
S6K2 in Focus: Signaling Pathways, Post-Translational Modifications, and Computational Analysis

 ,
,  ,
,  , , , and
, , , and 
Abstract
1. Introduction
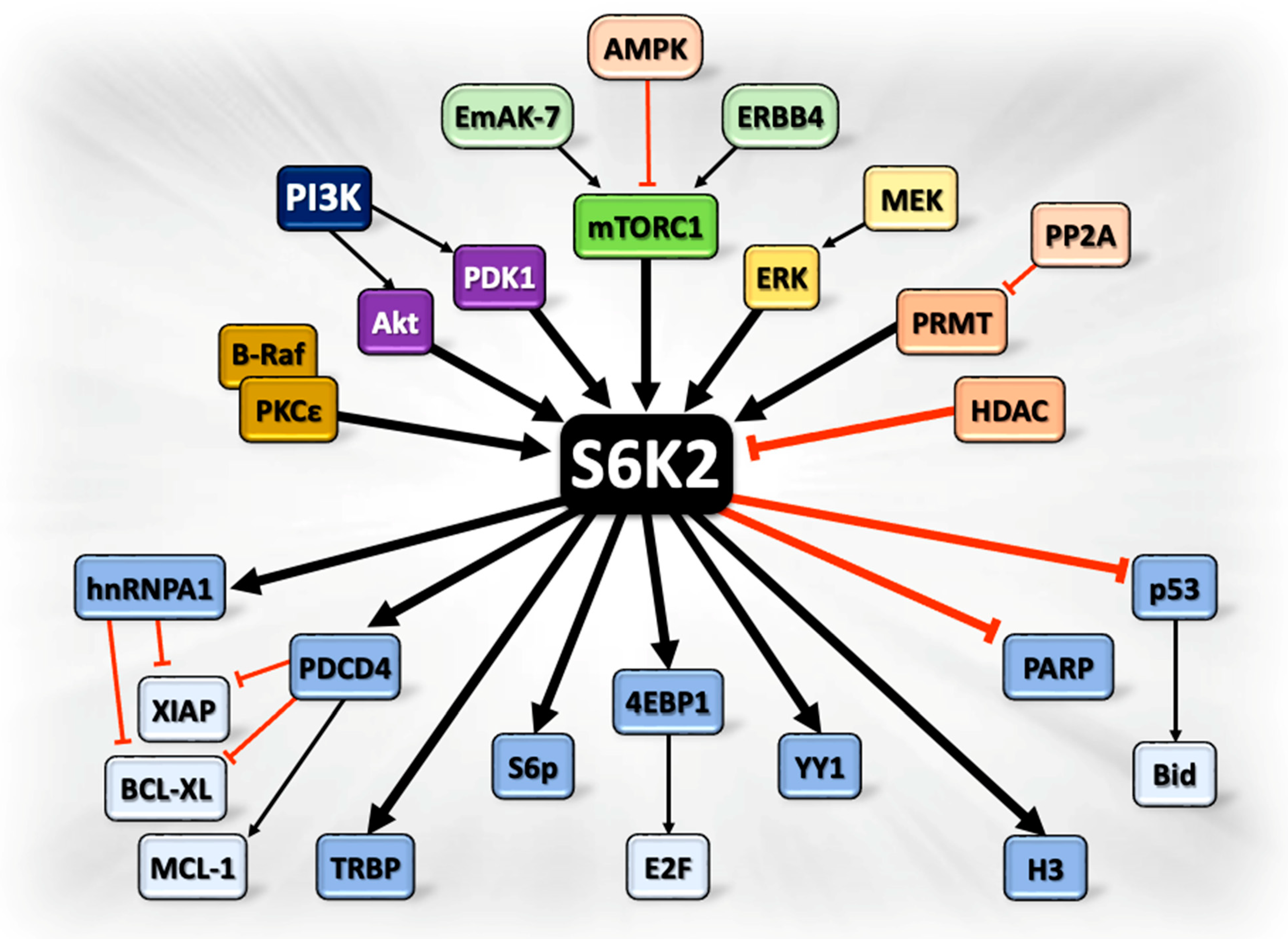
2. S6K2 and Cancer Progression: Signaling Pathways at a Glance
2.1. Upstream Cancer-Promoting Regulators of S6K2
2.1.1. mTORC1 Signaling Pathway and S6K2
2.1.2. Regulation of S6K2 Activity by Phosphoinositide-Dependent Kinase-1 (PDK1)
2.1.3. Regulation of S6K2 Signaling by Akt
2.1.4. Fibroblast Growth Factor (FGF2)-S6K2 Signaling Pathway
2.1.5. Inhibition of S6K2 Activity by Histone Deacetylase (HDAC)
2.1.6. Regulation of S6K2 by Protein Arginine Methyltransferases (PRMTs)
2.2. Downstream Effectors of S6K2 Promoting Cancer Progression
2.2.1. Regulation of the Activity of Ribosomal S6 Protein by S6K2
2.2.2. Regulation of Histone H3 Phosphorylation by S6K2 and Associated Cellular Functions
2.2.3. S6K2-Mediated Phosphorylation of Heterogeneous Nuclear Ribonucleoprotein A1 (hnRNPA1)
2.2.4. Phosphorylation of PDCD4 and Upregulation of Anti-Apoptotic Proteins by S6K2
2.2.5. S6K2 Controls the Transactivation Response RNA-Binding Protein (TRBP) Through mTOR Pathway
2.2.6. Regulation of the Transcription Factor YY1 by S6K2
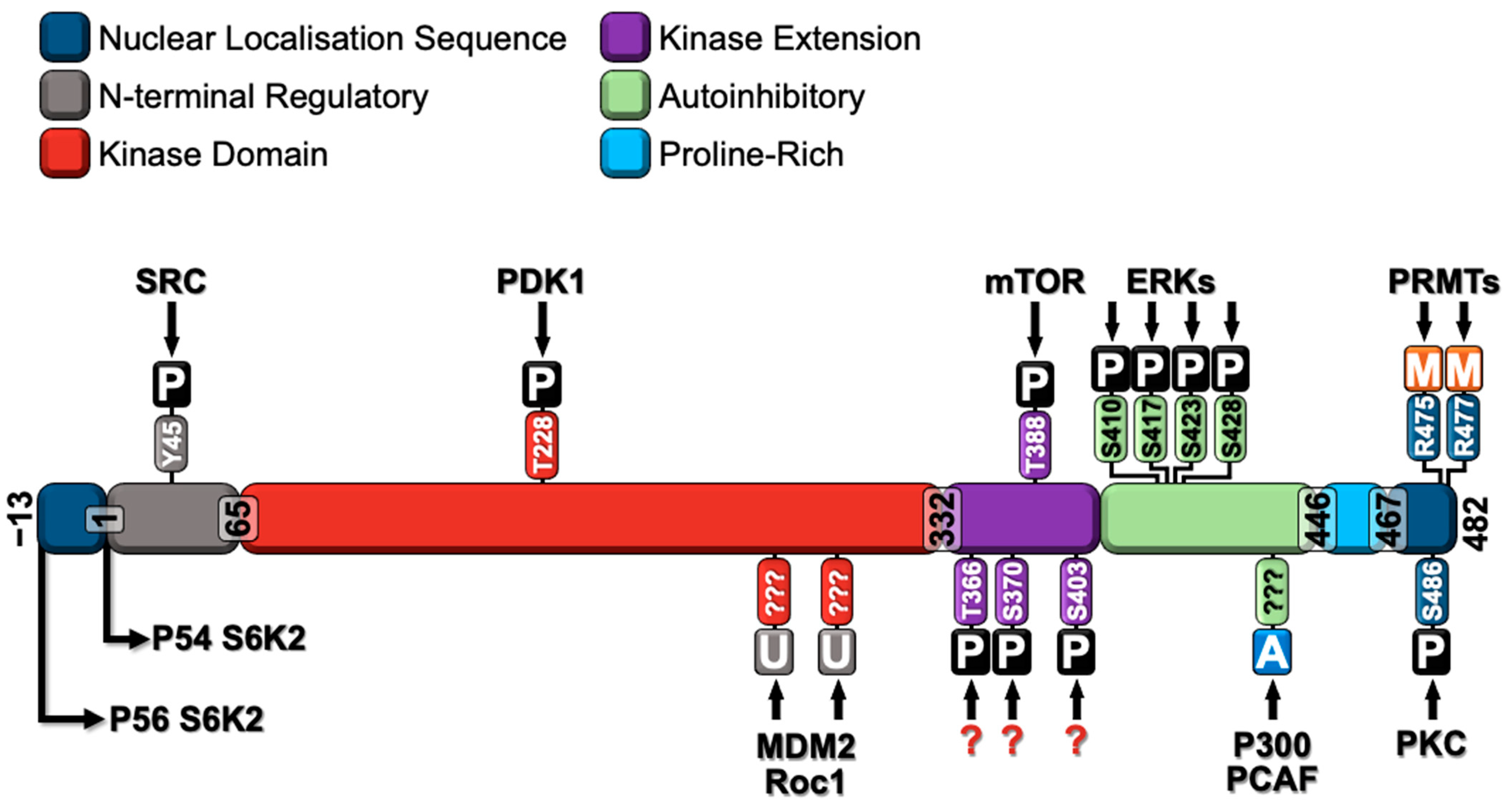
3. Major Post-Translational Modifications of S6K2
3.1. S6K2 Phosphorylation
3.2. S6K2 Methylation
3.3. S6K2 Acetylation
3.4. S6K2 Ubiquitination
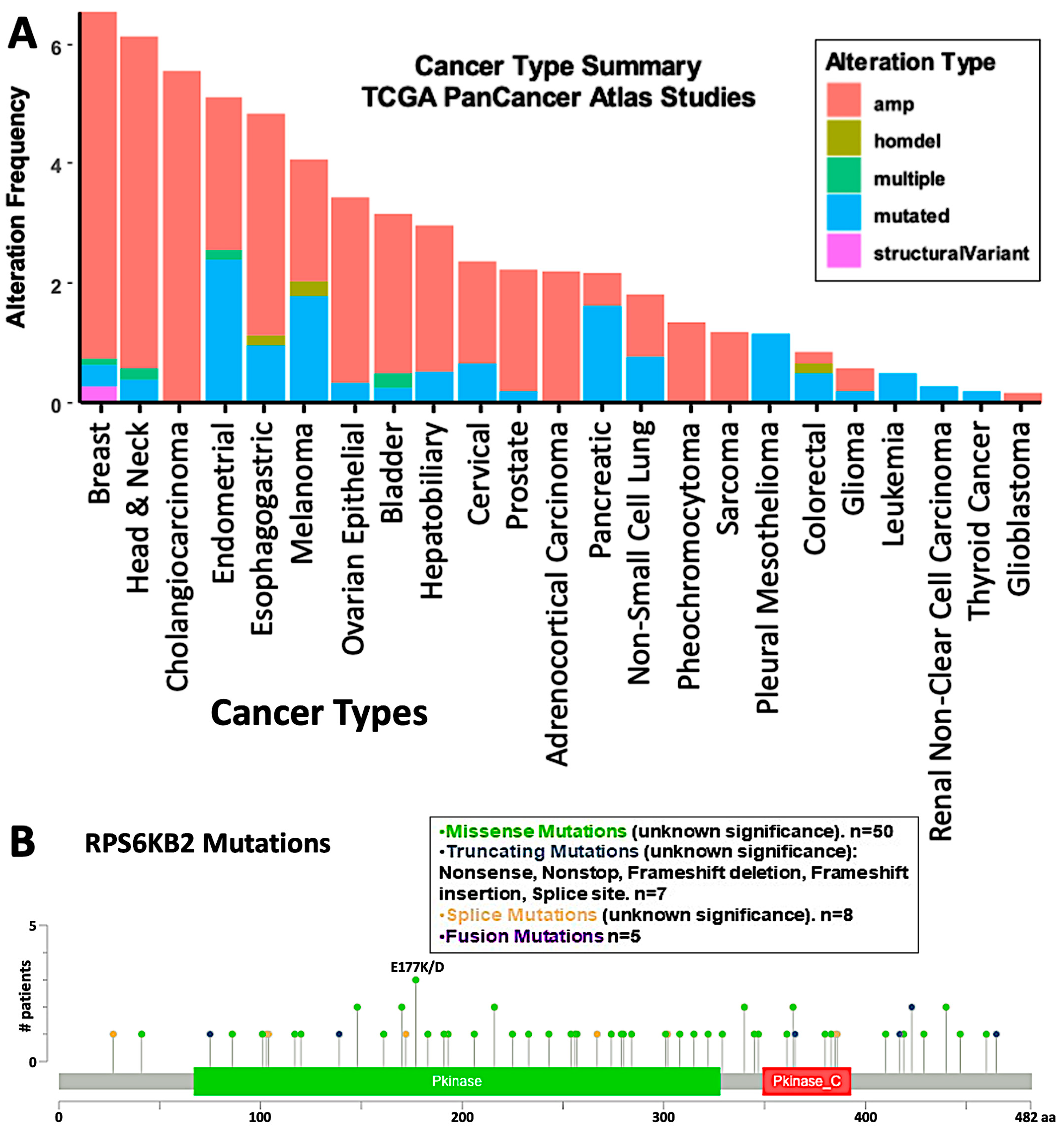
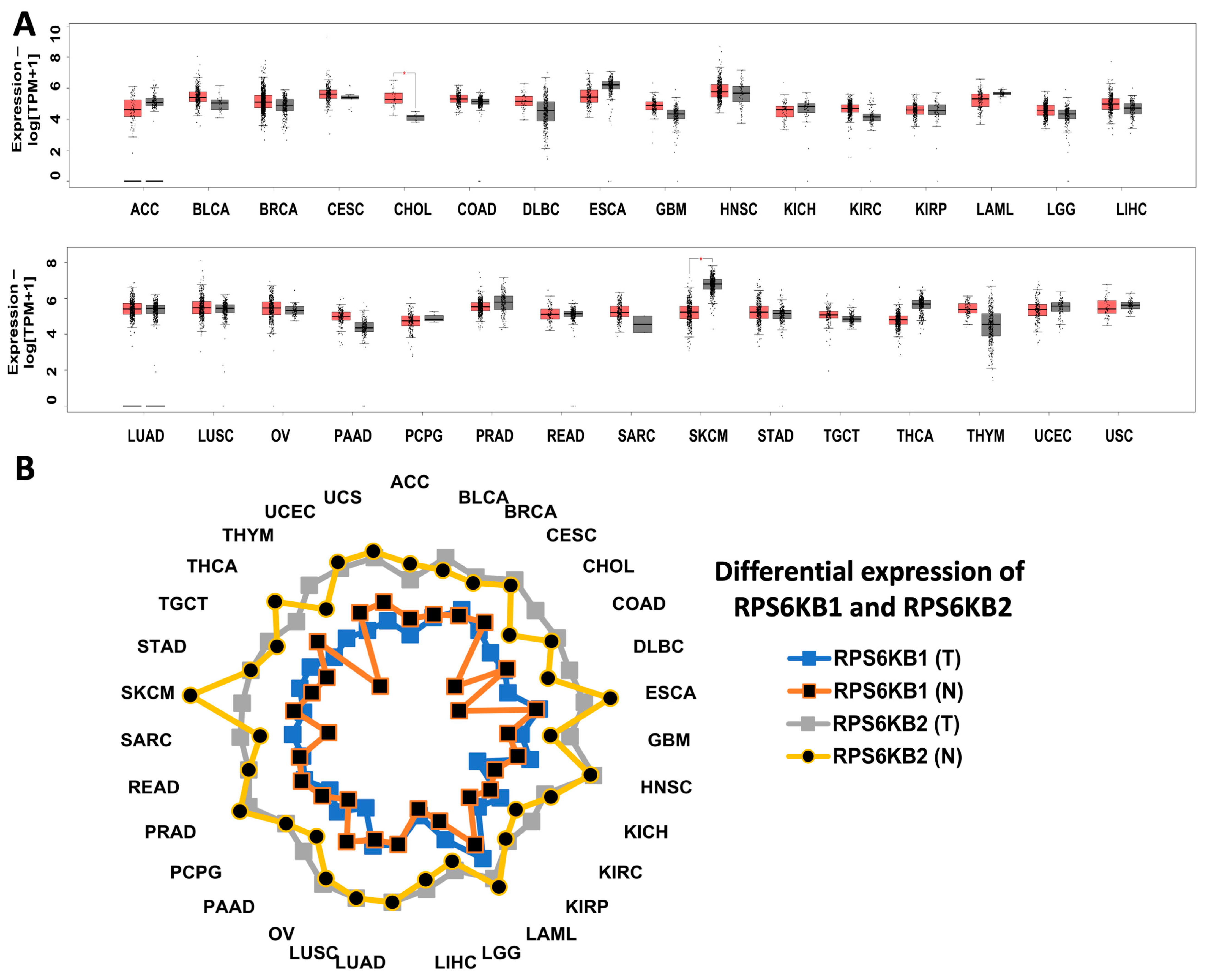
4. Inhibition of S6K2 in Cancer
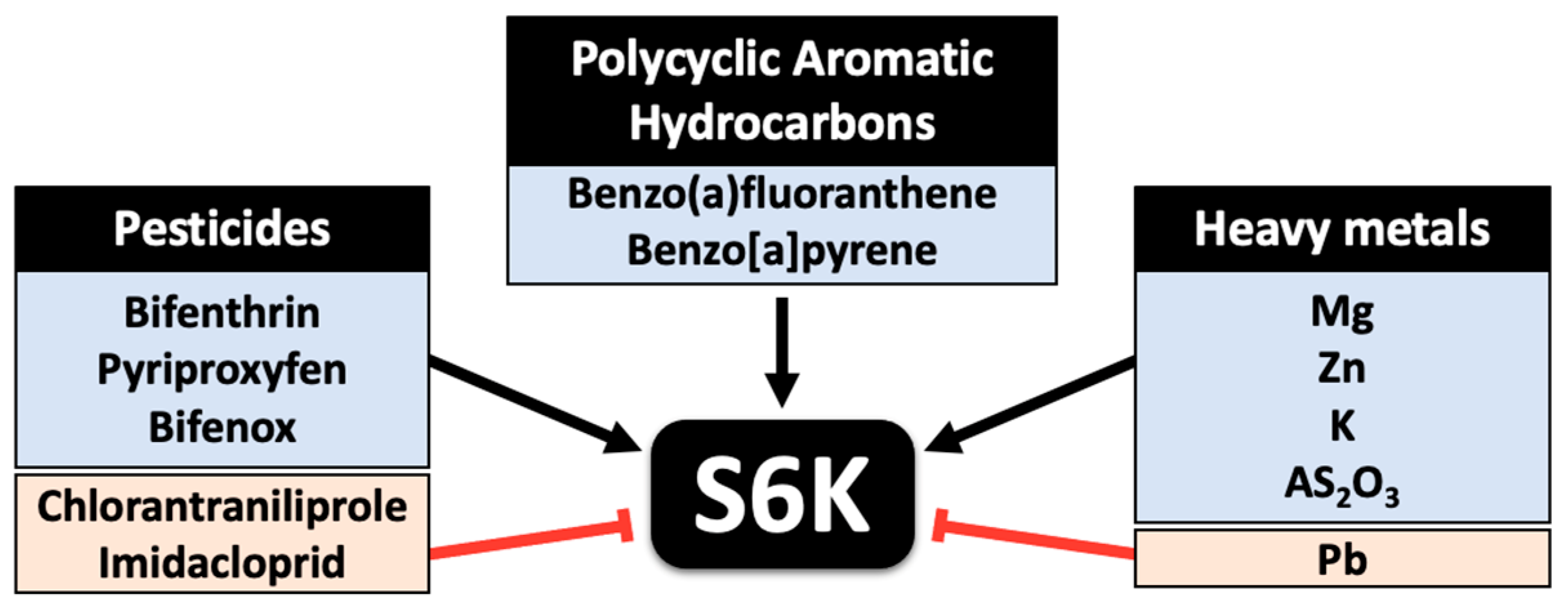
5. Impact of Environmental Contaminants on Ribosomal Protein Kinase Activity
6. Homology Modeling and Dynamics Simulations of S6K2 Protein
7. Structure-Based Design of Ribosomal S6 Kinase Beta 2 (S6K2) Inhibitors
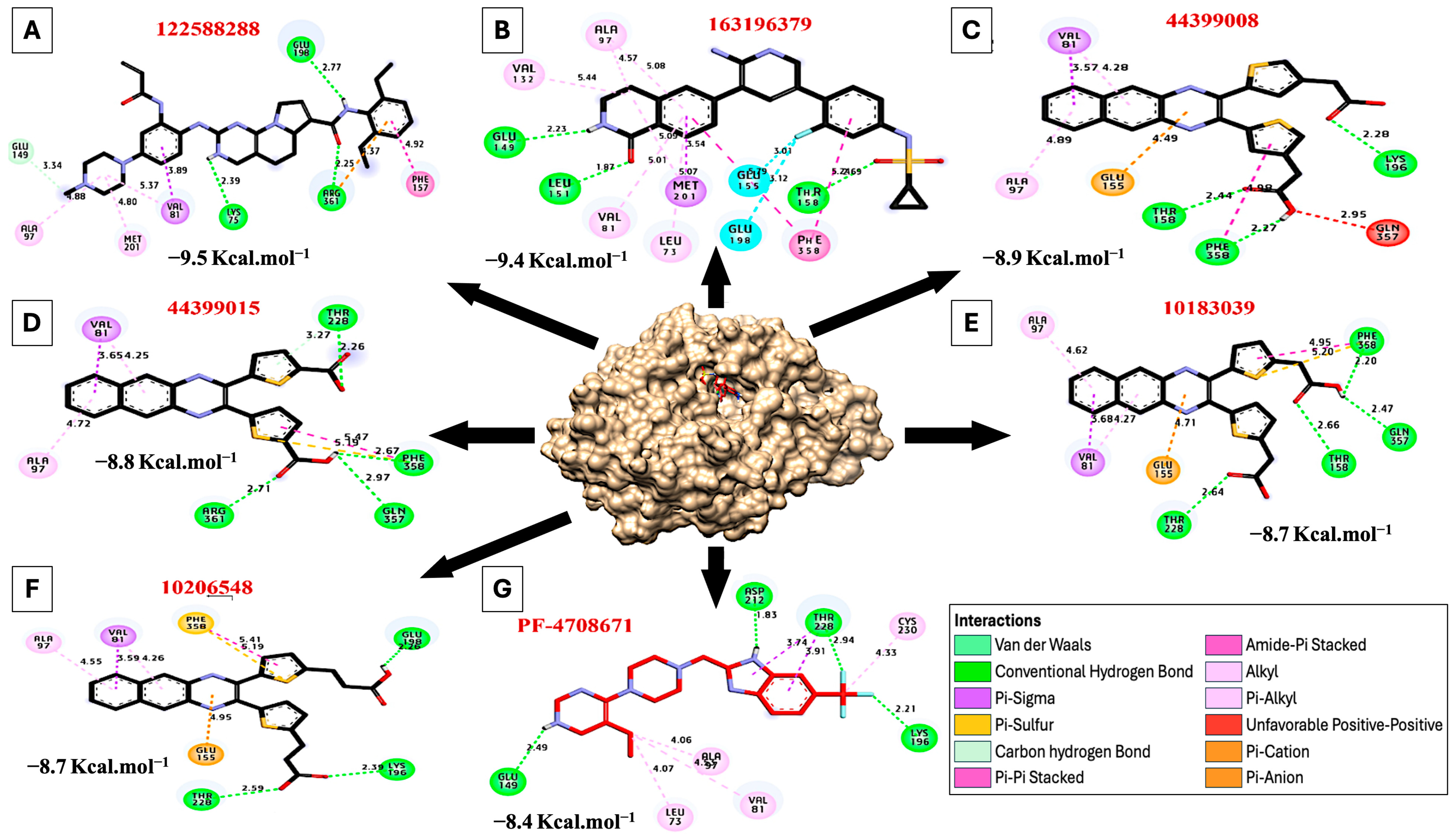
7.1. Molecular Docking of S6K2 Protein with Active Compounds from the PubChem Database
7.2. Molecular Docking of S6K2 Protein with Environmental Contaminants
8. Conclusions
9. Future Perspectives
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Gerstenecker, S.; Haarer, L.; Schroder, M.; Kudolo, M.; Schwalm, M.P.; Wydra, V.; Serafim, R.A.M.; Chaikuad, A.; Knapp, S.; Laufer, S.; et al. Discovery of a Potent and Highly Isoform-Selective Inhibitor of the Neglected Ribosomal Protein S6 Kinase Beta 2 (S6K2). Cancers 2021, 13, 5133. [Google Scholar] [CrossRef] [PubMed]
- Sridharan, S.; Basu, A. Distinct Roles of mTOR Targets S6K1 and S6K2 in Breast Cancer. Int. J. Mol. Sci. 2020, 21, 1199. [Google Scholar] [CrossRef]
- Lyzogubov, V.V.; Lytvyn, D.I.; Dudchenko, T.M.; Lubchenko, N.V.; Pogrybniy, P.V.; Nespryadko, S.V.; Vinnitska, A.B.; Usenko, V.S.; Gout, I.T.; Filonenko, V.V. Immunohistochemical analysis of S6K1 and S6K2 expression in endometrial adenocarcinomas. Exp. Oncol. 2004, 26, 287–293. [Google Scholar] [PubMed]
- Pavan, I.C.; Yokoo, S.; Granato, D.C.; Meneguello, L.; Carnielli, C.M.; Tavares, M.R.; do Amaral, C.L.; de Freitas, L.B.; Paes Leme, A.F.; Luchessi, A.D.; et al. Different interactomes for p70-S6K1 and p54-S6K2 revealed by proteomic analysis. Proteomics 2016, 16, 2650–2666. [Google Scholar] [CrossRef]
- Karlsson, E.; Magic, I.; Bostner, J.; Dyrager, C.; Lysholm, F.; Hallbeck, A.L.; Stal, O.; Lundstrom, P. Revealing Different Roles of the mTOR-Targets S6K1 and S6K2 in Breast Cancer by Expression Profiling and Structural Analysis. PLoS ONE 2015, 10, e0145013. [Google Scholar] [CrossRef]
- Gout, I.; Minami, T.; Hara, K.; Tsujishita, Y.; Filonenko, V.; Waterfield, M.D.; Yonezawa, K. Molecular cloning and characterization of a novel p70 S6 kinase, p70 S6 kinase β containing a proline-rich region. J. Biol. Chem. 1998, 273, 30061–30064. [Google Scholar] [CrossRef]
- Saitoh, M.; ten Dijke, P.; Miyazono, K.; Ichijo, H. Cloning and characterization of p70S6KβDefines a novel family of p70 S6 kinases. Biochem. Biophys. Res. Commun. 1998, 253, 470–476. [Google Scholar] [CrossRef] [PubMed]
- Lee-Fruman, K.K.; Kuo, C.J.; Lippincott, J.; Terada, N.; Blenis, J. Characterization of S6K2, a novel kinase homologous to S6K1. Oncogene 1999, 18, 5108–5114. [Google Scholar] [CrossRef]
- Martin, K.A.; Schalm, S.S.; Richardson, C.; Romanelli, A.; Keon, K.L.; Blenis, J. Regulation of ribosomal S6 kinase 2 by effectors of the phosphoinositide 3-kinase pathway. J. Biol. Chem. 2001, 276, 7884–7891. [Google Scholar] [CrossRef] [PubMed]
- Martin, K.A.; Schalm, S.S.; Romanelli, A.; Keon, K.L.; Blenis, J. Ribosomal S6 kinase 2 inhibition by a potent C-terminal repressor domain is relieved by mitogen-activated protein-extracellular signal-regulated kinase kinase-regulated phosphorylation. J. Biol. Chem. 2001, 276, 7892–7898. [Google Scholar] [CrossRef]
- Park, I.-H.; Bachmann, R.; Shirazi, H.; Chen, J. Regulation of ribosomal S6 kinase 2 by mammalian target of rapamycin. J. Biol. Chem. 2002, 277, 31423–31429. [Google Scholar] [CrossRef]
- Filonenko, V.V.; Tytarenko, R.; Azatjan, S.K.; Savinska, L.O.; Gaydar, Y.A.; Gout, I.T.; Usenko, V.S.; Lyzogubov, V.V. Immunohistochemical analysis of S6K1 and S6K2 localization in human breast tumors. Exp. Oncol. 2004, 26, 294–299. [Google Scholar] [PubMed]
- Cruz, R.; Hedden, L.; Boyer, D.; Kharas, M.G.; Fruman, D.A.; Lee-Fruman, K.K. S6 kinase 2 potentiates interleukin-3-driven cell proliferation. J. Leukoc. Biol. 2005, 78, 1378–1385. [Google Scholar] [CrossRef] [PubMed]
- Valovka, T.; Verdier, F.; Cramer, R.; Zhyvoloup, A.; Fenton, T.R.; Rebholz, H.; Wang, M.-L.; Gzhegotsky, M.; Lutsyk, A.D.; Matsuka, G.; et al. Protein Kinase C Phosphorylates Ribosomal Protein S6 Kinase βII and Regulates Its Subcellular Localization. Mol. Cell. Biol. 2003, 23, 852–863. [Google Scholar] [CrossRef] [PubMed]
- Pardo, O.E.; Wellbrock, C.; Khanzada, U.K.; Aubert, M.; Arozarena, I.; Davidson, S.; Bowen, F.; Parker, P.J.; Filonenko, V.V.; Gout, I.T.; et al. FGF-2 protects small cell lung cancer cells from apoptosis through a complex involving PKCepsilon, B-Raf and S6K2. EMBO J. 2006, 25, 3078–3088. [Google Scholar] [CrossRef] [PubMed]
- Rossi, R.; Pester, J.M.; McDowell, M.; Soza, S.; Montecucco, A.; Lee-Fruman, K.K. Identification of S6K2 as a centrosome-located kinase. FEBS Lett. 2007, 581, 4058–4064. [Google Scholar] [CrossRef] [PubMed]
- Boyer, D.; Quintanilla, R.; Lee-Fruman, K.K. Regulation of catalytic activity of S6 kinase 2 during cell cycle. Mol. Cell. Biochem. 2008, 307, 59–64. [Google Scholar] [CrossRef]
- Fenton, T.R.; Gwalter, J.; Ericsson, J.; Gout, I.T. Histone acetyltransferases interact with and acetylate p70 ribosomal S6 kinases in vitro and in vivo. Int. J. Biochem. Cell Biol. 2010, 42, 359–366. [Google Scholar] [CrossRef]
- Goh, E.T.; Pardo, O.E.; Michael, N.; Niewiarowski, A.; Totty, N.; Volkova, D.; Tsaneva, I.R.; Seckl, M.J.; Gout, I. Involvement of heterogeneous ribonucleoprotein F in the regulation of cell proliferation via the mammalian target of rapamycin/S6 kinase 2 pathway. J. Biol. Chem. 2010, 285, 17065–17076. [Google Scholar] [CrossRef]
- Sridharan, S.; Basu, A. S6 kinase 2 promotes breast cancer cell survival via Akt. Cancer Res. 2011, 71, 2590–2599. [Google Scholar] [CrossRef]
- Savinska, L.; Skorokhod, O.; Klipa, O.; Gout, I.; Filonenko, V. Development of monoclonal antibodies specific to ribosomal protein S6 kinase 2. Hybridoma 2012, 31, 289–294. [Google Scholar] [CrossRef] [PubMed]
- Liwak, U.; Thakor, N.; Jordan, L.E.; Roy, R.; Lewis, S.M.; Pardo, O.E.; Seckl, M.; Holcik, M. Tumor suppressor PDCD4 represses internal ribosome entry site-mediated translation of antiapoptotic proteins and is regulated by S6 kinase 2. Mol. Cell Biol. 2012, 32, 1818–1829. [Google Scholar] [CrossRef] [PubMed]
- Kurebayashi, Y.; Nagai, S.; Ikejiri, A.; Ohtani, M.; Ichiyama, K.; Baba, Y.; Yamada, T.; Egami, S.; Hoshii, T.; Hirao, A. PI3K-Akt-mTORC1-S6K1/2 axis controls Th17 differentiation by regulating Gfi1 expression and nuclear translocation of RORγ. Cell Rep. 2012, 1, 360–373. [Google Scholar] [CrossRef] [PubMed]
- Karlsson, E.; Perez-Tenorio, G.; Amin, R.; Bostner, J.; Skoog, L.; Fornander, T.; Sgroi, D.C.; Nordenskjold, B.; Hallbeck, A.L.; Stal, O. The mTOR effectors 4EBP1 and S6K2 are frequently coexpressed, and associated with a poor prognosis and endocrine resistance in breast cancer: A retrospective study including patients from the randomised Stockholm tamoxifen trials. Breast Cancer Res. 2013, 15, R96. [Google Scholar] [CrossRef] [PubMed]
- Roy, R.; Durie, D.; Li, H.; Liu, B.Q.; Skehel, J.M.; Mauri, F.; Cuorvo, L.V.; Barbareschi, M.; Guo, L.; Holcik, M.; et al. hnRNPA1 couples nuclear export and translation of specific mRNAs downstream of FGF-2/S6K2 signalling. Nucleic Acids Res. 2014, 42, 12483–12497. [Google Scholar] [CrossRef] [PubMed]
- Bostner, J.; Karlsson, E.; Eding, C.B.; Perez-Tenorio, G.; Franzén, H.; Konstantinell, A.; Fornander, T.; Nordenskjöld, B.; Stål, O. S6 kinase signaling: Tamoxifen response and prognostic indication in two breast cancer cohorts. Endocr. Relat. Cancer 2015, 22, 331–343. [Google Scholar] [CrossRef]
- Amaral, C.L.; Freitas, L.B.; Tamura, R.E.; Tavares, M.R.; Pavan, I.C.; Bajgelman, M.C.; Simabuco, F.M. S6Ks isoforms contribute to viability, migration, docetaxel resistance and tumor formation of prostate cancer cells. BMC Cancer 2016, 16, 602. [Google Scholar] [CrossRef]
- Sun, Y.; Luo, M.; Chang, G.; Ren, W.; Wu, K.; Li, X.; Shen, J.; Zhao, X.; Hu, Y. Phosphorylation of Ser6 in hnRNPA1 by S6K2 regulates glucose metabolism and cell growth in colorectal cancer. Oncol. Lett. 2017, 14, 7323–7331. [Google Scholar] [CrossRef] [PubMed]
- Basu, A.; Sridharan, S. Regulation of anti-apoptotic Bcl-2 family protein Mcl-1 by S6 kinase 2. PLoS ONE 2017, 12, e0173854. [Google Scholar] [CrossRef] [PubMed]
- Sfakianos, A.P.; Mellor, L.E.; Pang, Y.F.; Kritsiligkou, P.; Needs, H.; Abou-Hamdan, H.; Desaubry, L.; Poulin, G.B.; Ashe, M.P.; Whitmarsh, A.J. Correction to: The mTOR-S6 kinase pathway promotes stress granule assembly. Cell Death Differ. 2020, 27, 1744. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, J.T.; Haidar, F.S.; Fox, A.L.; Ray, C.; Mendonca, D.B.; Kim, J.K.; Krebsbach, P.H. mEAK-7 Forms an Alternative mTOR Complex with DNA-PKcs in Human Cancer. iScience 2019, 17, 190–207. [Google Scholar] [CrossRef]
- Tavares, M.R.; Lemes, S.F.; de Fante, T.; de Miera, C.S.; Pavan, I.C.B.; Bezerra, R.M.N.; Prada, P.O.; Torsoni, M.A.; Elias, C.F.; Simabuco, F.M. Modulation of hypothalamic S6K1 and S6K2 alters feeding behavior and systemic glucose metabolism. J. Endocrinol. 2020, 244, 71–82. [Google Scholar] [CrossRef] [PubMed]
- Khalil, M.I.; Ismail, H.M.; Panasyuk, G.; Bdzhola, A.; Filonenko, V.; Gout, I.; Pardo, O.E. Asymmetric Dimethylation of Ribosomal S6 Kinase 2 Regulates Its Cellular Localisation and Pro-Survival Function. Int. J. Mol. Sci. 2023, 24, 8806. [Google Scholar] [CrossRef]
- Saxton, R.A.; Sabatini, D.M. mTOR Signaling in Growth, Metabolism, and Disease. Cell 2017, 169, 361–371. [Google Scholar] [CrossRef] [PubMed]
- Aleksandrova, K.V.; Vorobev, M.L.; Suvorova, I.I. mTOR pathway occupies a central role in the emergence of latent cancer cells. Cell Death Dis. 2024, 15, 176. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Manning, B.D. The TSC1-TSC2 complex: A molecular switchboard controlling cell growth. Biochem. J. 2008, 412, 179–190. [Google Scholar] [CrossRef]
- Khalil, M.; Gout, I. Overexpression or downregulation of mTOR in mammalian cells. Methods Mol. Biol. 2012, 821, 87–103. [Google Scholar] [CrossRef]
- Ismail, H.M.; Hurd, P.J.; Khalil, M.I.; Kouzarides, T.; Bannister, A.; Gout, I. S6 kinase 2 is bound to chromatin-nuclear matrix cellular fractions and is able to phosphorylate histone H3 at threonine 45 in vitro and in vivo. J. Cell Biochem. 2014, 115, 1048–1062. [Google Scholar] [CrossRef]
- Nguyen, J.T.; Ray, C.; Fox, A.L.; Mendonca, D.B.; Kim, J.K.; Krebsbach, P.H. Mammalian EAK-7 activates alternative mTOR signaling to regulate cell proliferation and migration. Sci. Adv. 2018, 4, eaao5838. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Lin, Y.; Jin, X.; Zhang, W.; Guo, W.; Chen, L.; Chen, M.; Li, Y.; Fu, F.; Wang, C. Integrative proteomic and phosphoproteomic profiling of invasive micropapillary breast carcinoma. J. Proteom. 2022, 257, 104511. [Google Scholar] [CrossRef]
- Tavares, M.R.; Pavan, I.C.; Amaral, C.L.; Meneguello, L.; Luchessi, A.D.; Simabuco, F.M. The S6K protein family in health and disease. Life Sci. 2015, 131, 1–10. [Google Scholar] [CrossRef]
- Perez-Tenorio, G.; Karlsson, E.; Waltersson, M.A.; Olsson, B.; Holmlund, B.; Nordenskjold, B.; Fornander, T.; Skoog, L.; Stal, O. Clinical potential of the mTOR targets S6K1 and S6K2 in breast cancer. Breast Cancer Res. Treat. 2011, 128, 713–723. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Qian, X.; Zhu, M.; Li, A.; Fang, M.; Zhu, Y.; Zhang, J. miR-1273g-3p promotes proliferation, migration and invasion of LoVo cells via cannabinoid receptor 1 through activation of ERBB4/PIK3R3/mTOR/S6K2 signaling pathway. Mol. Med. Rep. 2018, 17, 4619–4626. [Google Scholar] [CrossRef]
- Schwaederle, M.; Daniels, G.A.; Piccioni, D.E.; Fanta, P.T.; Schwab, R.B.; Shimabukuro, K.A.; Parker, B.A.; Kurzrock, R. Cyclin alterations in diverse cancers: Outcome and co-amplification network. Oncotarget 2015, 6, 3033–3042. [Google Scholar] [CrossRef]
- Thedieck, K.; Holzwarth, B.; Prentzell, M.T.; Boehlke, C.; Klasener, K.; Ruf, S.; Sonntag, A.G.; Maerz, L.; Grellscheid, S.N.; Kremmer, E.; et al. Inhibition of mTORC1 by astrin and stress granules prevents apoptosis in cancer cells. Cell 2013, 154, 859–874. [Google Scholar] [CrossRef]
- Riou, P.; Saffroy, R.; Comoy, J.; Gross-Goupil, M.; Thiery, J.P.; Emile, J.F.; Azoulay, D.; Piatier-Tonneau, D.; Lemoine, A.; Debuire, B. Investigation in liver tissues and cell lines of the transcription of 13 genes mapping to the 16q24 region that are frequently deleted in hepatocellular carcinoma. Clin. Cancer Res. 2002, 8, 3178–3186. [Google Scholar] [PubMed]
- Ellsworth, R.E.; Field, L.A.; Love, B.; Kane, J.L.; Hooke, J.A.; Shriver, C.D. Differential gene expression in primary breast tumors associated with lymph node metastasis. Int. J. Breast Cancer 2011, 2011, 142763. [Google Scholar] [CrossRef]
- Zhang, W.; Liu, H.T. MAPK signal pathways in the regulation of cell proliferation in mammalian cells. Cell Res. 2002, 12, 9–18. [Google Scholar] [CrossRef]
- Armengol, G.; Rojo, F.; Castellvi, J.; Iglesias, C.; Cuatrecasas, M.; Pons, B.; Baselga, J.; Ramon y Cajal, S. 4E-binding protein 1: A key molecular “funnel factor” in human cancer with clinical implications. Cancer Res. 2007, 67, 7551–7555. [Google Scholar] [CrossRef]
- Magnuson, B.; Ekim, B.; Fingar, D.C. Regulation and function of ribosomal protein S6 kinase (S6K) within mTOR signalling networks. Biochem. J. 2012, 441, 1–21. [Google Scholar] [CrossRef]
- Phin, S.; Kupferwasser, D.; Lam, J.; Lee-Fruman, K.K. Mutational analysis of ribosomal S6 kinase 2 shows differential regulation of its kinase activity from that of ribosomal S6 kinase 1. Biochem. J. 2003, 373, 583–591. [Google Scholar] [CrossRef]
- Song, G.; Ouyang, G.; Bao, S. The activation of Akt/PKB signaling pathway and cell survival. J. Cell Mol. Med. 2005, 9, 59–71. [Google Scholar] [CrossRef] [PubMed]
- Shankar, E.; Sivaprasad, U.; Basu, A. Protein kinase C epsilon confers resistance of MCF-7 cells to TRAIL by Akt-dependent activation of Hdm2 and downregulation of p53. Oncogene 2008, 27, 3957–3966. [Google Scholar] [CrossRef]
- Alama, A.; Orengo, A.M.; Ferrini, S.; Gangemi, R. Targeting cancer-initiating cell drug-resistance: A roadmap to a new-generation of cancer therapies? Drug Discov. Today 2012, 17, 435–442. [Google Scholar] [CrossRef] [PubMed]
- Pardo, O.E.; Lesay, A.; Arcaro, A.; Lopes, R.; Ng, B.L.; Warne, P.H.; McNeish, I.A.; Tetley, T.D.; Lemoine, N.R.; Mehmet, H.; et al. Fibroblast growth factor 2-mediated translational control of IAPs blocks mitochondrial release of Smac/DIABLO and apoptosis in small cell lung cancer cells. Mol. Cell Biol. 2003, 23, 7600–7610. [Google Scholar] [CrossRef] [PubMed]
- Pardo, O.E.; Seckl, M.J. S6K2: The Neglected S6 Kinase Family Member. Front. Oncol. 2013, 3, 191. [Google Scholar] [CrossRef] [PubMed]
- Jarrold, J.; Davies, C.C. PRMTs and Arginine Methylation: Cancer’s Best-Kept Secret? Trends Mol. Med. 2019, 25, 993–1009. [Google Scholar] [CrossRef] [PubMed]
- Ratovitski, T.; Jiang, M.; O’Meally, R.N.; Rauniyar, P.; Chighladze, E.; Farago, A.; Kamath, S.V.; Jin, J.; Shevelkin, A.V.; Cole, R.N.; et al. Interaction of huntingtin with PRMTs and its subsequent arginine methylation affects HTT solubility, phase transition behavior and neuronal toxicity. Hum. Mol. Genet. 2022, 31, 1651–1672. [Google Scholar] [CrossRef] [PubMed]
- Duong, F.H.; Christen, V.; Berke, J.M.; Penna, S.H.; Moradpour, D.; Heim, M.H. Upregulation of protein phosphatase 2Ac by hepatitis C virus modulates NS3 helicase activity through inhibition of protein arginine methyltransferase 1. J. Virol. 2005, 79, 15342–15350. [Google Scholar] [CrossRef]
- Pende, M.; Um, S.H.; Mieulet, V.; Sticker, M.; Goss, V.L.; Mestan, J.; Mueller, M.; Fumagalli, S.; Kozma, S.C.; Thomas, G. S6K1(-/-)/S6K2(-/-) mice exhibit perinatal lethality and rapamycin-sensitive 5’-terminal oligopyrimidine mRNA translation and reveal a mitogen-activated protein kinase-dependent S6 kinase pathway. Mol. Cell Biol. 2004, 24, 3112–3124. [Google Scholar] [CrossRef] [PubMed]
- Atefi, M.; von Euw, E.; Attar, N.; Ng, C.; Chu, C.; Guo, D.; Nazarian, R.; Chmielowski, B.; Glaspy, J.A.; Comin-Anduix, B.; et al. Reversing melanoma cross-resistance to BRAF and MEK inhibitors by co-targeting the AKT/mTOR pathway. PLoS ONE 2011, 6, e28973. [Google Scholar] [CrossRef] [PubMed]
- Qin, J.; Rajaratnam, R.; Feng, L.; Salami, J.; Barber-Rotenberg, J.S.; Domsic, J.; Reyes-Uribe, P.; Liu, H.; Dang, W.; Berger, S.L.; et al. Development of organometallic S6K1 inhibitors. J. Med. Chem. 2015, 58, 305–314. [Google Scholar] [CrossRef] [PubMed]
- Madala, S.K.; Sontake, V.; Edukulla, R.; Davidson, C.R.; Schmidt, S.; Hardie, W.D. Unique and Redundant Functions of p70 Ribosomal S6 Kinase Isoforms Regulate Mesenchymal Cell Proliferation and Migration in Pulmonary Fibrosis. Am. J. Respir. Cell Mol. Biol. 2016, 55, 792–803. [Google Scholar] [CrossRef]
- Juan, G.; Traganos, F.; Darzynkiewicz, Z. Histone H3 phosphorylation in human monocytes and during HL-60 cell differentiation. Exp. Cell Res. 1999, 246, 212–220. [Google Scholar] [CrossRef]
- Carmo, C.R.; Lyons-Lewis, J.; Seckl, M.J.; Costa-Pereira, A.P. A novel requirement for Janus kinases as mediators of drug resistance induced by fibroblast growth factor-2 in human cancer cells. PLoS ONE 2011, 6, e19861. [Google Scholar] [CrossRef] [PubMed]
- Warner, M.J.; Bridge, K.S.; Hewitson, J.P.; Hodgkinson, M.R.; Heyam, A.; Massa, B.C.; Haslam, J.C.; Chatzifrangkeskou, M.; Evans, G.J.; Plevin, M.J.; et al. S6K2-mediated regulation of TRBP as a determinant of miRNA expression in human primary lymphatic endothelial cells. Nucleic Acids Res. 2016, 44, 9942–9955. [Google Scholar] [CrossRef] [PubMed]
- Ismail, H.M.; Myronova, O.; Tsuchiya, Y.; Niewiarowski, A.; Tsaneva, I.; Gout, I. Identification of the general transcription factor Yin Yang 1 as a novel and specific binding partner for S6 Kinase 2. Cell. Signal. 2013, 25, 1054–1063. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Xie, W.; Xie, W.; Wei, W.; Guo, J. Beyond controlling cell size: Functional analyses of S6K in tumorigenesis. Cell Death Dis. 2022, 13, 646. [Google Scholar] [CrossRef] [PubMed]
- Chauvin, C.; Koka, V.; Nouschi, A.; Mieulet, V.; Hoareau-Aveilla, C.; Dreazen, A.; Cagnard, N.; Carpentier, W.; Kiss, T.; Meyuhas, O.; et al. Ribosomal protein S6 kinase activity controls the ribosome biogenesis transcriptional program. Oncogene 2014, 33, 474–483. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.L.; Panasyuk, G.; Gwalter, J.; Nemazanyy, I.; Fenton, T.; Filonenko, V.; Gout, I. Regulation of ribosomal protein S6 kinases by ubiquitination. Biochem. Biophys. Res. Commun. 2008, 369, 382–387. [Google Scholar] [CrossRef] [PubMed]
- Gwalter, J.; Wang, M.L.; Gout, I. The ubiquitination of ribosomal S6 kinases is independent from the mitogen-induced phosphorylation/activation of the kinase. Int. J. Biochem. Cell Biol. 2009, 41, 828–833. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Gout, I.; Proud, C.G. Cross-talk between the ERK and p70 S6 kinase (S6K) signaling pathways. MEK-dependent activation of S6K2 in cardiomyocytes. J. Biol. Chem. 2001, 276, 32670–32677. [Google Scholar] [CrossRef] [PubMed]
- Pardo, O.E.; Arcaro, A.; Salerno, G.; Tetley, T.D.; Valovka, T.; Gout, I.; Seckl, M.J. Novel cross talk between MEK and S6K2 in FGF-2 induced proliferation of SCLC cells. Oncogene 2001, 20, 7658–7667. [Google Scholar] [CrossRef]
- Sato, S.; Fujita, N.; Tsuruo, T. Involvement of 3-phosphoinositide-dependent protein kinase-1 in the MEK/MAPK signal transduction pathway. J. Biol. Chem. 2004, 279, 33759–33767. [Google Scholar] [CrossRef]
- Rebholz, H.; Panasyuk, G.; Fenton, T.; Nemazanyy, I.; Valovka, T.; Flajolet, M.; Ronnstrand, L.; Stephens, L.; West, A.; Gout, I.T. Receptor association and tyrosine phosphorylation of S6 kinases. FEBS J. 2006, 273, 2023–2036. [Google Scholar] [CrossRef]
- Han, Y.; Jin, Y.H.; Kim, Y.J.; Kang, B.Y.; Choi, H.J.; Kim, D.W.; Yeo, C.Y.; Lee, K.Y. Acetylation of Sirt2 by p300 attenuates its deacetylase activity. Biochem. Biophys. Res. Commun. 2008, 375, 576–580. [Google Scholar] [CrossRef] [PubMed]
- Brunet, A.; Sweeney, L.B.; Sturgill, J.F.; Chua, K.F.; Greer, P.L.; Lin, Y.; Tran, H.; Ross, S.E.; Mostoslavsky, R.; Cohen, H.Y. Stress-dependent regulation of FOXO transcription factors by the SIRT1 deacetylase. Science 2004, 303, 2011–2015. [Google Scholar] [CrossRef] [PubMed]
- Blagosklonny, M.V. Cancer prevention with rapamycin. Oncotarget 2023, 14, 342–350. [Google Scholar] [CrossRef]
- Tolcher, A.; Goldman, J.; Patnaik, A.; Papadopoulos, K.P.; Westwood, P.; Kelly, C.S.; Bumgardner, W.; Sams, L.; Geeganage, S.; Wang, T.; et al. A phase I trial of LY2584702 tosylate, a p70 S6 kinase inhibitor, in patients with advanced solid tumours. Eur. J. Cancer 2014, 50, 867–875. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Brown, M.S.; Goldstein, J.L. Bifurcation of insulin signaling pathway in rat liver: mTORC1 required for stimulation of lipogenesis, but not inhibition of gluconeogenesis. Proc. Natl. Acad. Sci. USA 2010, 107, 3441–3446. [Google Scholar] [CrossRef] [PubMed]
- Pearce, L.R.; Alton, G.R.; Richter, D.T.; Kath, J.C.; Lingardo, L.; Chapman, J.; Hwang, C.; Alessi, D.R. Characterization of PF-4708671, a novel and highly specific inhibitor of p70 ribosomal S6 kinase (S6K1). Biochem. J. 2010, 431, 245–255. [Google Scholar] [CrossRef] [PubMed]
- Hong, S.E.; Kim, E.K.; Jin, H.O.; Kim, H.A.; Lee, J.K.; Koh, J.S.; Seol, H.; Kim, J.I.; Park, I.C.; Noh, W.C. S6K1 inhibition enhances tamoxifen-induced cell death in MCF-7 cells through translational inhibition of Mcl-1 and survivin. Cell Biol. Toxicol. 2013, 29, 273–282. [Google Scholar] [CrossRef]
- Choi, H.N.; Jin, H.O.; Kim, J.H.; Hong, S.E.; Kim, H.A.; Kim, E.K.; Lee, J.K.; Park, I.C.; Noh, W.C. Inhibition of S6K1 enhances glucose deprivation-induced cell death via downregulation of anti-apoptotic proteins in MCF-7 breast cancer cells. Biochem. Biophys. Res. Commun. 2013, 432, 123–128. [Google Scholar] [CrossRef] [PubMed]
- Qiu, Z.X.; Sun, R.F.; Mo, X.M.; Li, W.M. The p70S6K Specific Inhibitor PF-4708671 Impedes Non-Small Cell Lung Cancer Growth. PLoS ONE 2016, 11, e0147185. [Google Scholar] [CrossRef] [PubMed]
- Lipchick, B.; Guterres, A.N.; Chen, H.-Y.; Zundell, D.M.; Aguila, S.D.; Reyes-Uribe, P.I.; Basu, S.; Yin, X.; Kossenkov, A.V.; Lu, Y.; et al. Selective abrogation of S6K2 maps lipid homeostasis as a survival vulnerability in MAPKi-resistant NRASmut melanoma. bioRxiv 2024. [Google Scholar] [CrossRef]
- Wu, C.E.; Chen, M.H.; Yeh, C.N. mTOR Inhibitors in Advanced Biliary Tract Cancers. Int. J. Mol. Sci. 2019, 20, 500. [Google Scholar] [CrossRef] [PubMed]
- Patel, J.; Nguyen, S.A.; Ogretmen, B.; Gutkind, J.S.; Nathan, C.A.; Day, T. mTOR inhibitor use in head and neck squamous cell carcinoma: A meta-analysis on survival, tumor response, and toxicity. Laryngoscope Investig. Otolaryngol. 2020, 5, 243–255. [Google Scholar] [CrossRef]
- Kojima, T.; Kato, K.; Hara, H.; Takahashi, S.; Muro, K.; Nishina, T.; Wakabayashi, M.; Nomura, S.; Sato, A.; Ohtsu, A.; et al. Phase II study of BKM120 in patients with advanced esophageal squamous cell carcinoma (EPOC1303). Esophagus 2022, 19, 702–710. [Google Scholar] [CrossRef] [PubMed]
- Massarweh, S.; Romond, E.; Black, E.P.; Van Meter, E.; Shelton, B.; Kadamyan-Melkumian, V.; Stevens, M.; Elledge, R. A phase II study of combined fulvestrant and everolimus in patients with metastatic estrogen receptor (ER)-positive breast cancer after aromatase inhibitor (AI) failure. Breast Cancer Res. Treat. 2014, 143, 325–332. [Google Scholar] [CrossRef] [PubMed]
- Mohamed, H.; Sara, G.; Amany, E.-S. Impact of PCBs, Furan and Dioxin on Hepatocarcinogenesis. In Persistent Organic Pollutants (POPs); Mohamed Nageeb, R., Ed.; IntechOpen: Rijeka, Croatia, 2022; p. Ch. 3. [Google Scholar]
- Boffetta, P.; Nyberg, F. Contribution of environmental factors to cancer risk. Br. Med. Bull. 2003, 68, 71–94. [Google Scholar] [CrossRef]
- Labib, S.; Williams, A.; Guo, C.H.; Leingartner, K.; Arlt, V.M.; Schmeiser, H.H.; Yauk, C.L.; White, P.A.; Halappanavar, S. Comparative transcriptomic analyses to scrutinize the assumption that genotoxic PAHs exert effects via a common mode of action. Arch. Toxicol. 2016, 90, 2461–2480. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Yan, Y.; Li, J.; Ma, Q.; Stoner, G.D.; Ye, J.; Huang, C. Differential requirement of signal pathways for benzo[a]pyrene (B[a]P)-induced nitric oxide synthase (iNOS) in rat esophageal epithelial cells. Carcinogenesis 2005, 26, 1035–1043. [Google Scholar] [CrossRef] [PubMed]
- Ferrari, S.; Bandi, H.R.; Hofsteenge, J.; Bussian, B.M.; Thomas, G. Mitogen-activated 70K S6 kinase. Identification of in vitro 40 S ribosomal S6 phosphorylation sites. J. Biol. Chem. 1991, 266, 22770–22775. [Google Scholar] [CrossRef]
- Kim, S.; Jung, Y.; Kim, D.; Koh, H.; Chung, J. Extracellular zinc activates p70 S6 kinase through the phosphatidylinositol 3-kinase signaling pathway. J. Biol. Chem. 2000, 275, 25979–25984. [Google Scholar] [CrossRef]
- Avila-Rojas, S.H.; Lira-León, A.; Aparicio-Trejo, O.E.; Reyes-Fermín, L.M.; Pedraza-Chaverri, J. Role of Autophagy on Heavy Metal-Induced Renal Damage and the Protective Effects of Curcumin in Autophagy and Kidney Preservation. Medicina 2019, 55, 360. [Google Scholar] [CrossRef]
- Vergadi, E.; Ieronymaki, E.; Lyroni, K.; Vaporidi, K.; Tsatsanis, C. Akt Signaling Pathway in Macrophage Activation and M1/M2 Polarization. J. Immunol. 2017, 198, 1006–1014. [Google Scholar] [CrossRef] [PubMed]
- Song, X.; Li, Z.; Liu, F.; Wang, Z.; Wang, L. Restoration of autophagy by puerarin in lead-exposed primary rat proximal tubular cells via regulating AMPK-mTOR signaling. J. Biochem. Mol. Toxicol. 2017, 31. [Google Scholar] [CrossRef]
- Song, M.F.; Yang, Y.; Yi, Z.W.; Zhang, Z.Q.; Shen, X.D.; Hu, G.H.; Zhu, Y.F. Sema 3A as a biomarker of the activated mTOR pathway during hexavalent chromium-induced acute kidney injury. Toxicol. Lett. 2018, 299, 226–235. [Google Scholar] [CrossRef] [PubMed]
- Altman, J.K.; Yoon, P.; Katsoulidis, E.; Kroczynska, B.; Sassano, A.; Redig, A.J.; Glaser, H.; Jordan, A.; Tallman, M.S.; Hay, N.; et al. Regulatory effects of mammalian target of rapamycin-mediated signals in the generation of arsenic trioxide responses. J. Biol. Chem. 2008, 283, 1992–2001. [Google Scholar] [CrossRef]
- Flotow, H.; Thomas, G. Substrate recognition determinants of the mitogen-activated 70K S6 kinase from rat liver. J. Biol. Chem. 1992, 267, 3074–3078. [Google Scholar] [CrossRef] [PubMed]
- Frank, D.F.; Miller, G.W.; Harvey, D.J.; Brander, S.M.; Geist, J.; Connon, R.E.; Lein, P.J. Bifenthrin causes transcriptomic alterations in mTOR and ryanodine receptor-dependent signaling and delayed hyperactivity in developing zebrafish (Danio rerio). Aquat. Toxicol. 2018, 200, 50–61. [Google Scholar] [CrossRef] [PubMed]
- Zhao, G.; Guo, H.; Zhang, H.; Zhang, X.; Qian, H.; Li, G.; Xu, A. Effects of pyriproxyfen exposure on immune signaling pathway and transcription of detoxification enzyme genes in fat body of silkworm, Bombyx mori. Pestic. Biochem. Physiol. 2020, 168, 104621. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Guan, D.; Wei, J.; Ge, H.; Cao, X.; Lv, S.; Zhou, X.; Zheng, Y.; Meng, X.; Wang, J.; et al. Mechanisms underlying the effects of low concentrations of chlorantraniliprole on development and reproduction of the fall armyworm, Spodoptera frugiperda. Pestic. Biochem. Physiol. 2023, 191, 105362. [Google Scholar] [CrossRef] [PubMed]
- You, H.; An, G.; Lee, H.; Lim, W.; Song, G. Bifenox induces programmed cell death in bovine mammary epithelial cells by impairing calcium homeostasis, triggering ER stress, and altering the signaling cascades of PI3K/AKT and MAPK. Pestic. Biochem. Physiol. 2023, 196, 105626. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Park, Y.; Yoon, K.S.; Clark, J.M.; Park, Y. Imidacloprid, a neonicotinoid insecticide, induces insulin resistance. J. Toxicol. Sci. 2013, 38, 655–660. [Google Scholar] [CrossRef]
- Rascio, F.; Spadaccino, F.; Rocchetti, M.T.; Castellano, G.; Stallone, G.; Netti, G.S.; Ranieri, E. The Pathogenic Role of PI3K/AKT Pathway in Cancer Onset and Drug Resistance: An Updated Review. Cancers 2021, 13, 3949. [Google Scholar] [CrossRef] [PubMed]
- Zou, Z.; Tao, T.; Li, H.; Zhu, X. mTOR signaling pathway and mTOR inhibitors in cancer: Progress and challenges. Cell Biosci. 2020, 10, 31. [Google Scholar] [CrossRef]
- Debnath, J.; Gammoh, N.; Ryan, K.M. Autophagy and autophagy-related pathways in cancer. Nat. Rev. Mol. Cell Biol. 2023, 24, 560–575. [Google Scholar] [CrossRef]
- Almalki, S.G. The pathophysiology of the cell cycle in cancer and treatment strategies using various cell cycle checkpoint inhibitors. Pathol. Res. Pract. 2023, 251, 154854. [Google Scholar] [CrossRef] [PubMed]
- Lagunas-Rangel, F.A.; Linnea-Niemi, J.V.; Kudłak, B.; Williams, M.J.; Jönsson, J.; Schiöth, H.B. Role of the synergistic interactions of environmental pollutants in the development of cancer. GeoHealth 2022, 6, e2021GH000552. [Google Scholar] [CrossRef]
- Lagunas-Rangel, F.A.; Liu, W.; Schiöth, H.B. Can Exposure to Environmental Pollutants Be Associated with Less Effective Chemotherapy in Cancer Patients? Int. J. Environ. Res. Public Health 2022, 19, 2064. [Google Scholar] [CrossRef] [PubMed]
- Furue, M.; Ishii, Y.; Tsukimori, K.; Tsuji, G. Aryl Hydrocarbon Receptor and Dioxin-Related Health Hazards-Lessons from Yusho. Int. J. Mol. Sci. 2021, 22, 708. [Google Scholar] [CrossRef] [PubMed]
- Greathouse, K.L.; Bredfeldt, T.; Everitt, J.I.; Lin, K.; Berry, T.; Kannan, K.; Mittelstadt, M.L.; Ho, S.-m.; Walker, C.L. Environmental estrogens differentially engage the histone methyltransferase EZH2 to increase risk of uterine tumorigenesis. Mol. Cancer Res. 2012, 10, 546–557. [Google Scholar] [CrossRef]
- Yin, L.; Yu, X. Arsenic-induced apoptosis in the p53-proficient and p53-deficient cells through differential modulation of NFkB pathway. Food Chem. Toxicol. 2018, 118, 849–860. [Google Scholar] [CrossRef]
- Gagic, Z.; Ruzic, D.; Djokovic, N.; Djikic, T.; Nikolic, K. In silico methods for design of kinase inhibitors as anticancer drugs. Front. Chem. 2020, 7, 873. [Google Scholar] [CrossRef]
- Alam, A.; Khan, M.S.; Mathur, Y.; Sulaimani, M.N.; Farooqui, N.; Ahmad, S.F.; Nadeem, A.; Yadav, D.K.; Mohammad, T. Structure-based identification of potential inhibitors of ribosomal protein S6 kinase 1, targeting cancer therapy: A combined docking and molecular dynamics simulations approach. J. Biomol. Struct. Dyn. 2024, 42, 5758–5769. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Xiang, M.-L.; Liang, J.-Y.; Zeng, T.; Zhang, X.-N.; Zhang, J.; Yang, S.-Y. Combination of pharmacophore hypothesis, genetic function approximation model, and molecular docking to identify novel inhibitors of S6K1. Mol. Divers. 2013, 17, 767–772. [Google Scholar] [CrossRef]
- Zhang, H.; Zhang, H.-R.; Zhang, J.; Hu, M.-L.; Ren, L.; Luo, Q.-Q.; Qi, H.-Z. Discovery of novel S6K1 inhibitors by an ensemble-based virtual screening method and molecular dynamics simulation. J. Mol. Model. 2023, 29, 102. [Google Scholar] [CrossRef]
- Yu, F.; Wu, X.; Chen, W.; Yan, F.; Li, W. Computer-assisted discovery and evaluation of potential ribosomal protein S6 kinase beta 2 inhibitors. Comput. Biol. Med. 2024, 172, 108204. [Google Scholar] [CrossRef]
- Snezhkina, A.V.; Kudryavtseva, A.V.; Kardymon, O.L.; Savvateeva, M.V.; Melnikova, N.V.; Krasnov, G.S.; Dmitriev, A.A. ROS generation and antioxidant defense systems in normal and malignant cells. Oxidative Med. Cell. Longev. 2019, 2019, 6175804. [Google Scholar] [CrossRef]
- Bond, P. Regulation of mTORC1 by growth factors, energy status, amino acids and mechanical stimuli at a glance. J. Int. Soc. Sports Nutr. 2016, 13, 8. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| N | ID | Compound | PubChem Reference | Ac_Value # | Ac_Name # | SwissTarget Prediction ## | Other S6K SwissTarget Prediction ## |
|---|---|---|---|---|---|---|---|
| 1 | 9950039 | 2,3-Di-thiophen-2-yl-benzo[g]quinoxaline | bioassay/242021 | 1.36 µM | IC50 * | 0.91 | NA |
| 2 | 10164781 | 3-(5-(3-[5-(2-Methoxycarbonyl-ethyl)-thiophen-2-yl]-benzo[g]quinoxalin-2-yl)-thiophen-2-yl)-propionic acid methyl ester | bioassay/242021 | 6.13 µM | IC50 * | 0.72 | NA |
| 3 | 10183039 | (5-[3-(5-Carboxymethyl-thiophen-2-yl)-benzo[g]quinoxalin-2-yl]-thiophen-2-yl)-acetic acid | bioassay/242021 | 1.28 µM | IC50 * | 1.0 | NA |
| 4 | 10206548 | 3-(5-(3-[5-(2-Carboxy-ethyl)-thiophen-2-yl]-benzo[g]quinoxalin-2-yl)-thiophen-2-yl)-propionic acid | bioassay/242021 | 0.87 µM | IC50 * | 0.81 | NA |
| 5 | 44399002 | 6,7-Di-thiophen-2-yl-2,3-dihydro-1,4-dioxa-5,8-diaza-anthracene-2-carboxylic acid | bioassay/242021 | 0.95 µM | IC50 * | 1.0 | NA |
| 6 | 44399008 | (5-[3-(4-Carboxymethyl-thiophen-2-yl)-benzo[g]quinoxalin-2-yl]-thiophen-3-yl)-acetic acid | bioassay/242021 | 0.64 µM | IC50 * | 1.0 | NA |
| 7 | 44399015 | 5-[3-(5-Carboxythien-2-yl)benzo[g]quinoxalin-2-yl]thiophene-2-carboxylic acid | bioassay/242021 | 10 µM | IC50 * | 0.1 | NA |
| 8 | 44399109 | 4-(5-(3-[5-(3-Carboxy-propyl)-thiophen-2-yl]-benzo[g]quinoxalin-2-yl)-thiophen-2-yl)-butyric acid | bioassay/242021 | 0.82 µM | IC50 * | 0.91 | NA |
| 9 | 44399200 | 7,8-Dimethoxy-2,3-di-thiophen-2-yl-pyrazino [2,3-b]quinoxaline | bioassay/242021 | 1.14 µM | IC50 * | 1.0 | RPS6KA2; 0.12 |
| 10 | 135964360 | 2,3-Di-thiophen-2-yl-quinoxaline-6,7-diol | bioassay/242021 | 9.61 µM | IC50 * | 1.0 | RPS6KA3; 0.11 |
| 11 | 122588288 | N-(4-(6-amino-5-(1-oxo-1,2,3,4-tetrahydroisoquinolin-6-yl)pyridin-3-yl)-3-fluorophenyl) cyclopropanesulfonamide | bioassay/1746035 | 5.5 µM | %Inhibition ** | N/A | RPS6KB1; 0.1 |
| 12 | 163196379 | N-(2,6-diethylphenyl)-2-[4-(4-methylpiperazin-1-yl)-2-(prop-2-enoylamino)anilino]-5,6-dihydropyrimido [4,5-e]indolizine-7-carboxamide | bioassay/1819637 | 1.65 µM | IC50 *** | N/A | RPS6KA1, RPS6KA2, RPS6KA4, RPS6KA5, RPS6KA6; 0.06 |
| PubChem CID | Ligand Name | Binding Affinity |
|---|---|---|
| 122588288 | N-(4-(6-amino-5-(1-oxo-1-2-3, 4-tetrahydroisoquinoline-6-yl) pyridine-3-yl)-3-fluorophenyl) cyclopropane sulfonamide | −9.5 |
| 163196379 | N-(2,6-diethyl phenyl)-2- [4-(4-methyl piperazine-1-yl)-2-(prop-2-enoylamino) anilino]-5-6-dihydropyrimido [45-e] indolizine-7-carboxamide | −9.4 |
| 44399008 | 5-[3-(4-Carboxymethyl-thiophene-2-yl)-benzo[g]quinoxalin-2-yl]-thiophen-3-yl)-acetic_acid | −8.9 |
| 44399015 | 5-[3-(5-Carboxythien-2-yl) benzo[g]quinoxalin-2-yl] thiophene-2-carboxylic_acid | −8.8 |
| 10183039 | 5-[3-(5-Carboxymethyl-thiophene-2-yl)-benzo[g]quinoxalin-2-yl]-thiophen-2-yl)-acetic_acid | −8.7 |
| 10206548 | 3-(5-(3-[5-(2-Carboxy-ethyl)-thiophen-2-yl]-benzo[g]quinoxalin-2-yl)-thiophene-2-yl)-propionic_acid | −8.7 |
| PubChem CID | 3D Structure | Hydrophilic Interactions | Hydrophobic Contacts | No. of H-Bonds | No. of Total Bonds | Affinity kcal mol−1 | |||
|---|---|---|---|---|---|---|---|---|---|
| Residue (H-Bond) | Length | Residue (Bond Type) | Length | ||||||
| 1 | 122588288 |  | Glu198, (H-Bond) Arg361, (H-Bond) Lys75, (H-Bond) | 2.77 2.25 2.39 | Met201, (Pi-alkyl) Ala97, (Pi-alkyl) Val81, (Pi-alkyl) Val81, (Pi-Sigma) Glu149, (Carbon H-bond) Arg361, (Pi-cation) Phe157, (Pi stacked bond) | 4.80 4.88 5.37 3.89 3.34 4.37 4.92 | 3 | 10 | −9.50 |
| 2 | 163196379 |  | Thr158, (H-Bond) Leu151, (H-Bond) Glu149, (H-Bond) | 4.69 1.87 2.23 | Val132, (Pi-alkyl) Ala97, (Pi-alkyl) Met201, (Pi-alkyl) Leu73, (Pi-alkyl) Val81, (Pi-alkyl) Met201, (Pi-Sigma) Phe358, (Pi stacked bond) Glu155, (Halogen) Glu198, (Halogen) | 5.44 4.57 5.09 5.07 5.01 3.54 5.22 3.12 3.01 | 3 | 12 | −9.40 |
| 3 | 44399008 | 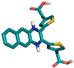 | Thr158, (H-Bond) Phe358, (H-Bond) Lys196, (H-Bond) | 2.44 2.27 2.28 | Ala97, (Pi-alkyl) Val81, (Pi-alkyl) Val81, (Pi-sigma) Glu155, (Pi-Cation) Phe358, (Pi stacked bond) Gln357, (Unfavorable bond) | 4.89 4.28 3.57 4.49 4.98 2.95 | 3 | 9 | −8.90 |
| 4 | 44399015 | 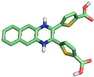 | Arg361, (H-Bond) Thr228, (H-Bond) Phe358, (H-Bond) Glu357, (H-Bond) | 2.71 2.26 2.67 2.97 | Ala97, (Pi-alkyl) Val81, (Pi-alkyl) Val81, (Pi-sigma) Phe358, (Sulfur) Phe358, (Pi stacked bond) | 4.72 4.25 3.65 5.19 5.47 | 4 | 9 | −8.80 |
| 5 | 10183039 | 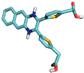 | Thr158, (H-Bond) Thr228, (H-Bond) Phe358, (H-Bond) Glu357, (H-Bond) | 2.66 2.64 2.20 2.47 | Ala97, (Pi-alkyl) Val81, (Pi-alkyl) Val81, (Pi-sigma) Phe358, (Sulfur) Phe358, (Pi stacked bond) Glu155, (Pi-Cation) | 4.62 4.27 3.68 5.20 4.95 4.71 | 4 | 10 | −8.70 |
| 6 | 10206548 | 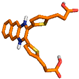 | Thr228, (H-Bond) Lys196, (H-Bond) Glu198, (H-Bond) | 2.59 2.39 2.26 | Ala97, (Pi-alkyl) Val81, (Pi-alkyl) Val81, (Pi-sigma) Phe358, (Sulfur) Phe358, (Pi stacked bond) Glu155, (Pi-Cation) | 4.55 4.26 3.59 5.19 5.41 4.95 | 3 | 9 | −8.70 |
| 7 | PF-4708671 |  | Asp212, (H-Bond) Thr228, (H-Bond) Lys196, (H-Bond) Glu149, (H-Bond) | 1.83 2.94 2.21 2.49 | Leu73, (Pi-alkyl) Ala97, (Pi-alkyl) Val81, (Pi-alkyl) Cys230, (Pi-alkyl) Thr228, (Pi-sigma) Thr228, (Pi-sigma) | 4.07 4.06 4.53 4.33 3.74 3.91 | 4 | 10 | −8.40 |
| Ligand (PubChem CID) | 3D Structure | Hydrophilic Interactions | Hydrophobic Contacts | No. of H-Bonds | No. of Total Bonds | Affinity kcal mol−1 | |||
|---|---|---|---|---|---|---|---|---|---|
| Residue (H-Bond) | Length | Residue (Bond Type) | Length | ||||||
| 1 | Benzo-a-pyrene (2336) |  | - | - | Lys99, (Pi-alkyl) Ala97, (Pi-alkyl) Ala97, (Pi-alkyl) Met201, (Pi-Sigma) Leu73, (Pi-Sigma) Val81, (Pi-Sigma) Val81, (Pi-Sigma) Ala97, (Pi-alkyl) Met201, (Pi-alkyl) Met201, (Pi-cation) Leu73, (Pi-alkyl) Val81, (Pi-alkyl) Val81, (Pi-alkyl) | 5.12 4.20 4.02 3.40 3.97 3.70 3.97 4.22 3.30 4.22 5.35 5.45 4.42 | 0 | 13 | −9.50 |
| 2 | Chlorantraniliprole (11271640) |  | Glu198, (H-Bond) Glu198, (H-Bond) | 2.61 2.20 | Leu73, (Pi-alkyl) Leu148, (Pi-alkyl) Ala97, (Pi-alkyl) Met201, (Pi-alkyl) Phe358, (Pi-alkyl) Val81, (Pi-alkyl) Val81, (Pi-Sigma) Asn199, (Carbon H-Bond) Asp212, (Carbon H-Bond) Phe358, (Pi-alkyl) Ala97, (Pi-alkyl) Val81, (Pi-alkyl) | 4.03 4.07 3.27 5.00 5.15 5.21 3.51 3.52 3.41 4.69 4.96 3.51 | 2 | 14 | −8.70 |
| 3 | Bifenthrin (6442842) |  | - | - | Leu73, (Pi-alkyl) Leu148, (Pi-alkyl) Ala97, (Pi-alkyl) Val81, (Pi-alkyl) Phe358, (Pi-sigma) Thr228, (Pi-sigma) Lys99, (Pi-Anion) Asp212, (Pi-Cation) Thr211, (Halogen) Leu73, (Pi-alkyl) Val81, (Pi-alkyl) | 4.94 5.07 4.33 3.74 3.73 3.47 4.13 3.66 3.56 3.03 4.59 | 0 | 11 | −8.50 |
| 4 | PF-4708671 (51371303) |  | Asp212, (H-Bond) Thr228, (H-Bond) Lys196, (H-Bond) Glu149, (H-Bond) | 1.83 2.94 2.21 2.49 | Leu73, (Pi-alkyl) Ala97, (Pi-alkyl) Val81, (Pi-alkyl) Cys230, (Pi-alkyl) Thr228, (Pi-sigma) Thr228, (Pi-sigma) | 4.07 4.06 4.53 4.33 3.74 3.91 | 4 | 10 | −8.40 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Khalil, M.I.; Helal, M.; El-Sayed, A.F.; El Hajj, R.; Holail, J.; Houssein, M.; Waraky, A.; Pardo, O.E. S6K2 in Focus: Signaling Pathways, Post-Translational Modifications, and Computational Analysis. Int. J. Mol. Sci. 2025, 26, 176. https://doi.org/10.3390/ijms26010176
Khalil MI, Helal M, El-Sayed AF, El Hajj R, Holail J, Houssein M, Waraky A, Pardo OE. S6K2 in Focus: Signaling Pathways, Post-Translational Modifications, and Computational Analysis. International Journal of Molecular Sciences. 2025; 26(1):176. https://doi.org/10.3390/ijms26010176
Chicago/Turabian StyleKhalil, Mahmoud I., Mohamed Helal, Ahmed F. El-Sayed, Rana El Hajj, Jasmine Holail, Marwa Houssein, Ahmed Waraky, and Olivier E. Pardo. 2025. "S6K2 in Focus: Signaling Pathways, Post-Translational Modifications, and Computational Analysis" International Journal of Molecular Sciences 26, no. 1: 176. https://doi.org/10.3390/ijms26010176
APA StyleKhalil, M. I., Helal, M., El-Sayed, A. F., El Hajj, R., Holail, J., Houssein, M., Waraky, A., & Pardo, O. E. (2025). S6K2 in Focus: Signaling Pathways, Post-Translational Modifications, and Computational Analysis. International Journal of Molecular Sciences, 26(1), 176. https://doi.org/10.3390/ijms26010176