Abstract
Parkinson’s disease (PD) stands as the most prevalent degenerative movement disorder, marked by the degeneration of dopaminergic neurons in the substantia nigra of the midbrain. In this study, we conducted a transcriptome analysis utilizing post mortem mRNA extracted from the substantia nigra of both PD patients and healthy control (CTRL) individuals. Specifically, we acquired eight samples from individuals with PD and six samples from CTRL individuals, with no discernible pathology detected in the latter group. RNA sequencing was conducted using the TapeStation 4200 system from Agilent Technologies. A total of 16,148 transcripts were identified, with 92 mRNAs displaying differential expression between the PD and control groups. Specifically, 33 mRNAs were significantly up-regulated, while 59 mRNAs were down-regulated in PD compared to the controls. The identification of statistically significant signaling pathways, with an adjusted p-value threshold of 0.05, unveiled noteworthy insights. Specifically, the enriched categories included cardiac muscle contraction (involving genes such as ATPase Na+/K+ transporting subunit beta 2 (ATP1B2), solute carrier family 8 member A1 (SLC8A1), and cytochrome c oxidase subunit II (COX2)), GABAergic synapse (involving GABA type A receptor-associated protein-like 1 (GABARAPL1), G protein subunit beta 5 (GNB5), and solute carrier family 38 member 2 (SLC38A2), autophagy (involving GABARAPL1 and tumor protein p53-inducible nuclear protein 2 (TP53INP2)), and Fc gamma receptor (FcγR) mediated phagocytosis (involving amphiphysin (AMPH)). These findings uncover new pathophysiological dimensions underlying PD, implicating genes associated with heart muscle contraction. This knowledge enhances diagnostic accuracy and contributes to the advancement of targeted therapies.
1. Introduction
PD stands out as the most prevalent movement disorder and neurodegenerative disease after Alzheimer’s dementia, affecting approximately seven million people globally [1,2]. Clinically, PD is a heterogeneous condition primarily characterized by a resting tremor, bradykinesia, and rigidity, commonly regarded as the “core motor symptoms”. However, PD is clinically diverse, presenting with various causes and clinical manifestations. While a resting tremor is often a prominent and traditionally associated sign of PD, its clinical variability can result in its absence in some cases.
Despite the multifaceted clinical presentation, current diagnosis remains primarily clinical, with laboratory tests, such as genetic testing, and instrumental exams, including structural and/or functional neuroimaging, reserved for patients with atypical presentations [3]. Diagnostic criteria define PD as the presence of bradykinesia combined with either a rest tremor, rigidity, or both [4]. However, the clinical spectrum encompasses additional motor symptoms and non-otor symptoms, some of which precede motor manifestations and are equally debilitating [5,6,7,8].
While non-specific symptoms, such as constipation, apathy, fatigue, or mild cognitive changes, are challenging to promptly link to PD onset or its preclinical/prodromal phase, others, like hyposmia, adult onset depression/anxiety, or REM sleep behavior disorder, are considered strong predictors of neurodegeneration. These symptoms play a crucial role in the early diagnosis of PD, even in the absence of common motor symptoms [9].
Among synucleinopathies, multiple system atrophy (MSA) and dementia with Lewy bodies (DLB) represent significant differential diagnoses. MSA is a sporadic neurodegenerative disease clinically characterized by parkinsonism or cerebellar ataxia, both combined with dysautonomia and a poor response to levodopa. Magnetic resonance imaging may reveal specific abnormalities, such as the “hot cross bun” sign and bilateral putaminal hyperintensity [10]. DLB is characterized by dementia preceding or developing alongside parkinsonism, with core features including fluctuating cognition, recurrent visual hallucinations, dysautonomia, and marked sensitivity to D2 receptor-blocking agents. Functional studies may show typical hypometabolism within the occipital cortex [11].
A comprehensive brain transcriptome study of subjects with MSA revealed the down-regulation of oligodendrocyte genes associated with a loss of myelination, particularly the QKI gene as a master regulator of this gene network. Additionally, they demonstrated the up-regulation of monomeric α-synuclein gene expression in neurons [12].
Despite considerable research efforts, encompassing both preclinical and clinical studies, PD remains incurable. The progressive loss of dopaminergic (DA) neurons in the substantia nigra pars compacta (SNpc), a crucial part of the midbrain regulating movement tone and velocity, persists without effective intervention, and the complete set of pathomechanisms behind this degeneration remains incompletely elucidated [1,13]. Pathophysiologically, PD exhibits a multifactorial origin, involving intricate interactions between various genetic and environmental factors, a phenomenon observed in other complex diseases, such as tumors [14]. While some factors exert their influence on the elderly population in general, others appear to be more specifically associated with this disorder [15]. It is noteworthy that post mortem studies consistently report that over 60% of DA neurons in the SNpc are already degenerated when overt clinical signs manifest. This indicates that PD is neuropathologically evident long before its clinical onset, suggesting a period during which the human brain can compensate for dopaminergic loss until reaching a “clinical threshold” for PD [16,17,18].
Histological studies reveal that PD is characterized by the abnormal deposition of the insoluble protein α-synuclein, forming aggregates known as Lewy bodies [19]. These protein aggregates progressively accumulate throughout the brainstem and various neocortical and limbic regions, reflecting the progressive degeneration of the entire central nervous system (CNS) [20]. More recently, a neuroinflammatory state has been observed in the brains of PD patients, particularly evident in the SNpc [21,22]. Both neuroinflammation and dysfunctional activation of the immune system within the CNS significantly contribute to PD pathology and pathophysiology [23]. The inflammasome, a crucial complex of immune-modulating receptors and sensors, plays a role in recruiting proteins associated with apoptotic mechanisms through caspase-1 activation [24,25]. Caspase-1, in turn, activates the proinflammatory cytokines interleukin (IL)-1β and IL-18, perpetuating the neuroinflammatory state in PD brains [25,26,27,28,29]. A recently proposed model suggests that alpha-synuclein activates the inflammasome in the SNpc, leading to IL-activated proinflammatory profiles, neuronal death, and clinical symptoms [30,31,32].
Genetically, several genes or gene variants, including leucine-rich repeat kinase 2 (LRRK2), synuclein alpha (SNCA), glucosylceramidase beta-1 (GBA1), Parkin RBR E3 ubiquitin protein ligase (PARKIN), and PTEN-induced kinase 1 (PINK1), have been implicated in causing PD [33,34,35,36]. Molecular profiling studies of post mortem SNpc samples, aimed at identifying differential molecular expression changes specific to PD compared to controls, have been conducted [37]. For instance, Simunovic et al. [38] used RNA microarrays to analyze SNpc gene expression in PD samples, identifying the dysregulation of known molecular regulatory pathways in PD, including dysfunction in mitochondrial and oxidative-stress-induced cellular responses [39,40]. In a recent comparative gene expression analysis on laser-dissected neurons from SNpc, Zaccaria et al. [41] revealed 52 dysregulated genes in PD samples compared to controls.
We recognize that synucleinopathies are neurodegenerative disorders associated with the misfolding and aggregation of Alpha-synuclein (α-Syn) [42]. Real-time tremor-induced conversion technology (RT-QuIC) is an in vitro amplification method initially developed for the analysis of the prion protein (PrP) [42]. Today, it is also employed for the biochemical assessment of different strains of α-Syn within synucleinopathies [42].
Although PD is encompassed within this family of pathologies (synucleinopathies), our study focused on transcriptome analysis by examining post mortem mRNA. This approach allows the targeted identification of gene expression, a critical condition for comprehending this well-known yet enigmatic pathology present in the global population. Transcriptomics enables the unveiling of microscopic changes, offering the potential to identify individuals at risk of developing the disease, even in the presence of non-motor symptoms, or to anticipate the evolution of the pathology.
The review by Rike and Stern [43] cites proteomic and transcriptomic studies involving post mortem brain samples from individuals with PD compared to controls. Some authors of these studies concentrated on the transcriptome in the substantia nigra [44,45]. The data obtained revealed that the dysregulated groups of genes/pathways/biological processes in individuals with PD, as opposed to controls, are more associated with extracellular matrix (ECM)-receptor interaction and cell adhesion molecules.
In our study, we conducted mRNA analysis and subsequent enrichment using the Kyoto Encyclopedia of Genes and Genomes (KEGG) and Gene Ontology (GO) to assess mRNAs extracted post mortem from the SN of subjects with PD and healthy controls. Utilizing samples obtained from Parkinson’s UK Brain Bank (Imperial College London, London, UK), we specifically addressed a case history of PD at Braak LB Stage 6.
2. Results
In the examination of mRNA deregulation in the SNpc of PD patients, we conducted gene expression profiling on eight post mortem SNpc samples from PD patients and six from healthy CTRL subjects using next-generation sequencing (RNA-Seq). The aim was to identify specific and differential changes in molecular expression. Following the removal of low-quality reads and adapter sequences, the high-quality reads were aligned against the human genome reference (hg38). In detail, we identified a total of 16,148 transcripts (Supplementary Table S1, sheet A), with 92 mRNAs differentially expressed (DEGs) between the two groups (PD vs. CTRL). Among these, 33 mRNAs were significantly up-regulated (Table 1, Figure 1), while 59 mRNAs were significantly down-regulated in PD compared to CTRL (Table 2, Figure 1). The normalized count of mRNAs is available at ArrayExpress (E-MTAB-13295).

Table 1.
mRNAs down-expressed in PD subjects compared to controls (padj ≤ 0.05 and |FC| ≥ 1.5).
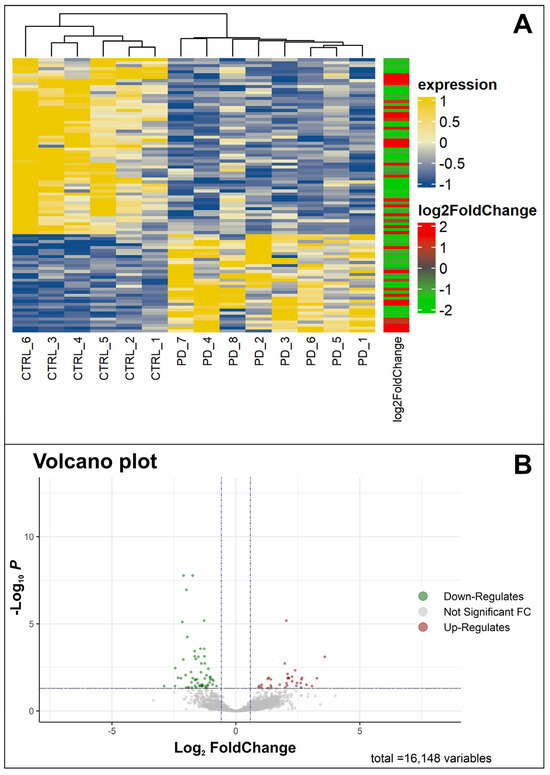
Figure 1.
Visualization of differentially expressed genes (DEGs). (A) Heatmap of significant DEGs in patients with Parkinson’s disease (PD) and healthy control (CTRL) individuals. In yellow, we see the genes with an up-normalized expression level, whereas in blue, we see the down genes. The log2 (foldChange) bar indicates, in red and in green, the up- and down-regulated genes, respectively. (B) Volcano plot of significant DEGs based on fold changes and p-values. The green color shows the down-regulated genes, whereas the red color shows the up-regulated genes.
Table 2.
mRNAs over-expressed in PD subjects compared to controls (padj ≤ 0.05 and |FC| ≥ 1.5).
The heatmap (Figure 1A) illustrates statistically significant differences in mRNA expression profiles between PD and CTRL. The volcano plot (Figure 1B) depicts the distribution of differentially expressed transcripts by their fold change and p-values. The most-up-regulated genes are toward the right, the most-down-regulated are toward the left, and the most statistically significant genes are at the top.
We employed the pathfindR tool to analyze significant DEGs in PD based on the KEGG pathway database. Enrichment analysis was performed to explore functional variations between the two groups and investigate pathways potentially associated with PD. Statistically significant signaling pathways included (hsa04260) cardiac muscle contraction, (hsa04727) GABAergic synapse, (hsa04140) autophagy, and (hsa04666) Fc gamma R-mediated phagocytosis (Figure 2A and Supplementary Table S1).
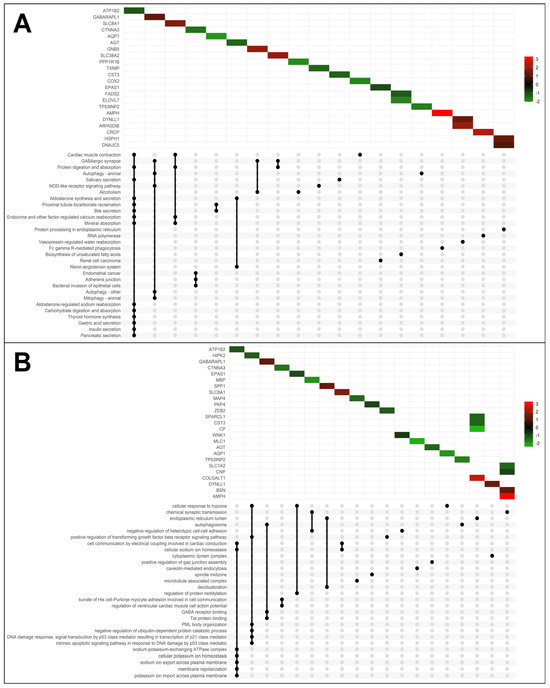
Figure 2.
KEGG and GO enrichment analysis of differentially expressed genes. (A) UpSet Plot shows the intersections of significant genes and top 30 enriched KEGG pathway and (B) GO terms, with the Log2FoldChange scale.
For GO enrichment, we identified both down-regulated and up-regulated genes involved in various molecular functions or biological processes (Figure 2B and Supplementary Table S2). Interestingly, among them, we found the enrichment of (GO:0007268) chemical synaptic transmission (AMPH, bassoon presynaptic cytomatrix protein (BSN), 2′,3′-cyclic nucleotide 3′ phosphodiesterase (CNP), myelin basic protein (MBP), solute carrier family 1 member 2 (SLC1A2), (GO:0071456) cellular response to hypoxia (aquaporin 1 (AQP1), endothelial PAS domain protein 1 (EPAS1), homeodomain-interacting protein kinase 2 (HIPK2), (GO:0000045) autophagosome assembly (GABARAPL1, TP53INP2), (GO:0050811) GABA type A receptor binding (GABARAPL1), and (GO:0086064) cell communication via electrical coupling involved in cardiac conduction (SLC8A1, ATP1B2).
To explore the Diseases and Biological Functions significantly enriched in DEGs and assess potential associations with PD susceptibility in the SNpc, we employed Ingenuity Pathway Analysis (IPA). The analysis revealed significant enrichment in Neurological Disease, with multiple annotations related to disorders of the basal ganglia, movement disorders, neuromuscular disease, dyskinesia, progressive motor neuropathy, familial neurological disorders, Parkinson’s disease, progressive neurological disorders, abnormal morphology of the nervous system, and tauopathy (Figure 3 and Supplementary Table S3). The DEG IPA Network Analysis identified seven networks with nodes and interactions associated with the top Diseases or Function Annotations, such as Neurological Disease, Organismal Injury and Abnormalities, and Psychological Disorders (Figure 4 and Table 3).
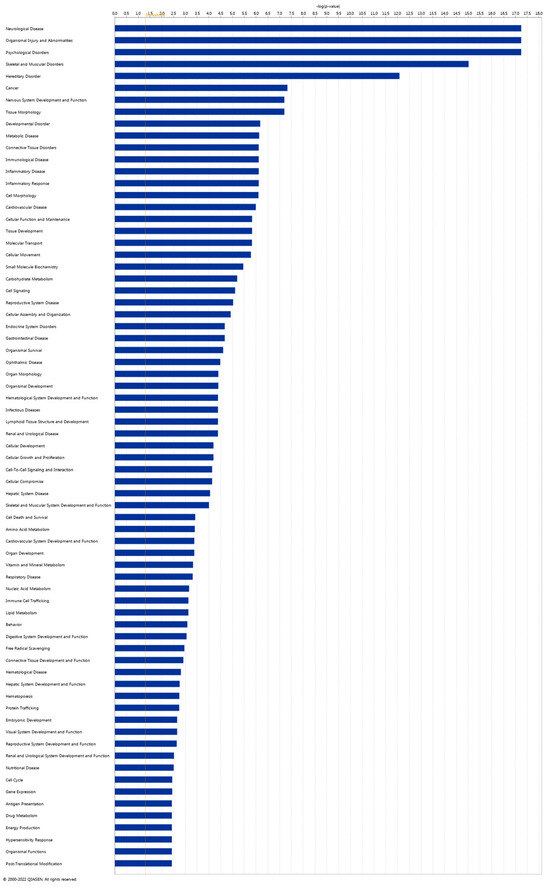
Figure 3.
Ingenuity Pathway Analysis (IPA). Disease and Function analysis of differentially expressed genes in the IPA software (Prism version 9.4.1, 2022) with Log2 (p-value) scale.
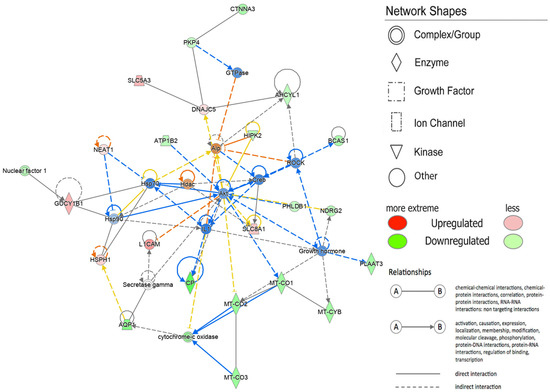
Figure 4.
Network by Ingenuity Pathway Analysis (IPA). Neurological Disease, Organismal Injury and Abnormalities, Psychological Disorders. Red indicates the up-regulated transcripts, whereas green indicates the down-regulated transcripts. The color of the lines indicates the different relationship states. In particular, the orange lines show an activation, the blue lines show an inhibition, gray indicates non-predicted effects, and yellow indicates findings inconsistent with the state of the downstream molecule.
Table 3.
Molecule networks obtained via Ingenuity Pathway Analysis (IPA).
3. Discussion
While PD was initially described by James Parkinson in 1817, it remains a disease with several knowledge gaps, spanning the ethnic divide, gender differences, and the field of PD genetics [46]. Numerous studies in the field of omics sciences (genomics, transcriptomics, proteomics, metabolomics) have contributed to the identification of new genes, pathways, and proteins, providing insight into the extensive variability of PD in the context of current trends in personalized medicine [47].
Referring to the review by Rike and Stern [43], our study, at the pathway analysis level, reveals some overlaps with interaction mechanisms and cell adhesion molecules. Importantly, we have identified data that were not previously highlighted in the literature.
The results obtained indicate 33 significantly up-regulated mRNAs (padj ≤ 0.05 and |FC| ≥ 1.5) and 59 significantly down-regulated mRNAs (padj ≤ 0.05 and |FC| ≤ −1.5) in PD subjects compared with CTRLs.
Among these, the genes most involved in the highlighted pathways, functions, and networks include ATP1B2, GABARAPL, SLC38A, mitochondrially encoded cytochrome c oxidase I (MT-CO1), mitochondrially encoded cytochrome c oxidase II (MT-CO2), mitochondrially encoded cytochrome c oxidase III (MT-CO3), mitochondrially encoded cytochrome b (MT-CYB), and cytochrome oxidase.
ATP1B2, a plasma membrane pump with diverse functions, including homeostasis in cell differentiation and apoptosis, is particularly expressed in the brain [48]. In our study, the ATP1B2 gene was under-expressed in PD brains compared to CTRLs (Table 1), confirming its peculiar expression in normal brains, as previously reported [49]. Following KEGG and GO enrichment analysis, ATP1B2 correlated with most of the relevant pathways observed (Figure 2A,B). Notably, in KEGG analysis, one pathway is “contraction of cardiac muscles”, while in GO analysis, ATP1B2 correlates with the “cellular communication via electrical coupling involved in cardiac conduction” pathway (Figure 2A,B).
The cardiac sodium/calcium exchanger (SLC8A1), found to be overexpressed (Table 2) in our data, is a bidirectional calcium transporter contributing to the electrical activity of the heart. It is abundantly expressed in the heart and less so in the brain, retina, and skeletal muscle [50,51]. Enriched categories included different genes involved in cardiac muscle contraction, highlighting the known clinical and experimental evidence linking cardiac changes and PD. Although most dysautonomic symptoms in PD arise from alterations in the peripheral nerves of the autonomic nervous system [52], a direct role of myocardial cell pathology in PD patients cannot be excluded.
Recent evidence shows a close link between PD and cardiac cell dysfunction, with mitophagy likely playing a crucial role [53,54]. Proteins like those encoded by the ATP1B2 gene, integral membrane proteins responsible for establishing and maintaining electrochemical gradients, may connect cardiac dysfunction with the PD patients studied here [55].
Notably, Bardutz et al. [56] recently suggested alterations in systolic function in PD patients, possibly related to dysfunctional cardiac muscle contraction. Oleksakova et al. [57] highlighted differences in baroreflex function and the baroreflex-mediated vasoconstriction response to orthostasis in PD patients. While data in the literature associate cardiac dysfunction with PD, our study directly underscores the dysregulation of genes related to cardiac contraction in the brain of PD patients. The specific role of these dysregulated genes in brain function remains unclear, but we cannot exclude their potential contribution to the neurodegeneration observed in PD.
Among the genes of interest, GABARAPL1 was found to be overexpressed, as indicated via both KEGG and GO analyses, involving a distinct set of pathways (five and three, respectively): GABAergic synapse, autophagy—animal, the nucleotide-oligomerization-domain (NOD)-like receptor signaling pathway (a specialized group of intracellular proteins that plays a critical role in the regulation of the host innate immune response), autophagy—other, and mitophagy—animal (according to KEGG), as well as autophagosome, GABA type A receptor binding, and Tat protein binding (according to GO). The Tat protein serves as a nuclear transactive regulator of viral gene expression, either for human immunodeficiency virus (HIV) or the equivalent protein of another virus (https://www.uniprot.org/uniprotkb/P04610/entry; accessed on 10 December 2023). Nagel et al. [58] demonstrated that the Tat-heat shock protein 70 (Hsp70) complex effectively prevents neuronal cell death in both in vitro and in vivo models of PD.
Autophagy, a highly conserved cellular degradation process regulated by specific autophagy-related (Atg) factors, entails the formation of double-membrane autophagosomes that engulf cytoplasmic components for degradation. In mammals, this process is complex due to the presence of six Atg8 homologues, categorized into the GABA type A receptor-associated protein (GABARAP) and microtubule-associated protein 1 light-chain 3 (MAP1LC3) subfamilies [59]. GABARAPL1/GEC1, a member of the GABARAP subfamily, exhibits the highest mRNA expression among Atg8 homologues in the CNS. Notably, GABARAPL1 brain expression is observable as early as embryonic day 11, increasing progressively to peak in adulthood. Significantly, GABARAPL1 expression in the adult brain is particularly intense in neurons involved in motor and neuroendocrine functions, notably in the SNpc [60]. A dysregulation of Atg8 homologues has been observed in other synucleinopathies, such as Lewy-body dementia and multiple-system atrophy [61]. In PD, alterations in autophagic mechanisms are evident, as demonstrated by transcript levels of several autophagy genes in blood cells. A recent study found the overexpression of autophagy-related genes, including MAP1LC3B, GABARAP, GABARAPL1, GABARAPL2, and sequestosome 1 (P62/SQSTM1), in PD patients, with potential implications for predicting markers and therapeutic responses [62].
Among the highlighted genes, AMPH stands out, as it was found to be overexpressed, as indicated via both the KEGG and GO analyses. This overexpression involves pathways such as phagocytosis mediated by Fc gamma R (according to KEGG) and chemical synaptic transmission (according to GO). The crystallizable fragment receptor (FcR) is a receptor with a specific binding capacity for the Fc region of the antibody tail and is responsible for inducing phagocytosis. When Fc gamma R on monocyte-macrophages or neutrophils combines with IgG through its Fc region, it triggers phagocytosis [63,64]. FcRs play a crucial role in normal responses to infection or tissue injury and are implicated in immune-related diseases and increasingly observed in neurodegenerative diseases [65]. The aberrant activation of FcRs in neural cells may contribute to the pathogenesis of major neurodegenerative conditions, including Alzheimer’s disease, Parkinson’s disease, ischemic stroke, and multiple sclerosis [65]. This observation aligns with the pathway expression found in our study sample.
Similarly, the SLC38A2 gene was identified as overexpressed and associated with two relevant pathways according to KEGG: GABAergic synapse and protein digestion and absorption. This finding strengthens the connection between GABA overexpression, PD pathology, and neural degeneration. In a recent study using a rotenone-induced PD rat model [66], nardosinone, a biochemical compound enhancing NGF-mediated neurite outgrowth and synaptogenesis, demonstrated anti-PD efficacy. Transcriptome and proteome analyses suggested that the anti-PD target of nardosinone is the SLC38A2 gene, potentially involving the GABAergic synaptic pathway. This underscores the SLC38A2 gene as a potential target for PD treatment and emphasizes its modulatory effects as an anti-PD agent through the GABA system [66]. Diseases and Function analysis in the IPA software highlighted that DEGs correlated with Neurological Disease (Figure 3 and Figure 4). Network IPA Analysis (Figure 4) revealed the down-regulation of many mitochondrial genes in the SN of PD subjects, including MT-CO1, MT-CO2, MT-CO3, MT-CYB, and cytochrome oxidase. This further supports mitochondrial dysfunction in PD, where mitochondria, involved in crucial functions, primarily energy generation, are essential for nearly all cellular activities. Alterations in mitochondrial functioning lead to insufficient energy production, particularly affecting the CNS [67]. Evidence indicates that mitochondrial respiratory-chain dysfunction plays a primary role in various neurodegenerative diseases, including PD [67,68]. Impaired elements of the respiratory chain, such as defects in complex I, have been associated with PD and frontal cortex dysfunction [69,70,71,72]. Damage to the electron transport chain increases oxidative stress and neuronal dysfunction, potentially contributing to the onset and progression of PD. Progressive mitochondrial damage results in the accumulation of non-functional mitochondria, further contributing to neuronal degeneration [73].
Looking at the down-regulated genes in the SN of PD patients, a group of genes involved in maintaining the structure and function of glial cells was noted. These genes include MBP, CPN (myelin protein cyclic nucleotide phosphodiesterase), CTNNA3 (alpha-T-catenin), AQP1, and glial fibrillary acidic protein (GFAP). Among the up-regulated genes, SLC8A1 is involved in linking trans-plasmalemmal gradients of sodium and calcium ions to the membrane potential of astrocytes. This outcome suggests the emerging important role of glial cells in neurodegeneration and PD pathogenesis [74,75,76,77,78,79].
4. Materials and Methods
4.1. Human Post Mortem Midbrain Samples
Human midbrain samples were generously provided by the Parkinson’s UK Brain Bank (Imperial College London, London, UK). A total of 8 PD (6 males and 2 females; mean age 81.125 and SD of 5.693) and 6 CTRL (1 male and 5 females; mean age 78.166 and SD of 10.381) samples were acquired, and the specimens were histologically sectioned at the midbrain, encompassing the human SN in all slides. Each section had a thickness of 4 µm. Supplementary Table S4 presents a concise summary of the clinical characteristics of PD subjects and controls, with the data sourced from the Parkinson’s UK Brain Bank (Imperial College London, London, UK). It is noteworthy that all PD cases included in the study are classified at Braak LB Stage 6. The study adhered to the principles of the Declaration of Helsinki of 1964 and its subsequent amendments. The Ethics Committee of the Oasi Research Institute—IRCCS of Troina (Italy) granted approval for the protocol on 5 April 2022 (approval code: 2022/04/05/CE-IRCCS-OASI/52).
4.2. RNA Isolation from Human Midbrain Samples
RNA was extracted from 4 µm of Formalin-fixed paraffin-embedded (FFPE) slide-mounted sections using the RecoverAll Total Nucleic Acid Isolation Protocol (Thermo Fisher Scientific Inc., Waltham, MA, USA), following the manufacturer’s instructions. Subsequently, the RNA was stored at −80 °C until further processing.
4.3. RNA Sequencing and Functional Analysis
RNA sequencing and subsequent data analysis were conducted by Genomix4Life Srl (Baronissi, Italy). The quality and quantity of RNA were assessed using a Qubit fluorometer (Thermo Fisher Scientific Inc.) and a TapeStation 4200 (Agilent Technologies, 5301 Stevens Creek Blvd), respectively.
Indexed libraries were prepared from 50 ng of purified RNA each, employing the Illumina Stranded Total RNA with Ribo-Zero Plus Kit (Illumina Inc., San Diego, CA, USA), following the manufacturer’s guidelines. Library quantification was performed using the TapeStation 4200 (Agilent Technologies, Santa Clara, CA, USA) and Qubit. Subsequently, the indexed libraries were pooled in equimolar amounts, resulting in a final concentration of 2 nM. The Illumina NovaSeq 6000 System was utilized to sequence the pooled samples in a 2 × 75 paired-end format.
Trimming of short reads (<25 bp) and removal of adapter sequences were carried out with cutadapt (v.2.8) [80]. Using STAR [81] software (version 2.7.3a), the trimmed fastq files were mapped to the reference genome (GenCode (HG38-Release 37 (GRCh38.p13)). [https://www.gencodegenes.org/human/ accessed on 1 August 2023]) STAR [81] was used with standard parameters to create paired-end fastq files and wrote several output files, such as alignments (SAM/BAM). The next step of RNA-seq workflow, involved the gene quantification per sample, was performed using the featureCounts (version 2.0) tool. FeatureCounts is a highly efficient read summarization program that counts mapped reads (SAM/BAM files) for genomic features such as genes [https://bioweb.pasteur.fr/docs/modules/subread/1.4.6-p3/SubreadUsersGuide, accessed on 29 December 2023]. A Custom R script was employed for data normalization and differential expression analysis using the Bioconductor DESeq2 [82] package, based on negative binomial generalized linear models and on the estimates of dispersion and logarithmic fold changes incorporated in data-driven prior distributions. The threshold for considering genes as differentially expressed was set at Fold-Change ≥ 1.50 or ≤−1.50 (|FC| ≥ 1.50) with adjusted p-values ≤ 0.05 (padj). Volcano plots and heatmaps were generated using the En-hancedVolcano (10.18129/B9.bioc.EnhancedVolcano) and ComplexHeatmap [83] packages in R.
For functional analysis, KEGG pathway and GO database analyses were conducted using the R package pathfinder. Additionally, to gain a deeper understanding of the complex transcriptomics data, Ingenuity Pathway Analysis (IPA) (Prism version 9.4.1, 2022) [84] was performed, specifically for investigating diseases and function analysis.
The raw data (.fastq files) and the normalized count of identified mRNAs are available on ArrayExpress (E-MTAB-13295).
5. Conclusions
One of the study’s limitations is undoubtably related to the number of cases examined, which stems from the necessity to select samples with a high yield of extracted mRNA. A future objective includes expanding the sample size of PD subjects to enhance clinical heterogeneity and explore the pathological anatomy further. Another limitation is the absence of an expression study on non-coding RNAs (ncRNAs), which could be a focus of subsequent research.
The in-depth analysis conducted on the transcriptome of our samples revealed statistically significant findings pertaining to various signaling pathways, including cardiac muscle contraction, GABAergic synapse, autophagy, the Fc gamma R-mediated phagocytosis signaling pathway, the cellular response to chemical synaptic transmission, hypoxia, autophagosome assembly, GABA type A receptor binding, and cellular communication via electrical coupling involved in cardiac conduction. These results were obtained through KEGG and GO enrichment analyses. Notably, we highlight gene expression alterations affecting “cardiac muscle contraction” and “cellular communication via electrical coupling involved in cardiac conduction” in PD brain tissues compared to CTRLs. This leads us to speculate that genes conventionally associated with electrical conduction mechanisms in cardiac muscle might also play a role in the brain, with their function still unclear. We cannot exclude the involvement of differentially expressed genes, such as SLC8A1 and ATP1B2, in various tissues within the context of PD.
Another significant finding is the altered expression of mitochondrial genes in the SN of subjects with PD. While this has been discussed in the literature, our study reaffirms it, emphasizing the central role of mitochondrial genes in PD. These discoveries may pave the way for improved diagnostic precision and the development of novel targeted therapies.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/ijms25020707/s1.
Author Contributions
Concept and design, M.S., G.C. and R.F.; next-generation sequencing analysis, M.R., G.M. and G.M.V.; acquisition of data or analysis, M.R., G.M., G.M.V. and M.S.; writing—original draft preparation, M.S., G.L., M.R., G.M., F.A.S., M.G.S., G.C. and R.F.; final approval, M.S., G.L., M.R. and R.F. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by Italian Ministry of Health, grant number RC-2779777.
Institutional Review Board Statement
The study was conducted in accordance with the Declaration of Helsinki of 1964 and its later amendments, and the Ethics Committee of the Oasi Research Institute–IRCCS of Troina (Italy) approved the protocol on 5 April 2022 (approval code: 2022/04/05/CE-IRCCS-OASI/52).
Informed Consent Statement
The Centre for Blast Injury Studies fully complies with The Human Tissue Act 2004 (which replaced the Human Tissue Act 1961, the Anatomy Act 1984, and the Human Organ Transplants Act 1989), governed by the Human Tissue Authority, which outlines the use of human tissue for scientific purposes in the UK. Although the Human Tissue Act does not cover the use of tissues that are sourced outside the UK, the Centre also complies with its standards in its dealings with human tissue from overseas. Imperial College London also holds an institutional license from the Human Tissue Authority to collect, store, and use human tissue. As provided for in the Human Tissue Act, and depending on the nature of the research, organizations other than the Human Tissue Authority regulate the actual research on human tissue and, in the case of this study, this has been via the Ethics Committee of the Oasi Research Institute—IRCCS of Troina (Italy).
Data Availability Statement
https://www.bioinformatics.babraham.ac.uk/projects/fastqc/ accessed on 29 December 2023 (E-MTAB-13295).
Acknowledgments
We thank Parkinson’s UK Brain Bank (Imperial College London, London, UK) for providing human midbrain samples.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Kalia, L.V.; Lang, A.E. Parkinson’s Disease. Lancet 2015, 386, 896–912. [Google Scholar] [CrossRef]
- Quik, M. Smoking, Nicotine and Parkinson’s Disease. Trends Neurosci. 2004, 27, 561–568. [Google Scholar] [CrossRef] [PubMed]
- Bloem, B.R.; Okun, M.S.; Klein, C. Parkinson’s Disease. Lancet 2021, 397, 2284–2303. [Google Scholar] [CrossRef] [PubMed]
- Postuma, R.B.; Berg, D.; Stern, M.; Poewe, W.; Olanow, C.W.; Oertel, W.; Obeso, J.; Marek, K.; Litvan, I.; Lang, A.E.; et al. MDS Clinical Diagnostic Criteria for Parkinson’s Disease. Mov. Disord. 2015, 30, 1591–1601. [Google Scholar] [CrossRef] [PubMed]
- Lang, A.E.; Obeso, J.A. Time to Move beyond Nigrostriatal Dopamine Deficiency in Parkinson’s Disease. Ann. Neurol. 2004, 55, 761–765. [Google Scholar] [CrossRef] [PubMed]
- Obeso, J.A.; Stamelou, M.; Goetz, C.G.; Poewe, W.; Lang, A.E.; Weintraub, D.; Burn, D.; Halliday, G.M.; Bezard, E.; Przedborski, S.; et al. Past, Present, and Future of Parkinson’s Disease: A Special Essay on the 200th Anniversary of the Shaking Palsy. Mov. Disord. 2017, 32, 1264–1310. [Google Scholar] [CrossRef] [PubMed]
- Figorilli, M.; Lanza, G.; Congiu, P.; Lecca, R.; Casaglia, E.; Mogavero, M.P.; Puligheddu, M.; Ferri, R. Neurophysiological Aspects of REM Sleep Behavior Disorder (RBD): A Narrative Review. Brain Sci. 2021, 11, 1588. [Google Scholar] [CrossRef] [PubMed]
- Joza, S.; Hu, M.T.; Jung, K.-Y.; Kunz, D.; Stefani, A.; Dušek, P.; Terzaghi, M.; Arnaldi, D.; Videnovic, A.; Schiess, M.C.; et al. Progression of Clinical Markers in Prodromal Parkinson’s Disease and Dementia with Lewy Bodies: A Multicentre Study. Brain 2023, 146, 3258–3272. [Google Scholar] [CrossRef]
- Jankovic, J. Parkinson’s Disease: Clinical Features and Diagnosis. J. Neurol. Neurosurg. Psychiatry 2008, 79, 368–376. [Google Scholar] [CrossRef]
- Bhidayasiri, R.; Sringean, J.; Reich, S.G.; Colosimo, C. Red Flags Phenotyping: A Systematic Review on Clinical Features in Atypical Parkinsonian Disorders. Park. Relat. Disord. 2019, 59, 82–92. [Google Scholar] [CrossRef]
- Caproni, S.; Colosimo, C. Diagnosis and Differential Diagnosis of Parkinson Disease. Clin. Geriatr. Med. 2020, 36, 13–24. [Google Scholar] [CrossRef] [PubMed]
- Piras, I.S.; Bleul, C.; Schrauwen, I.; Talboom, J.; Llaci, L.; De Both, M.D.; Naymik, M.A.; Halliday, G.; Bettencourt, C.; Holton, J.L.; et al. Transcriptional Profiling of Multiple System Atrophy Cerebellar Tissue Highlights Differences between the Parkinsonian and Cerebellar Sub-Types of the Disease. Acta Neuropathol. Commun. 2020, 8, 76. [Google Scholar] [CrossRef] [PubMed]
- Lang, A.E. A Critical Appraisal of the Premotor Symptoms of Parkinson’s Disease: Potential Usefulness in Early Diagnosis and Design of Neuroprotective Trials. Mov. Disord. 2011, 26, 775–783. [Google Scholar] [CrossRef] [PubMed]
- Salemi, M.; Mogavero, M.P.; Lanza, G.; Mongioì, L.M.; Calogero, A.E.; Ferri, R. Examples of Inverse Comorbidity between Cancer and Neurodegenerative Diseases: A Possible Role for Noncoding RNA. Cells 2022, 11, 1930. [Google Scholar] [CrossRef] [PubMed]
- Fisicaro, F.; Lanza, G.; Cantone, M.; Ferri, R.; Pennisi, G.; Nicoletti, A.; Zappia, M.; Bella, R.; Pennisi, M. Clinical and Electrophysiological Hints to TMS in De Novo Patients with Parkinson’s Disease and Progressive Supranuclear Palsy. J. Pers. Med. 2020, 10, 274. [Google Scholar] [CrossRef] [PubMed]
- Cheng, H.-C.; Ulane, C.M.; Burke, R.E. Clinical Progression in Parkinson Disease and the Neurobiology of Axons. Ann. Neurol. 2010, 67, 715–725. [Google Scholar] [CrossRef] [PubMed]
- Burke, R.E.; O’Malley, K. Axon Degeneration in Parkinson’s Disease. Exp. Neurol. 2013, 246, 72–83. [Google Scholar] [CrossRef]
- Dauer, W.; Przedborski, S. Parkinson’s Disease: Mechanisms and Models. Neuron 2003, 39, 889–909. [Google Scholar] [CrossRef]
- Braak, H.; Sandmann-Keil, D.; Gai, W.; Braak, E. Extensive Axonal Lewy Neurites in Parkinson’s Disease: A Novel Pathological Feature Revealed by Alpha-Synuclein Immunocytochemistry. Neurosci. Lett. 1999, 265, 67–69. [Google Scholar] [CrossRef]
- Braak, H.; Del Tredici, K.; Rüb, U.; de Vos, R.A.I.; Jansen Steur, E.N.H.; Braak, E. Staging of Brain Pathology Related to Sporadic Parkinson’s Disease. Neurobiol. Aging 2003, 24, 197–211. [Google Scholar] [CrossRef]
- Fearnley, J.M.; Lees, A.J. Ageing and Parkinson’s Disease: Substantia Nigra Regional Selectivity. Brain 1991, 114 Pt 5, 2283–2301. [Google Scholar] [CrossRef]
- Hirsch, E.C.; Breidert, T.; Rousselet, E.; Hunot, S.; Hartmann, A.; Michel, P.P. The Role of Glial Reaction and Inflammation in Parkinson’s Disease. Ann. N. Y. Acad. Sci. 2003, 991, 214–228. [Google Scholar] [CrossRef]
- Haque, M.E.; Akther, M.; Jakaria, M.; Kim, I.-S.; Azam, S.; Choi, D.-K. Targeting the Microglial NLRP3 Inflammasome and Its Role in Parkinson’s Disease. Mov. Disord. 2020, 35, 20–33. [Google Scholar] [CrossRef]
- De Rivero Vaccari, J.P.; Dietrich, W.D.; Keane, R.W. Therapeutics Targeting the Inflammasome after Central Nervous System Injury. Transl. Res. 2016, 167, 35–45. [Google Scholar] [CrossRef]
- Martinon, F.; Burns, K.; Tschopp, J. The Inflammasome: A Molecular Platform Triggering Activation of Inflammatory Caspases and Processing of proIL-Beta. Mol. Cell 2002, 10, 417–426. [Google Scholar] [CrossRef]
- Broz, P.; Dixit, V.M. Inflammasomes: Mechanism of Assembly, Regulation and Signalling. Nat. Rev. Immunol. 2016, 16, 407–420. [Google Scholar] [CrossRef]
- Malik, A.; Kanneganti, T.-D. Inflammasome Activation and Assembly at a Glance. J. Cell Sci. 2017, 130, 3955–3963. [Google Scholar] [CrossRef]
- Van Opdenbosch, N.; Lamkanfi, M. Caspases in Cell Death, Inflammation, and Disease. Immunity 2019, 50, 1352–1364. [Google Scholar] [CrossRef]
- Franklin, B.S.; Bossaller, L.; De Nardo, D.; Ratter, J.M.; Stutz, A.; Engels, G.; Brenker, C.; Nordhoff, M.; Mirandola, S.R.; Al-Amoudi, A.; et al. The Adaptor ASC Has Extracellular and “prionoid” Activities That Propagate Inflammation. Nat. Immunol. 2014, 15, 727–737. [Google Scholar] [CrossRef]
- Cabrera Ranaldi, E.D.L.R.M.; Nuytemans, K.; Martinez, A.; Luca, C.C.; Keane, R.W.; de Rivero Vaccari, J.P. Proof-of-Principle Study of Inflammasome Signaling Proteins as Diagnostic Biomarkers of the Inflammatory Response in Parkinson’s Disease. Pharmaceuticals 2023, 16, 883. [Google Scholar] [CrossRef]
- Codolo, G.; Plotegher, N.; Pozzobon, T.; Brucale, M.; Tessari, I.; Bubacco, L.; de Bernard, M. Triggering of Inflammasome by Aggregated α-Synuclein, an Inflammatory Response in Synucleinopathies. PLoS ONE 2013, 8, e55375. [Google Scholar] [CrossRef]
- De Rivero Vaccari, J.P.; Dietrich, W.D.; Keane, R.W. Activation and Regulation of Cellular Inflammasomes: Gaps in Our Knowledge for Central Nervous System Injury. J. Cereb. Blood. Flow Metab. 2014, 34, 369–375. [Google Scholar] [CrossRef]
- Funayama, M.; Nishioka, K.; Li, Y.; Hattori, N. Molecular Genetics of Parkinson’s Disease: Contributions and Global Trends. J. Hum. Genet. 2023, 68, 125–130. [Google Scholar] [CrossRef]
- Nalls, M.A.; Pankratz, N.; Lill, C.M.; Do, C.B.; Hernandez, D.G.; Saad, M.; DeStefano, A.L.; Kara, E.; Bras, J.; Sharma, M.; et al. Large-Scale Meta-Analysis of Genome-Wide Association Data Identifies Six New Risk Loci for Parkinson’s Disease. Nat. Genet. 2014, 46, 989–993. [Google Scholar] [CrossRef]
- Bandres-Ciga, S.; Diez-Fairen, M.; Kim, J.J.; Singleton, A.B. Genetics of Parkinson’s Disease: An Introspection of Its Journey towards Precision Medicine. Neurobiol. Dis. 2020, 137, 104782. [Google Scholar] [CrossRef]
- Dulski, J.; Uitti, R.J.; Ross, O.A.; Wszolek, Z.K. Genetic Architecture of Parkinson’s Disease Subtypes—Review of the Literature. Front. Aging Neurosci. 2022, 14, 1023574. [Google Scholar] [CrossRef]
- Redenšek, S.; Dolžan, V.; Kunej, T. From Genomics to Omics Landscapes of Parkinson’s Disease: Revealing the Molecular Mechanisms. OMICS 2018, 22, 1–16. [Google Scholar] [CrossRef]
- Simunovic, F.; Yi, M.; Wang, Y.; Macey, L.; Brown, L.T.; Krichevsky, A.M.; Andersen, S.L.; Stephens, R.M.; Benes, F.M.; Sonntag, K.C. Gene Expression Profiling of Substantia Nigra Dopamine Neurons: Further Insights into Parkinson’s Disease Pathology. Brain 2009, 132, 1795–1809. [Google Scholar] [CrossRef]
- Salemi, M.; Cosentino, F.; Lanza, G.; Cantone, M.; Salluzzo, M.G.; Giurato, G.; Borgione, E.; Marchese, G.; Santa Paola, S.; Lanuzza, B.; et al. MRNA Expression Profiling of Mitochondrial Subunits in Subjects with Parkinson’s Disease. Arch. Med. Sci. 2023, 19, 678–686. [Google Scholar] [CrossRef]
- Salemi, M.; Lanza, G.; Mogavero, M.P.; Cosentino, F.I.I.; Borgione, E.; Iorio, R.; Ventola, G.M.; Marchese, G.; Salluzzo, M.G.; Ravo, M.; et al. A Transcriptome Analysis of MRNAs and Long Non-Coding RNAs in Patients with Parkinson’s Disease. Int. J. Mol. Sci. 2022, 23, 1535. [Google Scholar] [CrossRef] [PubMed]
- Zaccaria, A.; Antinori, P.; Licker, V.; Kövari, E.; Lobrinus, J.A.; Burkhard, P.R. Multiomic Analyses of Dopaminergic Neurons Isolated from Human Substantia Nigra in Parkinson’s Disease: A Descriptive and Exploratory Study. Cell Mol. Neurobiol. 2022, 42, 2805–2818. [Google Scholar] [CrossRef]
- Okuzumi, A.; Hatano, T.; Fukuhara, T.; Ueno, S.; Nukina, N.; Imai, Y.; Hattori, N. α-Synuclein Seeding Assay Using RT-QuIC. Methods Mol. Biol. 2021, 2322, 3–16. [Google Scholar] [CrossRef]
- Rike, W.A.; Stern, S. Proteins and Transcriptional Dysregulation of the Brain Extracellular Matrix in Parkinson’s Disease: A Systematic Review. Int. J. Mol. Sci. 2023, 24, 7435. [Google Scholar] [CrossRef]
- Liu, Y.; Zhou, Q.; Tang, M.; Fu, N.; Shao, W.; Zhang, S.; Yin, Y.; Zeng, R.; Wang, X.; Hu, G.; et al. Upregulation of alphaB-Crystallin Expression in the Substantia Nigra of Patients with Parkinson’s Disease. Neurobiol. Aging 2015, 36, 1686–1691. [Google Scholar] [CrossRef]
- Dumitriu, A.; Golji, J.; Labadorf, A.T.; Gao, B.; Beach, T.G.; Myers, R.H.; Longo, K.A.; Latourelle, J.C. Integrative Analyses of Proteomics and RNA Transcriptomics Implicate Mitochondrial Processes, Protein Folding Pathways and GWAS Loci in Parkinson Disease. BMC Med. Genom. 2016, 9, 5. [Google Scholar] [CrossRef]
- González-Casacuberta, I.; Juárez-Flores, D.L.; Morén, C.; Garrabou, G. Bioenergetics and Autophagic Imbalance in Patients-Derived Cell Models of Parkinson Disease Supports Systemic Dysfunction in Neurodegeneration. Front. Neurosci. 2019, 13, 894. [Google Scholar] [CrossRef]
- Gilbert, R.M.; Standaert, D.G. Bridging the Gaps: More Inclusive Research Needed to Fully Understand Parkinson’s Disease. Mov. Disord. 2020, 35, 231–234. [Google Scholar] [CrossRef]
- Li, Y.; Yang, J.; Li, S.; Zhang, J.; Zheng, J.; Hou, W.; Zhao, H.; Guo, Y.; Liu, X.; Dou, K.; et al. N-Myc Downstream-Regulated Gene 2, a Novel Estrogen-Targeted Gene, Is Involved in the Regulation of Na+/K+-ATPase. J. Biol. Chem. 2011, 286, 32289–32299. [Google Scholar] [CrossRef]
- Mauri, N.; Kleiter, M.; Dietschi, E.; Leschnik, M.; Högler, S.; Wiedmer, M.; Dietrich, J.; Henke, D.; Steffen, F.; Schuller, S.; et al. A SINE Insertion in ATP1B2 in Belgian Shepherd Dogs Affected by Spongy Degeneration with Cerebellar Ataxia (SDCA2). G3 2017, 7, 2729–2737. [Google Scholar] [CrossRef] [PubMed]
- Marchiano, S.; Nakamura, K.; Reinecke, H.; Neidig, L.; Lai, M.; Kadota, S.; Perbellini, F.; Yang, X.; Klaiman, J.M.; Blakely, L.P.; et al. Gene Editing to Prevent Ventricular Arrhythmias Associated with Cardiomyocyte Cell Therapy. Cell Stem. Cell 2023, 30, 396–414.e9. [Google Scholar] [CrossRef] [PubMed]
- Khananshvili, D. The SLC8 Gene Family of Sodium-Calcium Exchangers (NCX)—Structure, Function, and Regulation in Health and Disease. Mol. Asp. Med. 2013, 34, 220–235. [Google Scholar] [CrossRef] [PubMed]
- Cuenca-Bermejo, L.; Almela, P.; Navarro-Zaragoza, J.; Fernández Villalba, E.; González-Cuello, A.-M.; Laorden, M.-L.; Herrero, M.-T. Cardiac Changes in Parkinson’s Disease: Lessons from Clinical and Experimental Evidence. Int. J. Mol. Sci. 2021, 22, 13488. [Google Scholar] [CrossRef] [PubMed]
- Yu, W.; Wang, L.; Ren, W.-Y.; Xu, H.-X.; Wu, N.N.; Yu, D.-H.; Reiter, R.J.; Zha, W.-L.; Guo, Q.-D.; Ren, J. SGLT2 Inhibitor Empagliflozin Alleviates Cardiac Remodeling and Contractile Anomalies in a FUNDC1-Dependent Manner in Experimental Parkinson’s Disease. Acta Pharmacol. Sin. 2023. [Google Scholar] [CrossRef] [PubMed]
- Ren, J.; Porter, J.E.; Wold, L.E.; Aberle, N.S.; Muralikrishnan, D.; Haselton, J.R. Depressed Contractile Function and Adrenergic Responsiveness of Cardiac Myocytes in an Experimental Model of Parkinson Disease, the MPTP-Treated Mouse. Neurobiol. Aging 2004, 25, 131–138. [Google Scholar] [CrossRef]
- Piqueras-Flores, J.; López-García, A.; Moreno-Reig, Á.; González-Martínez, A.; Hernández-González, A.; Vaamonde-Gamo, J.; Jurado-Román, A. Structural and Functional Alterations of the Heart in Parkinson’s Disease. Neurol. Res. 2018, 40, 53–61. [Google Scholar] [CrossRef]
- Bardutz, H.; Singh, J.; Rehman, Z.; Bernat, P. Parkinson’s Disease and the Cardiac Cycle: A Rapid Literature Review and Case Series. Life 2023, 13, 1003. [Google Scholar] [CrossRef]
- Oleksakova, J.; Javorka, M.; Czippelova, B.; Mazgutova, N.; Grofik, M.; Babalova, L.; Skacik, P.; Kurca, E. Autonomic Control of Heart and Vessels in Patients with Very Early Stage of Parkinson Disease. Physiol. Meas. 2023, 44, 054002. [Google Scholar] [CrossRef]
- Nagel, F.; Falkenburger, B.H.; Tönges, L.; Kowsky, S.; Pöppelmeyer, C.; Schulz, J.B.; Bähr, M.; Dietz, G.P.H. Tat-Hsp70 Protects Dopaminergic Neurons in Midbrain Cultures and in the Substantia Nigra in Models of Parkinson’s Disease. J. Neurochem. 2008, 105, 853–864. [Google Scholar] [CrossRef]
- Schaaf, M.B.E.; Keulers, T.G.; Vooijs, M.A.; Rouschop, K.M.A. LC3/GABARAP Family Proteins: Autophagy-(Un)Related Functions. FASEB J. 2016, 30, 3961–3978. [Google Scholar] [CrossRef]
- Le Grand, J.N.; Bon, K.; Fraichard, A.; Zhang, J.; Jouvenot, M.; Risold, P.-Y.; Boyer-Guittaut, M.; Delage-Mourroux, R. Specific Distribution of the Autophagic Protein GABARAPL1/GEC1 in the Developing and Adult Mouse Brain and Identification of Neuronal Populations Expressing GABARAPL1/GEC1. PLoS ONE 2013, 8, e63133. [Google Scholar] [CrossRef]
- Wakabayashi, K.; Tanji, K. Multiple system atrophy and autophagy. Rinsho Shinkeigaku 2014, 54, 966–968. [Google Scholar] [CrossRef] [PubMed]
- El Haddad, S.; Serrano, A.; Moal, F.; Normand, T.; Robin, C.; Charpentier, S.; Valery, A.; Brulé-Morabito, F.; Auzou, P.; Mollet, L.; et al. Disturbed Expression of Autophagy Genes in Blood of Parkinson’s Disease Patients. Gene 2020, 738, 144454. [Google Scholar] [CrossRef] [PubMed]
- Fanger, N.A.; Wardwell, K.; Shen, L.; Tedder, T.F.; Guyre, P.M. Type I (CD64) and Type II (CD32) Fc Gamma Receptor-Mediated Phagocytosis by Human Blood Dendritic Cells. J. Immunol. 1996, 157, 541–548. [Google Scholar] [CrossRef] [PubMed]
- Joshi, T.; Butchar, J.P.; Tridandapani, S. Fcγ Receptor Signaling in Phagocytes. Int. J. Hematol. 2006, 84, 210–216. [Google Scholar] [CrossRef] [PubMed]
- Okun, E.; Mattson, M.P.; Arumugam, T.V. Involvement of Fc Receptors in Disorders of the Central Nervous System. Neuromolecular Med. 2010, 12, 164–178. [Google Scholar] [CrossRef] [PubMed]
- Bian, L.-H.; Yao, Z.-W.; Wang, Z.-Y.; Wang, X.-M.; Li, Q.-Y.; Yang, X.; Li, J.-Y.; Wei, X.-J.; Wan, G.-H.; Wang, Y.-Q.; et al. Nardosinone Regulates the Slc38a2 Gene to Alleviate Parkinson’s Symptoms in Rats through the GABAergic Synaptic and cAMP Pathways. Biomed. Pharmacother. 2022, 153, 113269. [Google Scholar] [CrossRef] [PubMed]
- Lezi, E.; Swerdlow, R.H. Mitochondria in Neurodegeneration. Adv. Exp. Med. Biol. 2012, 942, 269–286. [Google Scholar] [CrossRef] [PubMed]
- Power, J.H.T.; Barnes, O.L.; Chegini, F. Lewy Bodies and the Mechanisms of Neuronal Cell Death in Parkinson’s Disease and Dementia with Lewy Bodies. Brain Pathol. 2017, 27, 3–12. [Google Scholar] [CrossRef]
- Parker, W.D.; Boyson, S.J.; Parks, J.K. Abnormalities of the Electron Transport Chain in Idiopathic Parkinson’s Disease. Ann. Neurol. 1989, 26, 719–723. [Google Scholar] [CrossRef]
- Schapira, A.H.; Cooper, J.M.; Dexter, D.; Jenner, P.; Clark, J.B.; Marsden, C.D. Mitochondrial Complex I Deficiency in Parkinson’s Disease. Lancet 1989, 1, 1269. [Google Scholar] [CrossRef]
- Bindoff, L.A.; Birch-Machin, M.; Cartlidge, N.E.; Parker, W.D.; Turnbull, D.M. Mitochondrial Function in Parkinson’s Disease. Lancet 1989, 2, 49. [Google Scholar] [CrossRef] [PubMed]
- Parker, W.D.; Parks, J.K.; Swerdlow, R.H. Complex I Deficiency in Parkinson’s Disease Frontal Cortex. Brain Res. 2008, 1189, 215–218. [Google Scholar] [CrossRef]
- McCoy, M.K.; Cookson, M.R. Mitochondrial Quality Control and Dynamics in Parkinson’s Disease. Antioxid. Redox Signal. 2012, 16, 869–882. [Google Scholar] [CrossRef] [PubMed]
- Nave, K.-A.; Trapp, B.D. Axon-Glial Signaling and the Glial Support of Axon Function. Annu. Rev. Neurosci. 2008, 31, 535–561. [Google Scholar] [CrossRef] [PubMed]
- Weng, A.; Rabin, E.E.; Flozak, A.S.; Chiarella, S.E.; Aillon, R.P.; Gottardi, C.J. Alpha-T-Catenin Is Expressed in Peripheral Nerves as a Constituent of Schwann Cell Adherens Junctions. Biol. Open 2022, 11, bio059634. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Z.; Zhan, J.; Cai, Q.; Xu, F.; Chai, R.; Lam, K.; Luan, Z.; Zhou, G.; Tsang, S.; Kipp, M.; et al. The Water Transport System in Astrocytes-Aquaporins. Cells 2022, 11, 2564. [Google Scholar] [CrossRef] [PubMed]
- Phillips, C.L.; Faridounnia, M.; Armao, D.; Snider, N.T. Stability Dynamics of Neurofilament and GFAP Networks and Protein Fragments. Curr. Opin. Cell Biol. 2023, 85, 102266. [Google Scholar] [CrossRef]
- Rose, C.R.; Ziemens, D.; Verkhratsky, A. On the Special Role of NCX in Astrocytes: Translating Na+-Transients into Intracellular Ca2+ Signals. Cell Calcium 2020, 86, 102154. [Google Scholar] [CrossRef]
- Valori, C.F.; Guidotti, G.; Brambilla, L.; Rossi, D. Astrocytes: Emerging Therapeutic Targets in Neurological Disorders. Trends Mol. Med. 2019, 25, 750–759. [Google Scholar] [CrossRef]
- Kechin, A.; Boyarskikh, U.; Kel, A.; Filipenko, M. cutPrimers: A New Tool for Accurate Cutting of Primers from Reads of Targeted Next Generation Sequencing. J. Comput. Biol. 2017, 24, 1138–1143. [Google Scholar] [CrossRef]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast Universal RNA-Seq Aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef] [PubMed]
- Love, M.I.; Huber, W.; Anders, S. Moderated Estimation of Fold Change and Dispersion for RNA-Seq Data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [PubMed]
- Gu, Z.; Eils, R.; Schlesner, M. Complex Heatmaps Reveal Patterns and Correlations in Multidimensional Genomic Data. Bioinformatics 2016, 32, 2847–2849. [Google Scholar] [CrossRef] [PubMed]
- Krämer, A.; Green, J.; Pollard, J.; Tugendreich, S. Causal Analysis Approaches in Ingenuity Pathway Analysis. Bioinformatics 2014, 30, 523–530. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).