
UBC9-Mediated SUMO Pathway Drives Prohibitin-1 Nuclear Accumulation and PITX1 Repression in Primary Osteoarthritis

 , and
, and
Abstract
1. Introduction
2. Results
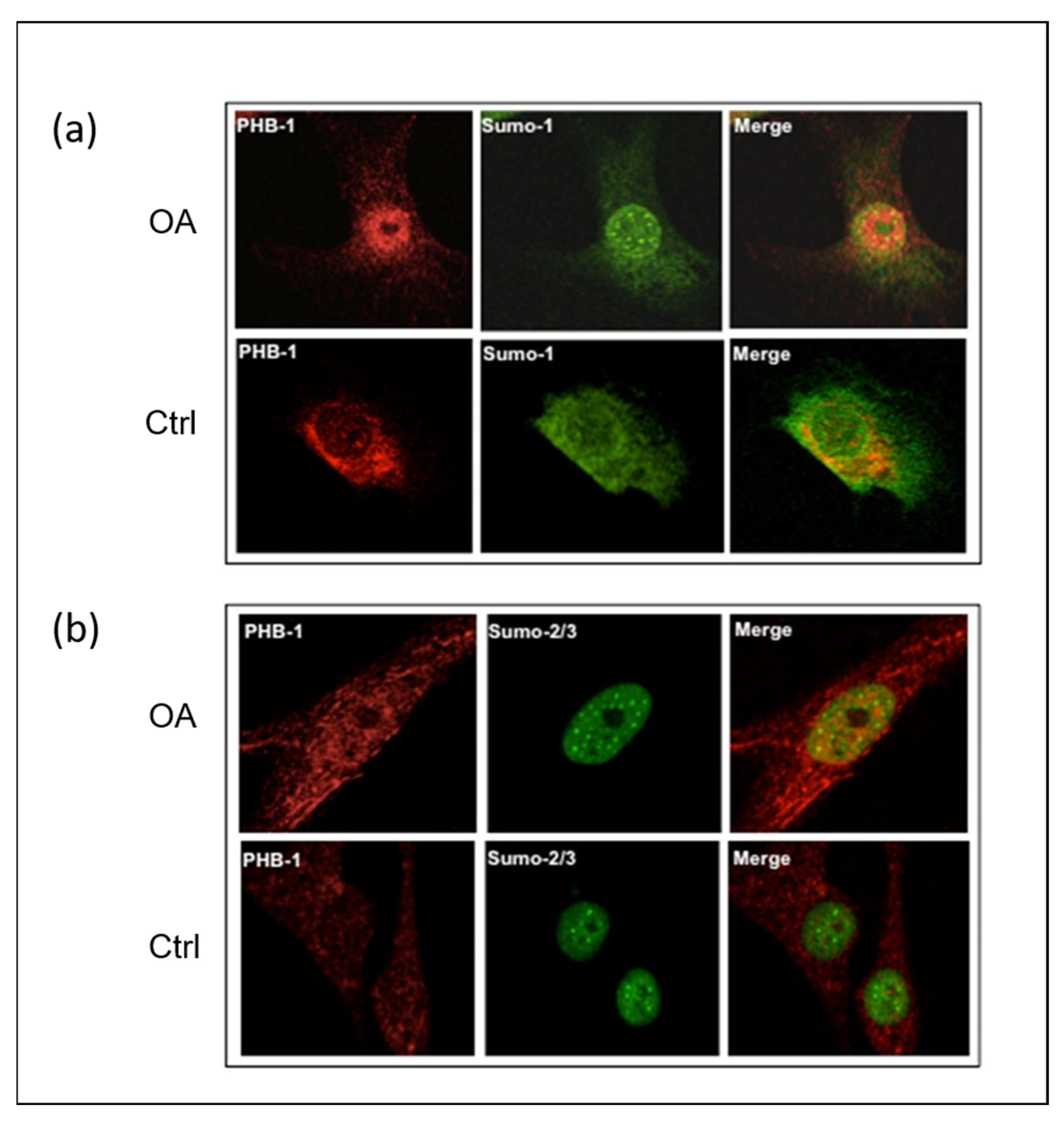
2.1. SUMO-1 Proteins Accumulate in the Nuclei of OA Articular Chondrocytes and Co-Localize with PHB1
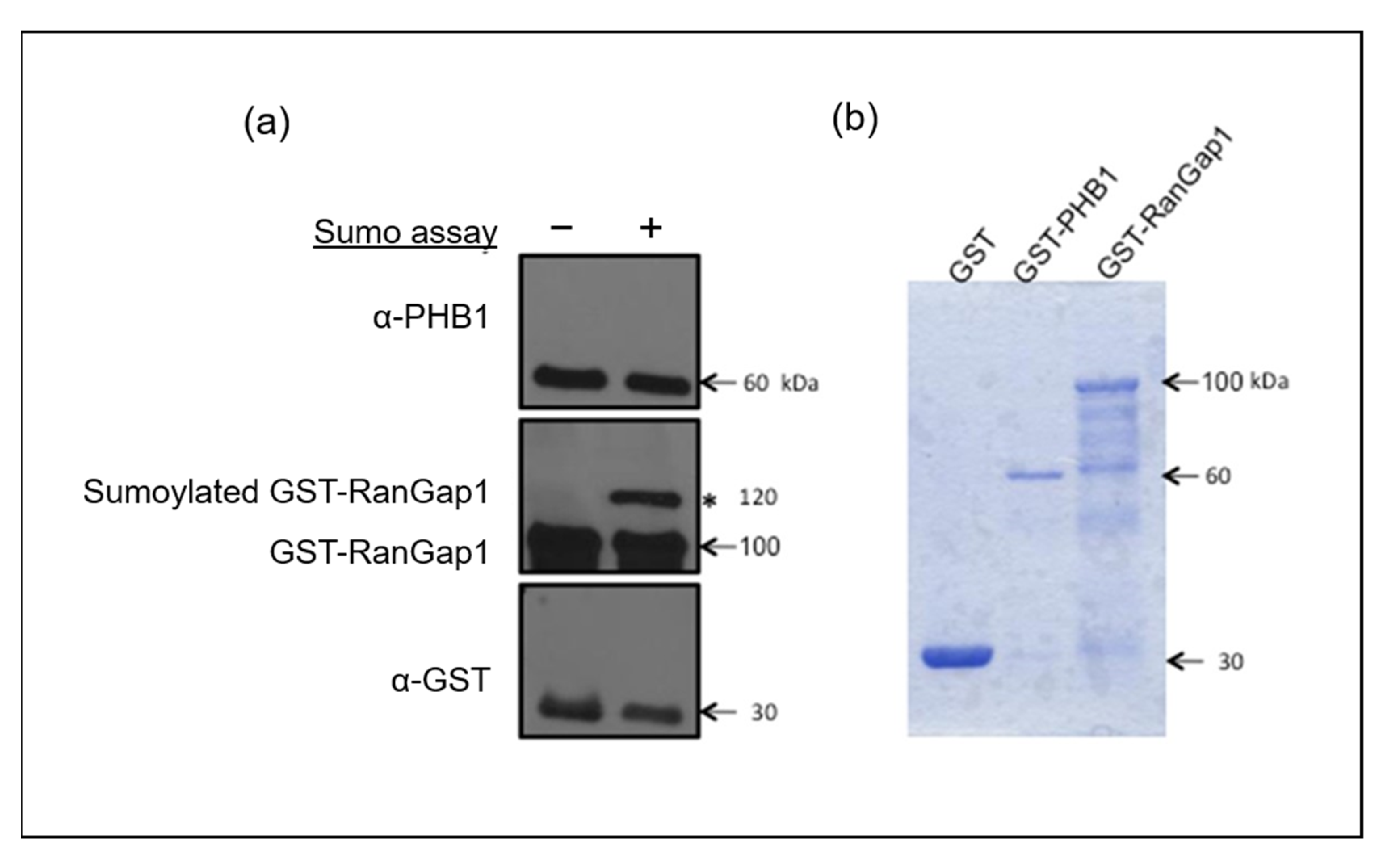
2.2. Is PHB1 Directly SUMOylated or Does It Interact with SUMOylated Partners in Primary OA?
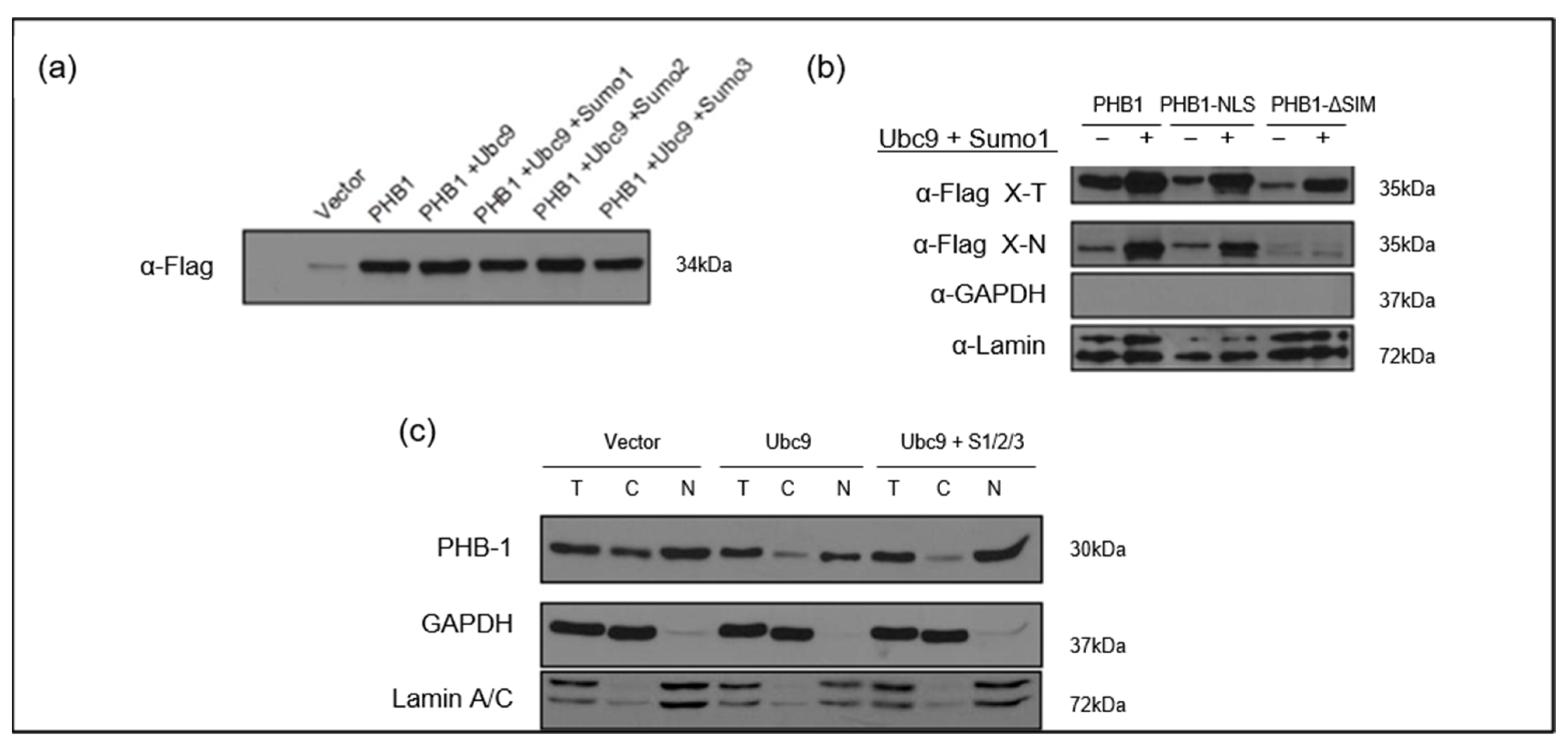
2.3. UBC9-Mediated SUMOylation Promotes Nuclear Accumulation of PHB1 in Primary OA
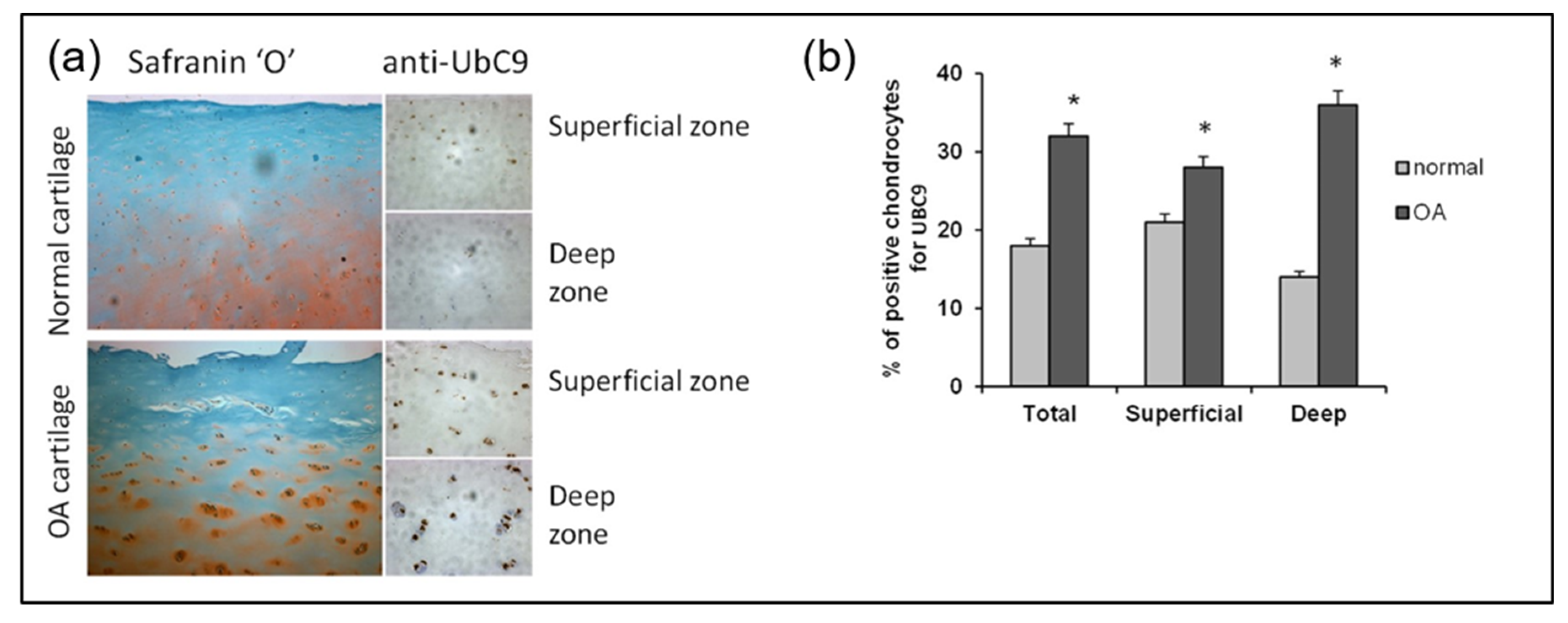
2.4. UBC9 Overexpression in Mice Induces Structural Joint Alterations Characteristic of Primary OA
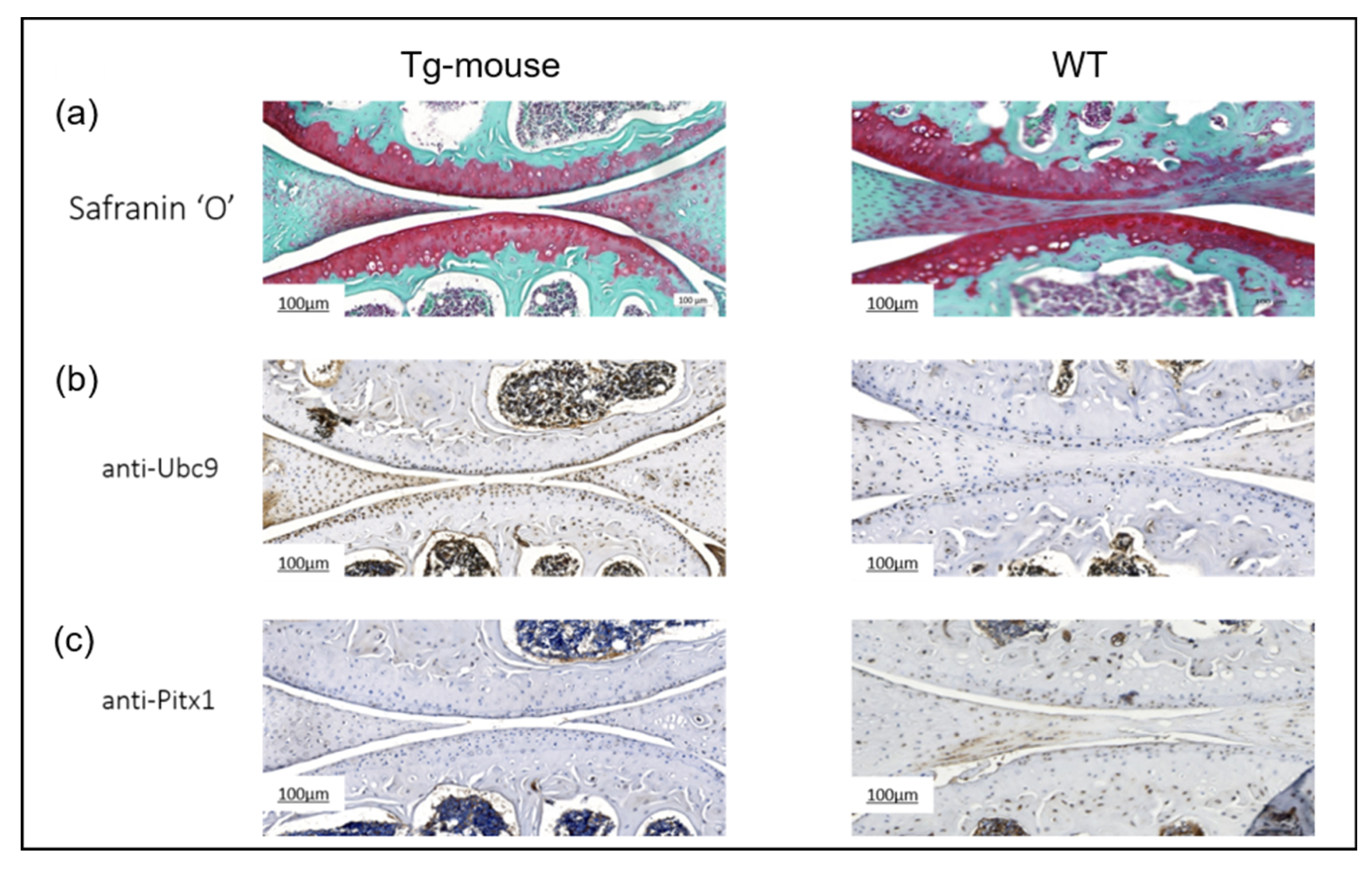
2.5. UBC9 Overexpression Impairs Cartilage Integrity and Suppresses Pitx1 Expression in Mouse Knee Joints
3. Discussion
4. Materials and Methods
4.1. Study Design
4.2. Human Primary Chondrocyte Cell Preparation
4.3. Cell Line
4.4. Antibodies and Reagents
4.5. Vector Constructs
4.6. Nuclear and Cytoplasmic Protein Isolation
4.7. Co-Immunoprecipitation Assays
4.8. GST-PHB1 and GST-RanGap1 Purification
4.9. In Vitro SUMOylation Assay
4.10. Immunohistochemistry Analysis
4.11. Immunofluorescence Analysis
4.12. In Silico Analysis
4.13. Animal Model
4.14. Radiographic Evaluation of Knee Joints in the Mice
4.15. Immunohistochemistry of Articular Cartilage
4.16. Safranin-O Staining of Articular Cartilage
4.17. Statistical Analysis
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| ATP | Adenosine triphosphate |
| BSA | Bovine serum albumin |
| C28/I2 | Human chondrocyte cell line |
| C57BL/6 | Strain of mice |
| CAG | CMV enhancer/chicken β-actin |
| CHU | Centre Hospitalier Universitaire |
| CIBPAR | Comité institutionnel des bonnes pratiques animales en recherche |
| CMV | Cytomegalovirus |
| c-Myc | Cellular myc, a family of transcription factors |
| CO2 | Carbon dioxide |
| CRCHUM | Centre de Recherche Centre Hospitalier de l’Université de Montréal |
| DAB | 3,3′-diaminobenzidine |
| DI water | Deionized water |
| DlacZ | Deleted lacZ gene which encodes b-galactosidase |
| DMEM | Dulbecco’s Modified Eagle Medium |
| DNA | Deoxyribonucleic acid |
| DTT | Dithiothreitol |
| E1 enzyme | Ubiquitin-activating enzymes |
| E2 | Ubiquitin-conjugating enzyme |
| E2F | Family of transcription factors |
| EDTA | Ethylenediaminetetraacetic acid |
| EGTA | Ethylene glycol tetraacetic acid |
| FBS | Fetal bovine serum |
| FLAG | Short peptide sequence used to label proteins |
| GAG | Glycosaminoglycan |
| Gal4 | Galactose-responsive transcription factor |
| GAPDH | Glyceraldehyde-3-phosphate dehydrogenase |
| GST | Glutathione S-transferase |
| IgG | Immunoglobulin G |
| JSN | Joint space narrowing |
| KCl | Potassium chloride |
| Kpn1 | Klebsiella pneumoniae OK8 restriction enzyme |
| MgCl2 | Magnesium chloride |
| NaCl | Sodium chloride |
| NCBI | National Center for Biotechnology Information |
| NEM | N-ethylmaleimide |
| NES | Nuclear export signal |
| NLS | Nuclear localization signal |
| NP-40 | Nonidet P-40 |
| OA | Osteoarthritis |
| p53 | Tumor protein p53 |
| PBS | Phosphate-buffered saline |
| pCCALL2 | Plasmid Conditional CAG- CAG-βgeo-Lox-STOP-Lox-2 |
| pcDNA | Plasmid for cloning, DNA |
| PCR | Polymerase chain reaction |
| PFA | Paraformaldehyde |
| PHB | Prohibitin |
| PHB1_NLS | PHB1 construct in with deleted nuclear export signal replaced with a nuclear localization signal |
| PHB1_ΔSIM | PHB1 construct lacking the identified consensus SUMO-interacting motif |
| PITX1 | Paired-like homeodomain transcription factor 1 |
| PML | Promyelocytic leukemia |
| PML-NBs | Promyelocytic leukemia-nuclear bodies |
| PMSF | Phenylmethylsulfonyl fluoride |
| RanGap1 | Ran GTPase-activating protein 1 |
| RB | Retinoblastoma protein |
| SDS-PAGE | Sodium dodecyl sulfate-polyacrylamide gel electrophoresis |
| SEM | Standard error of the mean |
| SIM | SUMO-interacting motifs |
| SUMO | Small ubiquitin-like modifier proteins |
| SUMOylation | Process where SUMO proteins are covalently attached to target proteins |
| Tg | Transgenic |
| TOM-20 | Translocase of outer mitochondrial membrane 20 |
| Tris-HCl | Tris (hydroxymethyl)aminomethane hydrochloride |
| U2OS | Cell line from human osteosarcoma |
| UBC9 | Ubiquitin-conjugating enzyme 9 |
| UBE2I | Ubiquitin-conjugating enzyme E2 I |
| WT | Wild type |
| Xho1 | Restriction enzyme from Escherichia coli strain that carries Xanthomonas holcicola gene |
| [V/I]-[V/I]-x-[V/I] | Sequence valine/isoleucine- valine/isoleucine- unknown- valine/isoleucine |
| [V/I]-x-[V/I]-[V/I] | Sequence valine/isoleucine-unknown- valine/isoleucine- valine/isoleucine |
References
- Tang, S.; Zhang, C.; Oo, W.M.; Fu, K.; Risberg, M.A.; Bierma-Zeinstra, S.M.; Neogi, T.; Atukorala, I.; Malfait, A.-M.; Ding, C.; et al. Osteoarthritis. Nat. Rev. Dis. Primers 2025, 11, 10. [Google Scholar] [CrossRef]
- Primorac, D.; Molnar, V.; Rod, E.; Jeleč, Ž.; Čukelj, F.; Matišić, V.; Vrdoljak, T.; Hudetz, D.; Hajsok, H.; Borić, I. Knee Osteoarthritis: A Review of Pathogenesis and State-Of-The-Art Non-Operative Therapeutic Considerations. Genes 2020, 11, 854. [Google Scholar] [CrossRef]
- Vidal-Bralo, L.; Lopez-Golan, Y.; Mera-Varela, A.; Rego-Perez, I.; Horvath, S.; Zhang, Y.; del Real, Á.; Zhai, G.; Blanco, F.J.; Riancho, J.A.; et al. Specific premature epigenetic aging of cartilage in osteoarthritis. Aging 2016, 8, 2222–2231. [Google Scholar] [CrossRef]
- Tardio, L.; Andrés-Bergós, J.; Zachara, N.E.; Larrañaga-Vera, A.; Rodriguez-Villar, C.; Herrero-Beaumont, G.; Largo, R. O-linked N-acetylglucosamine (O-GlcNAc) protein modification is increased in the cartilage of patients with knee osteoarthritis. Osteoarthr. Cartil. 2014, 22, 256–263. [Google Scholar] [CrossRef] [PubMed]
- Catterall, J.B.; Hsueh, M.F.; Stabler, T.V.; McCudden, C.R.; Bolognesi, M.; Zura, R.; Jordan, J.M.; Renner, J.B.; Feng, S.; Kraus, V.B. Protein modification by deamidation indicates variations in joint extracellular matrix turnover. J. Biol. Chem. 2012, 287, 4640–4651. [Google Scholar] [CrossRef] [PubMed]
- Zahan, O.; Serban, O.; Gherman, C.; Fodor, D. The evaluation of oxidative stress in osteoarthritis. Med. Pharm. Rep. 2020, 93, 12–22. [Google Scholar] [CrossRef] [PubMed]
- Cillero-Pastor, B.; Caramés, B.; Lires-Deán, M.; Vaamonde-García, C.; Blanco, F.J.; López-Armada, M.J. Mitochondrial dysfunction activates cyclooxygenase 2 expression in cultured normal human chondrocytes. Arthritis Rheum. 2008, 58, 2409–2419. [Google Scholar] [CrossRef] [PubMed]
- Blanco, F.J.; Rego, I.; Ruiz-Romero, C. The role of mitochondria in osteoarthritis. Nat. Rev. Rheumatol. 2011, 7, 161–169. [Google Scholar] [CrossRef]
- Loeser, R.F. Aging and osteoarthritis: The role of chondrocyte senescence and aging changes in the cartilage matrix. Osteoarthr. Cartil. 2009, 17, 971–979. [Google Scholar] [CrossRef]
- Signorile, A.; Sgaramella, G.; Bellomo, F.; De Rasmo, D. Prohibitins: A Critical Role in Mitochondrial Functions and Implication in Diseases. Cells 2019, 8, 71. [Google Scholar] [CrossRef]
- Oyang, L.; Li, J.; Jiang, X.; Lin, J.; Xia, L.; Yang, L.; Tan, S.; Wu, N.; Han, Y.; Yang, Y.; et al. The function of prohibitins in mitochondria and the clinical potentials. Cancer Cell Int. 2022, 22, 343. [Google Scholar] [CrossRef] [PubMed]
- Artal-Sanz, M.; Tavernarakis, N. Prohibitin couples diapause signalling to mitochondrial metabolism during ageing in C. elegans. Nature 2009, 461, 793–797. [Google Scholar] [CrossRef] [PubMed]
- Koushyar, S.; Jiang, W.G.; Dart, D.A. Unveiling the potential of prohibitin in cancer. Cancer Lett. 2015, 369, 316–322. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; McDonald, D.; Blain, A.; Mossman, E.; Atkin, K.; Marusich, M.F.; Capaldi, R.; Bone, L.; Smith, A.; Filby, A.; et al. Parkinson’s disease neurons exhibit alterations in mitochondrial quality control proteins. NPJ Park. Dis. 2023, 9, 120. [Google Scholar] [CrossRef]
- Dutta, D.; Ali, N.; Banerjee, E.; Singh, R.; Naskar, A.; Paidi, R.K.; Mohanakumar, K.P. Low Levels of Prohibitin in Substantia Nigra Makes Dopaminergic Neurons Vulnerable in Parkinson’s Disease. Mol. Neurobiol. 2018, 55, 804–821. [Google Scholar] [CrossRef]
- Supale, S.; Thorel, F.; Merkwirth, C.; Gjinovci, A.; Herrera, P.L.; Scorrano, L.; Meda, P.; Langer, T.; Maechler, P. Loss of prohibitin induces mitochondrial damages altering β-cell function and survival and is responsible for gradual diabetes development. Diabetes 2013, 62, 3488–3499. [Google Scholar] [CrossRef]
- Ising, C.; Koehler, S.; Brähler, S.; Merkwirth, C.; Höhne, M.; Baris, O.R.; Hagmann, H.; Kann, M.; Fabretti, F.; Dafinger, C.; et al. Inhibition of insulin/IGF-1 receptor signaling protects from mitochondria-mediated kidney failure. EMBO Mol. Med. 2015, 7, 275–287. [Google Scholar] [CrossRef]
- Thuaud, F.; Ribeiro, N.; Nebigil, C.G.; Désaubry, L. Prohibitin ligands in cell death and survival: Mode of action and therapeutic potential. Chem. Biol. 2013, 20, 316–331. [Google Scholar] [CrossRef]
- Picard, C.; Azeddine, B.; Moldovan, F.; Martel-Pelletier, J.; Moreau, A. New emerging role of Pitx1 transcription factor in osteoarthritis pathogenesis. Clin. Orthop. Relat. Res. 2007, 462, 59–66. [Google Scholar] [CrossRef]
- Schleicher, M.; Shepherd, B.R.; Suarez, Y.; Fernandez-Hernando, C.; Yu, J.; Pan, Y.; Acevedo, L.M.; Shadel, G.S.; Sessa, W.C. Prohibitin-1 maintains the angiogenic capacity of endothelial cells by regulating mitochondrial function and senescence. J. Cell Biol. 2008, 180, 101–112. [Google Scholar] [CrossRef]
- Wang, S.; Zhang, B.; Faller, D.V. Prohibitin requires Brg-1 and Brm for the repression of E2F and cell growth. EMBO J. 2002, 21, 3019–3028. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Nath, N.; Fusaro, G.; Chellappan, S. Rb and Prohibitin Target Distinct Regions of E2F1 for Repression and Respond to Different Upstream Signals. Mol. Cell. Biol. 1999, 19, 7447–7460. [Google Scholar] [CrossRef] [PubMed]
- Müller, H.; Bracken, A.P.; Vernell, R.; Moroni, M.C.; Christians, F.; Grassilli, E.; Prosperini, E.; Vigo, E.; Oliner, J.D.; Helin, K. E2Fs regulate the expression of genes involved in differentiation, development, proliferation, and apoptosis. Genes Dev. 2001, 15, 267–285. [Google Scholar] [CrossRef]
- Gareau, J.R.; Lima, C.D. The SUMO pathway: Emerging mechanisms that shape specificity, conjugation and recognition. Nat. Rev. Mol. Cell Biol. 2010, 11, 861–871. [Google Scholar] [CrossRef]
- Nüñez-O’Mara, A.; Berra, E. Deciphering the emerging role of SUMO conjugation in the hypoxia-signaling cascade. Biol. Chem. 2013, 394, 459–469. [Google Scholar] [CrossRef]
- Johnson, E.S. Protein modification by SUMO. Annu. Rev. Biochem. 2004, 73, 355–382. [Google Scholar] [CrossRef]
- Princz, A.; Tavernarakis, N. The role of SUMOylation in ageing and senescent decline. Mech. Ageing Dev. 2017, 162, 85–90. [Google Scholar] [CrossRef] [PubMed]
- Hickey, C.M.; Wilson, N.R.; Hochstrasser, M. Function and regulation of SUMO proteases. Nat. Rev. Mol. Cell Biol. 2012, 13, 755–766. [Google Scholar] [CrossRef]
- Da Silva-Ferrada, E.; Ribeiro-Rodrigues, T.M.; Rodríguez, M.S.; Girão, H. Proteostasis and SUMO in the heart. Int. J. Biochem. Cell Biol. 2016, 79, 443–450. [Google Scholar] [CrossRef]
- Mun, M.-J.; Kim, J.-H.; Choi, J.-Y.; Kim, M.-S.; Jang, W.-C.; Lee, J.J.; Eun, Y.L.; Kwak, S.-J.; Kim, K.W.; Lee, S.B. Polymorphisms of small ubiquitin-related modifier genes are associated with risk of Alzheimer’s disease in Korean: A case-control study. J. Neurol. Sci. 2016, 364, 122–127. [Google Scholar] [CrossRef]
- Seeler, J.S.; Dejean, A. SUMO and the robustness of cancer. Nat. Rev. Cancer 2017, 17, 184–197. [Google Scholar] [CrossRef]
- Huang, C.-H.; Yang, T.-T.; Lin, K.-I. Mechanisms and functions of SUMOylation in health and disease: A review focusing on immune cells. J. Biomed. Sci. 2024, 31, 16. [Google Scholar] [CrossRef] [PubMed]
- Enserink, J.M. Sumo and the cellular stress response. Cell Div. 2015, 10, 4. [Google Scholar] [CrossRef] [PubMed]
- Liang, Y.-C.; Lee, C.-C.; Yao, Y.-L.; Lai, C.-C.; Schmitz, M.L.; Yang, W.-M. SUMO5, a novel poly-SUMO isoform, regulates PML nuclear bodies. Sci. Rep. 2016, 6, 26509. [Google Scholar] [CrossRef]
- Celen, A.B.; Sahin, U. SUMOylation on its 25th anniversary: Mechanisms, pathology, and emerging concepts. FEBS J. 2020, 287, 3110–3140. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.H.; Baek, S.H. Emerging roles of desumoylating enzymes. Biochim. Biophys. Acta-Mol. Basis Dis. 2009, 1792, 155–162. [Google Scholar] [CrossRef]
- Seufert, W.; Futcher, B.; Jentsch, S. Role of a ubiquitin-conjugating enzyme in degradation of S- and M-phase cyclins. Nature 1995, 373, 78–81. [Google Scholar] [CrossRef]
- Sahin, U.; Ferhi, O.; Jeanne, M.; Benhenda, S.; Berthier, C.; Jollivet, F.; Niwa-Kawakita, M.; Faklaris, O.; Setterblad, N.; De Thé, H.; et al. Oxidative stress-induced assembly of PML nuclear bodies controls SUMOylation of partner proteins. J. Cell Biol. 2014, 204, 931–945. [Google Scholar] [CrossRef]
- Sahin, U.; de Thé, H.; Lallemand-Breitenbach, V. SUMOylation in Physiology, Pathology and Therapy. Cells 2022, 11, 814. [Google Scholar] [CrossRef]
- Chang, H.R.; Munkhjargal, A.; Kim, M.-J.; Park, S.Y.; Jung, E.; Ryu, J.-H.; Yang, Y.; Lim, J.-S.; Kim, Y. The functional roles of PML nuclear bodies in genome maintenance. Mutat. Res. 2018, 809, 99–107. [Google Scholar] [CrossRef]
- Rie Lallemand-Breitenbach, V.; De Thé, H. PML nuclear bodies: From architecture to function. Curr. Opin. Cell Biol. 2018, 52, 154–161. [Google Scholar] [CrossRef] [PubMed]
- Lång, A.; Lång, E.; Bøe, S.O. PML Bodies in Mitosis. Cells 2019, 8, 893. [Google Scholar] [CrossRef]
- Corpet, A.; Kleijwegt, C.; Roubille, S.; Juillard, F.; Jacquet, K.; Texier, P.; Lomonte, P. PML nuclear bodies and chromatin dynamics: Catch me if you can! Nucleic Acids Res. 2020, 48, 11890–11912. [Google Scholar] [CrossRef] [PubMed]
- Miteva, M.; Keusekotten, K.; Hofmann, K.; Praefcke, G.J.K.; Dohmen, R.J. SUMOylation as a Signal for Polyubiquitylation and Proteasomal Degradation in Conjugation and Deconjugation of Ubiquitin Family Modifiers. Subcell. Biochem. 2010, 54, 195–214. [Google Scholar] [CrossRef] [PubMed]
- Husnjak, K.; Keiten-Schmitz, J.; Müller, S. Identification and Characterization of SUMO-SIM Interactions. Methods Mol. Biol. 2016, 1475, 79–98. [Google Scholar] [CrossRef]
- Takahashi, H.; Hatakeyama, S.; Saitoh, H.; Nakayama, K.I. Noncovalent SUMO-1 Binding Activity of Thymine DNA Glycosylase (TDG) Is Required for Its SUMO-1 Modification and Colocalization with the Promyelocytic Leukemia Protein. J. Biol. Chem. 2005, 280, 5611–5621. [Google Scholar] [CrossRef]
- Picard, C.; Pellicelli, M.; Taheri, M.; Lavoie, J.; Doucet, R.; Wang, D.; Bernard, L.; Bouhanik, S.; Lavigne, P.; Moreau, A. Nuclear accumulation of prohibitin 1 in osteoarthritic chondrocytes down-regulates PITX1 expression. Arthritis Rheum. 2013, 65, 993–1003. [Google Scholar] [CrossRef]
- Merkwirth, C.; Langer, T. Prohibitin function within mitochondria: Essential roles for cell proliferation and cristae morphogenesis. Biochim. Biophys. Acta-Mol. Cell Res. 2009, 1793, 27–32. [Google Scholar] [CrossRef]
- Coates, P.; Nenutil, R.; McGregor, A.; Picksley, S.; Crouch, D.; Hall, P.; Wright, E. Mammalian prohibitin proteins respond to mitochondrial stress and decrease during cellular senescence. Exp. Cell Res. 2001, 265, 262–273. [Google Scholar] [CrossRef]
- Blanco, F.J.; López-Armada, M.J.; Maneiro, E. Mitochondrial dysfunction in osteoarthritis. Mitochondrion 2004, 4, 715–728. [Google Scholar] [CrossRef]
- Fernández-Moreno, M.; Rego-Pérez, I.; Blanco, F.J. Is osteoarthritis a mitochondrial disease? What is the evidence. Curr. Opin. Rheumatol. 2022, 34, 46–53. [Google Scholar] [CrossRef] [PubMed]
- Mao, X.; Fu, P.; Wang, L.; Xiang, C. Mitochondria: Potential Targets for Osteoarthritis. Front. Med. 2020, 7, 581402. [Google Scholar] [CrossRef]
- Sarge, K.D.; Park-Sarge, O.K. SUMOylation and human disease pathogenesis. Trends Biochem. Sci. 2009, 34, 200–205. [Google Scholar] [CrossRef]
- Liu, H.; Craig, S.E.L.; Molchanov, V.; Floramo, J.S.; Zhao, Y.; Yang, T. SUMOylation in Skeletal Development, Homeostasis, and Disease. Cells 2022, 11, 2710. [Google Scholar] [CrossRef]
- Pichler, A.; Fatouros, C.; Lee, H.; Eisenhardt, N. SUMO conjugation—A mechanistic view. Biomol. Concepts 2017, 8, 13–36. [Google Scholar] [CrossRef]
- Meinecke, I.; Cinski, A.; Baier, A.; Peters, M.A.; Dankbar, B.; Wille, A.; Drynda, A.; Mendoza, H.; Gay, R.E.; Hay, R.T.; et al. Modification of nuclear PML protein by SUMO-1 regulates Fas-induced apoptosis in rheumatoid arthritis synovial fibroblasts. Proc. Natl. Acad. Sci. USA 2007, 104, 5073–5078. [Google Scholar] [CrossRef] [PubMed]
- Fu, C.; Ahmed, K.; Ding, H.; Ding, X.; Lan, J.; Yang, Z.; Miao, Y.; Zhu, Y.; Shi, Y.; Zhu, J.; et al. Stabilization of PML nuclear localization by conjugation and oligomerization of SUMO-3. Oncogene 2005, 24, 5401–5413. [Google Scholar] [CrossRef] [PubMed]
- Johnson, K.; Reynard, L.N.; Loughlin, J. Functional characterisation of the osteoarthritis susceptibility locus at chromosome 6q14.1 marked by the polymorphism rs9350591. BMC Med. Genet. 2015, 16, 81. [Google Scholar] [CrossRef]
- Hattersley, N.; Shen, L.; Jaffray, E.G.; Hay, R.T. The SUMO protease SENP6 is a direct regulator of PML nuclear bodies. Mol. Biol. Cell 2011, 22, 78–90. [Google Scholar] [CrossRef]
- Theiss, A.L.; Sitaraman, S.V. The role and therapeutic potential of prohibitin in disease. Biochim. Biophys. Acta-Mol. Cell Res. 2011, 1813, 1137–1143. [Google Scholar] [CrossRef]
- Matunis, M.J.; Rodriguez, M.S. Concepts and methodologies to study protein SUMOylation: An overview. Methods Mol. Biol. 2016, 1475, 3–22. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Zhao, D.; Qiu, F.; Zhang, L.-L.; Liu, S.-K.; Li, Y.-Y.; Liu, M.-T.; Wu, D.; Wang, J.-X.; Ding, X.-Q.; et al. Manipulating PML SUMOylation via Silencing UBC9 and RNF4 Regulates Cardiac Fibrosis. Mol. Ther. 2017, 25, 666–678. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.-J.; Mou, Y.; Maric, D.; Klimanis, D.; Auh, S. Elevated Global SUMOylation in UBC9 Transgenic Mice Protects Their Brains against Focal Cerebral Ischemic Damage. PLoS ONE 2011, 6, e25852. [Google Scholar] [CrossRef] [PubMed]
- Pap, T.; Korb-Pap, A. Cartilage damage in osteoarthritis and rheumatoid arthritis--two unequal siblings. Nat. Rev. Rheumatol. 2015, 11, 606–615. [Google Scholar] [CrossRef]
- Li, F.; Li, X.; Kou, L.; Li, Y.; Meng, F.; Ma, F. SUMO-conjugating enzyme UBC9 promotes proliferation and migration of fibroblast-like synoviocytes in rheumatoid arthritis. Inflammation 2014, 37, 1134–1141. [Google Scholar] [CrossRef]
- Lavoie, J.-F.; Picard, C.; Moreau, A. La Prohibitine: Un nouveau bio-marqueur sanguin pour le diagnostic de l’arthrose [Prohibitin: Novel blood-based biomarker for the diagnosis of osteoarthritis]. Méd./Sci. Am. 2012, 1, 8–14. [Google Scholar]
- Altman, R.D.; Bloch, D.A.; Bole, G.G.; Brandt, K.D.; Cooke, D.V.; Greenwald, R.A.; Hochberg, M.C.; Howell, D.S.; Kaplan, D.; Koopman, W.J. Development of clinical criteria for osteoarthritis. J. Rheumatol. 1987, 14, 3–6. [Google Scholar]
- Finger, F.; Schörle, C.; Zien, A.; Gebhard, P.; Goldring, M.B.; Aigner, T. Molecular phenotyping of human chondrocyte cell lines T/C-28a2, T/C-28a4, and C-28/I2. Arthritis Rheum. 2003, 48, 3395–3403. [Google Scholar] [CrossRef]
- Calabrese, V.; Mallette, F.A.; Deschênes-Simard, X.; Ramanathan, S.; Gagnon, J.; Moores, A.; Ilangumaran, S.; Ferbeyre, G. SOCS1 Links Cytokine Signaling to p53 and Senescence. Mol. Cell 2009, 36, 754–767. [Google Scholar] [CrossRef]
- Ren, J.; Gao, X.; Jin, C.; Zhu, M.; Wang, X.; Shaw, A.; Wen, L.; Yao, X.; Xue, Y. Systematic study of protein SUMOylation: Development of a site-specific predictor of SUMOsp 2.0. Proteimics 2009, 9, 3409–3412. [Google Scholar] [CrossRef]
- Zhao, Q.; Xie, Y.; Zheng, Y.; Jiang, S.; Liu, W.; Mu, W.; Liu, Z.; Zhao, Y.; Xue, Y.; Ren, J. GPS-SUMO: A tool for the prediction of SUMOylation sites and SUMO-interaction motifs. Nucleic Acids Res. 2014, 42, W325–W330. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| OA | Matched Non-OA Controls (Trauma Cases) | |
|---|---|---|
| N | 27 | 4 |
| Women/men | 17/10 | 2/2 |
| Age | 66 ± 2.8 | 43 ± 13.7 |
| Characteristic | UBC9 Group (n = 12) | C57BL/6/Wild Type Group (n = 12) |
|---|---|---|
| Average Age (Weeks) | 38 ± 1.1 | 33 ± 2.4 |
| Sex Ratio (M:F) | 6:6 | 6:6 |
| Average Weight (g) | 30 ± 2.1 | 33 ± 2.3 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Doucet, R.; Elseoudi, A.; Rostami-Afshari, B.; Elbakry, M.; Taheri, M.; Pellicelli, M.; Picard, C.; Lavoie, J.-F.; Wang, D.S.; Lavigne, P.; et al. UBC9-Mediated SUMO Pathway Drives Prohibitin-1 Nuclear Accumulation and PITX1 Repression in Primary Osteoarthritis. Int. J. Mol. Sci. 2025, 26, 6281. https://doi.org/10.3390/ijms26136281
Doucet R, Elseoudi A, Rostami-Afshari B, Elbakry M, Taheri M, Pellicelli M, Picard C, Lavoie J-F, Wang DS, Lavigne P, et al. UBC9-Mediated SUMO Pathway Drives Prohibitin-1 Nuclear Accumulation and PITX1 Repression in Primary Osteoarthritis. International Journal of Molecular Sciences. 2025; 26(13):6281. https://doi.org/10.3390/ijms26136281
Chicago/Turabian StyleDoucet, Roxanne, Abdellatif Elseoudi, Bita Rostami-Afshari, Mohamed Elbakry, Maryam Taheri, Martin Pellicelli, Cynthia Picard, Jean-François Lavoie, Da Shen Wang, Patrick Lavigne, and et al. 2025. "UBC9-Mediated SUMO Pathway Drives Prohibitin-1 Nuclear Accumulation and PITX1 Repression in Primary Osteoarthritis" International Journal of Molecular Sciences 26, no. 13: 6281. https://doi.org/10.3390/ijms26136281
APA StyleDoucet, R., Elseoudi, A., Rostami-Afshari, B., Elbakry, M., Taheri, M., Pellicelli, M., Picard, C., Lavoie, J.-F., Wang, D. S., Lavigne, P., Gorman, K. F., Elremaly, W., & Moreau, A. (2025). UBC9-Mediated SUMO Pathway Drives Prohibitin-1 Nuclear Accumulation and PITX1 Repression in Primary Osteoarthritis. International Journal of Molecular Sciences, 26(13), 6281. https://doi.org/10.3390/ijms26136281