
HTK vs. HTK-N for Coronary Endothelial Protection during Hypothermic, Oxygenated Perfusion of Hearts Donated after Circulatory Death

 , and
, and
Abstract
1. Introduction
2. Results
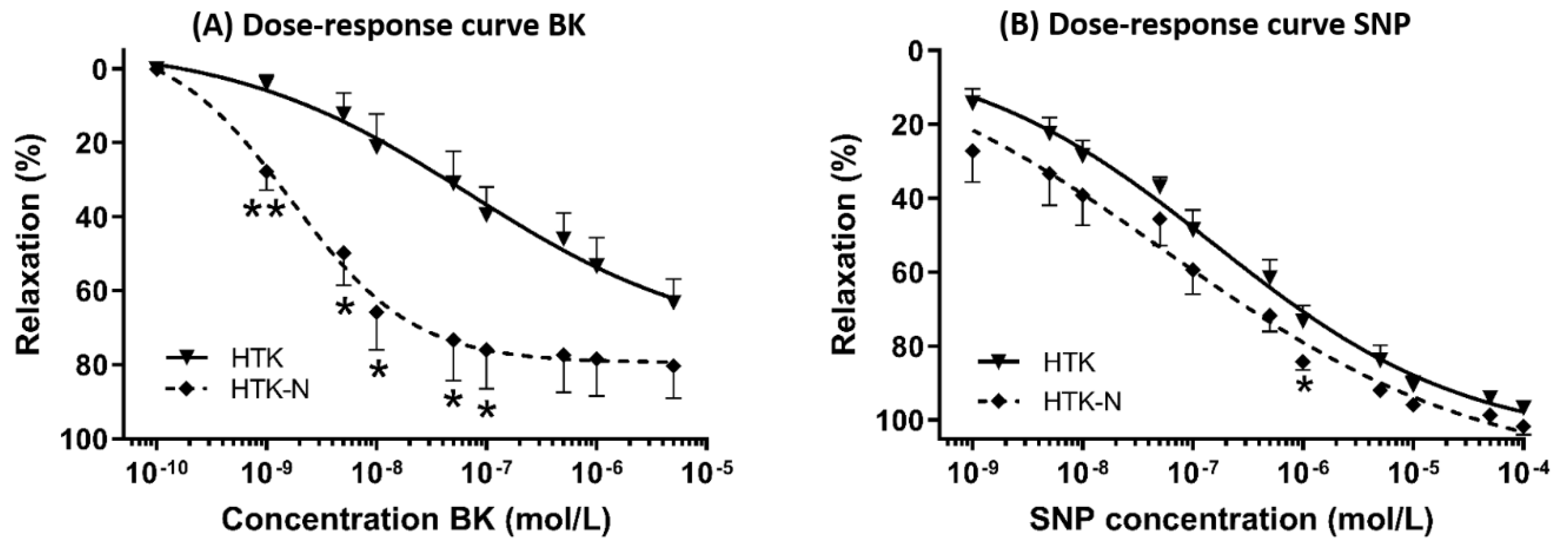
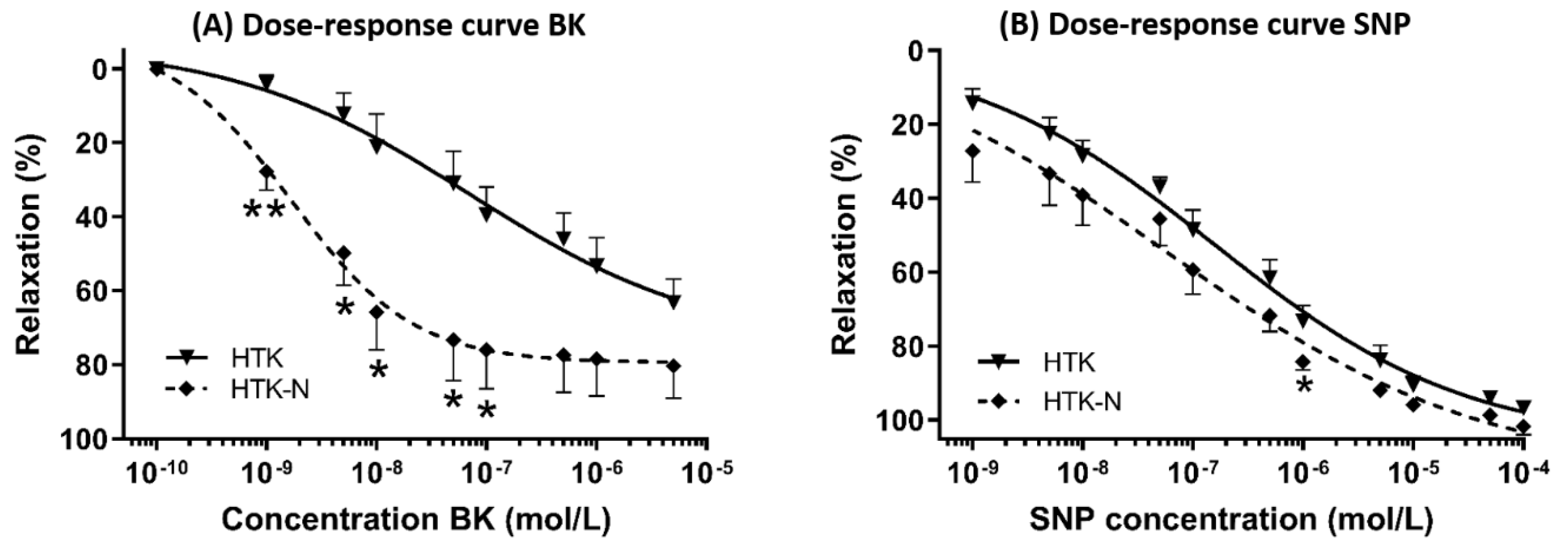
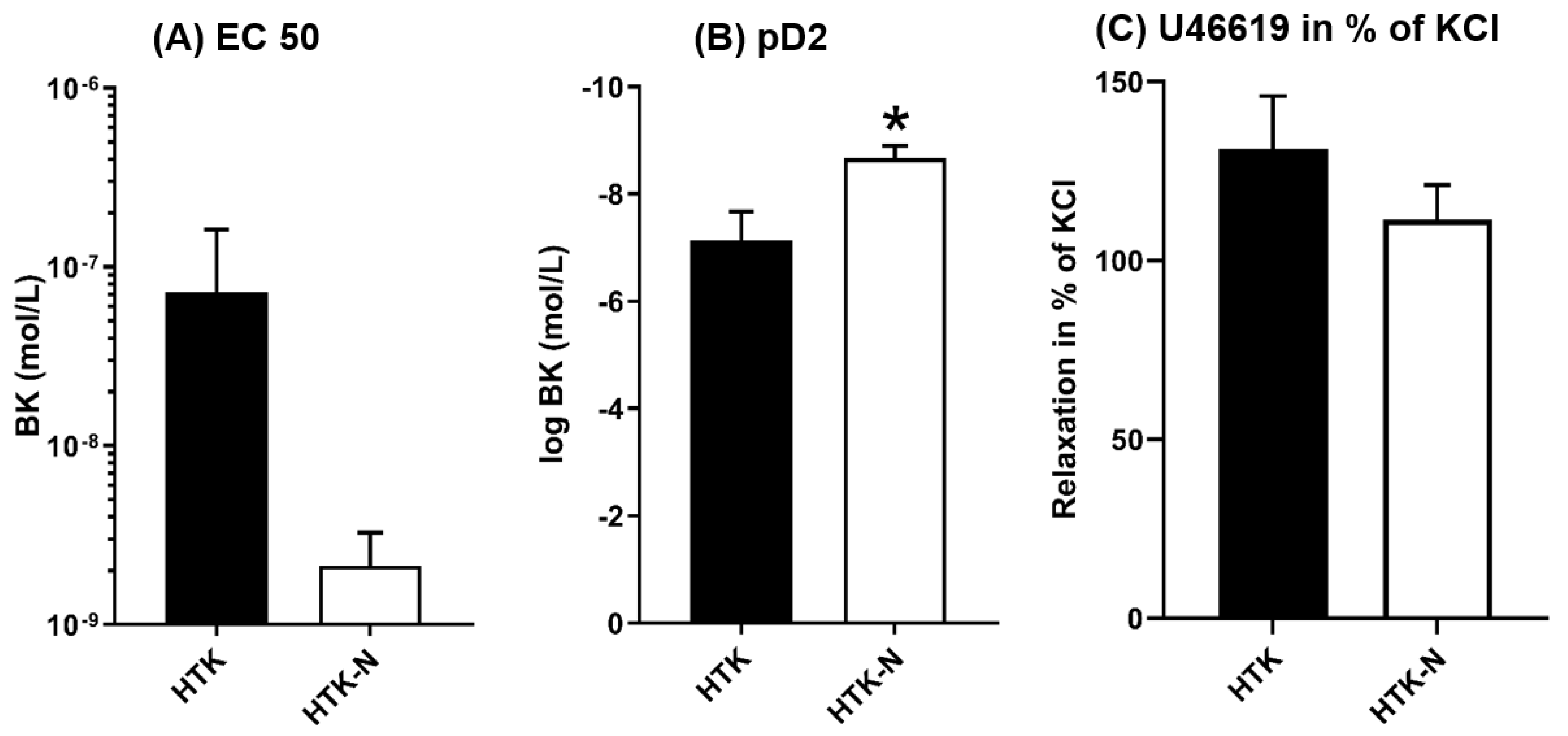
2.1. Vasomotor Function
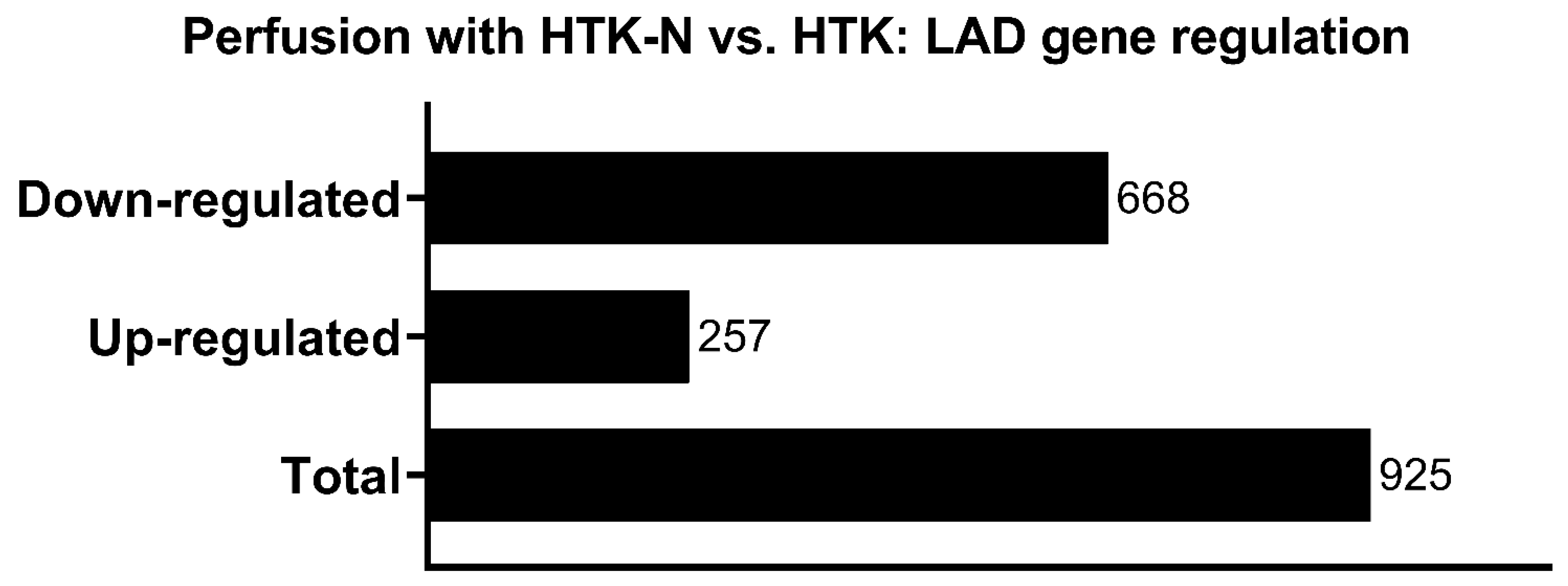
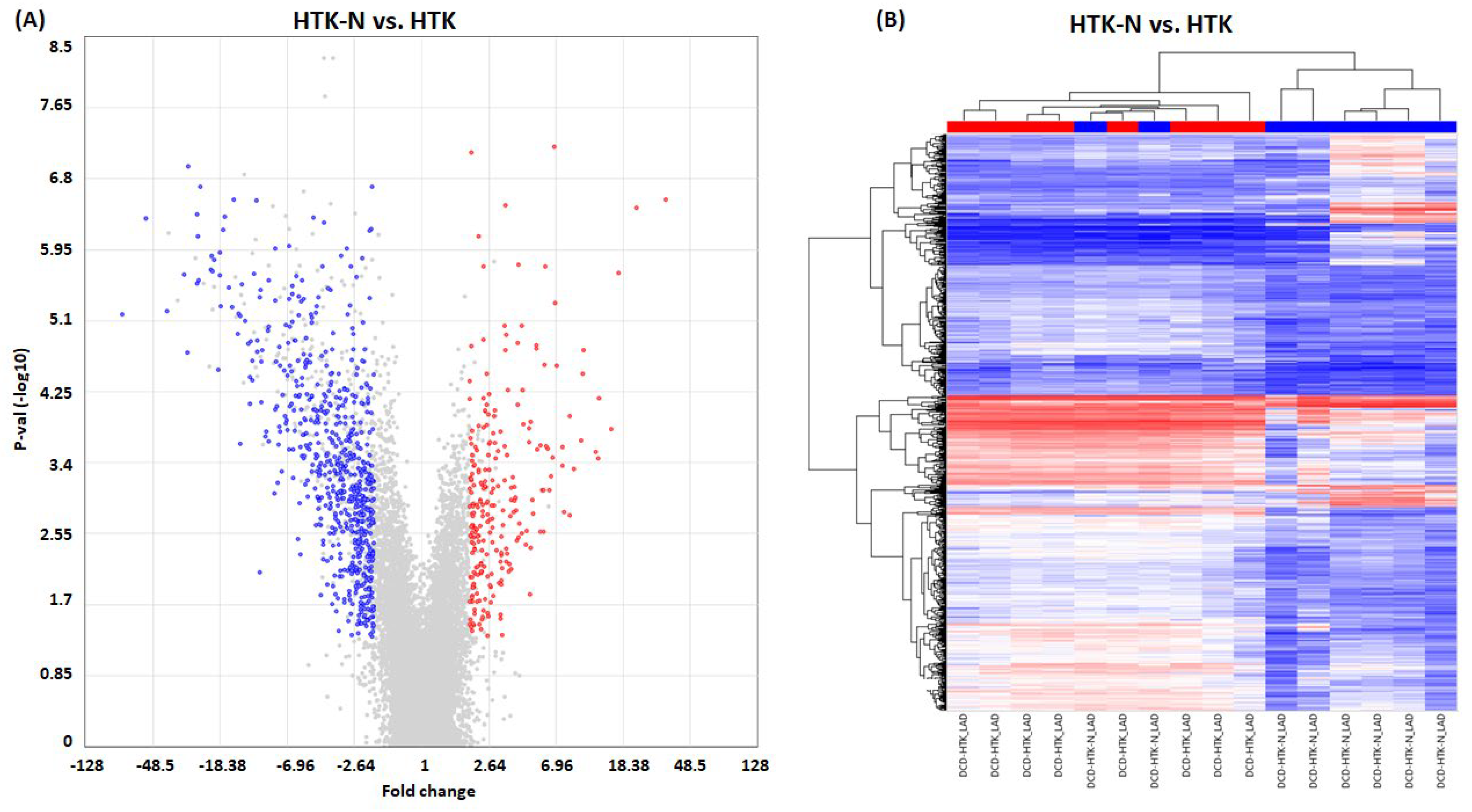
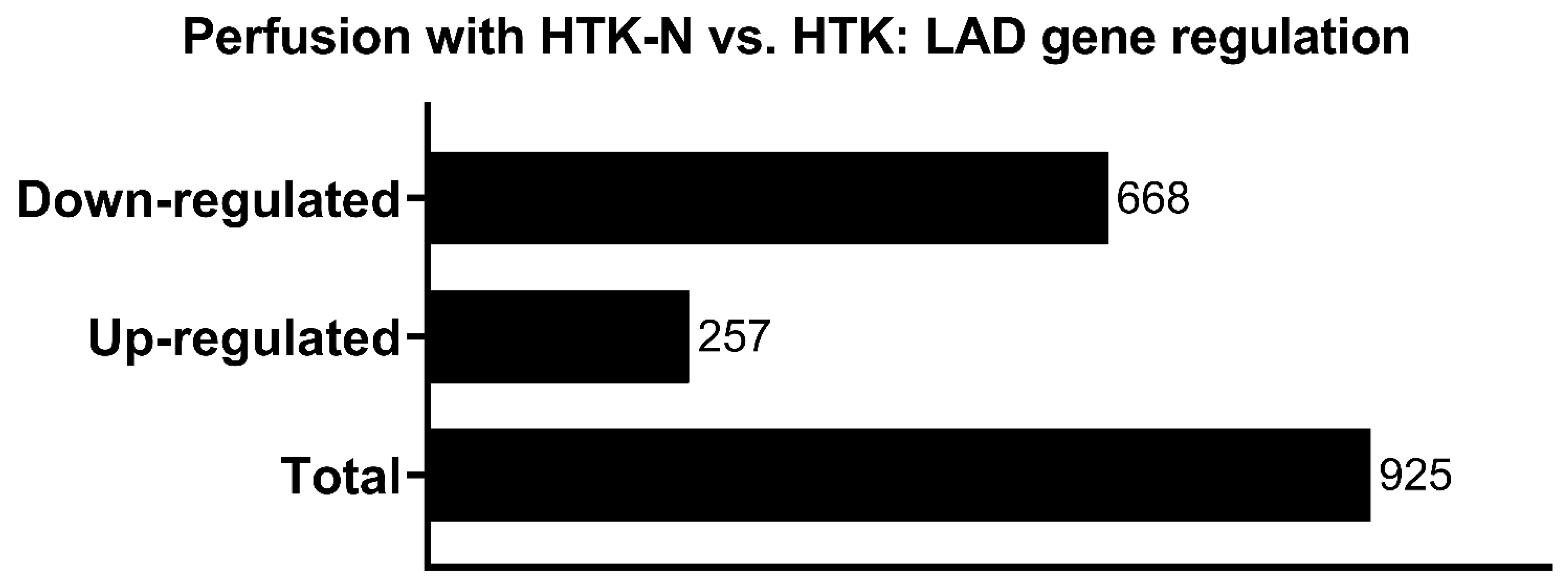
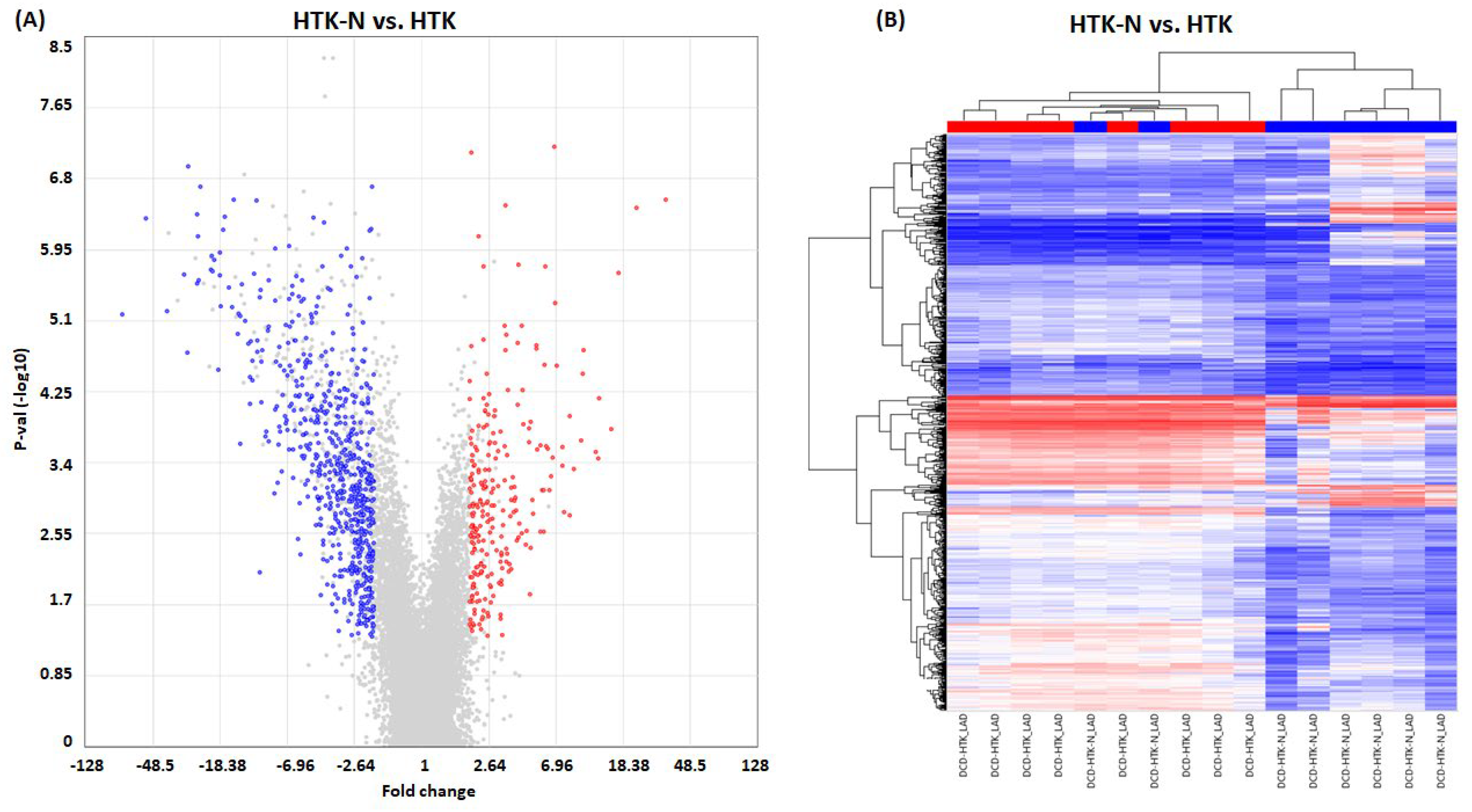
2.2. Transcriptome
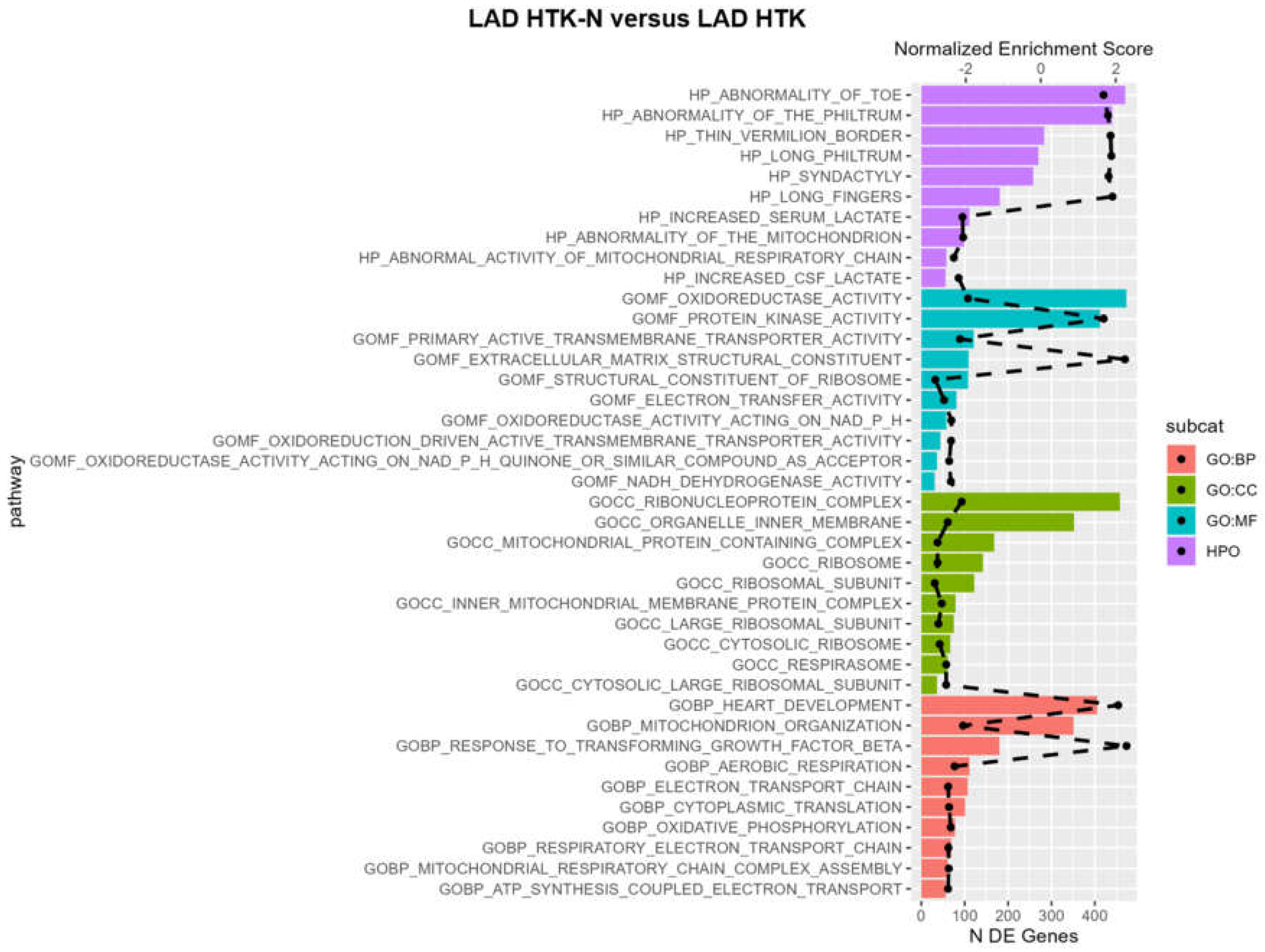
2.3. Pathways
3. Discussion
3.1. Functional Effects
3.2. Transcriptome Effects
3.2.1. Endothelial Cell Functionality
3.2.2. Ischemia/Reperfusion and Oxidative Stress
3.2.3. Immune Reaction and Survival
3.2.4. Regeneration
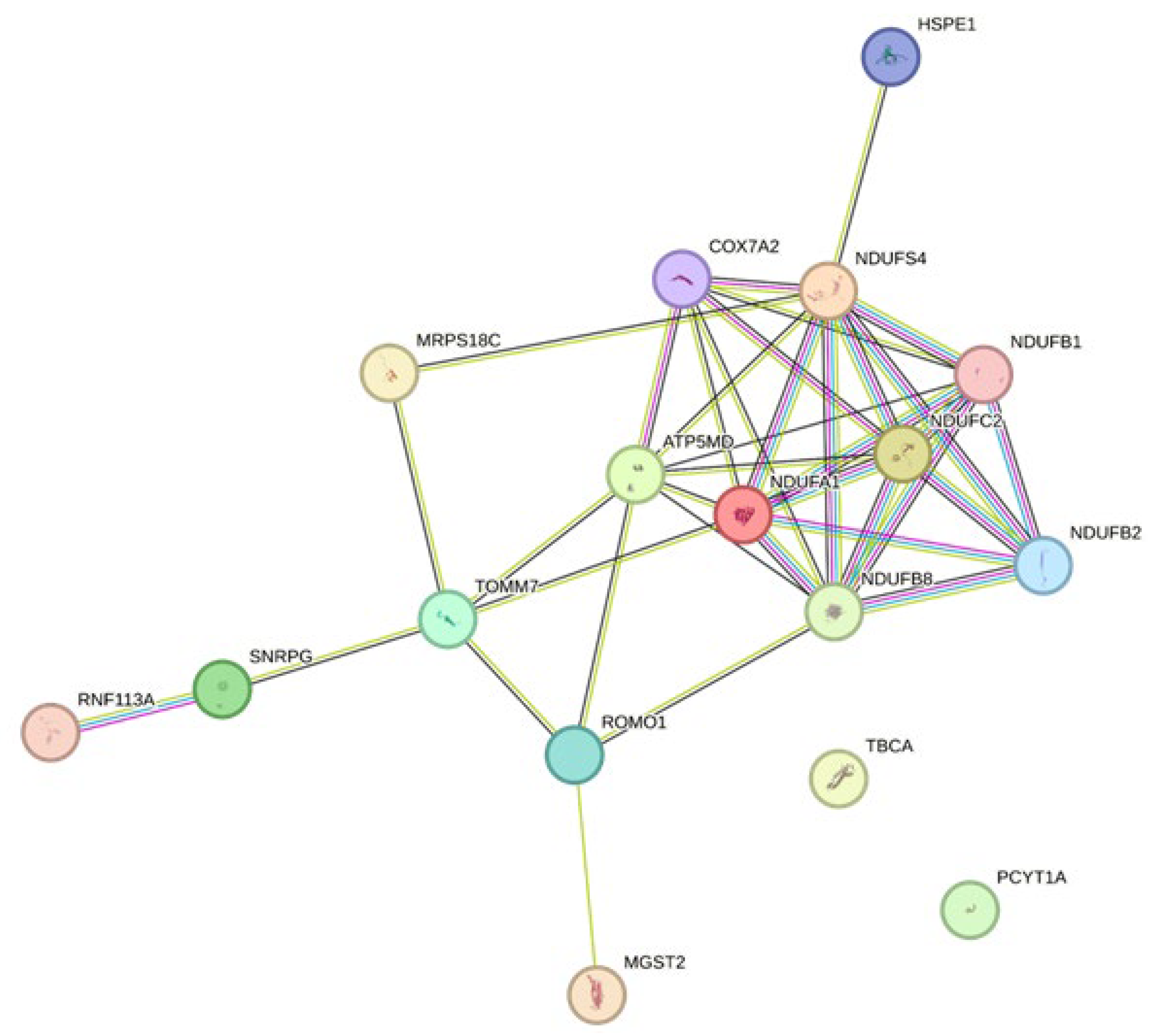
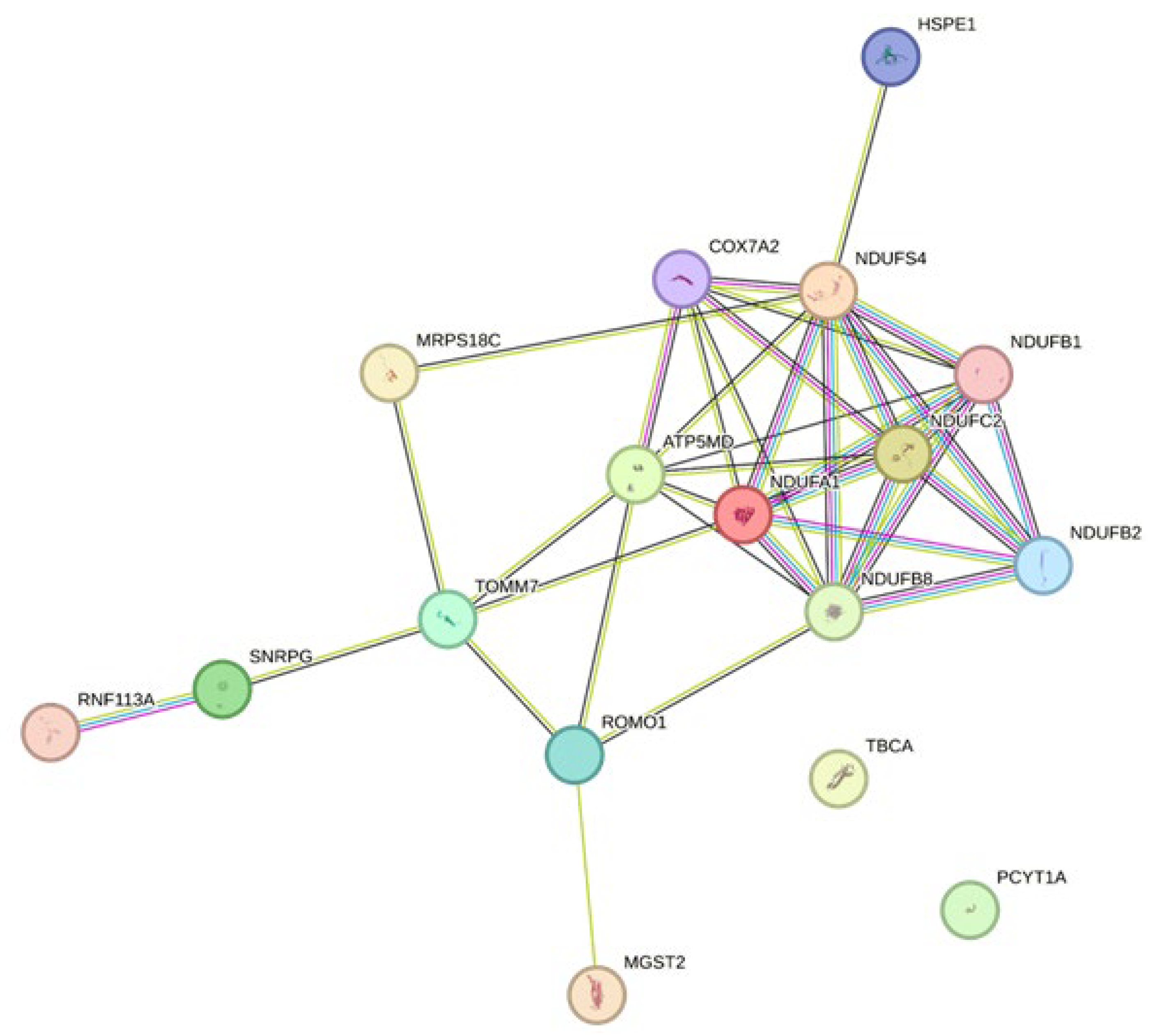
3.2.5. Mitochondrial Relevance
3.2.6. Coronary Artery Disease Associations
3.3. Associations with Graft Dysfunction
3.4. Pathways
3.5. Comparison of Perfusion Solutions
3.6. Limitations
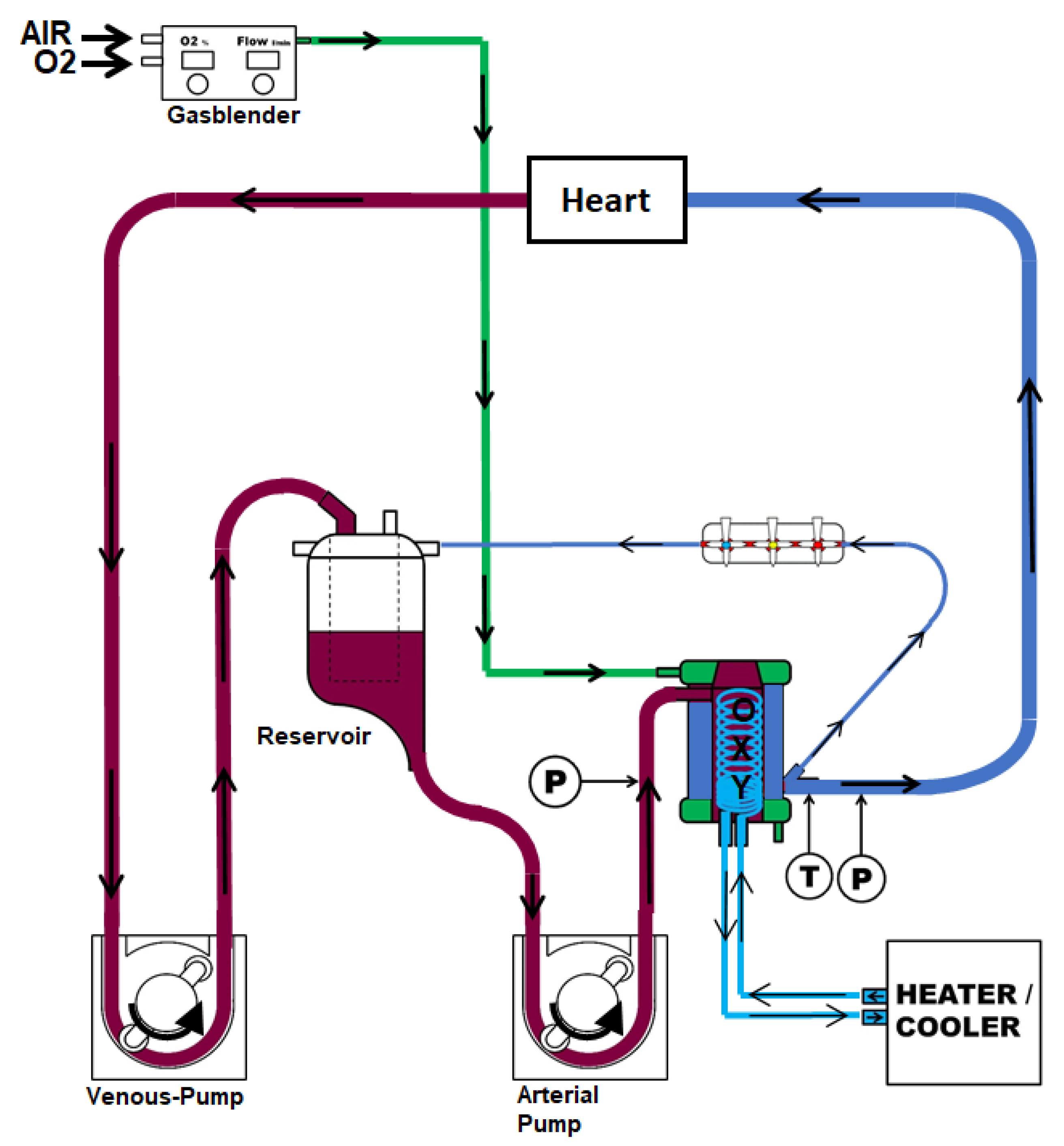
4. Materials and Methods
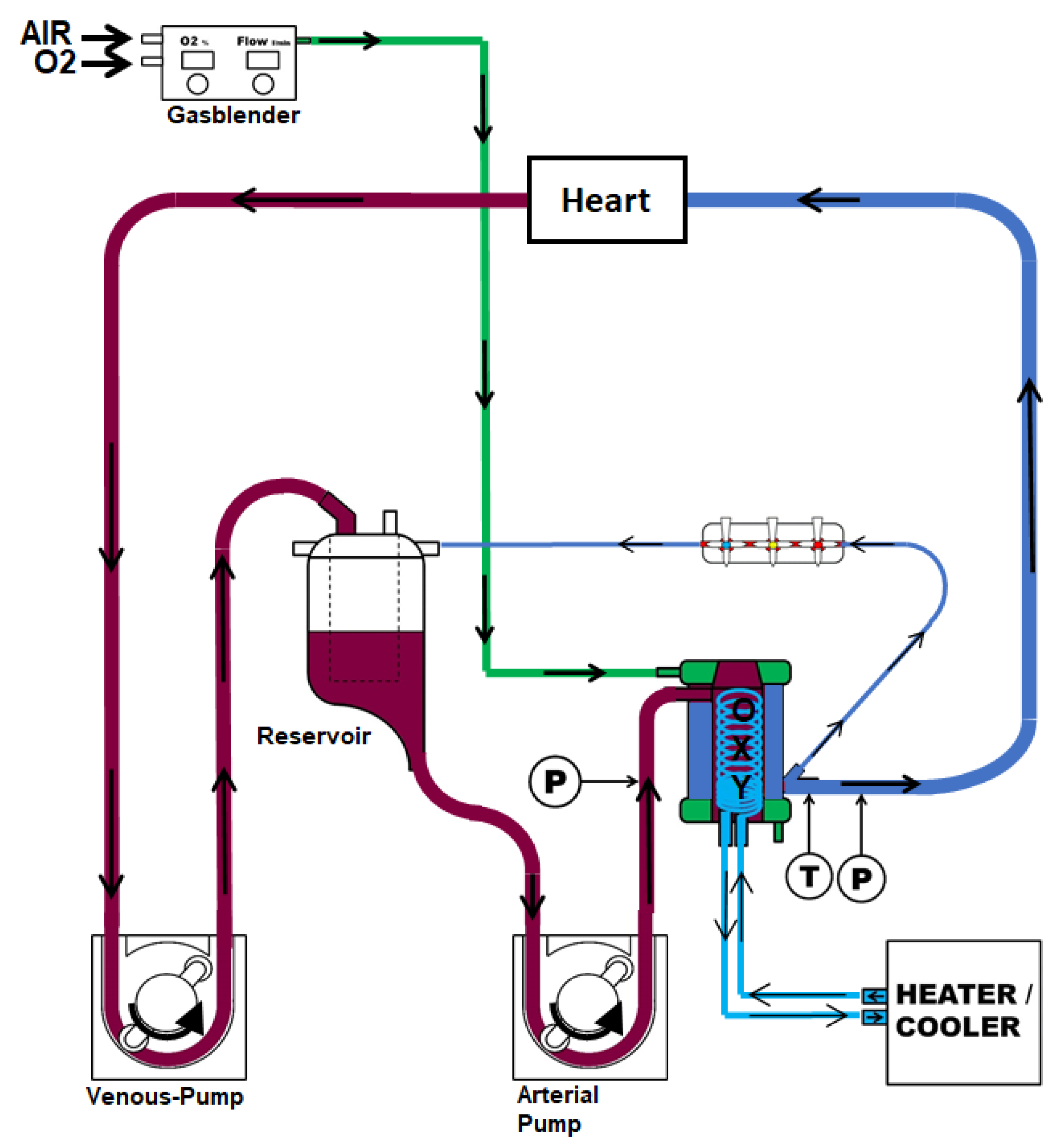
4.1. Animals and Anesthesia
4.2. Surgical Procedure and Study Groups
4.3. Reperfusion and Tissue Collection
4.4. Coronary Artery Vasomotor Analysis
4.5. RNA Preparation
4.6. Microarrays
4.7. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Moeslund, N.; Ertugrul, I.A.; Hu, M.A.; Dalsgaard, F.F.; Ilkjaer, L.B.; Ryhammer, P.; Pedersen, M.; Erasmus, M.E.; Eiskjaer, H. Ex-situ oxygenated hypothermic machine perfusion in donation after circulatory death heart transplantation following either direct procurement or in-situ normothermic regional perfusion. J. Heart Lung Transplant. 2023, 42, 730–740. [Google Scholar] [CrossRef]
- Nilsson, J.; Jernryd, V.; Qin, G.; Paskevicius, A.; Metzsch, C.; Sjoberg, T.; Steen, S. A nonrandomized open-label phase 2 trial of nonischemic heart preservation for human heart transplantation. Nat. Commun. 2020, 11, 2976. [Google Scholar] [CrossRef]
- Louca, J.O.; Manara, A.; Messer, S.; Öchsner, M.; McGiffin, D.; Austin, I.; Bell, E.; Leboff, S.; Large, S. Getting out of the box: The future of the UK donation after circulatory determination of death heart programme. EClinicalMedicine 2023, 66, 102320. [Google Scholar] [CrossRef]
- Ramzy, D.; Rao, V.; Brahm, J.; Miriuka, S.; Delgado, D.; Ross, H.J. Cardiac allograft vasculopathy: A review. Can. J. Surg. 2005, 48, 319–327. [Google Scholar]
- Loganathan, S.; Radovits, T.; Hirschberg, K.; Korkmaz, S.; Koch, A.; Karck, M.; Szabó, G. Effects of Custodiol-N, a novel organ preservation solution, on ischemia/reperfusion injury. J. Thorac. Cardiovasc. Surg. 2010, 139, 1048–1056. [Google Scholar] [CrossRef] [PubMed]
- Mohr, A.; Brockmann, J.G.; Becker, F. HTK-N: Modified histidine-tryptophan-ketoglutarate solution—A promising new tool in solid organ preservation. Int. J. Mol. Sci. 2020, 21, 6468. [Google Scholar] [CrossRef] [PubMed]
- Saemann, L.; Georgevici, A.-I.; Hoorn, F.; Gharpure, N.; Veres, G.; Korkmaz-Icöz, S.; Karck, M.; Simm, A.; Wenzel, F.; Szabó, G. Improving diastolic and microvascular function in heart transplantation with donation after circulatory death. Int. J. Mol. Sci. 2023, 24, 11562. [Google Scholar] [CrossRef]
- Zimmer, R.J.; Lee, M.S. Transplant coronary artery disease. JACC Cardiovasc. Interv. 2010, 3, 367–377. [Google Scholar] [CrossRef] [PubMed]
- Dugina, V.B.; Shagieva, G.S.; Shakhov, A.S.; Alieva, I.B. The cytoplasmic actins in the regulation of endothelial cell function. Int. J. Mol. Sci. 2021, 22, 7836. [Google Scholar] [CrossRef]
- Ruter, D.L.; Liu, Z.; Ngo, K.M.; Marvin, A.; Buglak, D.B.; Kidder, E.J.; Bautch, V.L. SMAD6 transduces endothelial cell flow responses required for blood vessel homeostasis. Angiogenesis 2021, 24, 387–398. [Google Scholar] [CrossRef] [PubMed]
- Wylie, L.A.; Mouillesseaux, K.P.; Chong, D.C.; Bautch, V.L. Developmental SMAD6 loss leads to blood vessel hemorrhage and disrupted endothelial cell junctions. Dev. Biol. 2018, 442, 199–209. [Google Scholar] [CrossRef]
- Li, Y.; Yan, H.; Guo, J.; Han, Y.; Zhang, C.; Liu, X.; Du, J.; Tian, X.-L. Down-regulated RGS5 by genetic variants impairs endothelial cell function and contributes to coronary artery disease. Cardiovasc. Res. 2019, 117, 240–255. [Google Scholar] [CrossRef]
- Munshaw, S.; Redpath, A.N.; Pike, B.T.; Smart, N. Thymosin β4 preserves vascular smooth muscle phenotype in atherosclerosis via regulation of low density lipoprotein related protein 1 (LRP1). Int. Immunopharmacol. 2023, 115, 109702. [Google Scholar] [CrossRef] [PubMed]
- Wu, F.; Huang, W.; Tan, Q.; Guo, Y.; Cao, Y.; Shang, J.; Ping, F.; Wang, W.; Li, Y. ZFP36L2 regulates myocardial ischemia/reperfusion injury and attenuates mitochondrial fusion and fission by LncRNA PVT1. Cell Death Dis. 2021, 12, 614. [Google Scholar] [CrossRef] [PubMed]
- He, D.; Hu, J.; Lu, Y.; Jia, W.; Wei, M.; Zeng, X.; Wang, H. The Effect of miR-505-5p on Inhibition of serum uromodulin ameliorates myocardial inflammation and apoptosis induced by ischemia-reperfusion. Oxidative Med. Cell. Longev. 2022, 2022, 3521971. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Gong, G.; Wang, P.; Zhang, Z.; Kolwicz, S.C.; Rabinovitch, P.S.; Tian, R.; Wang, W. Heart specific knockout of Ndufs4 ameliorates ischemia reperfusion injury. J. Mol. Cell. Cardiol. 2018, 123, 38–45. [Google Scholar] [CrossRef] [PubMed]
- Heger, J.; Abdallah, Y.; Shahzad, T.; Klumpe, I.; Piper, H.; Schultheiss, H.-P.; Schlüter, K.-D.; Schulz, R.; Euler, G.; Dörner, A. Transgenic overexpression of the adenine nucleotide translocase 1 protects cardiomyocytes against TGFβ1-induced apoptosis by stabilization of the mitochondrial permeability transition pore. J. Mol. Cell. Cardiol. 2012, 53, 73–81. [Google Scholar] [CrossRef] [PubMed]
- Winter, J.; Hammer, E.; Heger, J.; Schultheiss, H.-P.; Rauch, U.; Landmesser, U.; Dörner, A. Adenine nucleotide translocase 1 expression is coupled to the HSP27-mediated TLR4 signaling in cardiomyocytes. Cells 2019, 8, 1588. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Hu, J.; Zhang, R.; Deng, J. CEBPD regulates oxidative stress and inflammatory responses in hypertensive cardiac remodeling. Shock 2023, 60, 713–723. [Google Scholar] [CrossRef] [PubMed]
- Tang, C.; Deng, L.; Luo, Q.; He, G. Identification of oxidative stress-related genes and potential mechanisms in atherosclerosis. Front. Genet. 2022, 13, 998954. [Google Scholar] [CrossRef]
- Chen, J.; Liu, X.; Bi, R.; Liu, P.; Gong, W. NDUFC2 deficiency exacerbates endothelial mesenchymal transformation during ischemia-reperfusion via NLRP3. NeuroReport 2023, 34, 670–676. [Google Scholar] [CrossRef] [PubMed]
- Saemann, L.; Korkmaz-Icöz, S.; Hoorn, F.; Veres, G.; Kraft, P.; Georgevici, A.-I.; Brune, M.; Guo, Y.; Loganathan, S.; Wenzel, F.; et al. Reconditioning of circulatory death hearts by ex-vivo machine perfusion with a novel HTK-N preservation solution. J. Heart Lung Transplant. 2021, 40, 1135–1144. [Google Scholar] [CrossRef]
- Zhang, H.; Taylor, W.R.; Joseph, G.; Caracciolo, V.; Gonzales, D.M.; Sidell, N.; Seli, E.; Blackshear, P.J.; Kallen, C.B.; MacLean, P.S.; et al. mRNA-binding protein ZFP36 is expressed in atherosclerotic lesions and reduces inflammation in aortic endothelial cells. Arterioscler. Thromb. Vasc. Biol. 2013, 33, 1212–1220. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Xu, H.; Li, Y.; Zhang, X.; Cui, J.; Zou, Y.; Yu, J.; Wu, J.; Xia, J. Single-Cell RNA sequencing reveals immune cell dynamics and local intercellular communication in acute murine cardiac allograft rejection. Theranostics 2022, 12, 6242–6257. [Google Scholar] [CrossRef]
- Arlt, A.; Schäfer, H. Role of the immediate early response 3 (IER3) gene in cellular stress response, inflammation and tumorigenesis. Eur. J. Cell Biol. 2011, 90, 545–552. [Google Scholar] [CrossRef]
- Chen, M.; Yi, B.; Zhu, N.; Wei, X.; Zhang, G.-X.; Huang, S.; Sun, J. Pim1 kinase promotes angiogenesis through phosphorylation of endothelial nitric oxide synthase at Ser-633. Cardiovasc. Res. 2016, 109, 141–150. [Google Scholar] [CrossRef] [PubMed]
- Mohsin, S.; Khan, M.; Toko, H.; Bailey, B.; Cottage, C.T.; Wallach, K.; Nag, D.; Lee, A.; Siddiqi, S.; Lan, F.; et al. Human cardiac progenitor cells engineered with Pim-I kinase enhance myocardial repair. J. Am. Coll. Cardiol. 2012, 60, 1278–1287. [Google Scholar] [CrossRef]
- Zou, R.; Zhang, M.; Zou, Z.; Shi, W.; Tan, S.; Wang, C.; Xu, W.; Jin, J.; Milton, S.; Chen, Y.; et al. Single-cell transcriptomics reveals zinc and copper ions homeostasis in epicardial adipose tissue of heart failure. Int. J. Biol. Sci. 2023, 19, 4036–4051. [Google Scholar] [CrossRef]
- Kume, T. Foxc2 transcription factor: A newly described regulator of angiogenesis. Trends Cardiovasc. Med. 2008, 18, 224–228. [Google Scholar] [CrossRef]
- Ranjbarvaziri, S.; Kooiker, K.B.; Ellenberger, M.; Fajardo, G.; Zhao, M.; Vander Roest, A.S.; Woldeyes, R.A.; Koyano, T.T.; Fong, R.; Ma, N.; et al. Altered cardiac energetics and mitochondrial dysfunction in hypertrophic cardiomyopathy. Circulation 2021, 144, 1714–1731. [Google Scholar] [CrossRef]
- Norton, M.; Ng, A.C.-H.; Baird, S.; Dumoulin, A.; Shutt, T.; Mah, N.; Andrade-Navarro, M.A.; McBride, H.M.; Screaton, R.A. ROMO1 is an essential redox-dependent regulator of mitochondrial dynamics. Sci. Signal. 2014, 7, ra10. [Google Scholar] [CrossRef]
- Taurino, C.; Miller, W.H.; McBride, M.W.; McClure, J.D.; Khanin, R.; Moreno, M.U.; Dymott, J.A.; Delles, C.; Dominiczak, A. Gene expression profiling in whole blood of patients with coronary artery disease. Clin. Sci. 2010, 119, 335–343. [Google Scholar] [CrossRef] [PubMed]
- Jones, R.E.; Gruszczyk, A.V.; Schmidt, C.; Hammersley, D.J.; Mach, L.; Lee, M.; Wong, J.; Yang, M.; Hatipoglu, S.; Lota, A.S.; et al. Assessment of left ventricular tissue mitochondrial bioenergetics in patients with stable coronary artery disease. Nat. Cardiovasc. Res. 2023, 2, 733–745. [Google Scholar] [CrossRef]
- Halloran, P.F.; Madill-Thomsen, K.; Mackova, M.; Aliabadi-Zuckermann, A.Z.; Cadeiras, M.; Crespo-Leiro, M.G.; Depasquale, E.C.; Deng, M.; Gökler, J.; Hall, S.A.; et al. Molecular states associated with dysfunction and graft loss in heart transplants. J. Heart Lung Transplant. 2023; in press. [Google Scholar] [CrossRef]
- Giarraputo, A.; Fedrigo, M.; Tona, F.; Rossi, E.; Barison, I.; Castellani, C.; Bottio, T.; Toscano, G.; Gerosa, G.; Mandruzzato, S.; et al. Gene network analysis of cardiac allograft vasculopathy in heart transplantation through messanger RNA expression profile. J. Heart Lung Transplant. 2021, 40, S236–S237. [Google Scholar] [CrossRef]
- Dambrova, M.; Zuurbier, C.J.; Borutaite, V.; Liepinsh, E.; Makrecka-Kuka, M. Energy substrate metabolism and mitochondrial oxidative stress in cardiac ischemia/reperfusion injury. Free. Radic. Biol. Med. 2021, 165, 24–37. [Google Scholar] [CrossRef]
- Verma, S.; Fedak, P.W.; Weisel, R.D.; Butany, J.; Rao, V.; Maitland, A.; Li, R.-K.; Dhillon, B.; Yau, T.M. Fundamentals of reperfusion injury for the clinical cardiologist. Circulation 2002, 105, 2332–2336. [Google Scholar] [CrossRef] [PubMed]
- Rauen, U.; Kerkweg, U.; de Groot, H. Iron-dependent vs. iron-independent cold-induced injury to cultured rat hepatocytes: A comparative study in physiological media and organ preservation solutions. Cryobiology 2007, 54, 77–86. [Google Scholar] [CrossRef]
- Grosser, N.; Oberle, S.; Berndt, G.; Erdmann, K.; Hemmerle, A.; Schröder, H. Antioxidant action of l-alanine: Heme oxygenase-1 and ferritin as possible mediators. Biochem. Biophys. Res. Commun. 2004, 314, 351–355. [Google Scholar] [CrossRef]
- Veres, G.; Radovits, T.; Merkely, B.; Karck, M.; Szabó, G. Custodiol-N, the novel cardioplegic solution reduces ischemia/reperfusion injury after cardiopulmonary bypass. J. Cardiothorac. Surg. 2015, 10, 27. [Google Scholar] [CrossRef]
- Saemann, L.; Wenzel, F.; Kohl, M.; Korkmaz-Icöz, S.; Hoorn, F.; Loganathan, S.; Guo, Y.; Ding, Q.; Zhou, P.; Veres, G.; et al. Monitoring of perfusion quality and prediction of donor heart function during ex-vivo machine perfusion by myocardial microcirculation versus surrogate parameters. J. Heart Lung Transplant. 2021, 40, 387–391. [Google Scholar] [CrossRef] [PubMed]
- Szklarczyk, D.; Kirsch, R.; Koutrouli, M.; Nastou, K.; Mehryary, F.; Hachilif, R.; Gable, A.L.; Fang, T.; Doncheva, N.T.; Pyysalo, S.; et al. The STRING database in 2023: Protein–protein association networks and functional enrichment analyses for any sequenced genome of interest. Nucleic Acids Res. 2023, 51, D638–D646. [Google Scholar] [CrossRef] [PubMed]
- Herwig, R.; Hardt, C.; Lienhard, M.; Kamburov, A. Analyzing and interpreting genome data at the network level with ConsensusPathDB. Nat. Protoc. 2016, 11, 1889–1907. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Fold Change | p-Value | FDR p-Value | Gene Symbol | Description |
|---|---|---|---|---|
| 10.36 | 1.78 × 10−5 | 0.0024 | CEBPD | CCAAT/enhancer binding protein (C/EBP), delta |
| 7 | 2.72 × 10−5 | 0.0029 | CCL2 | chemokine (C-C motif) ligand 2 |
| 6.89 | 4.92 × 10−6 | 0.0011 | IER3 | immediate early response 3 |
| 6.18 | 0.0003 | 0.0113 | ZFP36L2 | ZFP36 ring finger protein-like 2 |
| 5.87 | 2.68 × 10−5 | 0.0029 | ACTB; ACTG1 | actin, beta; actin gamma 1 |
| 5.31 | 0.0003 | 0.0114 | FOXC2 | forkhead box C2 |
| 5.25 | 1.56 × 10−5 | 0.0023 | ZFP36 | zinc finger protein 36, C3H type, homolog (mouse); ZFP36 ring finger protein |
| 4.73 | 0.0002 | 0.0081 | ATN1 | atrophin 1 |
| 4.36 | 0.0001 | 0.0075 | TMSB4X | thymosin beta 4, X-linked |
| 4.24 | 9.23 × 10−6 | 0.0016 | GJA1 | gap junction protein, alpha 1, 43kDa |
| 4.05 | 1.71 × 10−6 | 0.0008 | SNAI2 | snail homolog 2 (Drosophila); snail family zinc finger 2 |
| 4.02 | 1.46 × 10−5 | 0.0022 | PNRC1 | proline-rich nuclear receptor coactivator 1 |
| 3.98 | 7.96 × 10−5 | 0.0055 | SMAD6 | SMAD family member 6 |
| 3.86 | 0.0011 | 0.0258 | RNF149 | ring finger protein 149 |
| 3.84 | 0.0008 | 0.0211 | BTG2 | BTG family, member 2 |
| 3.62 | 0.0011 | 0.0262 | PIM1 | Pim-1 proto-oncogene, serine/threonine kinase |
| 3.61 | 0.0017 | 0.0344 | COL3A1 | collagen, type III, alpha 1 |
| 3.56 | 0.0066 | 0.0783 | MIR505 | microRNA mir-505 |
| 3.53 | 0.0080 | 0.0874 | RGS5 | regulator of G-protein signaling 5 |
| 3.47 | 5.39 × 10−5 | 0.0044 | SLC39A14 | solute carrier family 39 (zinc transporter), member 14 |
| Fold Change | p-Value | FDR p-Value | Gene Symbol | Description |
|---|---|---|---|---|
| −39.77 | 6.06 × 10−6 | 0.0013 | NDUFA1 | NADH dehydrogenase (ubiquinone) 1 alpha subcomplex, 1, 7.5 kDa |
| −30.89 | 2.24 × 10−6 | 0.0008 | TOMM7 | translocase of outer mitochondrial membrane 7 homolog (yeast) |
| −29.51 | 1.93 × 10−5 | 0.0025 | NDUFB2 | NADH dehydrogenase (ubiquinone) 1 beta subcomplex, 2, 8 kDa |
| −25.59 | 2.87 × 10−6 | 0.0009 | NDUFB1 | NADH dehydrogenase (ubiquinone) 1 beta subcomplex, 1, 7 kDa |
| −25.48 | 4.18 × 10−7 | 0.0005 | LOC100626068; IGIP | IgA-inducing protein |
| −24.94 | 2.62 × 10−6 | 0.0008 | NDUFS4 | NADH dehydrogenase (ubiquinone) Fe-S protein 4, 18 kDa (NADH-coenzyme Q reductase) |
| −24.47 | 1.98 × 10−7 | 0.0004 | TBCA | tubulin folding cofactor A |
| −18.96 | 3.04 × 10−5 | 0.0032 | NDUFC2 | NADH dehydrogenase (ubiquinone) 1, subcomplex unknown, 2, 14.5 kDa |
| −18.39 | 2.29 × 10−6 | 0.0008 | COX7A2 | COX7A2 protein |
| −18.36 | 1.20 × 10−6 | 0.0007 | USMG5 | upregulated during skeletal muscle growth 5 homolog (mouse) |
| −17.63 | 6.49 × 10−7 | 0.0005 | MGST2 | microsomal glutathione S-transferase 2 |
| −17.12 | 4.49 × 10−7 | 0.0005 | CHCHD7 | coiled-coil-helix-coiled-coil-helix domain containing 7 |
| −16.26 | 3.19 × 10−6 | 0.0009 | HSPE1 | heat shock 10 kDa protein 1 |
| −15.06 | 2.84 × 10−7 | 0.0004 | PCYT1A | phosphate cytidylyltransferase 1, choline, alpha |
| −14.3 | 1.14 × 10−5 | 0.0019 | NDUFB8 | NADH dehydrogenase (ubiquinone) 1 beta subcomplex, 8, 19 kDa |
| −14.11 | 6.51 × 10−6 | 0.0013 | ROMO1 | reactive oxygen species modulator 1 |
| −13.77 | 6.97 × 10−6 | 0.0014 | RPL31; LOC100520127 | ribosomal protein L31; 60S ribosomal protein L31 |
| −13.42 | 2.89 × 10−6 | 0.0009 | SNRPG | small nuclear ribonucleoprotein polypeptide G |
| −11.99 | 7.81 × 10−5 | 0.0054 | RNF113A | ring finger protein 113A |
| −11.94 | 3.75 × 10−5 | 0.0035 | MRPS18C | mitochondrial ribosomal protein S18C |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Saemann, L.; Wächter, K.; Gharpure, N.; Pohl, S.; Hoorn, F.; Korkmaz-Icöz, S.; Karck, M.; Veres, G.; Simm, A.; Szabó, G. HTK vs. HTK-N for Coronary Endothelial Protection during Hypothermic, Oxygenated Perfusion of Hearts Donated after Circulatory Death. Int. J. Mol. Sci. 2024, 25, 2262. https://doi.org/10.3390/ijms25042262
Saemann L, Wächter K, Gharpure N, Pohl S, Hoorn F, Korkmaz-Icöz S, Karck M, Veres G, Simm A, Szabó G. HTK vs. HTK-N for Coronary Endothelial Protection during Hypothermic, Oxygenated Perfusion of Hearts Donated after Circulatory Death. International Journal of Molecular Sciences. 2024; 25(4):2262. https://doi.org/10.3390/ijms25042262
Chicago/Turabian StyleSaemann, Lars, Kristin Wächter, Nitin Gharpure, Sabine Pohl, Fabio Hoorn, Sevil Korkmaz-Icöz, Matthias Karck, Gábor Veres, Andreas Simm, and Gábor Szabó. 2024. "HTK vs. HTK-N for Coronary Endothelial Protection during Hypothermic, Oxygenated Perfusion of Hearts Donated after Circulatory Death" International Journal of Molecular Sciences 25, no. 4: 2262. https://doi.org/10.3390/ijms25042262
APA StyleSaemann, L., Wächter, K., Gharpure, N., Pohl, S., Hoorn, F., Korkmaz-Icöz, S., Karck, M., Veres, G., Simm, A., & Szabó, G. (2024). HTK vs. HTK-N for Coronary Endothelial Protection during Hypothermic, Oxygenated Perfusion of Hearts Donated after Circulatory Death. International Journal of Molecular Sciences, 25(4), 2262. https://doi.org/10.3390/ijms25042262