
Functional Roles of CD26/DPP4 in Bleomycin-Induced Pulmonary Hypertension Associated with Interstitial Lung Disease


 , ,
, ,  , and
, and
Abstract
1. Introduction
2. Results
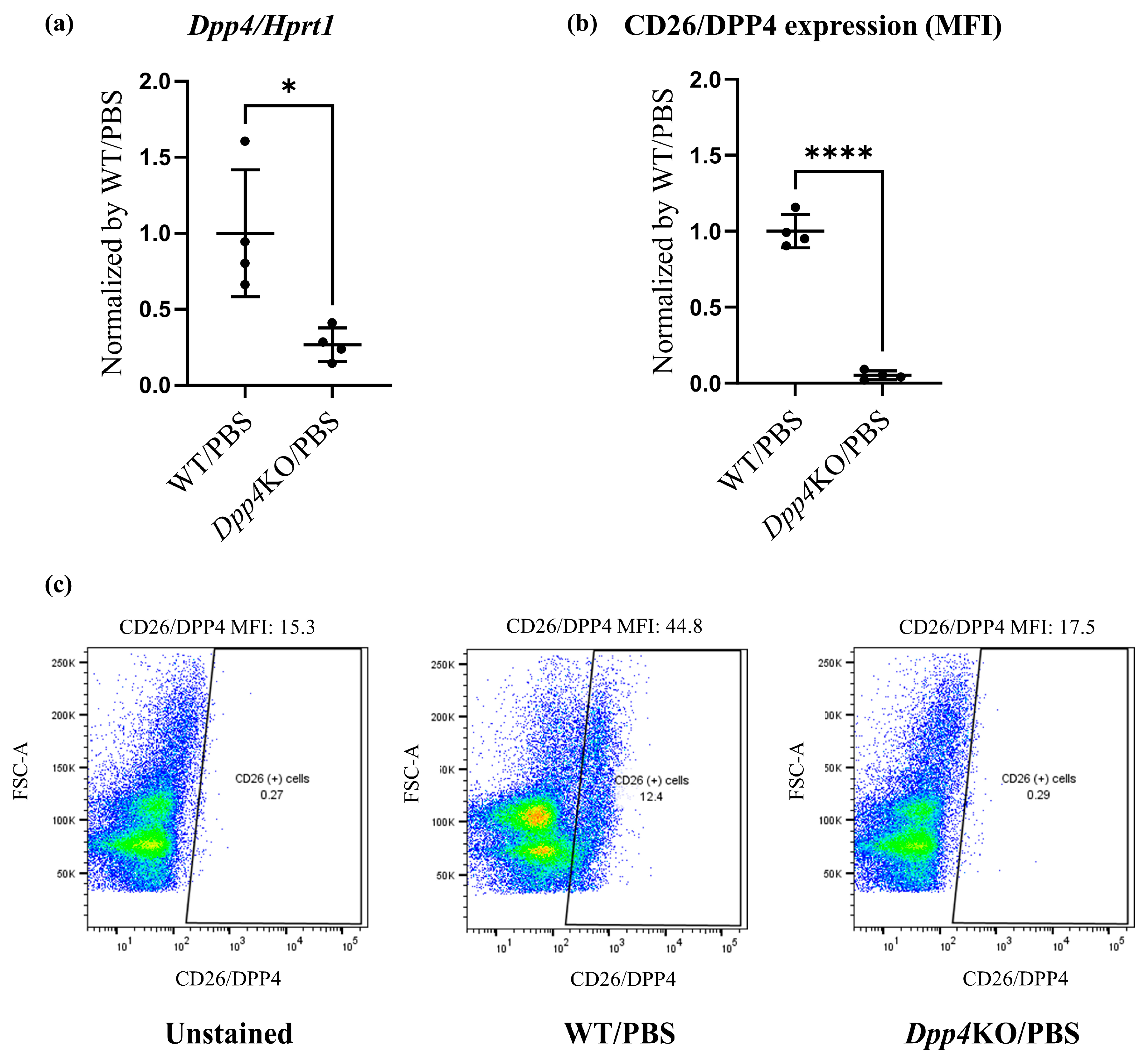
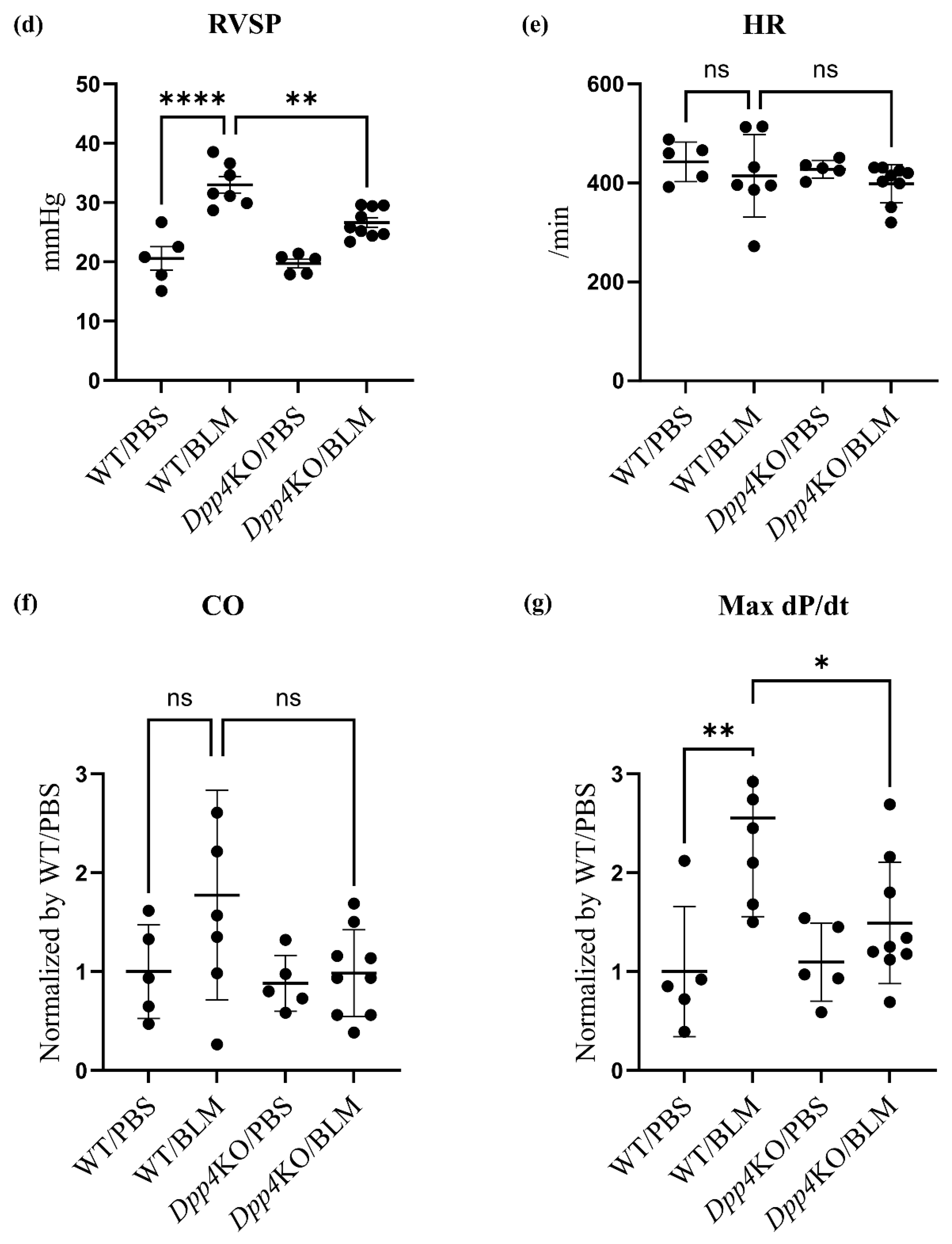
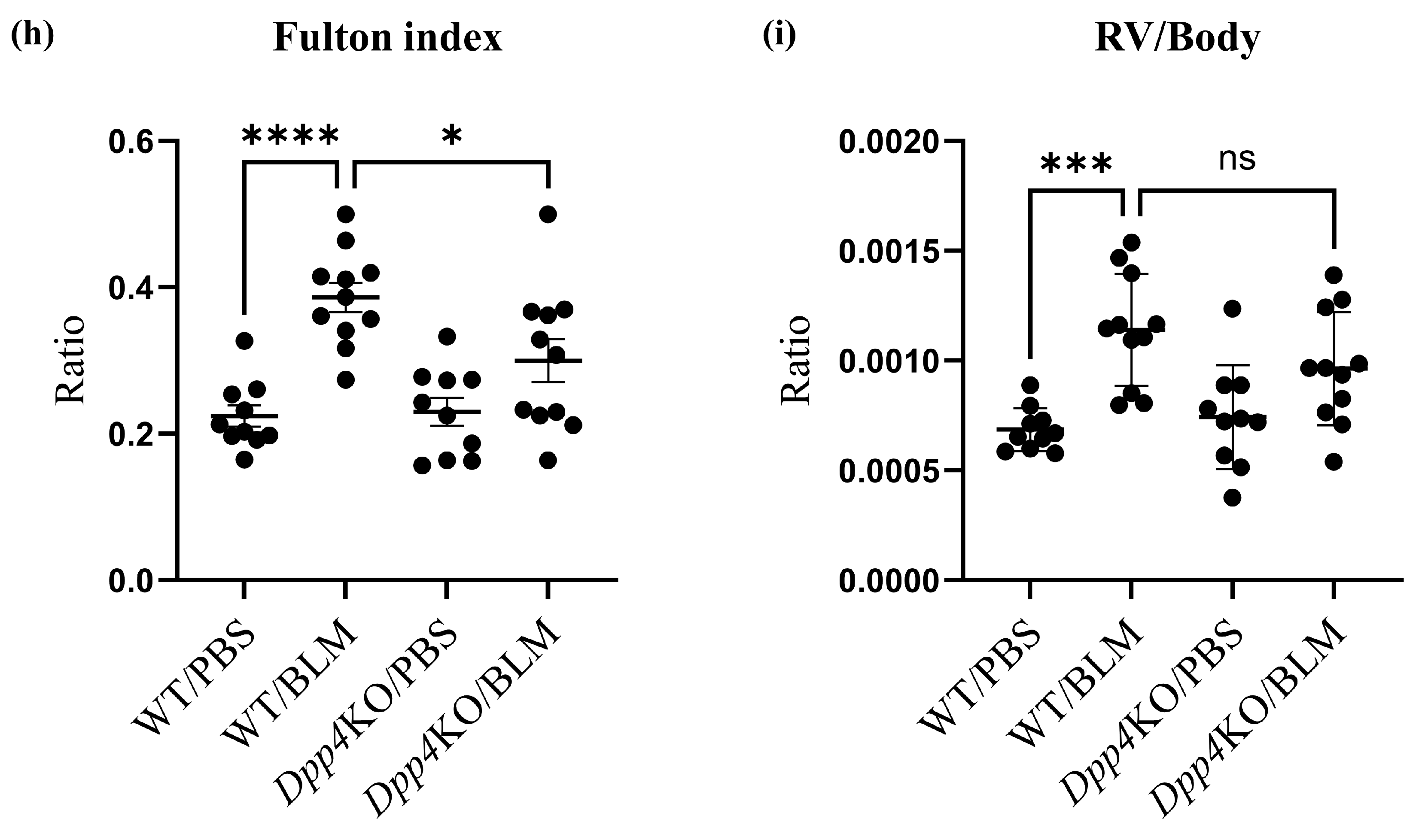
2.1. BLM-Induced Pulmonary Hypertension Was Attenuated in Dpp4KO Mice
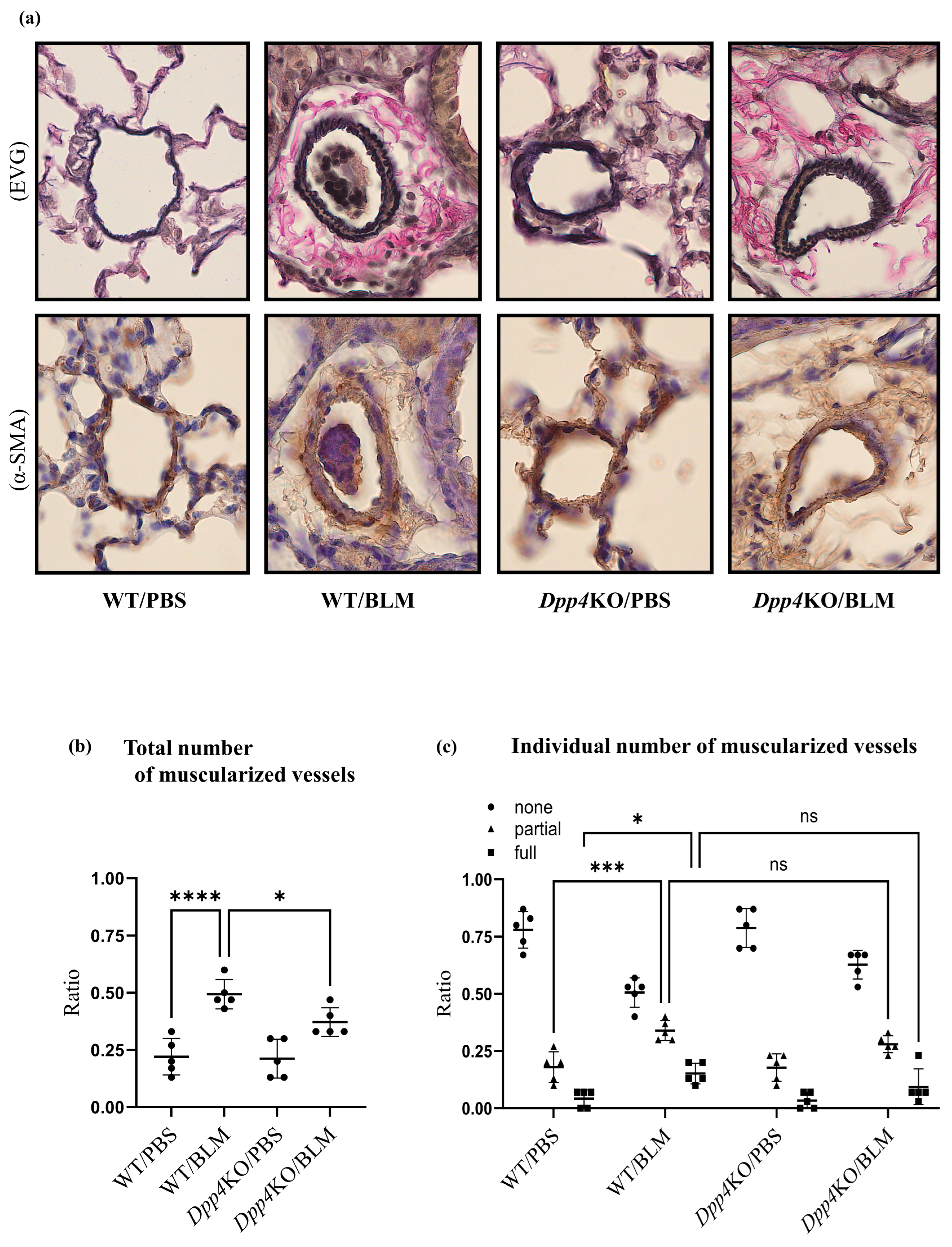
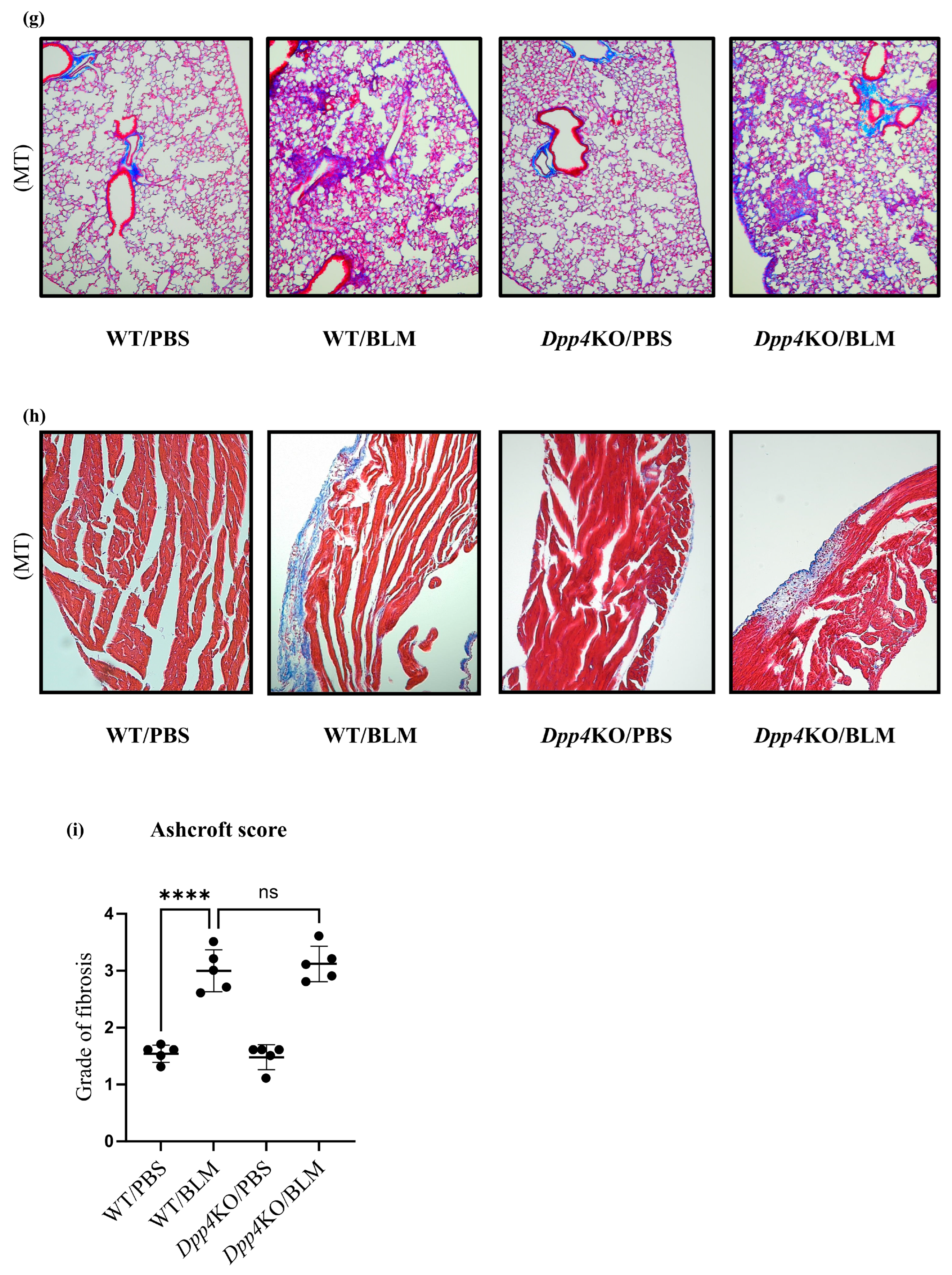
2.2. Media Thickening in Small Pulmonary Vessels Was Attenuated in Dpp4KO Mice
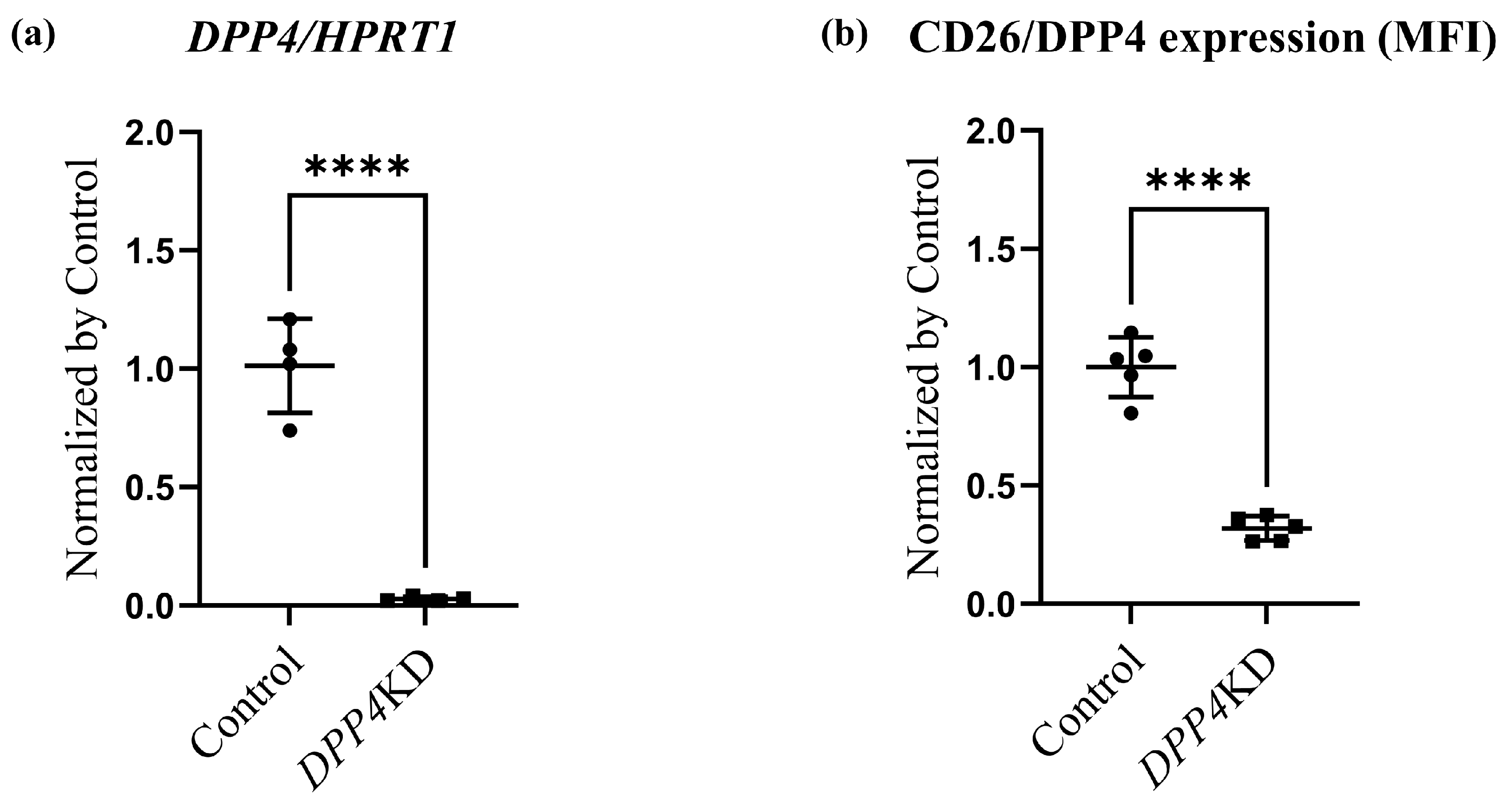
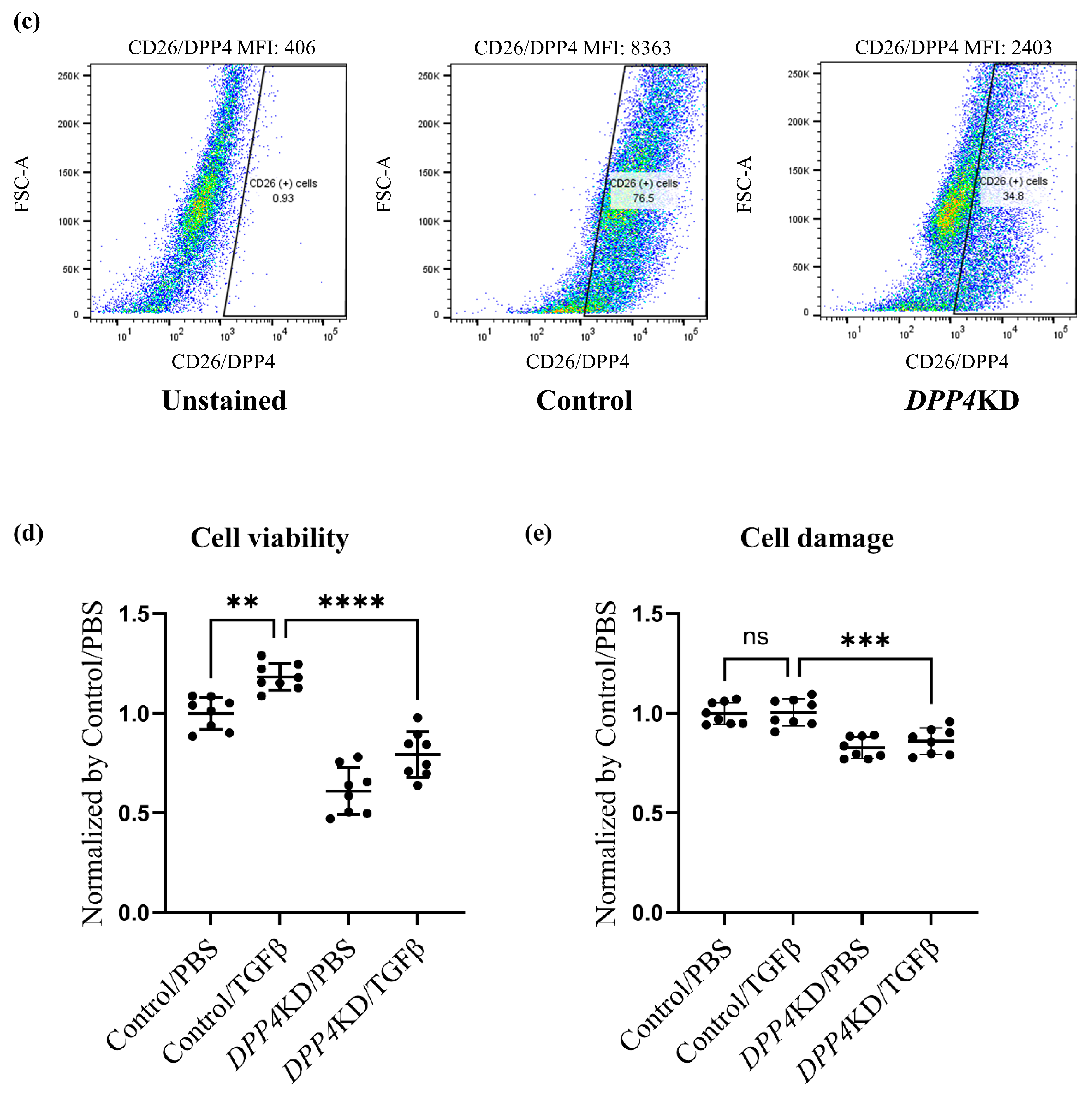
2.3. Cell Proliferation and Cytotoxicity Were Reduced by DPP4-siRNA Treatment in Cultured hPASMCs
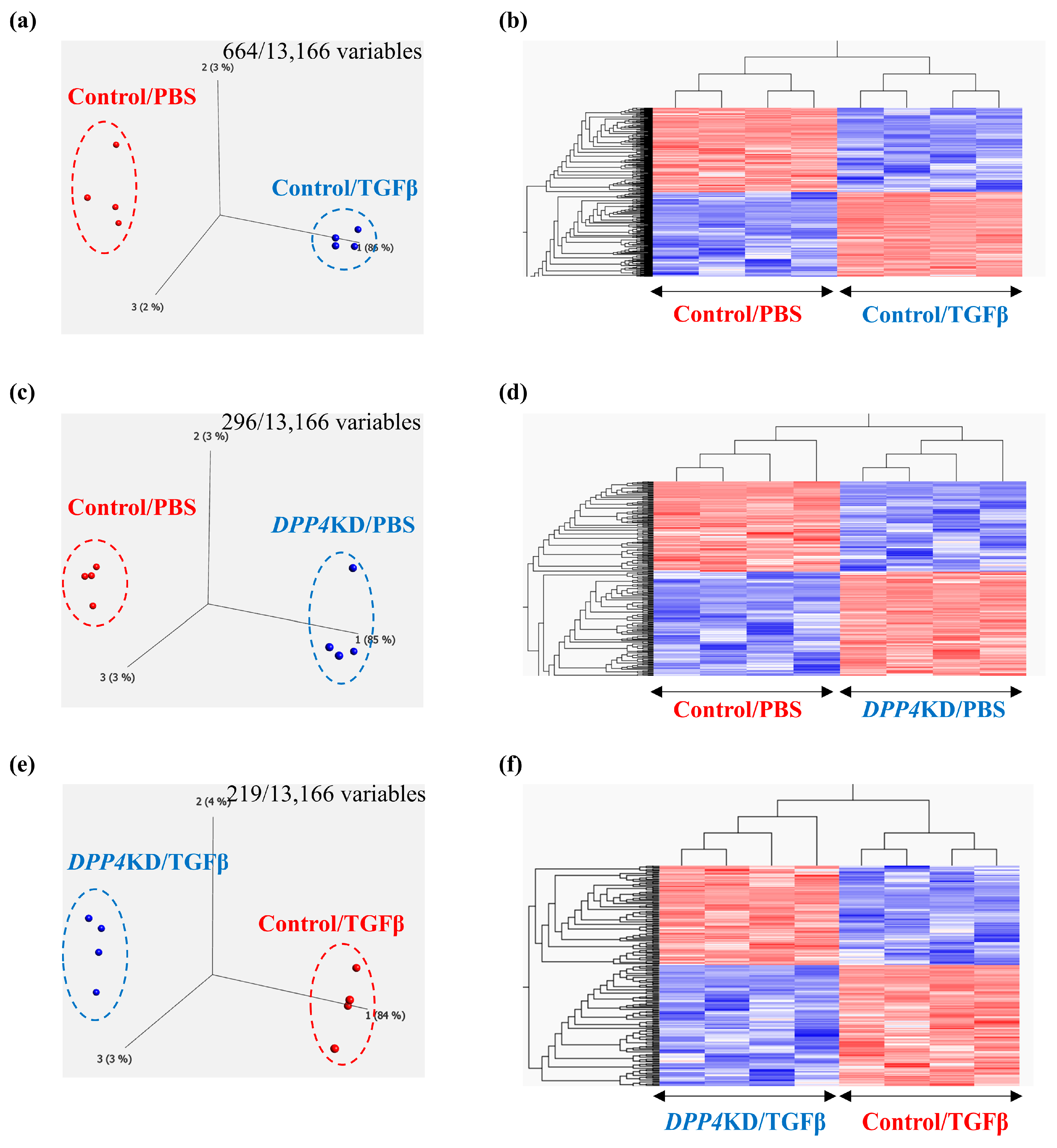
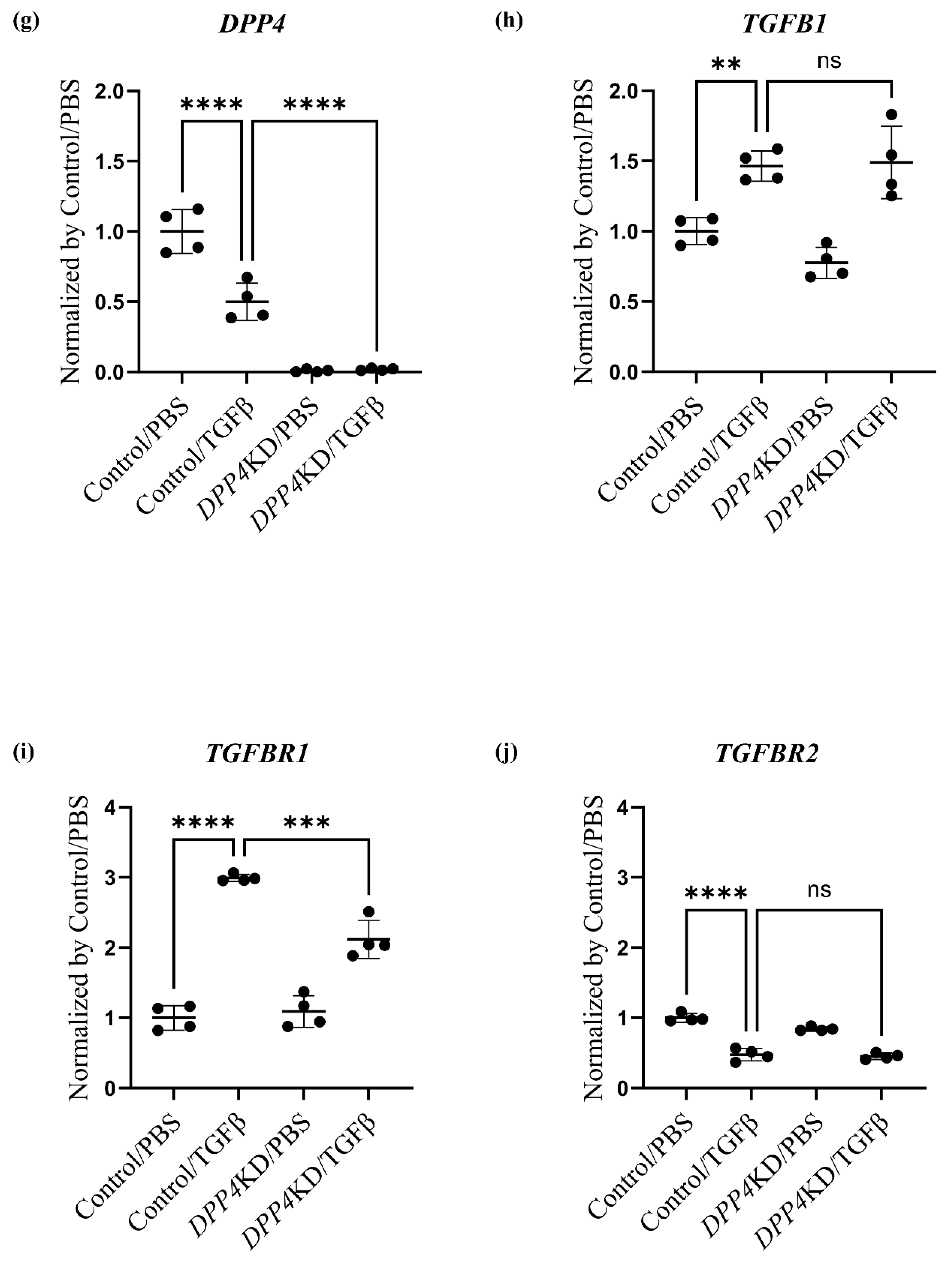
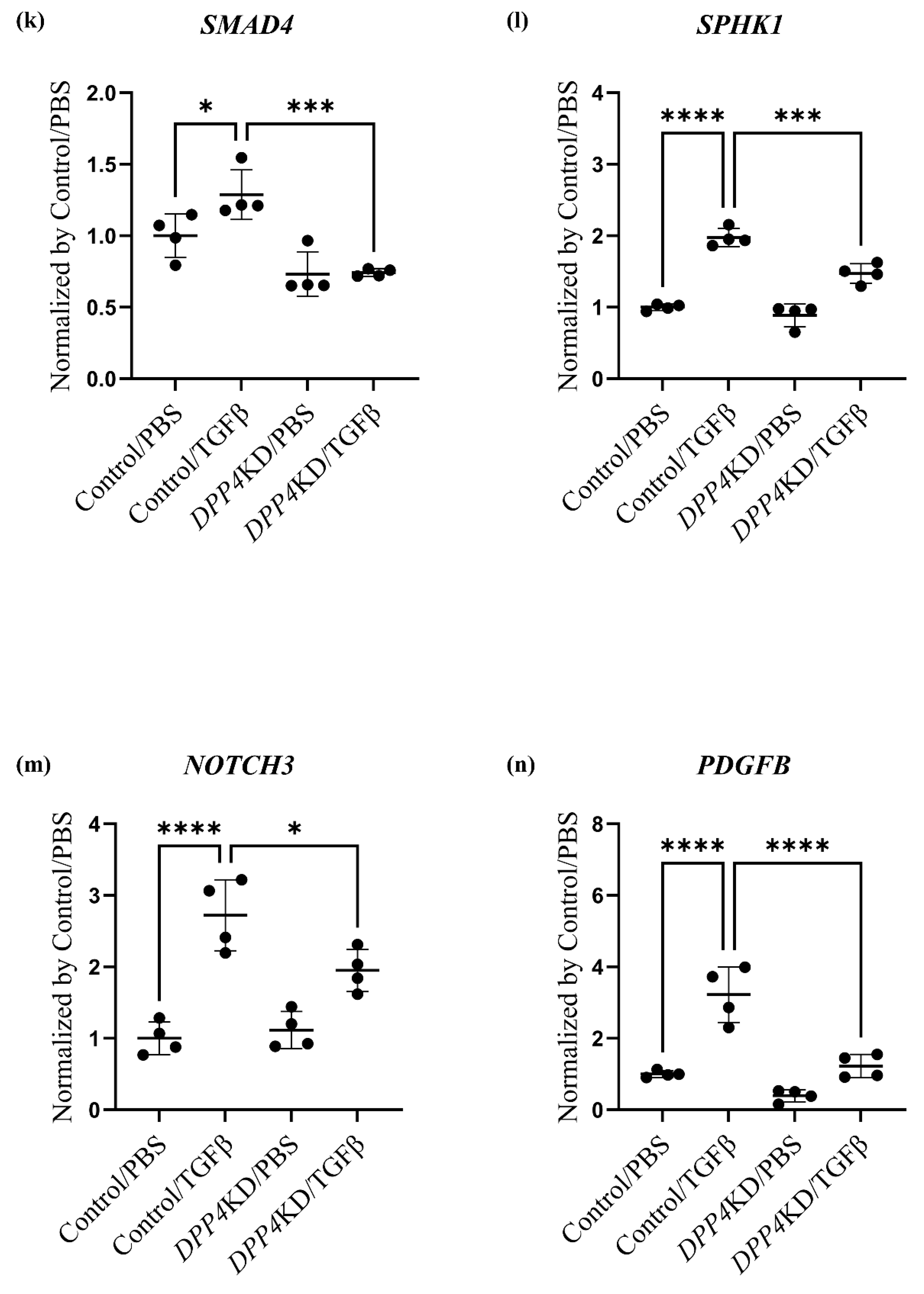
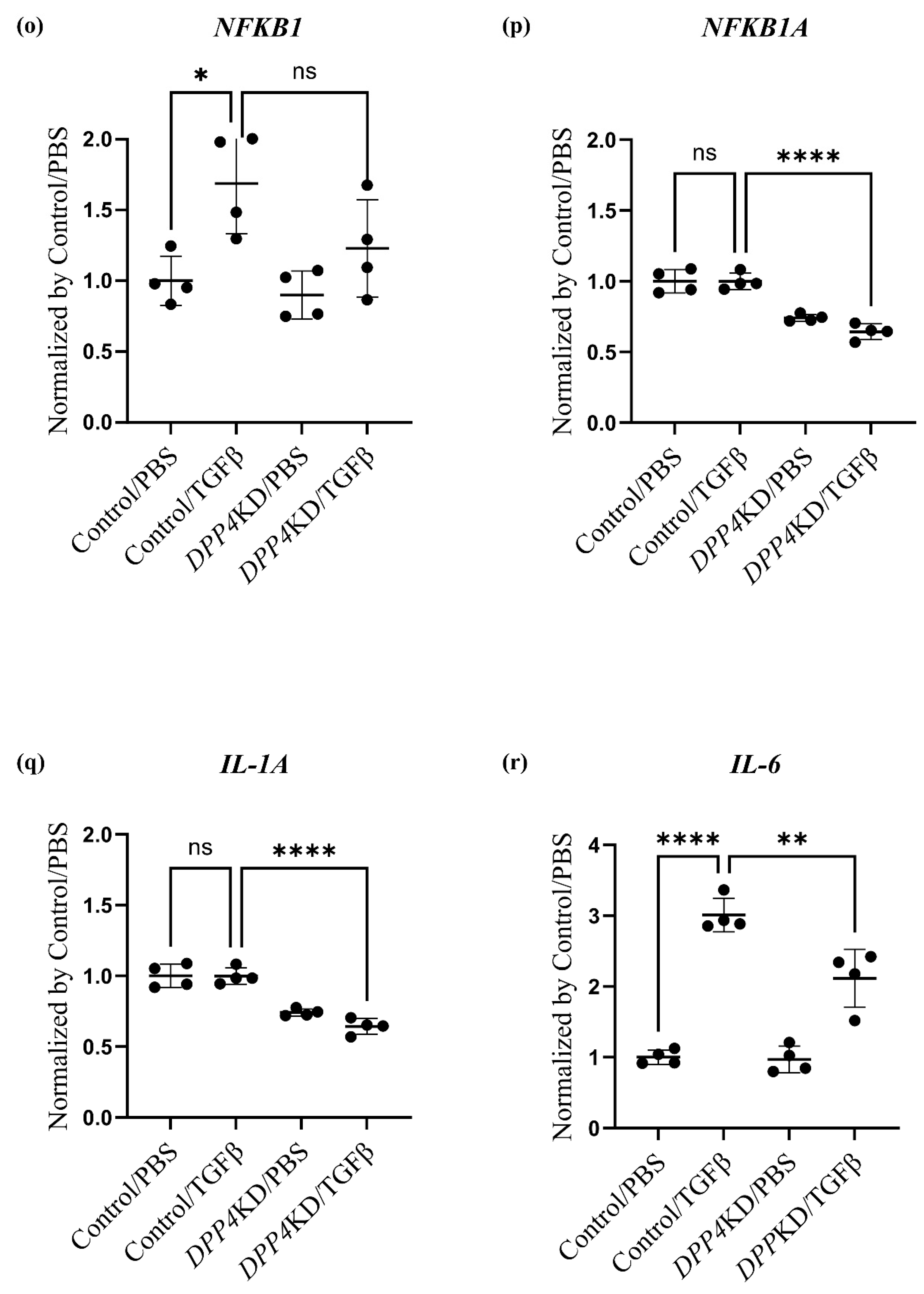
2.4. Transcriptome Analysis of Cultured hPASMCs after Treatment with TGFβ and DPP4-siRNA
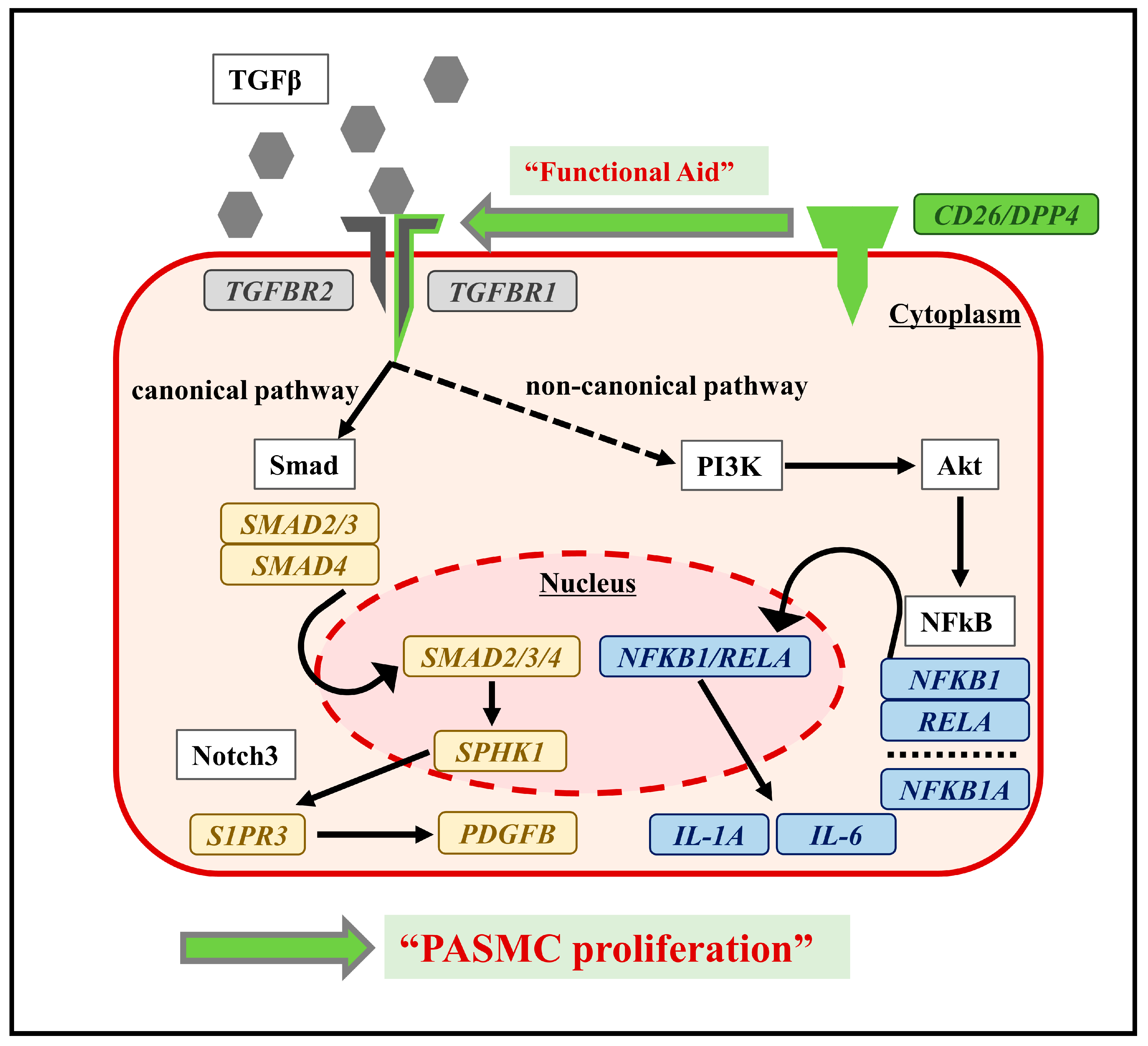
3. Discussion
4. Materials and Methods
4.1. Animal Model of Pulmonary Hypertension with Interstitial Pneumonia
4.2. Hemodynamic Analysis
4.3. Histological Analysis
4.4. Cell Culture and Treatments of Small Interfering RNA and TGF-β1
4.5. Proliferation and Cytotoxicity Assay
4.6. Real-Time Quantitative PCR Analysis
4.7. Flow Cytometry Analysis
4.8. Transcriptome Analysis
4.9. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Humbert, M.; Kovacs, G.; Hoeper, M.M.; Badagliacca, R.; Berger, R.M.F.; Brida, M.; Carlsen, J.; Coats, A.J.S.; Escribano-Subias, P.; Ferrari, P.; et al. 2022 ESC/ERS Guidelines for the diagnosis and treatment of pulmonary hypertension. G. Ital. Cardiol. 2023, 24, 1e–116e. [Google Scholar] [CrossRef] [PubMed]
- Gall, H.; Felix, J.F.; Schneck, F.K.; Milger, K.; Sommer, N.; Voswinckel, R.; Franco, O.H.; Hofman, A.; Schermuly, R.T.; Weissmann, N.; et al. The Giessen Pulmonary Hypertension Registry: Survival in pulmonary hypertension subgroups. J. Heart Lung Transplant. 2017, 36, 957–967. [Google Scholar] [CrossRef] [PubMed]
- Gomberg-Maitland, M.; Olschewski, H. Prostacyclin therapies for the treatment of pulmonary arterial hypertension. Eur. Respir. J. 2008, 31, 891–901. [Google Scholar] [CrossRef]
- Boucly, A.; Savale, L.; Jais, X.; Bauer, F.; Bergot, E.; Bertoletti, L.; Beurnier, A.; Bourdin, A.; Bouvaist, H.; Bulifon, S.; et al. Association between Initial Treatment Strategy and Long-Term Survival in Pulmonary Arterial Hypertension. Am. J. Respir. Crit. Care Med. 2021, 204, 842–854. [Google Scholar] [CrossRef] [PubMed]
- Nathan, S.D.; Barbera, J.A.; Gaine, S.P.; Harari, S.; Martinez, F.J.; Olschewski, H.; Olsson, K.M.; Peacock, A.J.; Pepke-Zaba, J.; Provencher, S.; et al. Pulmonary hypertension in chronic lung disease and hypoxia. Eur. Respir. J. 2019, 53, 1801914. [Google Scholar] [CrossRef] [PubMed]
- Prins, K.W.; Duval, S.; Markowitz, J.; Pritzker, M.; Thenappan, T. Chronic use of PAH-specific therapy in World Health Organization Group III Pulmonary Hypertension: A systematic review and meta-analysis. Pulm. Circ. 2017, 7, 145–155. [Google Scholar] [CrossRef] [PubMed]
- Raghu, G.; Nathan, S.D.; Behr, J.; Brown, K.K.; Egan, J.J.; Kawut, S.M.; Flaherty, K.R.; Martinez, F.J.; Wells, A.U.; Shao, L.; et al. Pulmonary hypertension in idiopathic pulmonary fibrosis with mild-to-moderate restriction. Eur. Respir. J. 2015, 46, 1370–1377. [Google Scholar] [CrossRef]
- Nathan, S.D.; Behr, J.; Collard, H.R.; Cottin, V.; Hoeper, M.M.; Martinez, F.J.; Corte, T.J.; Keogh, A.M.; Leuchte, H.; Mogulkoc, N.; et al. Riociguat for idiopathic interstitial pneumonia-associated pulmonary hypertension (RISE-IIP): A randomised, placebo-controlled phase 2b study. Lancet Respir. Med. 2019, 7, 780–790. [Google Scholar] [CrossRef]
- Waxman, A.; Restrepo-Jaramillo, R.; Thenappan, T.; Ravichandran, A.; Engel, P.; Bajwa, A.; Allen, R.; Feldman, J.; Argula, R.; Smith, P.; et al. Inhaled Treprostinil in Pulmonary Hypertension Due to Interstitial Lung Disease. N. Engl. J. Med. 2021, 384, 325–334. [Google Scholar] [CrossRef]
- Tanabe, N.; Kumamaru, H.; Tamura, Y.; Taniguchi, H.; Emoto, N.; Yamada, Y.; Nishiyama, O.; Tsujino, I.; Kuraishi, H.; Nishimura, Y.; et al. Multi-Institutional Prospective Cohort Study of Patients With Pulmonary Hypertension Associated With Respiratory Diseases. Circ. J. 2021, 85, 333–342. [Google Scholar] [CrossRef]
- Heath, D.; Edwards, J.E. The pathology of hypertensive pulmonary vascular disease; a description of six grades of structural changes in the pulmonary arteries with special reference to congenital cardiac septal defects. Circulation 1958, 18, 533–547. [Google Scholar] [CrossRef] [PubMed]
- Eddahibi, S.; Chaouat, A.; Morrell, N.; Fadel, E.; Fuhrman, C.; Bugnet, A.S.; Dartevelle, P.; Housset, B.; Hamon, M.; Weitzenblum, E.; et al. Polymorphism of the serotonin transporter gene and pulmonary hypertension in chronic obstructive pulmonary disease. Circulation 2003, 108, 1839–1844. [Google Scholar] [CrossRef] [PubMed]
- Della Latta, V.; Cecchettini, A.; Del Ry, S.; Morales, M.A. Bleomycin in the setting of lung fibrosis induction: From biological mechanisms to counteractions. Pharmacol. Res. 2015, 97, 122–130. [Google Scholar] [CrossRef] [PubMed]
- Moore, B.B.; Hogaboam, C.M. Murine models of pulmonary fibrosis. Am. J. Physiol. Lung Cell. Mol. Physiol. 2008, 294, L152–L160. [Google Scholar] [CrossRef] [PubMed]
- Santos-Ribeiro, D.; Lecocq, M.; de Beukelaer, M.; Bouzin, C.; Palmai-Pallag, M.; Yakoub, Y.; Huaux, F.; Horman, S.; Perros, F.; Pilette, C.; et al. Bleomycin-induced lung injury: Revisiting an old tool to model group III PH associated with pulmonary fibrosis. Pulm. Circ. 2023, 13, e12177. [Google Scholar] [CrossRef] [PubMed]
- Mulvihill, E.E.; Drucker, D.J. Pharmacology, physiology, and mechanisms of action of dipeptidyl peptidase-4 inhibitors. Endocr. Rev. 2014, 35, 992–1019. [Google Scholar] [CrossRef] [PubMed]
- Green, B.D.; Flatt, P.R.; Bailey, C.J. Dipeptidyl peptidase IV (DPP IV) inhibitors: A newly emerging drug class for the treatment of type 2 diabetes. Diab. Vasc. Dis. Res. 2006, 3, 159–165. [Google Scholar] [CrossRef] [PubMed]
- Morimoto, C.; Schlossman, S.F. The structure and function of CD26 in the T-cell immune response. Immunol. Rev. 1998, 161, 55–70. [Google Scholar] [CrossRef]
- Klemann, C.; Wagner, L.; Stephan, M.; von Horsten, S. Cut to the chase: A review of CD26/dipeptidyl peptidase-4’s (DPP4) entanglement in the immune system. Clin. Exp. Immunol. 2016, 185, 1–21. [Google Scholar] [CrossRef]
- Zhang, T.; Tong, X.; Zhang, S.; Wang, D.; Wang, L.; Wang, Q.; Fan, H. The Roles of Dipeptidyl Peptidase 4 (DPP4) and DPP4 Inhibitors in Different Lung Diseases: New Evidence. Front. Pharmacol. 2021, 12, 731453. [Google Scholar] [CrossRef]
- Kawasaki, T.; Chen, W.; Htwe, Y.M.; Tatsumi, K.; Dudek, S.M. DPP4 inhibition by sitagliptin attenuates LPS-induced lung injury in mice. Am. J. Physiol. Lung Cell. Mol. Physiol. 2018, 315, L834–L845. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Qi, Y. Vildagliptin, a CD26/DPP4 inhibitor, ameliorates bleomycin-induced pulmonary fibrosis via regulating the extracellular matrix. Int. Immunopharmacol. 2020, 87, 106774. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.Y.; Kong, X.Q.; Zhang, K.F.; Luo, S.; Wang, F.; Zhang, J.J. DPP4 as a Potential Candidate in Cardiovascular Disease. J. Inflamm. Res. 2022, 15, 5457–5469. [Google Scholar] [CrossRef] [PubMed]
- Bostick, B.; Habibi, J.; Ma, L.; Aroor, A.; Rehmer, N.; Hayden, M.R.; Sowers, J.R. Dipeptidyl peptidase inhibition prevents diastolic dysfunction and reduces myocardial fibrosis in a mouse model of Western diet induced obesity. Metabolism 2014, 63, 1000–1011. [Google Scholar] [CrossRef] [PubMed]
- Shah, Z.; Kampfrath, T.; Deiuliis, J.A.; Zhong, J.; Pineda, C.; Ying, Z.; Xu, X.; Lu, B.; Moffatt-Bruce, S.; Durairaj, R.; et al. Long-term dipeptidyl-peptidase 4 inhibition reduces atherosclerosis and inflammation via effects on monocyte recruitment and chemotaxis. Circulation 2011, 124, 2338–2349. [Google Scholar] [CrossRef] [PubMed]
- Anderluh, M.; Kocic, G.; Tomovic, K.; Kocic, H.; Smelcerovic, A. DPP-4 inhibition: Capital A, Cyrillic novel therapeutic approach to the treatment of pulmonary hypertension? Pharmacol. Ther. 2019, 201, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Yang, L.; Dong, L.; Yang, Z.W.; Zhang, J.; Zhang, S.L.; Niu, M.J.; Xia, J.W.; Gong, Y.; Zhu, N.; et al. Crosstalk between the Akt/mTORC1 and NF-kappaB signaling pathways promotes hypoxia-induced pulmonary hypertension by increasing DPP4 expression in PASMCs. Acta Pharmacol. Sin. 2019, 40, 1322–1333. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Wang, J.; He, M.; Han, H.; Xie, W.; Wang, H.; Kong, H. Dipeptidyl peptidase IV (DPP-4) inhibition alleviates pulmonary arterial remodeling in experimental pulmonary hypertension. Lab. Invest. 2018, 98, 1333–1346. [Google Scholar] [CrossRef]
- Baggio, L.L.; Varin, E.M.; Koehler, J.A.; Cao, X.; Lokhnygina, Y.; Stevens, S.R.; Holman, R.R.; Drucker, D.J. Plasma levels of DPP4 activity and sDPP4 are dissociated from inflammation in mice and humans. Nat. Commun. 2020, 11, 3766. [Google Scholar] [CrossRef]
- Koyanagi, Y.; Kawasaki, T.; Kasuya, Y.; Hatano, R.; Sato, S.; Takahashi, Y.; Ohnuma, K.; Morimoto, C.; Dudek, S.M.; Tatsumi, K.; et al. Functional roles of CD26/DPP4 in bleomycin-induced pulmonary fibrosis. Physiol. Rep. 2023, 11, e15645. [Google Scholar] [CrossRef]
- Lindenschmidt, R.C.; Tryka, A.F.; Godfrey, G.A.; Frome, E.L.; Witschi, H. Intratracheal versus intravenous administration of bleomycin in mice: Acute effects. Toxicol. Appl. Pharmacol. 1986, 85, 69–77. [Google Scholar] [CrossRef] [PubMed]
- Cutroneo, K.R.; White, S.L.; Phan, S.H.; Ehrlich, H.P. Therapies for bleomycin induced lung fibrosis through regulation of TGF-beta1 induced collagen gene expression. J. Cell. Physiol. 2007, 211, 585–589. [Google Scholar] [CrossRef] [PubMed]
- Ohm, B.; Moneke, I.; Jungraithmayr, W. Targeting cluster of differentiation 26 / dipeptidyl peptidase 4 (CD26/DPP4) in organ fibrosis. Br. J. Pharmacol. 2023, 180, 2846–2861. [Google Scholar] [CrossRef] [PubMed]
- Shi, S.; Srivastava, S.P.; Kanasaki, M.; He, J.; Kitada, M.; Nagai, T.; Nitta, K.; Takagi, S.; Kanasaki, K.; Koya, D. Interactions of DPP-4 and integrin beta1 influences endothelial-to-mesenchymal transition. Kidney Int. 2015, 88, 479–489. [Google Scholar] [CrossRef] [PubMed]
- Soare, A.; Gyorfi, H.A.; Matei, A.E.; Dees, C.; Rauber, S.; Wohlfahrt, T.; Chen, C.W.; Ludolph, I.; Horch, R.E.; Bauerle, T.; et al. Dipeptidylpeptidase 4 as a Marker of Activated Fibroblasts and a Potential Target for the Treatment of Fibrosis in Systemic Sclerosis. Arthritis Rheumatol. 2020, 72, 137–149. [Google Scholar] [CrossRef] [PubMed]
- Zakrzewicz, A.; Kouri, F.M.; Nejman, B.; Kwapiszewska, G.; Hecker, M.; Sandu, R.; Dony, E.; Seeger, W.; Schermuly, R.T.; Eickelberg, O.; et al. The transforming growth factor-beta/Smad2,3 signalling axis is impaired in experimental pulmonary hypertension. Eur. Respir. J. 2007, 29, 1094–1104. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Feng, W.; Li, F.; Shi, W.; Zhai, C.; Li, S.; Zhu, Y.; Yan, X.; Wang, Q.; Liu, L.; et al. SphK1/S1P mediates TGF-beta1-induced proliferation of pulmonary artery smooth muscle cells and its potential mechanisms. Pulm. Circ. 2019, 9, 1–8. [Google Scholar] [CrossRef]
- Li, X.; Zhang, X.; Leathers, R.; Makino, A.; Huang, C.; Parsa, P.; Macias, J.; Yuan, J.X.; Jamieson, S.W.; Thistlethwaite, P.A. Notch3 signaling promotes the development of pulmonary arterial hypertension. Nat. Med. 2009, 15, 1289–1297. [Google Scholar] [CrossRef]
- Tzavlaki, K.; Moustakas, A. TGF-beta Signaling. Biomolecules 2020, 10, 487. [Google Scholar] [CrossRef]
- Yu, M.; Wu, X.; Wang, J.; He, M.; Han, H.; Hu, S.; Xu, J.; Yang, M.; Tan, Q.; Wang, Y.; et al. Paeoniflorin attenuates monocrotaline-induced pulmonary arterial hypertension in rats by suppressing TAK1-MAPK/NF-kappaB pathways. Int. J. Med. Sci. 2022, 19, 681–694. [Google Scholar] [CrossRef]
- Karmouty-Quintana, H.; Zhong, H.; Acero, L.; Weng, T.; Melicoff, E.; West, J.D.; Hemnes, A.; Grenz, A.; Eltzschig, H.K.; Blackwell, T.S.; et al. The A2B adenosine receptor modulates pulmonary hypertension associated with interstitial lung disease. FASEB J. 2012, 26, 2546–2557. [Google Scholar] [CrossRef] [PubMed]
- Hansen, T.S.; Bubb, K.J.; Schiattarella, G.G.; Ugander, M.; Tan, T.C.; Figtree, G.A. High-Resolution Transthoracic Echocardiography Accurately Detects Pulmonary Arterial Pressure and Decreased Right Ventricular Contractility in a Mouse Model of Pulmonary Fibrosis and Secondary Pulmonary Hypertension. J. Am. Heart Assoc. 2022, 11, e018353. [Google Scholar] [CrossRef] [PubMed]
- Ashcroft, T.; Simpson, J.M.; Timbrell, V. Simple method of estimating severity of pulmonary fibrosis on a numerical scale. J. Clin. Pathol. 1988, 41, 467–470. [Google Scholar] [CrossRef] [PubMed]
- Ciuclan, L.; Bonneau, O.; Hussey, M.; Duggan, N.; Holmes, A.M.; Good, R.; Stringer, R.; Jones, P.; Morrell, N.W.; Jarai, G.; et al. A novel murine model of severe pulmonary arterial hypertension. Am. J. Respir. Crit. Care Med. 2011, 184, 1171–1182. [Google Scholar] [CrossRef]
- Li, L.; Zhang, X.; Li, X.; Lv, C.; Yu, H.; Xu, M.; Zhang, M.; Fu, Y.; Meng, H.; Zhou, J. TGF-beta1 inhibits the apoptosis of pulmonary arterial smooth muscle cells and contributes to pulmonary vascular medial thickening via the PI3K/Akt pathway. Mol. Med. Rep. 2016, 13, 2751–2756. [Google Scholar] [CrossRef][Green Version]














{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| (A): GO: relevant terms were excerpted | |
| Terms with upregulated genes following TGFβ treatment | p-value |
| Regulation of SMC differentiation (GO: 0051150) | 0.0041 |
| Cellular response to growth factor stimulus (GO: 0071363) | 0.0051 |
| Regulation of vascular associated SMC migration (GO: 1904754) | 0.0057 |
| Regulation of SMC proliferation (GO: 0048660) | 0.013 |
| Pathway-restricted SMAD protein phosphorylation (GO: 0060393) | 0.020 |
| Response to TGF-beta (GO: 0071559) | 0.026 |
| Regulation of TGF-beta production (GO: 0071634) | 0.030 |
| Notch signaling pathway (GO: 0007219) | 0.030 |
| Regulation of TGF-beta receptor signaling pathway (GO: 0017015) | 0.041 |
| Regulation of vascular associated SMC proliferation (GO: 1904707) | 0.045 |
| (B): KEGG: relevant pathways were excerpted | |
| Pathways with upregulated genes following TGFβ treatment | p-value |
| PI3K-Akt signaling pathway | <0.0001 |
| Cytokine–cytokine receptor interaction | 0.013 |
| Notch signaling pathway | 0.018 |
| TGF-beta signaling pathway | 0.076 |
| NF-kappa B signaling pathway | 0.10 |
| (A): GO: relevant terms were excerpted | |
| Terms with downregulated genes following DPP4-siRNA treatment | p-value |
| Cellular response to growth factor stimulus (GO: 0071363) | <0.0001 |
| Cellular response to cytokine stimulus (GO: 0071345) | <0.0001 |
| Notch signaling pathway (GO: 0007219) | 0.0018 |
| Pathway-restricted SMAD protein phosphorylation (GO: 0060389) | 0.0031 |
| SMAD protein signal transduction (GO: 0060395) | 0.0036 |
| Regulation of TGF-beta receptor signaling pathway (GO: 0017015) | 0.029 |
| Regulation of vascular associated SMC differentiation (GO: 1905063) | 0.035 |
| Positive regulation of NIK/NF-kappa B signaling (GO: 1901224) | 0.040 |
| Regulation of vascular associated SMC migration (GO: 1904754) | 0.041 |
| Regulation of SMC proliferation (GO: 0048660) | 0.042 |
| (B): KEGG: relevant pathways were excerpted | |
| Pathways with downregulated genes following DPP4-siRNA treatment | p-value |
| Cytokine–cytokine receptor interaction | 0.0020 |
| PI3K-Akt signaling pathway | 0.020 |
| NF-kappa B signaling pathway | 0.0036 |
| TGF-beta signaling pathway | 0.019 |
| Notch signaling pathway | 0.049 |
| (A): Genes related to DPP4 and the TGFβ family | |||||
| Gene ID | Control /PBS | Control /TGFβ | DPP4KD /PBS | DPP4KD /TGFβ | p-value |
| DPP4 | 43.50 | 21.74 | 0.37 | 0.81 | <0.0001 |
| TGFB1 | 55.25 | 80.83 | 42.85 | 82.31 | <0.0001 |
| TGFBR1 | 25.78 | 77.10 | 28.13 | 54.63 | <0.0001 |
| TGFBR2 | 116.50 | 55.40 | 98.17 | 52.79 | <0.0001 |
| (B): Genes related to the canonical pathway (Notch pathway) | |||||
| Gene ID | Control /PBS | Control /TGFβ | DPP4KD /PBS | DPP4KD /TGFβ | p-value |
| SMAD2 | 52.51 | 39.64 | 46.69 | 44.30 | 0.064 |
| SMAD4 | 16.08 | 20.70 | 11.76 | 11.93 | 0.0003 |
| SPHK1 | 49.69 | 98.18 | 44.09 | 73.12 | <0.0001 |
| S1PR3 | 38.57 | 113.9 | 33.89 | 74.95 | <0.0001 |
| NOTCH3 | 21.81 | 59.39 | 24.31 | 42.56 | <0.0001 |
| PDGFB | 6.55 | 21.10 | 2.59 | 8.01 | <0.0001 |
| (C): Genes related to the non-canonical pathway (NFκB pathway) | |||||
| Gene ID | Control /PBS | Control /TGFβ | DPP4KD /PBS | DPP4KD /TGFβ | p-value |
| NFKB1 | 22.19 | 38.16 | 19.97 | 27.27 | 0.0077 |
| RELA | 44.16 | 49.64 | 46.41 | 50.99 | 0.56 |
| NFKB1A | 170.20 | 170.00 | 126.10 | 109.50 | <0.0001 |
| IL-1A | 38.41 | 46.95 | 20.57 | 18.31 | 0.0003 |
| IL-6 | 44.71 | 134.60 | 44.34 | 94.56 | <0.0001 |
| CXCL8 | 939.60 | 821.60 | 518.10 | 360.10 | <0.0001 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Okaya, T.; Kawasaki, T.; Sato, S.; Koyanagi, Y.; Tatsumi, K.; Hatano, R.; Ohnuma, K.; Morimoto, C.; Kasuya, Y.; Hasegawa, Y.; et al. Functional Roles of CD26/DPP4 in Bleomycin-Induced Pulmonary Hypertension Associated with Interstitial Lung Disease. Int. J. Mol. Sci. 2024, 25, 748. https://doi.org/10.3390/ijms25020748
Okaya T, Kawasaki T, Sato S, Koyanagi Y, Tatsumi K, Hatano R, Ohnuma K, Morimoto C, Kasuya Y, Hasegawa Y, et al. Functional Roles of CD26/DPP4 in Bleomycin-Induced Pulmonary Hypertension Associated with Interstitial Lung Disease. International Journal of Molecular Sciences. 2024; 25(2):748. https://doi.org/10.3390/ijms25020748
Chicago/Turabian StyleOkaya, Tadasu, Takeshi Kawasaki, Shun Sato, Yu Koyanagi, Koichiro Tatsumi, Ryo Hatano, Kei Ohnuma, Chikao Morimoto, Yoshitoshi Kasuya, Yoshinori Hasegawa, and et al. 2024. "Functional Roles of CD26/DPP4 in Bleomycin-Induced Pulmonary Hypertension Associated with Interstitial Lung Disease" International Journal of Molecular Sciences 25, no. 2: 748. https://doi.org/10.3390/ijms25020748
APA StyleOkaya, T., Kawasaki, T., Sato, S., Koyanagi, Y., Tatsumi, K., Hatano, R., Ohnuma, K., Morimoto, C., Kasuya, Y., Hasegawa, Y., Ohara, O., & Suzuki, T. (2024). Functional Roles of CD26/DPP4 in Bleomycin-Induced Pulmonary Hypertension Associated with Interstitial Lung Disease. International Journal of Molecular Sciences, 25(2), 748. https://doi.org/10.3390/ijms25020748