.; Paggi, J.M.; Park, C.; Bennett, C.; Salzberg, S.L. Graph-based genome alignment and genotyping with HISAT2 and HISAT-genotype. Nat. Biotechnol. 2019, 37, 907–915. [Google Scholar] [CrossRef]
- Nachtigall, P.G.; Kashiwabara, A.Y.; Durham, A.M. CodAn: Predictive models for precise identification of coding regions in eukaryotic transcripts. Brief. Bioinform. 2021, 22, bbaa045. [Google Scholar] [CrossRef] [PubMed]
- Li, H. Minimap2: Pairwise alignment for nucleotide sequences. Bioinformatics 2018, 34, 3094–3100. [Google Scholar] [CrossRef]
- Tang, A.D.; Soulette, C.M.; van Baren, M.J.; Hart, K.; Hrabeta-Robinson, E.; Wu, C.J.; Brooks, A.N. Full-length transcript characterization of SF3B1 mutation in chronic lymphocytic leukemia reveals down-regulation of retained introns. Nat. Commun. 2020, 11, 1438. [Google Scholar] [CrossRef]
- Zdobnov, E.M.; Apweiler, R. InterProScan—An integration platform for the signature-recognition methods in InterPro. Bioinformatics 2001, 17, 847–848. [Google Scholar] [CrossRef]
- Teufel, F.; Almagro Armenteros, J.J.; Johansen, A.R.; Gíslason, M.H.; Pihl, S.I.; Tsirigos, K.D.; Winther, O.; Brunak, S.; von Heijne, G.; Nielsen, H. SignalP 6.0 predicts all five types of signal peptides using protein language models. Nat. Biotechnol. 2022, 40, 1023–1025. [Google Scholar] [CrossRef] [PubMed]
- Krogh, A.; Larsson, B.; von Heijne, G.; Sonnhammer, E.L. Predicting transmembrane protein topology with a hidden Markov model: Application to complete genomes. J. Mol. Biol. 2001, 305, 567–580. [Google Scholar] [CrossRef]
- Pertea, M.; Pertea, G.M.; Antonescu, C.M.; Chang, T.C.; Mendell, J.T.; Salzberg, S.L. StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat. Biotechnol. 2015, 33, 290–295. [Google Scholar] [CrossRef]
- Edgar, R.C. MUSCLE: A multiple sequence alignment method with reduced time and space complexity. BMC Bioinform. 2004, 5, 113. [Google Scholar] [CrossRef]
- Darriba, D.; Posada, D.; Kozlov, A.M.; Stamatakis, A.; Morel, B.; Flouri, T. ModelTest-NG: A new and scalable tool for the selection of DNA and protein evolutionary models. Mol. Biol. Evol. 2020, 37, 291–294. [Google Scholar] [CrossRef] [PubMed]
- Ronquist, F.; Teslenko, M.; van der Mark, P.; Ayres, D.L.; Darling, A.; Höhna, S.; Larget, B.; Liu, L.; Suchard, M.A.; Huelsenbeck, J.P. MrBayes 3.2: Efficient Bayesian phylogenetic inference and model choice across a large model space. Syst. Biol. 2012, 61, 539–542. [Google Scholar] [CrossRef] [PubMed]
- Yu, G. Using ggtree to visualize data on tree-like structures. Curr. Protoc. Bioinform. 2020, 69, e96. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Ma, G.; Ang, C.; Korhonen, P.K.; Rong, X.; Nie, S.; Koehler, A.V.; Simpson, R.J.; Greening, D.W.; Reid, G.E.; et al. Somatic proteome of Haemonchus contortus. Int. J. Parasitol. 2019, 49, 311–320. [Google Scholar] [CrossRef]
- Wang, T.; Ma, G.; Ang, C.S.; Korhonen, P.K.; Koehler, A.V.; Young, N.D.; Nie, S.; Williamson, N.A.; Gasser, R.B. High throughput LC-MS/MS-based proteomic analysis of excretory-secretory products from short-term in vitro culture of Haemonchus contortus. J. Proteomics 2019, 204, 103375. [Google Scholar] [CrossRef]
- Tyanova, S.; Temu, T.; Cox, J. The MaxQuant computational platform for mass spectrometry-based shotgun proteomics. Nat. Protoc. 2016, 11, 2301–2319. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Assembly | Complete Assembly [ComA] a | Representative Assembly [RepA] a | Chromosomal Assembly [ChrA] a | Spirometra erinaceieuropaei b GCA_902702965.1 | Sparganum proliferum b GCA_902702955.1 |
|---|---|---|---|---|---|
| Number of scaffolds | 315 | 297 | 9 | 5723 | 7388 |
| Total size of scaffolds | 1,730,282,878 | 728,638,509 | 572,004,676 | 796,029,360 | 653,387,223 |
| Longest scaffold | 111,593,610 | 111,593,610 | 111,593,610 | 5,490,141 | 8,099,213 |
| Number of scaffolds: >100 K; 1 M; 10 M | 198; 78; 27 | 180; 60; 9 | 9; 9; 9 | 1304; 200; 0 | 724; 203; 0 |
| N50 scaffold length; L50 scaffold count | 68,120,031; 11 | 59,837,588; 5 | 68,120,031; 4 | 820,922; 271 | 1,241,503; 146 |
| Scaffold GC (%) | 45.4 | 45.4 | 45.4 | 40.7 | 41.8 |
| Scaffold N (%) | 0.02 | 0.02 | 0.02 | 9.8 | 7.8 |
| Number of contigs | 1793 | 854 | 495 | 50,415 | 41,218 |
| Longest contig | 11,343,096 | 11,343,096 | 11,343,096 | 471,202 | 304,998 |
| Number of contigs: >100 K; 1 M; 10 M | 1488; 591; 1 | 661; 256; 1 | 443; 204; 1 | 465; 0; 0 | 451; 0; 0 |
| N50 contig length; L50 contig count | 1,895,074; 271 | 1,793,361; 120 | 2,026,377; 85 | 29,662; 6600 | 32,650; 5100 |
| Contig GC (%) | 45.4 | 45.4 | 45.4 | 45.1 | 45.3 |
| Genome completeness and accuracy: | |||||
| Complete BUSCO c | 651 (68.2%) | 629 (65.9%) | 609 (63.8%) | 599 (62.8%) | 605 (63.4%) |
| Complete single-copy BUSCO | 49 (5.1%) | 569 (59.6%) | 604 (63.3%) | 529 (55.5%) | 584 (61.2%) |
| Complete and duplicated BUSCO | 602 (63.1%) | 60 (6.3%) | 5 (0.5%) | 70 (7.3%) | 21 (2.2%) |
| Fragmented BUSCO | 70 (7.3%) | 78 (8.2%) | 74 (7.8%) | 100 (10.5%) | 97 (10.2%) |
| Features | Complete Assembly [ComA] a | Representative Assembly [RepA] a | Chromosomal Assembly [ChrA] a | Spirometra erinaceieuropaeib GCA_902702965.1 | Sparganum proliferumb GCA_902702955.1 |
|---|---|---|---|---|---|
| Numbers of genes (mRNAs) | 27,172 (31,299) | 11,064 (12,750) | 9314 (10,734) | 20,774 (20,774) | 16,508 (16,508) |
| Gene length c | 14,596 ± 19,177 | 14,342 ± 18,657 | 14,654 ± 18,839 | 15,539 ± 20,390 | 16,017 ± 21,161 |
| mRNA length | 1490 ± 1400 | 1479 ± 1384 | 1504 ± 1414 | 1361 ± 1338 | 1395 ± 1422 |
| Coding domain length | 1489 ± 1397 | 1476 ± 1377 | 1504 ± 1414 | 1361 ± 1338 | 1395 ± 1422 |
| Number of exons | 6.6 ± 6.3 | 6.6 ± 6.2 | 6.7 ± 6.3 | 5.8 ± 5.5 | 6.2 ± 5.9 |
| Exon length | 222.3 ± 298.5 | 224.3 ± 302.2 | 222.7 ± 301.6 | 234.4 ± 290.0 | 225.8 ± 274.3 |
| Intron length | 2400 ± 3599 | 2388 ± 3576 | 2376 ± 3582 | 2925 ± 4573 | 2797 ± 4396 |
| Protein length | 495.3 ± 465.7 | 491.1 ± 459.1 | 500.4 ± 471.2 | 407.5 ± 405.9 | 401.5 ± 425.1 |
| Completeness: | |||||
| Complete BUSCO d | 618 (64.8%) | 572 (60.0%) | 549 (57.6%) | 617 (64.7%) | 560 (58.7%) |
| Complete single-copy BUSCO | 75 (7.9%) | 510 (53.5%) | 534 (56.0%) | 557 (58.4%) | 542 (56.8%) |
| Complete and duplicated BUSCO | 543 (56.9%) | 62 (6.5%) | 15 (1.6%) | 60(6.3%) | 18 (1.9%) |
| Fragmented BUSCO | 70 (7.3%) | 72 (7.5%) | 72 (7.5%) | 87 (9.1%) | 110 (11.5%) |
| Description | Complete Assembly [ComA] a (%) | Representative Assembly [RepA] a (%) | Chromosomal Assembly [ChrA] a (%) |
|---|---|---|---|
| Number of genes | 27,172 | 11,064 | 9314 |
| Evidence (short-read transcripts) b,c | 21,061 (77.5) | 8577 (77.5) | 7289 (78.3) |
| Evidence (long-read transcripts) c | 19,361 (71.3) | 7927 (71.7) | 6801 (73.0) |
| Evidence (both short- and long-read transcripts) c | 18,890 (69.5) | 7748 (70.0) | 6643 (71.3) |
| Evidence (protein expression) | 5549 (20.4) | 2223 (20.1) | 1921 (20.6) |
| Evidence (transcription and expression) d | 5482 (20.2) | 2195 (19.8) | 1901 (20.4) |
| Homology searches | |||
| eggNOG mapper b | 20,193 (74.3) | 8289 (74.9) | 6969 (74.8) |
| InterProScan domains | 20,005 (73.6) | 8183 (74.0) | 6904 (74.1) |
| PFAM domains | 17,833 (65.6) | 7293 (65.9) | 6154 (66.1) |
| Gene ontology (GO) results f | 15,032 (55.3) | 6083 (55.0) | 5180 (55.6) |
| KEGG orthologues | 15,925 (58.6) | 6456 (58.4) | 5474 (58.8) |
| Reactome pathways | 18,323 (67.4) | 7491 (67.7) | 6326 (67.9) |
| MetaCyc pathways | 14,043 (51.7) | 5711 (51.6) | 4848 (52.1) |
| Proteins with signal peptides | 2239 (8.2) | 902 (8.2) | 762 (8.2) |
| Transmembrane (TM) domains | 5018 (18.5) | 2040 (18.4) | 1743 (18.7) |
| Excretory/secretory (ES) proteins e | 1405 (5.2) | 565 (5.1) | 470 (5.0) |
| Transcribed ES protein genes | 1075 (3.6) | 433 (3.9) | 363 (3.9) |
| Transcribed and expressed ES protein genes d | 400 (1.5) | 146 (1.3) | 127 (1.4) |
| Syntenic Scaffolds | Length of Scaffolds with Bundled Links (Percentage of Genome Assembly) | Number of Syntenic Blocks | Number of Genes | |||
|---|---|---|---|---|---|---|
| RepA | ChrB | RepA | ChrB | |||
| Spiro_Aus1: RepA vs. ChrB | 18 | 9 | 612,663,739 (84.1%) | 538,366,406 (100%) | 23 | 7106 |
| RepA | ChrC | RepA | ChrC | |||
| Spiro_Aus1: RepA vs. ChrC | 16 | 9 | 622,304,671 (85.4%) | 463,277,963 (100%) | 23 | 5746 |
| RepA | SerJ | RepA | S. erinaceieuropaei | |||
| RepA vs. SerJ | 38 | 395 | 667,935,674 (91.7%) | 446,608,837 (56.1%) | 451 | 5197 |
| RepA | Spr | RepA | Spr | |||
| RepA vs. Spr | 43 | 333 | 685,813,185 (94.1%) | 473,742,847 (72.5%) | 403 | 6248 |
| Protein Groups | Total Number of KEGG Terms | Number of KEGG Terms for Highly Transcribed Genes (p-Value) | Number of KEGG Terms for Expressed ES Proteins (p-Value) | ||
|---|---|---|---|---|---|
| ko04147 Exosome | 324 | 74 | (0.0000) | 15 | (0.0368) |
| ko03011 Ribosome | 117 | 61 | (0.0000 | 0 | |
| ko04131 Membrane trafficking | 594 | 46 | (0.0280) | 12 | (0.0000) |
| ko04812 Cytoskeleton proteins | 187 | 28 | (0.0000) | 0 | |
| ko03110 Chaperones and folding catalysts | 124 | 27 | (0.0000) | 19 | (0.0002) |
| ko03019 Messenger RNA biogenesis | 228 | 20 | (0.0372) | 0 | |
| ko03036 Chromosome and associated proteins | 459 | 13 | (0.0001) | 0 | |
| ko03012 Translation factors | 60 | 11 | (0.0010) | 0 | |
| ko04031 GTP-binding proteins | 86 | 8 | (0.0887) | 0 | |
| ko03009 Ribosome biogenesis | 175 | 8 | (0.0795) | 0 | |
| ko03000 Transcription factors | 259 | 6 | (0.0011) | 0 | |
| ko00536 Glycosaminoglycan binding proteins | 35 | 6 | (0.0169) | 0 | |
| ko01007 Amino acid related enzymes | 33 | 6 | (0.0132) | 0 | |
| ko01009 Protein phosphatases and associated proteins | 177 | 5 | (0.0146) | 0 | |
| ko03037 Cilium and associated proteins | 131 | 4 | (0.0407) | 0 | |
| ko00537 Glycosylphosphatidylinositol (GPI)-anchored proteins | 13 | 3 | (0.0396) | 2 | (0.1567) |
| ko04091 Lectins | 16 | 3 | (0.0635) | 4 | (0.0142) |
| ko03021 Transcription machinery | 143 | 3 | (0.0103) | 0 | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Young, N.D.; Malik, R.; Brown, A.; Wang, T.; Ash, A.; Korhonen, P.K.; Gasser, R.B. Chromosome-Contiguous Reference Genome for Spirometra to Underpin Future Discovery Research. Int. J. Mol. Sci. 2025, 26, 6417. https://doi.org/10.3390/ijms26136417
Young ND, Malik R, Brown A, Wang T, Ash A, Korhonen PK, Gasser RB. Chromosome-Contiguous Reference Genome for Spirometra to Underpin Future Discovery Research. International Journal of Molecular Sciences. 2025; 26(13):6417. https://doi.org/10.3390/ijms26136417
Chicago/Turabian StyleYoung, Neil D., Richard Malik, Alexa Brown, Tao Wang, Amanda Ash, Pasi K. Korhonen, and Robin B. Gasser. 2025. "Chromosome-Contiguous Reference Genome for Spirometra to Underpin Future Discovery Research" International Journal of Molecular Sciences 26, no. 13: 6417. https://doi.org/10.3390/ijms26136417
APA StyleYoung, N. D., Malik, R., Brown, A., Wang, T., Ash, A., Korhonen, P. K., & Gasser, R. B. (2025). Chromosome-Contiguous Reference Genome for Spirometra to Underpin Future Discovery Research. International Journal of Molecular Sciences, 26(13), 6417. https://doi.org/10.3390/ijms26136417