
Advances and Challenges in Gene Therapy for Neurodegenerative Diseases: A Systematic Review

 , , , and
, , , and
Abstract
1. Introduction
1.1. Monogenic Diseases
- -
- Huntington’s disease (HD) is a hereditary degenerative disorder characterized by the expansion of CAG triplet repeats (>36) in the first exon of the HTT gene located on chromosome 4q. The repetition of amino acid glutamine causes the production of an abnormal huntingtin (HTT) protein [6]. This protein aggregates causing the extensive degeneration of the cortex and basal ganglia [4,16], with the severity of symptoms correlating with the number of glutamine repeats [17].
- -
- Spinal muscular atrophy (SMA) is an autosomal recessive neurodegenerative disease caused by a biallelic mutation of the neuronal survival gene SMN1 on chromosome 5q. It is the main cause of childhood mortality of genetic origin, due to a protein deficiency that results in the progressive degeneration of motor neurons, leading to significant weakness and muscle atrophy [1,18]. The severity of SMA depends on the number of functional SMN2 gene copies, which modulate the harmful effects of SMN1 deficiency, affecting the age of onset and prognosis [19,20,21].
1.2. Polygenic Diseases
- -
- Alzheimer’s disease (AD) is the leading cause of dementia [24]. Although the pathogenesis of the sporadic form is unknown, familial AD (<5% of cases) shows mutations in genes coding for amyloid-beta precursor protein (APP), presenilin 1 and 2 (PSEN1-2), and microtubule-associated protein Tau (MAPT). The accumulation of β-amyloid plaques and tangles of hyperphosphorylated tau is associated with memory loss and cognitive decline, with death typically occurring 5–12 years after diagnosis [43].
- -
- Parkinson’s disease (PD) is characterized by resting tremor, bradykinesia, and postural instability, along with cognitive impairment [33,44]. Motor dysfunction is produced by the degradation of dopaminergic neurons in the substantia nigra pars compacta [36,45,46]. PD can be sporadic (10–15%) or familial (85–90%). The average age of onset for PD is estimated to be around 60 years, though some patients experience significantly earlier onset. PD is linked to increased mortality and a reduced life expectancy compared to the general population, particularly when diagnosed before the age of 70 [47,48]. Alterations have been identified in the membrane-interacting gene α-synuclein (SNCA), [49,50]; in mitochondrial quality control genes, such as phosphatase and tensin homolog (PTEN) and leucine-rich repeat kinase 2 (LRRK2) [49,51]; in oxidative damage control genes, such as the gene that encodes the parkinsonism associated deglycase (PARK7); and the gene that encodes the lysosomal enzyme glucosylceramidase Beta 1 (GBA1) [52].
- -
- Amyotrophic lateral sclerosis (ALS) is a neurodegenerative disorder affecting motor neurons, with both familial (20%) and sporadic (80%) forms [53]. In both, protein aggregation leads to neuronal degeneration, which leads to progressive paralysis and, often, death within 3–5 years [54]. ALS is frequently associated with the mutation of the superoxide dismutase 1 (SOD1) gene, causing the accumulation of abnormal SOD1 protein [54], and with the mutation of the TAR DNA-binding protein 43 (TDP-43) gene. The TDP-43 protein plays a crucial role in the regulation of RNA splicing, which is essential for the proper expression of genes. Additionally, the dysregulation of TDP-43 has been linked to alterations in neurotransmitter systems, particularly affecting gamma-aminobutyric acid (GABA), an important inhibitory neurotransmitter in the brain. These changes can disrupt neural signaling and may contribute to various NDDs [42,55].
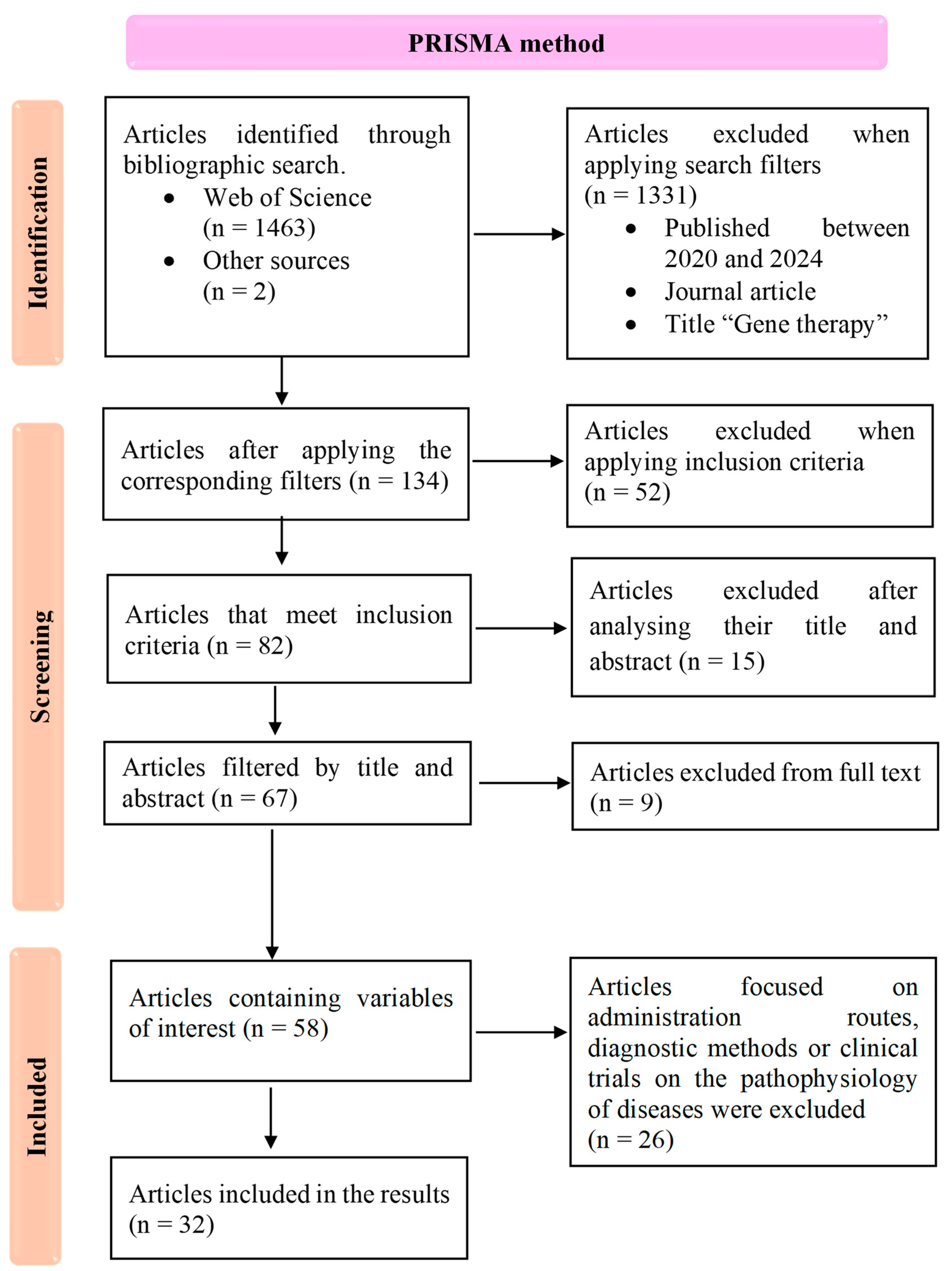
2. Methods
2.1. Search Strategy
2.2. Selection Process and Critical Appraisal
3. Results
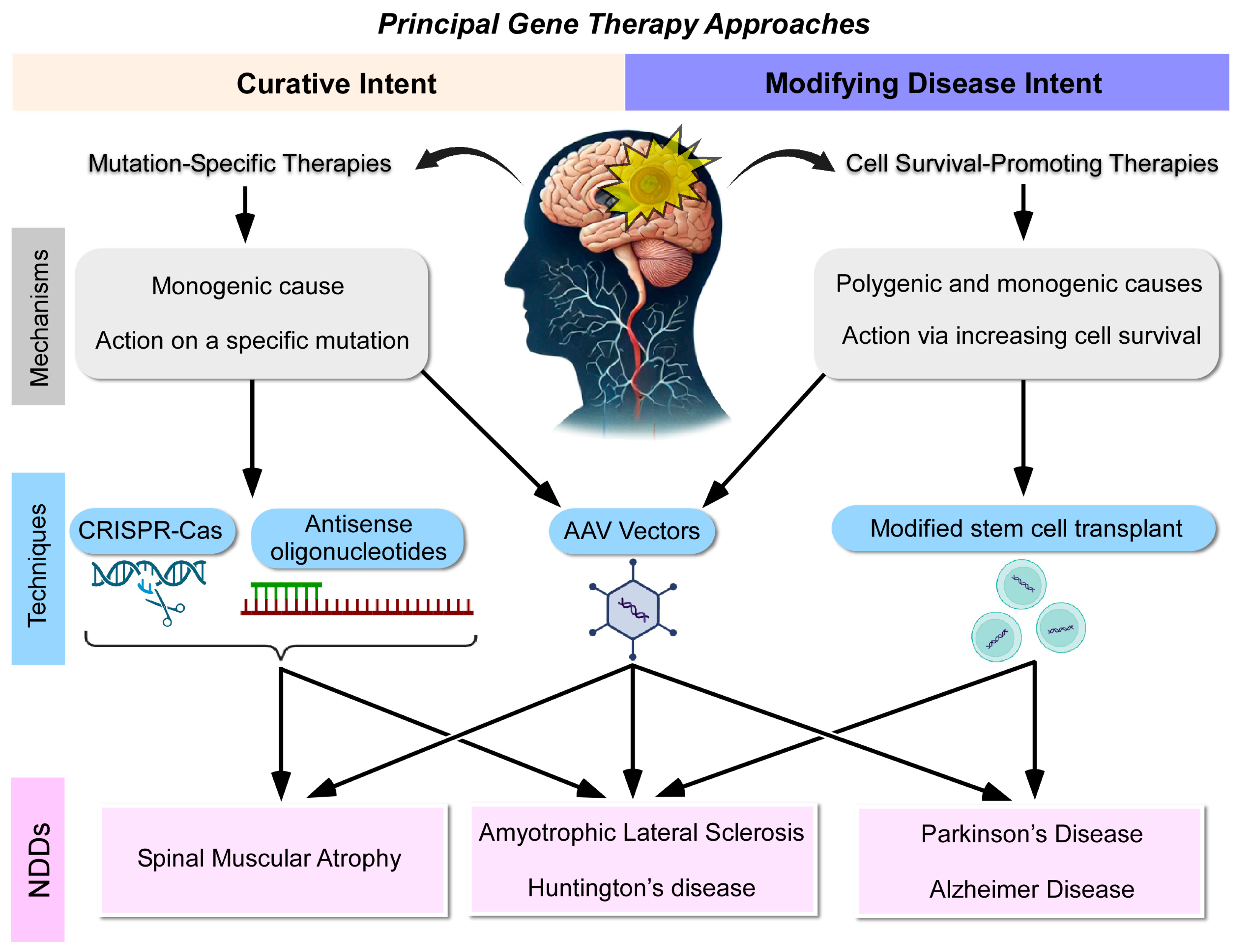
3.1. Therapies with Curative Intent
3.2. Therapies with Modifying Intent
4. Discussion
5. Conclusions
- -
- Gene therapy for NDDs offers significant promise for developing interventions that can either slow disease progression or, in some cases, provide curative outcomes;
- -
- Therapies with curative intent focus on addressing specific genetic mutations, while disease-modifying therapies aim to enhance cell survival through the administration of neurotrophic factors or neuroprotective agents;
- -
- Therapies with curative intent that have shown promising results in terms of clinical significance include the introduction of a gene or oligonucleotide, particularly for SMA and gene editing techniques for ALS. However, these approaches have not yet achieved meaningful clinical results for HD;
- -
- Therapies with modifying intent have demonstrated significant clinical results in PD, using both stem cells and the administration of a neurotrophic factor using vectors. Advances have also been made in HD with stem cell therapies and in ALS with the introduction of protective factors through viral vectors or oligonucleotides. However, in AD, the results obtained so far are more limited, highlighting the complexity of targeting polygenic diseases;
- -
- Our analysis identified shared pathogenic pathways among certain NDDs, which may present viable targets for gene therapy. This is particularly evident between PD and AD, as well as HD, ALS, and SMA;
- -
- According to the characteristics described of each administration route for gene therapy and based on their ability to effectively cross the BBB and target the central nervous system, the optimal routes for gene therapy administration in NDDs appear to be the following: intra-arterial, intravenous with ultrasound facilitation, as well as intrathecal delivery;
- -
- Overall, gene therapy offers a promising avenue for both curative and disease-modifying treatments, but further research is necessary to refine these approaches and achieve more widespread clinical success.
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- McMillan, H.J.; Proud, C.M.; Farrar, M.A.; Alexander, I.E.; Muntoni, F.; Servais, L. Onasemnogene abeparvovec for the treatment of spinal muscular atrophy. Expert. Opin. Biol. Ther. 2022, 22, 1075–1090. [Google Scholar] [CrossRef] [PubMed]
- Pauwels, E.K.J.; Boer, G.J. Parkinson’s Disease; a Tale of Many Players. Med. Princ. Pract. 2023, 32, 155–165. [Google Scholar] [CrossRef] [PubMed]
- Fischell, J.M.; Fishman, P.S. A Multifaceted Approach to Optimizing AAV Delivery to the Brain for the Treatment of Neurodegenerative Diseases. Front. Neurosci. 2021, 15, 747726. [Google Scholar] [CrossRef] [PubMed]
- Wenceslau, C.V.; de Souza, D.M.; Mambelli-Lisboa, N.C.; Ynoue, L.H.; Araldi, R.P.; da Silva, J.M.; Pagani, E.; Haddad, M.S.; Kerkis, I. Restoration of BDNF, DARPP32, and D2R Expression Following Intravenous Infusion of Human Immature Dental Pulp Stem Cells in Huntington’s Disease 3-NP Rat Model. Cells 2022, 11, 1664. [Google Scholar] [CrossRef] [PubMed]
- Macedo, J.; Pagani, E.; Wenceslau, C.V.; Ferrara, L.; Kerkis, I. A Phase I clinical trial on intravenous administration of immature human dental pulp stem cells (Nestacell HDTM) to Huntington’s Disease patients. Cytotherapy 2021, 23, 1. [Google Scholar] [CrossRef]
- Kotowska-Zimmer, A.; Przybyl, L.; Pewinska, M.; Suszynska-Zajczyk, J.; Wronka, D.; Figiel, M.; Olejniczak, M. A CAG repeat-targeting artificial miRNA lowers the mutant huntingtin level in the YAC128 model of Huntington’s disease. Mol. Ther.-Nucleic Acids 2022, 28, 702–715. [Google Scholar] [CrossRef]
- Ferlazzo, G.M.; Gambetta, A.M.; Amato, S.; Cannizzaro, N.; Angiolillo, S.; Arboit, M.; Diamante, L.; Carbognin, E.; Romani, P.; La Torre, F.; et al. Genome-wide screening in pluripotent cells identifies Mtf1 as a suppressor of mutant huntingtin toxicity. Nat. Commun. 2023, 14, 3962. [Google Scholar] [CrossRef]
- Mendell, J.R.; Al-Zaidy, S.A.; Lehman, K.J.; McColly, M.; Lowes, L.P.; Alfano, L.N.; Reash, N.F.; Iammarino, M.A.; Church, K.R.; Kleyn, A.; et al. Five-Year Extension Results of the Phase 1 START Trial of Onasemnogene Abeparvovec in Spinal Muscular Atrophy. JAMA Neurol. 2021, 78, 834–841. [Google Scholar] [CrossRef]
- Strauss, K.A.; Farrar, M.A.; Muntoni, F.; Saito, K.; Mendell, J.R.; Servais, L.; McMillan, H.J.; Finkel, R.S.; Swoboda, K.J.; Kwon, J.M.; et al. Onasemnogene abeparvovec for presymptomatic infants with two copies of SMN2 at risk for spinal muscular atrophy type 1: The Phase III SPR1NT trial. Nat. Med. 2022, 28, 1381. [Google Scholar] [CrossRef]
- Stettner, G.M.; Hasselmann, O.; Tscherter, A.; Galiart, E.; Jacquier, D.; Klein, A. Treatment of spinal muscular atrophy with Onasemnogene Abeparvovec in Switzerland: A prospective observational case series study. BMC Neurol. 2023, 23, 88. [Google Scholar] [CrossRef]
- Pane, M.; Coratti, G.; Pera, M.C.; Sansone, V.A.; Messina, S.; d’Amico, A.; Bruno, C.; Salmin, F.; Albamonte, E.; De Sanctis, R.; et al. Nusinersen efficacy data for 24-month in type 2 and 3 spinal muscular atrophy. Ann. Clin. Transl. Neurol. 2022, 9, 404–409. [Google Scholar] [CrossRef] [PubMed]
- Pane, M.; Berti, B.; Capasso, A.; Coratti, G.; Varone, A.; D’Amico, A.; Bruno, C.; Ricci, F.; Pini, A.; Gagliardi, D.; et al. Onasemnogene abeparvovec in spinal muscular atrophy: Predictors of efficacy and safety in naive patients with spinal muscular atrophy and following switch from other therapies. EClinicalMedicine 2023, 59, 101997. [Google Scholar] [CrossRef] [PubMed]
- Chand, D.H.; Mitchell, S.; Sun, R.; LaMarca, N.; Reyna, S.P.; Sutter, T. Research Paper Safety of Onasemnogene Abeparvovec for Patients With Spinal Muscular Atrophy 8.5 kg or Heavier in a Global Managed Access Program. Pediatr. Neurol. 2022, 132, 27–32. [Google Scholar] [CrossRef] [PubMed]
- Besse, A.; Astord, S.; Marais, T.; Roda, M.; Giroux, B.; Lejeune, F.X.; Relaix, F.; Smeriglio, P.; Barkats, M.; Biferi, M.G. AAV9- Mediated Expression of SMN Restricted to Neurons Does Not Rescue the Spinal Muscular Atrophy Phenotype in Mice. Mol. Ther. 2020, 28, 1887–1901. [Google Scholar] [CrossRef] [PubMed]
- Rich, K.A.; Wier, C.G.; Russo, J.; Kong, L.; Heilman, P.L.; Reynolds, A.; Knapp, A.; Pino, M.G.; Keckley, E.; Mattox, L.; et al. Premature delivery in the domestic sow in response to in utero delivery of AAV9 to fetal piglets. Gene Ther. 2022, 29, 513–519. [Google Scholar] [CrossRef]
- Morais, R.D.V.S.; Sogorb-Gonzalez, M.; Bar, C.; Timmer, N.C.; Van der Bent, M.L.; Wartel, M.; Vallès, A. Functional Intercellular Transmission of miHTT via Extracellular Vesicles: An In Vitro Proof-of-Mechanism Study. Cells 2022, 11, 2748. [Google Scholar] [CrossRef]
- Cummings, D.M.; Alaghband, Y.; Hickey, M.A.; Joshi, P.R.; Hong, S.C.; Zhu, C.; Ando, T.K.; André, V.M.; Cepeda, C.; Watson, J.B.; et al. A critical window of CAG repeat-length correlates with phenotype severity in the R6/2 mouse model of Huntington’s disease. J. Neurophysiol. 2012, 107, 677–691. [Google Scholar] [CrossRef]
- Day, J.W.; Mendell, J.R.; Mercuri, E.; Finkel, R.S.; Strauss, K.A.; Kleyn, A.; Tauscher-Wisniewski, S.; Tukov, F.F.; Reyna, S.P.; Chand, D.H. Clinical Trial and Postmarketing Safety of Onasemnogene Abeparvovec Therapy. Drug Saf. 2021, 44, 1109–1119. [Google Scholar] [CrossRef]
- Retson, L.; Tiwari, N.; Vaughn, J.; Bernes, S.; Adelson, P.D.; Mansfield, K.; Libertini, S.; Kuzmiski, B.; Alecu, I.; Gabriel, R. Epithelioid neoplasm of the spinal cord in a child with spinal muscular atrophy treated with onasemnogene abeparvovec. Mol. Ther. 2023, 31, 2991–2998. [Google Scholar] [CrossRef]
- Hirunagi, T.; Sahashi, K.; Meilleur, K.G.; Katsuno, M. Nucleic Acid-Based Therapeutic Approach for Spinal and Bulbar Muscular Atrophy and Related Neurological Disorders. Genes 2022, 13, 109. [Google Scholar] [CrossRef]
- Zhou, M.; Tang, S.; Duan, N.; Xie, M.; Li, Z.; Feng, M.; Wu, L.; Hu, Z.; Liang, D. Targeted-Deletion of a Tiny Sequence via Prime Editing to Restore SMN Expression. Int. J. Mol. Sci. 2022, 23, 7941. [Google Scholar] [CrossRef] [PubMed]
- Jeon, M.T.; Moon, G.J.; Kim, S.; Choi, M.; Oh, Y.S.; Kim, D.W.; Kim, H.J.; Lee, K.J.; Choe, Y.; Ha, C.M.; et al. Neurotrophic interactions between neurons and astrocytes following AAV1-Rheb(S16H) transduction in the hippocampus in vivo. Br. J. Pharmacol. 2020, 177, 668–686. [Google Scholar] [CrossRef] [PubMed]
- Capsoni, S.; Cattaneo, A. Getting Into the Brain: The Intranasal Approach to Enhance the Delivery of Nerve Growth Factor and Its Painless Derivative in Alzheimer’s Disease and Down Syndrome. Front. Neurosci. 2022, 16, 773347. [Google Scholar] [CrossRef] [PubMed]
- Mishra, P.; Silva, A.; Sharma, J.; Nguyen, J.; Pizzo, D.P.; Hinz, D.; Sahoo, D.; Cherqui, S. Rescue of Alzheimer’s disease phenotype in a mouse model by transplantation of wild-type hematopoietic stem and progenitor cells. Cell Rep. 2023, 42, 112956. [Google Scholar] [CrossRef] [PubMed]
- Rocco, M.T.; Akhter, A.S.; Ehrlich, D.J.; Scott, G.C.; Lungu, C.; Munjal, V.; Aquino, A.; Lonser, R.R.; Fiandaca, M.S.; Hallett, M.; et al. Long-term safety of MRI-guided administration of AAV2-GDNF and gadoteridol in the putamen of individuals with Parkinson’s disease. Mol. Therapy 2022, 30, 3632–3638. [Google Scholar] [CrossRef]
- Onuki, Y.; Ono, S.; Nakajima, T.; Kojima, K.; Taga, N.; Ikeda, T.; Kuwajima, M.; Kurokawa, Y.; Kato, M.; Kawai, K.; et al. Dopaminergic restoration of prefrontal cortico-putaminal network in gene therapy for aromatic l-amino acid decarboxylase deficiency. Brain Commun. 2021, 3, fcab078. [Google Scholar] [CrossRef]
- Tai, C.H.; Lee, N.C.; Chien, Y.H.; Byrne, B.J.; Muramatsu, S.H.; Tseng, S.H.; Hwu, W.L. Long-term efficacy and safety of eladocagene exuparvovec in patients with AADC deficiency. Mol. Ther. 2022, 30, 509–518. [Google Scholar] [CrossRef]
- Chen, C.; Guderyon, M.J.; Li, Y.; Ge, G.; Bhattacharjee, A.; Ballard, C.; He, Z.; Masliah, E.; Clark, R.A.; O’Connor, J.C.; et al. Non-toxic HSC Transplantation-Based Macrophage/Microglia-Mediated GDNF Delivery for Parkinson’s Disease. Mol. Ther.-Methods Clin. Dev. 2020, 17, 83–98. [Google Scholar] [CrossRef]
- Gupta, R.; Santiago-Lopez, A.J.; Berglund, K.; Gross, R.E.; Gutekunst, C.A.N. In Vivo Assessment of Cell Death and Nigrostriatal Pathway Integrity Following Continuous Expression of C3 Transferase. Neuroscience 2020, 442, 183–192. [Google Scholar] [CrossRef]
- Wang, F.; Li, N.; Hou, R.; Wang, L.; Zhang, L.; Li, C.; Zhang, Y.; Yin, Y.; Chang, L.; Cheng, Y.; et al. Treatment of Parkinson’s disease using focused ultrasound with GDNF retrovirus-loaded microbubbles to open the blood-brain barrier. Open Chem. 2020, 18, 882–889. [Google Scholar] [CrossRef]
- Cui, K.; Yang, F.; Tufan, T.; Raza, M.U.; Zhan, Y.; Fan, Y.; Zeng, F.; Brown, R.W.; Price, J.B.; Jones, T.C.; et al. Restoration of Noradrenergic Function in Parkinson’s Disease Model Mice. ASN Neuro 2021, 13, 17590914211009730. [Google Scholar] [CrossRef] [PubMed]
- Fernandez-Parrilla, M.A.; Reyes-Corona, D.; Flores-Martinez, Y.M.; Nadella, R.; Bannon, M.J.; Escobedo, L.; Maldonado-Berny, M.; Santoyo-Salazar, J.; Soto-Rojas, L.O.; Luna-Herrera, C.; et al. Cerebral dopamine neurotrophic factor transfection in dopamine neurons using neurotensin-polyplex nanoparticles reverses 6-hydroxydopamine-induced nigrostriatal neurodegeneration. Neural Regen. Res. 2022, 17, 854. [Google Scholar] [PubMed]
- Kip, E.; Bentall, L.; Underwood, C.F.; Hughes, S.M.; Parr-Brownlie, L.C. Patterned Stimulation of the Chrimson Opsin in Glutamatergic Motor Thalamus Neurons Improves Forelimb Akinesia in Parkinsonian Rats. Neuroscience 2022, 507, 64–78. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Li, N.; Wei, J.; Feng, C.; Chen, Y.; Chen, T.; Ai, Z.; Zhu, X.; Ji, W.; Li, T. Genetically engineered mesenchymal stem cells with dopamine synthesis for Parkinson’s disease in animal models. NPJ Park. Dis. 2022, 8, 175. [Google Scholar] [CrossRef] [PubMed]
- Azevedo, M.D.; Prince, N.; Humbert-Claude, M.; Mesa-Infante, V.; Jeanneret, C.; Golzne, V.; De Matos, K.; Jamot, B.B.; Magara, F.; Gonzalez-Hernandez, T.; et al. Oxidative stress induced by sustained supraphysiological intrastriatal GDNF delivery is prevented by dose regulation. Mol. Ther.-Methods Clin. Dev. 2023, 31, 101106. [Google Scholar] [CrossRef]
- Narvaez-Perez, L.F.; Paz-Bermudez, F.; Avalos-Fuentes, J.A.; Campos-Romo, A.; Floran-Garduno, B.; Segovia, J. CRISPR/sgRNA-directed synergistic activation mediator (SAM) as a therapeutic tool for Parkinson′s disease. Gene Ther. 2023, 31, 31–44. [Google Scholar] [CrossRef]
- Miller, T.; Cudkowicz, M.; Shaw, P.J.; Andersen, P.M.; Atassi, N.; Bucelli, R.C.; Genge, A.; Glass, J.; Ladha, S.; Ludolph, A.L.; et al. Phase 1–2 Trial of Antisense Oligonucleotide Tofersen for SOD1 ALS. N. Engl. J. Med. 2020, 383, 109–119. [Google Scholar] [CrossRef]
- Mueller, C.; Berry, J.D.; McKenna-Yasek, D.M.; Gernoux, G.; Owegi, M.A.; Pothier, L.M.; Douthwright, C.L.; Gelevski, D.; Luppino, S.D.; Blackwood, M.; et al. SOD1 Suppression with Adeno-Associated Virus and MicroRNA in Familial ALS. N. Engl. J. Med. 2020, 383, 151–158. [Google Scholar] [CrossRef]
- Lim, C.K.W.; Gapinske, M.; Brooks, A.K.; Woods, W.S.; Powell, J.E.; Zeballos, M.A.C.; Winter, J.; Perez-Pinera, P.; Gaj, T. Treatment of a Mouse Model of ALS by In Vivo Base Editing. Mol. Ther. 2020, 28, 1177–1189. [Google Scholar] [CrossRef]
- Nizzardo, M.; Taiana, M.; Rizzo, F.; Benitez, J.A.; Nijssen, J.; Allodi, I.; Melzi, V.; Bresolin, N.; Comi, G.P.; Hedlund, E.; et al. Synaptotagmin 13 is neuroprotective across motor neuron diseases. Acta Neuropathol. 2020, 139, 837–853. [Google Scholar] [CrossRef]
- Wang, S.; Ichinomiya, T.; Savchenko, P.; Wang, D.; Sawada, A.; Li, X.; Duong, T.; Li, W.; Bonds, J.A.; Kim, E.J.; et al. Subpial delivery of adeno-associated virus 9-synapsin- caveolin-1 (AAV9-SynCavl) preserves motor neuron and neuromuscular junction morphology, motor function, delays disease onset, and extends survival in hSOD1G93A mice. Theranostics 2022, 12, 5389–5403. [Google Scholar] [CrossRef] [PubMed]
- Tejwani, L.; Jung, Y.; Kokubu, H.; Sowmithra, S.; Ni, L.; Lee, C.; Sanders, B.; Lee, P.J.; Xiang, Y.; Luttik, K.; et al. Reduction of nemo-like kinase increases lysosome biogenesis and ameliorates TDP-43-related neurodegeneration. J. Clin. Investig. 2023, 133, e138207. [Google Scholar] [CrossRef] [PubMed]
- Zeng, L.; Jiang, H.L.; Ashraf, G.M.; Li, Z.R.; Liu, R. MicroRNA and mRNA profiling of cerebral cortex in a transgenic mouse model of Alzheimer’s disease by RNA sequencing. Neural Regen. Res. 2021, 16, 2099. [Google Scholar] [PubMed]
- Renko, J.M.; Mahato, A.K.; Visnapuu, T.; Valkonen, K.; Karelson, M.; Voutilainen, M.H.; Saarma, M.; Tuominen, R.K.; Sidorova, Y.A. Neuroprotective Potential of a Small Molecule RET Agonist in Cultured Dopamine Neurons and Hemiparkinsonian Rats. J. Park. Dis. 2021, 11, 1023–1046. [Google Scholar] [CrossRef] [PubMed]
- Soner, B.C.; Acikgoz, E.; Inan, S.Y.; Ayla, S.; Sahin, A.S.; Oktem, G. Neuroprotective Effect of Intrastriatal Caffeic Acid Phenethyl Ester Treatment in 6-OH Dopamine Model of Parkinson’s Disease in Rats. Park. Dis. 2021, 2021, 5553480. [Google Scholar] [CrossRef]
- Feher, M.; Marton, Z.; Szabo, A.; Kocsa, J.; Kormos, V.; Hunyady, A.; Kovács, L.A.; Ujvári, B.; Berta, G.; Farkas, J.; et al. Downregulation of PACAP and the PAC1 Receptor in the Basal Ganglia, Substantia Nigra and Centrally Projecting Edinger-Westphal Nucleus in the Rotenone model of Parkinson’s Disease. Int. J. Mol. Sci. 2023, 24, 11843. [Google Scholar] [CrossRef]
- Dommershuijsen, L.J.; Heshmatollah, A.; Darweesh, S.K.L.; Koudstaal, P.J.; Ikram, M.A.; Ikram, M.K. Life expectancy of parkinsonism patients in the general population. Park. Relat. Disord. 2020, 77, 94–99. [Google Scholar] [CrossRef]
- Ishihara, L.S.; Cheesbrough, A.; Brayne, C.; Schrag, A. Estimated life expectancy of Parkinson’s patients compared with the UK population. J. Neurol. Neurosurg. Psychiatry 2007, 78, 1304–1309. [Google Scholar] [CrossRef]
- Song, J.; Liu, L.; Li, Z.; Mao, T.; Zhang, J.; Zhou, L.; Chen, X.; Shang, Y.; Sun, T.; Luo, Y.; et al. Lycium barbarum polysaccharide improves dopamine metabolism and symptoms in an MPTP-induced model of Parkinson’s disease. BMC Med. 2022, 20, 412. [Google Scholar] [CrossRef]
- Challis, C.; Hori, A.; Sampson, T.R.; Yoo, B.B.; Challis, R.C.; Hamilton, A.M.; Mazmanian, S.K.; Volpicelli-Daley, L.A.; Gradinaru, V. Gut-seeded α-synuclein fibrils promote gut dysfunction and brain pathology specifically in aged mice. Nat. Neurosci. 2020, 23, 327. [Google Scholar] [CrossRef]
- Lee, J.H.; Han, J.H.; Joe, E.H.; Jou, I. Small heterodimer partner (SHP) aggravates ER stress in Parkinson’s disease-linked LRRK2 mutant astrocyte by regulating XBP1 SUMOylation. J. Biomed. Sci. 2021, 28, 51. [Google Scholar] [CrossRef] [PubMed]
- Kumar, M.; Srikanth, M.P.; Deleidi, M.; Hallett, P.J.; Isacson, O.; Feldman, R.A. Acid ceramidase involved in pathogenic cascade leading to accumulation of α-synuclein in iPSC model of GBA1-associated Parkinson’s disease. Hum. Mol. Genet. 2023, 32, 1888–1900. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Sanchez, S.; Valiente, N.; Sesena, S.; Cabrera-Pinto, M.; Rodriguez, A.; Aranda, A.; Palop, L.; Fernández-Martos, C.M. Ozone modified hypothalamic signaling enhancing thermogenesis in the TDP-43A315T transgenic model of Amyotrophic Lateral Sclerosis. Sci. Rep. 2022, 12, 20814. [Google Scholar] [CrossRef] [PubMed]
- Gao, T.; Huo, J.; Xin, C.; Yang, J.; Liu, Q.; Dong, H.; Li, R.; Liu, Y. Protective effects of intrathecal injection of AAV9-RabGGTB-GFP+ in SOD1G93A mice. Front. Aging Neurosci. 2023, 15, 1092607. [Google Scholar] [CrossRef] [PubMed]
- Romano, G.; Holodkov, N.; Klima, R.; Feiguin, F. TDP-43 regulates GAD1 mRNA splicing and GABA signaling in Drosophila CNS. Sci. Rep. 2021, 11, 18761. [Google Scholar] [CrossRef]
- Kay, M.A.; Glorioso, J.C.; Naldini, L. Viral vectors for gene therapy: The art of turning infectious agents into vehicles of therapeutics. Nat. Med. 2001, 7, 33–40. [Google Scholar] [CrossRef]
- Akkewar, A.; Mahajan, N.; Kharwade, R.; Gangane, P. Liposomes in the Targeted Gene Therapy of Cancer: A Critical Review. Curr. Drug Deliv. 2023, 20, 350–370. [Google Scholar] [CrossRef]
- Luiz, M.T.; Tofani, L.B.; Araújo, V.H.S.; Di Filippo, L.D.; Duarte, J.L.; Marchetti, J.M.; Chorilli, M. Gene Therapy Based on Lipid Nanoparticles as Non-viral Vectors for Glioma Treatment. Curr. Gene Ther. 2021, 21, 452–463. [Google Scholar] [CrossRef]
- Kojima, R.; Bojar, D.; Rizzi, G.; Hamri, G.C.; El-Baba, M.D.; Saxena, P.; Ausländer, S.; Tan, K.R.; Fussenegger, M. Designer exosomes produced by implanted cells intracerebrally deliver therapeutic cargo for Parkinson’s disease treatment. Nat. Commun. 2018, 9, 1305. [Google Scholar] [CrossRef]
- Hinderer, C.; Katz, N.; Dyer, C.; Goode, T.; Johansson, J.; Bell, P.; Richman, L.; Buza, E.; Wilson, J.M. Translational Feasibility of Lumbar Puncture for Intrathecal AAV Administration. Mol. Ther.-Methods Clin. Dev. 2020, 17, 969–974. [Google Scholar] [CrossRef]
- Salafutdinov, I.I.; Gatina, D.Z.; Markelova, M.I.; Garanina, E.E.; Malanin, S.Y.; Gazizov, I.M.; Izmailov, A.A.; Rizvanov, A.A.; Islamov, R.R.; Palotás, A.; et al. A Biosafety Study of Human Umbilical Cord Blood Mononuclear Cells Transduced with Adenoviral Vector Carrying Human Vascular Endothelial Growth Factor cDNA In Vitro. Biomedicines 2023, 11, 2020. [Google Scholar] [CrossRef] [PubMed]
- Plasschaert, R.N.; DeAndrade, M.P.; Hull, F.; Nguyen, Q.; Peterson, T.; Yan, A.; Loperfido, M.; Baricordi, C.; Barbarossa, L.; Yoon, J.K.; et al. High-throughput analysis of hematopoietic stem cell engraftment after intravenous and intracerebroventricular dosing. Mol. Ther. 2022, 30, 3209–3225. [Google Scholar] [CrossRef] [PubMed]
- Dowdy, S.F.; Setten, R.L.; Cui, X.S.; Jadhav, S.G. Delivery of RNA Therapeutics: The Great Endosomal Escape! Nucleic Acid. Ther. 2022, 32, 361–368. [Google Scholar] [CrossRef] [PubMed]
- Palfi, S.; Gurruchaga, J.M.; Ralph, G.S.; Lepetit, H.; Lavisse, S.; Buttery, P.C.; Watts, C.; Miskin, J.; Kelleher, M.; Deeley, S.; et al. Long-term safety and tolerability of ProSavin, a lentiviral vector-based gene therapy for Parkinson’s disease: A dose escalation, open-label, phase 1/2 trial. Lancet 2014, 383, 1138–1146. [Google Scholar] [CrossRef] [PubMed]
- Shirley, J.L.; de Jong, Y.P.; Terhorst, C.; Herzog, R.W. Immune Responses to Viral Gene Therapy Vectors. Mol. Ther. 2020, 28, 709–722. [Google Scholar] [CrossRef]
- Kakoty, V.; Sarathlal, K.C.; Dubey, S.K.; Yang, C.H. Lentiviral mediated gene delivery as an effective therapeutic approach for Parkinson disease. Neurosci. Lett. 2021, 750, 135769. [Google Scholar] [CrossRef]
- Weiss, A.R.; Liguore, W.A.; Domire, J.S.; Button, D.; McBride, J.L. Intra-striatal AAV2.retro administration leads to extensive retrograde transport in the rhesus macaque brain: Implications for disease modeling and therapeutic development. Sci. Rep. 2020, 10, 6970. [Google Scholar] [CrossRef]
- Jang, S.; Shen, H.K.; Ding, X.; Miles, T.F.; Gradinaru, V. Structural basis of receptor usage by the engineered capsid AAV-PHP.eB. Mol. Ther.-Methods Clin. Dev. 2022, 26, 343–354. [Google Scholar] [CrossRef]
- Blesa, J.; Pineda-Pardo, J.A.; Inoue, K.I.; Gasca-Salas, C.; Balzano, T.; Del Rey, N.L.G.; Reinares-Sebastián, A.; Esteban-García, N.; Rodríguez-Rojas, R.; Márquez, R.; et al. BBB opening with focused ultrasound in nonhuman primates and Parkinson’s disease patients: Targeted AAV vector delivery and PET imaging. Sci. Adv. 2023, 9, eadf4888. [Google Scholar] [CrossRef]
- Mendes, B.B.; Conniot, J.; Avital, A.; Yao, D.; Jiang, X.; Zhou, X.; Sharf-Pauker, N.; Xiao, Y.; Adir, O.; Liang, H.; et al. Nanodelivery of nucleic acids. Nat. Rev. Methods Primers 2022, 2, 24. [Google Scholar] [CrossRef]
- Ereej, N.; Hameed, H.; Khan, M.A.; Faheem, S.; Hameed, A. Nanoparticle-based Gene Therapy for Neurodegenerative Disorders. Mini-Rev. Med. Chem. 2024, 24, 1723–1745. [Google Scholar] [CrossRef] [PubMed]
- Soini, V.; Schreiber, G.; Wilken, B.; Hell, A.K. Early Development of Spinal Deformities in Children Severely Affected with Spinal Muscular Atrophy after Gene Therapy with Onasemnogene Abeparvovec-Preliminary Results. Child 2023, 10, 998. [Google Scholar] [CrossRef] [PubMed]
- Barbato, G.; Nistico, R.; Triaca, V. Exploiting Focused Ultrasound to Aid Intranasal Drug Delivery for Brain Therapy. Front. Pharmacol. 2022, 13, 786475. [Google Scholar] [CrossRef] [PubMed]
- PRISMA Statement. Available online: https://www.prisma-statement.org (accessed on 15 October 2024).
- McColgan, P.; Thobhani, A.; Boak, L.; Schobel, S.A.; Nicotra, A.; Palermo, G.; Trundell, D.; Zhou, J.; Schlegel, V.; Sanwald Ducray, P.; et al. Tominersen in Adults with Manifest Huntington’s Disease. N. Engl. J. Med. 2023, 389, 2203–2205. [Google Scholar] [CrossRef] [PubMed]
- Dawson, T.M.; Golde, T.E.; Lagier-Tourenne, C. Animal models of neurodegenerative diseases. Nat. Neurosci. 2018, 21, 1370–1379. [Google Scholar] [CrossRef] [PubMed]
- Xhima, K.; McMahon, D.; Ntiri, E.E.; Goubran, M.; Hynynen, K.; Aubert, I. Intravenous and Non-invasive Drug Delivery to the Mouse Basal Forebrain Using MRIguided Focused Ultrasound. Bio-Protoc. 2021, 11, e4056. [Google Scholar] [CrossRef]
- Christine, C.W.; Starr, P.A.; Larson, P.S.; Eberling, J.L.; Jagust, W.J.; Hawkins, R.A.; VanBrocklin, H.F.; Wright, J.F.; Bankiewicz, K.S.; Aminoff, M.J. Safety and tolerability of putaminal AADC gene therapy for Parkinson disease. Neurology 2009, 73, 1662–1669. [Google Scholar] [CrossRef]
- Zhou, K.; Han, J.; Wang, Y.; Zhang, Y.; Zhu, C. Routes of administration for adeno-associated viruses carrying gene therapies for brain diseases. Front. Mol. Neurosci. 2022, 15, 988914. [Google Scholar] [CrossRef]
- Bankiewicz, K.S.; Eberling, J.L.; Kohutnicka, M.; Jagust, W.; Pivirotto, P.; Bringas, J.; Cunningham, J.; Budinger, T.F.; Harvey-White, J. Convection-enhanced delivery of AAV vector in parkinsonian monkeys; in vivo detection of gene expression and restoration of dopaminergic function using pro-drug approach. Exp. Neurol. 2000, 164, 2–14. [Google Scholar] [CrossRef]
- Hadaczek, P.; Kohutnicka, M.; Krauze, M.T.; Bringas, J.; Pivirotto, P.; Cunningham, J.; Bankiewicz, K. Convection-enhanced delivery of adeno-associated virus type 2 (AAV2) into the striatum and transport of AAV2 within monkey brain. Hum. Gene Ther. 2006, 17, 291–302. [Google Scholar] [CrossRef]
- Lonser, R.R.; Sarntinoranont, M.; Morrison, P.F.; Oldfield, E.H. Convection-enhanced delivery to the central nervous system. J. Neurosurg. 2015, 122, 697–706. [Google Scholar] [CrossRef] [PubMed]
- Kofoed, H.; Aubert, I. Focused ultrasound gene delivery for the treatment of neurological disorders. Trends Mol. Med. 2024, 30, 263–277. [Google Scholar] [CrossRef] [PubMed]
- Hanson, L.R.; Frey, W.H. Intranasal delivery bypasses the blood-brain barrier to target therapeutic agents to the central nervous system and treat neurodegenerative disease. BMC Neurosci. 2008, 9 (Suppl. S3), S5. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Luo, K.; Li, J.; Liao, Y.; Liao, C.; Chen, W.S.; Chen, M.; Ao, L. Focused Ultrasound Promotes the Delivery of Gastrodin and Enhances the Protective Effect on Dopaminergic Neurons in a Mouse Model of Parkinson’s Disease. Front. Cell Neurosci. 2022, 16, 884788. [Google Scholar] [CrossRef]
- Bulcha, J.T.; Wang, Y.; Ma, H.; Tai, P.W.L.; Gao, G. Viral vector platforms within the gene therapy landscape. Signal Transduct. Target. Ther. 2021, 6, 53. [Google Scholar] [CrossRef]
- Scazzone, C.; Lo Sasso, B.; Ciaccio, M. Molecular Biomarkers of Neurodegenerative Disorders: A Practical Guide to Their Appropriate Use and Interpretation in Clinical Practice. Int. J. Mol. Sci. 2024, 25, 4323. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
| Reference Year and Country | Clinical/Preclinical | Design | Gene Therapy | Results | Side Effects |
|---|---|---|---|---|---|
| Mendell Jerry et al., (2021) [8] USA | Clinical trial; Descriptive study with historical cohort. | Patients with SMA type 1 (n = 15); Age avg: 6.3 months; 5-year follow-up; • Low dose (n = 3) (6.7 × 1013 vg/kg); • High dose (n = 10) (1.1 × 1014 vg/kg). | IV Onasemnogene Abeparvovec (AAV9-SMN). | Significant; Higher achievement of motor development milestones and survival of treatment, when compared to historical cohort. Better results for the high dose group. Long-term favorable safety profile. | Hypersalivation. |
| Strauss et al., (2022) [9] USA | Clinical trial; Descriptive study with historical cohort. | Presymptomatic patients with SMA type 1 or type 2, with 2 or 3 copies of SMN2 (n = 14); Age avg: 21 days (8–34); 18-month follow up; Dose: 1.1 × 1014 vg/kg. | IV Onasemnogene Abeparvovec (AAV9-SMN). | Significant; Early administration improves disease progression. | Transient hepatotoxicity, thrombocytopenia, cardiac events, thrombotic microangiopathy, and sensory abnormalities suggestive of ganglionopathy. |
| Stettner et al., (2023) [10] Switzerland | Clinical trial; Prospective observational study. | Patients with SMA (n = 9); Weight 5.9 ± 1.4 kg; Age avg: 160 days; Dose: 1.1 × 1014 vg/kg; • SMA type 1 (n = 6); • SMA type 2 (n = 1); • Presymptomatic (n = 2); Duration 2 years. | IV Onasemnogene Abeparvovec (AAV9-SMN). | Significant; Analysis of complications of the therapy. | Hepatotoxicity events; Increased troponin T levels. |
| Pane et al., (2022) [11] Italy | Case series; Descriptive study with historical Cohort. | Patients with SMA2-3; • SMA type 2: Age: 2.64 to 47.82 months (n = 46); • SMA type 3: Age: 3.21 to 47.82 months (n = 65); Standard nusinersen dosing; Duration: 2.56 years. | Intrathecal nusinersen ASO activator of SMN2 mRNA. | Significant; Improvement in motor function tests in both groups after 12 months, when compared to the historical cohort. | Hepatotoxicity events. |
| Pane et al., (2023) [12] Italy | Case series. | Patients with SMA type 1 (n = 67) (>6 months, >8.5 kg); Age avg: 20 months; 12-month follow up; Dose: 1.1 × 1014 vg/kg. | IV Onasemnogene Abeparvovec (AAV9-SMN). | Significant; Asymptomatic patients of younger age and lower age show better results. | Hepatotoxicity events. |
| Chand et al., (2022) [13] USA | Case series. | Patients with SMA type 1 with two copies of SMN2 (n = 102); • Weight < 8.5 kg (n = 97); • Weight < 13.5 kg (n = 5); Standard onasemnogene abeparvovec dosing. | IV Onasemnogene abeparvovec (AAV9-SMN) | Non-significant; Similar safety and presence of adverse events in both groups. | Acute respiratory events, hepatotoxicity, thrombotic microangiopathy, and thrombocytopenia. |
| Besse et al., (2020) [14] France | Preclinical study. | Transgenic mice; Age: PND1 80 days follow-up; • Control (n = 22); Low dose AAV9: 3 × 1013 vg/kg; High dose AAV9: 8 × 1014 vg/kg; • IV AAV9-SMN (n = 22); • ICV AAV9-SMN (n = 18). | IV or ICV AAV9-SMN. | Significant differences with the control group; No significant differences between IV and ICV in SMN expression; Significant increase in survival in ICV group. | Kyphosis and necrosis in the tail. |
| Rich et al., (2022) [15] USA | Preclinical study. | Pregnant sows; Gestational age 77 to 110 (corresponding to third trimester in humans) (4–5 piglets) (n = 13); • Treated (n = 11); Doses: 2.0 × 1013 vg/kg; 6.5 × 1012 vg/kg; • Control (n = 2). | IV shRNA-AAV9. | Non-significant; Not a viable model for the study of in utero viral delivery of gene therapy. | Premature birth. |
| Reference Year and Country | Clinical/Preclinical | Design | Gene Therapy | Results | Side Effects |
|---|---|---|---|---|---|
| Wencesalau et al., (2022) [4] Brazil | Preclinical study. | Male Wistar rats treated with 3-nitropropionic acid; 8 weeks old (n = 60); • Control groups; • Single or three doses of 1 × 106 or 1 × 107 hIDPSCs. | Intraperitoneal administration of hIDPSCs. | Significant; BDNF levels were restored with a low dose of cells; Based on these data, a phase I clinical trial showed that IV administration of hIDPSCs is well tolerated and improves HD motor dysfunction [5]; Phase II clinical trial is under analysis. | No side effects were observed. |
| Kotowska-Zimmer et al., (2022) [6] Poland | Preclinical study. | YAC128 mice; 12–16 weeks old; • Control group (n = 9); • miRNA group Dose: 1 × 1011 gc (n = 10); • shRNA group (n = 10); Dose: 3 × 1011 gc. | Intrastratial injection of AAV5-miRNA or AAV5-shRNA. | Significant mHTT reduction in both groups compared to control without affecting endogenous HTT; miRNA reduced spleen weight to values characteristic of wild-type mice and improved motor function. | Abnormal behavior of animals treated with AAV5-shRNAs, and some of them had to be killed before termination of the study; No signs of toxicity were observed in the miRNA group. |
| Ferlazzo et al., (2023) [7] Italy | Preclinical study. | Mice (R6/2) (n = 44); 4 weeks old; • Control; • CAG repeats; In vitro human neural precursor cells HD. | IV AAV9-MTF1. | Significant reduction in mHTT aggregate formation; Amelioration of motor defects. | No side effects were observed. |
| Reference Year and Country | Clinical/Preclinical | Design | Gene Therapy | Results | Side Effects |
|---|---|---|---|---|---|
| Miller et al., (2020) [37] USA | Phase I/II clinical trial; Descriptive study with historical cohort. | Adult patients with SOD1 mutations (n = 50); Four dose cohorts; Ascending-dose trial: 20 (n = 10), 40 (n = 9), 60 (n = 9), or 100 mg (n = 10) or placebo (n = 12); 31 weeks follow-up. | Intrathecal administration of tofersen. | Significant decrease in SOD1 concentrations in CSF at the highest concentration of tofersen at day 85; Neurofilament concentrations decreased; Non-significant improvement in clinical parameters. | Lumbar-puncture-related adverse events in most participants; Pleocytosis in some patients; Headache, procedural pain, post–lumbar puncture syndrome, and falls; Two died from pulmonary embolism and respiratory failure. |
| Mueller et al., (2020) [38] USA | Case report. | Two patients with familial ALS (SOD1 mutations); Patient 1: 22-year-old; Patient 2: 56-year-old; Dose: 4.2 × 1014 vg. | Intrathecal infusion of AAV encoding a microRNA targeting SOD1. | Patient 1: Lower SOD1 levels in spinal cord on autopsy; Levels of SOD1 in CSF were only transiently slightly lower; Transient improvement in the strength of his right leg, but no change in vital capacity; Patient 2: stable scores on a composite measure of ALS function and stable vital capacity during a 12-month period. | In Patient 1, meningoradiculitis developed after the infusion; Patient 2 was pretreated with immunosuppressive drugs and did not have this complication. |
| Lim et al., (2020) [39] USA | Preclinical study. | Male transgenic mice hSOD1 (n = 14); 8 weeks old; Dose: 8 × 1010 vg (10 µL). | Intrathecal delivery of AAV9-CRISPR-Cas9. | Significantly delayed progression in the final phase of the disease; Non-significant in the early stages. | No side effects were observed. |
| Nizzardo et al., (2020) [40] Italy | Preclinical study. | Male and female SOD1 transgenic mice at 80 days of age (n = 9); Dose: 11 × 1011 particles. | Intramuscular injection of AAV9-SYT13. | Significant; SYT13 overexpression increases survival and reduces motor neuron pathology. | Not mentioned. |
| S. Wang et al., (2022) [41] USA | Preclinical study. | Mice (n = 3–18 animals per experiment/group); • Control; • Transgenic SOD1; Dose: 2 × 1013 gc/mL (10 µL). | Subpial administration of AAV1-CAV-1. | Significant; Increase in survival (10%); Delayed disease onset and progression. | Adverse effects from the subpial surgery on body weight at the late stage of disease, with no effect on disease onset or survival. |
| Tejwani et al., (2023) [42] USA | Preclinical study. | Mice (age P1) divided in four groups (n = 3–28 animals per experiment/group): mutated (TDP-43) and nonmutated, treated and nontreated; Dose: 10 µg. | ICV delivery of ASO-Nlk. | Significant; Treatment reduces TDP-43 inclusions and related pathology, increases life span, and ameliorates motor behavior. | Not mentioned. |
| Reference Year and Country | Clinical/Preclinical | Design | Gene Therapy | Results | Side Effects |
|---|---|---|---|---|---|
| Rocco et al., (2022) [25] USA | Phase I clinical trial; Descriptive study with historical cohort. | Patients with advanced PD (n = 13); Age: 51–75 years (mean 65.1 ± 6.4); Three dose cohorts: 450 mL each side; 1.9 × 1010 vg (n = 6); 2.3 × 1011 vg (n = 6); 9.9 × 1011 vg (n = 1); 36–60 months follow-up. | Bilateral putaminal infusions with iMRI aid. AAV2-GDNF and gadoteridol. | Administration: Imaging platforms improve therapeutic distribution, increase safety, enable administration customization, and make it easier to measure dosage effectiveness and efficacy. | No evidence of significant, persistent localized trauma or an inflammatory response. |
| Onuki et al., (2021) [26] Japan | Clinical trial. Descriptive study with historical cohort. | Patients with AADC deficiency (n = 8); Age: 4–18 years (mean 9.5 ± 4.5); 2 × 1011 vg each side (200 µL); 2 years s follow-up. | Bilateral putamen AAV2-h AADC. | Significant disappearance of painful dystonia; Improvement of motor functions; The cortico-putaminal network is restored. | Not mentioned. |
| Tai et al., (2022) [27] Taiwan | Phase I/II clinical trial; Descriptive study with historical cohort. | Patients with AADC deficiency (n = 26); Age: 1.7–8.5 years (mean 5.4 ± 2.6); Phase I/II: 1.8 × 1011 vg (each side) (n = 10); Phase 2b: 2.4 × 1011 vg (each side) (n = 5); 5.4 years follow-up. | Bilateral putamen eladocagene exuparvovec (AAV2-h AADC). | Significant de novo dopamine production; Improvement in motor and cognitive function; Improved quality of life. | Potentially related to the surgical procedure; Temporary mild dyskinesia events. |
| Chen et al., (2020) [28] USA | Preclinical study. | MPTP-induced Neurodegeneration; Mice (n = 3–15 animals per experiment/group); 10–14 weeks old; Dose: 2.0 × 106 cells. | IV transplantation of HSC-GDNF. | Significant improvement of motor and nonmotor symptoms; Restoration of dopamine levels in SNpc. | No side effects were observed. |
| Gupta et al., (2020) [29] USA | Preclinical study. | Mice (n = 19); Aged 6–12 weeks: • Control; • Injection of C3 or Fluorescent; • 6-OHDA; Dose: ~2 × 109 vg (2 µL). | Administration of AAV2-C3 transferase. | No significant differences between C3 and control groups in behavioral and histological analyses groups. | Transduced dopaminergic neurons express C3 continuously without apparent adverse effects. |
| Wang et al., (2020) [30] China | Preclinical study. | 6-OHDA rats: • Control; • 3 groups treated with ultrasounds; One of them receives gene therapy; Dose: 4 × 107 microbubbles. | Ultrasound combined with lV administration of GDNF retrovirus-loaded microbubbles. | Significant increase in dopamine concentration and number of dopaminergic neurons in the treated group; Improvement of motor symptoms. | Not mentioned. |
| Cui et al., (2021) [31] USA | Preclinical study. | Transgenic VMAT2 lo mice in two groups (n = 5–6 animals per experiment/group): • 12 months of age; • 18 months of age; Dose: 1 × 108 TU (1–2 µL). | Intracerebral administration of lentivirus mRNA transcription Factors, Phox2a/2b, Hand2, and Gata3. | Significant improvement of motor symptoms and spatial memory by increasing norepinephrine and dopamine concentration. | Not mentioned. |
| Fernández-Parrilla et al., (2022) [32] Mexico | Preclinical study. | Male rats aged 2 months (n = 120): • Control (n = 20); • 6-OHDA (n = 40); • Treated group (n = 60); Dose: 30 nM; 60 days follow up. | Intracranial nanoparticle-mediated hCDNF gene delivery. | Significant re-establishment of the nigrostriatal circuits, motor, and nonmotor deficits. | No side effects were observed. |
| Kip et al., (2022) [33] USA | Preclinical study. | PD neurodegeneration rat model (6-OHDA) (n = 9); Adult rats; Dose: 1.4 × 1013 vg/mL (200 nL). | DBS. AAV5-opsins. | Significant improvement in the motor symptoms of PD. | Selective stimulation of target neurons may reduce side effects compared to electrical DBS. |
| J. Li et al., (2022) [34] China | Preclinical study. | •Male monkeys, 8–15 years old (MPTP/MPP+ model) (n = 12); • Male rats, 8–10 weeks (6-OHDA model) (n = 19); • Female mice, 6–8 weeks old (n = 8); Dose: 1 × 105 cells/µL. | Intrastriatal administration of MSCs-DOPA. | Significant; Intrastriatal delivery restores dopamine levels and improves motor deficit; Long-term benefit (up to 51 months). | Safety of MSC-DOPA was assessed in mice in detail; No side effects were observed. |
| Azevedo el al., (2023) [35] Netherlands | Preclinical study. | Female Wistar rats (n = 184), 7 weeks old; • Control (n1 = 20, n2 = 44); • Low dose (n = 32) 4.1 × 109 TU; • High dose (n = 88) 1.0 × 1010 TU; Applied either continuously or intermittently. | Intrastriatal administration of doxycycline inducible AAV vector, AAV2/5-V16-hGDNF. | Significant; A low-dose or an intermittent high-dose treatment resulted in neuron protection and motor symptom reduction, without inducing deleterious effects. | Continuous administration of high-dose GDNF caused a 50% increased level of oxidized DNA in the substantia nigra pars compacta and aberrant sprouting. |
| Narváez et al., (2023) [36] USA | Preclinical study. | Male Wistar rats (200–300 g) (6-OHDA) groups (n = 4 animals per experiment/group); Dose: 20,000 cells (3–4 µL). | Intrastriatal CRISP/Cas9-lentivirus; Activation of tyrosine hydroxylase gene. | Significant. Increased dopamine production by astrocytes in the striatum; Transplanted TH-producing astrocytes induced motor recovery. | Not mentioned. |
| Reference Year and Country | Clinical/Preclinical | Design | Gene Therapy | Results | Side Effects |
|---|---|---|---|---|---|
| Jeon et al., (2020) [22] South Korea | Preclinical study. | Female Sprague-Dawley rats, 8 weeks old; Dose: 2 × 1012 vg/mL (2 µL). | Intrahippocampal injection AAV1-Rheb (S16H). | Significantly increased expression of BDNF in hippocampal neurons but not in astrocytes. | Not mentioned. |
| Capsoni and Cattaneo et al. (2022) [23] Italy | Preclinical study. | Ts65Dn mice at 4 months of age with accumulation of APP (n = 6); Dose: not mentioned. | Intranasal delivery of hNGFp. | Significant; Treatment rescues astrogliosis, dystrophic microglia, and neurogenesis deficits. | Intranasal delivery reduces the possibility of side effects due to leakage of the protein in blood circulation. hNGFp has similar neurotrophic potency to wildtype NGF, while showing reduced pain sensitization potency. |
| Mishra et al., (2023) [24] USA | Preclinical study. | 5xFAD transgenic mice at 2 months of age (n = 46); Dose: 2 × 106 cells (100 µL). | IV transplantation of HSPCs. | Significant improvement in cognitive function, locomotor activity. Microglia activation and neuroinflammation reduction; Decrease in β-amyloid plaques. | No side effects were observed. |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
García-González, N.; Gonçalves-Sánchez, J.; Gómez-Nieto, R.; Gonçalves-Estella, J.M.; López, D.E. Advances and Challenges in Gene Therapy for Neurodegenerative Diseases: A Systematic Review. Int. J. Mol. Sci. 2024, 25, 12485. https://doi.org/10.3390/ijms252312485
García-González N, Gonçalves-Sánchez J, Gómez-Nieto R, Gonçalves-Estella JM, López DE. Advances and Challenges in Gene Therapy for Neurodegenerative Diseases: A Systematic Review. International Journal of Molecular Sciences. 2024; 25(23):12485. https://doi.org/10.3390/ijms252312485
Chicago/Turabian StyleGarcía-González, Nerea, Jaime Gonçalves-Sánchez, Ricardo Gómez-Nieto, Jesús M. Gonçalves-Estella, and Dolores E. López. 2024. "Advances and Challenges in Gene Therapy for Neurodegenerative Diseases: A Systematic Review" International Journal of Molecular Sciences 25, no. 23: 12485. https://doi.org/10.3390/ijms252312485
APA StyleGarcía-González, N., Gonçalves-Sánchez, J., Gómez-Nieto, R., Gonçalves-Estella, J. M., & López, D. E. (2024). Advances and Challenges in Gene Therapy for Neurodegenerative Diseases: A Systematic Review. International Journal of Molecular Sciences, 25(23), 12485. https://doi.org/10.3390/ijms252312485