
Compound Heterozygous Mutations of SACS in a Korean Cohort Study of Charcot-Marie-Tooth Disease Concurrent Cerebellar Ataxia and Spasticity

 , and
, and
Abstract
1. Introduction
2. Results
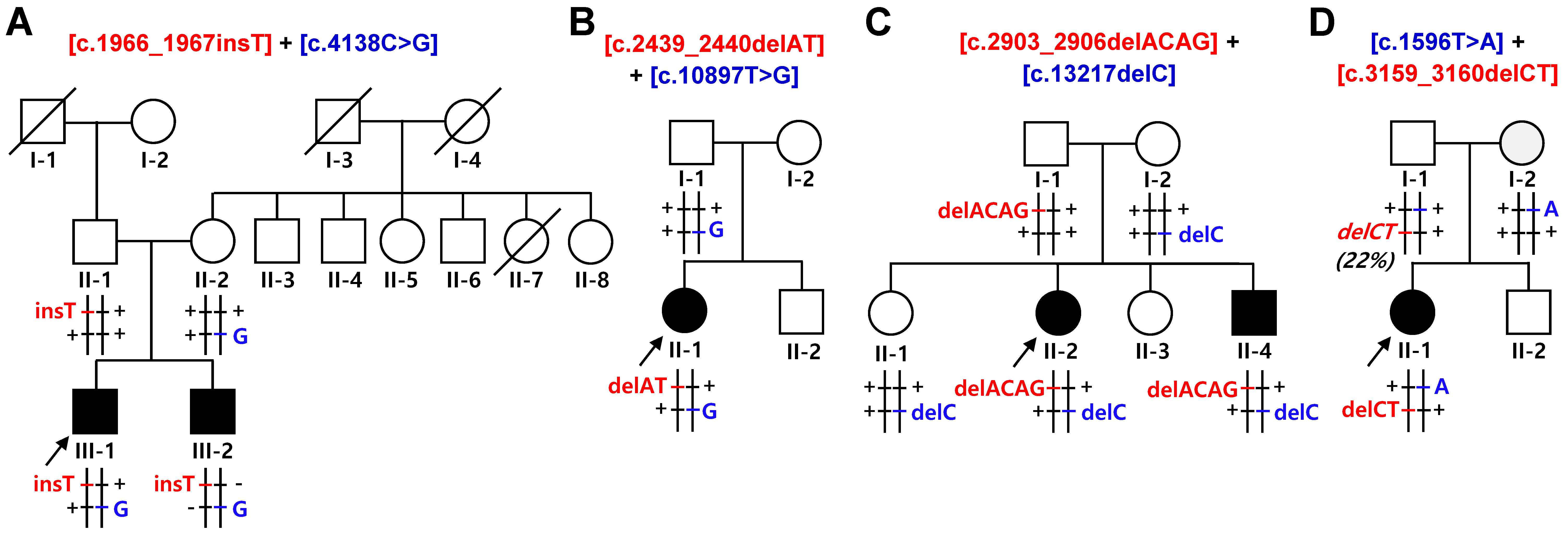
2.1. Identification of Compound Heterozygous SACS Mutations
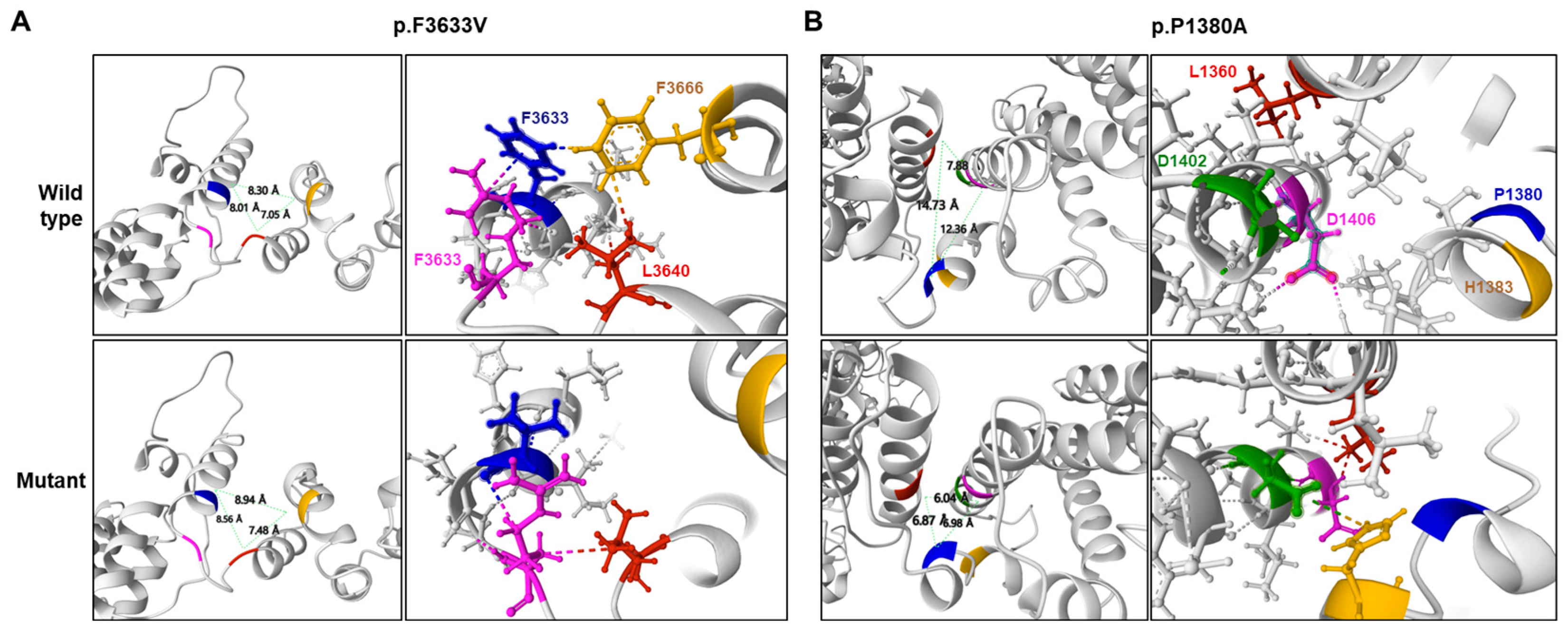
2.2. Prediction of Conformational Changes by Missense Mutations
2.3. Clinical Manifestation
2.4. Electrophysiological Findings
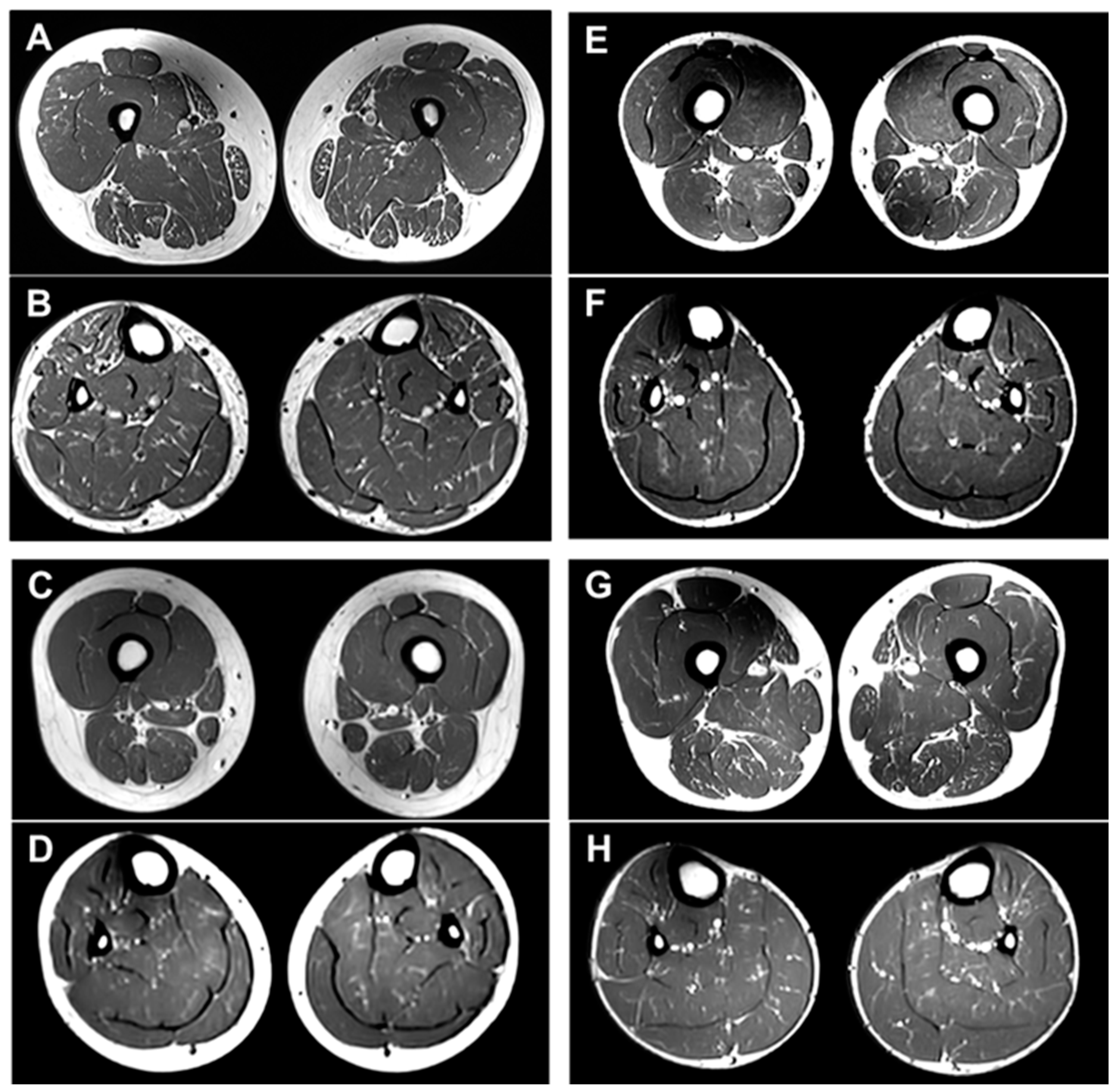
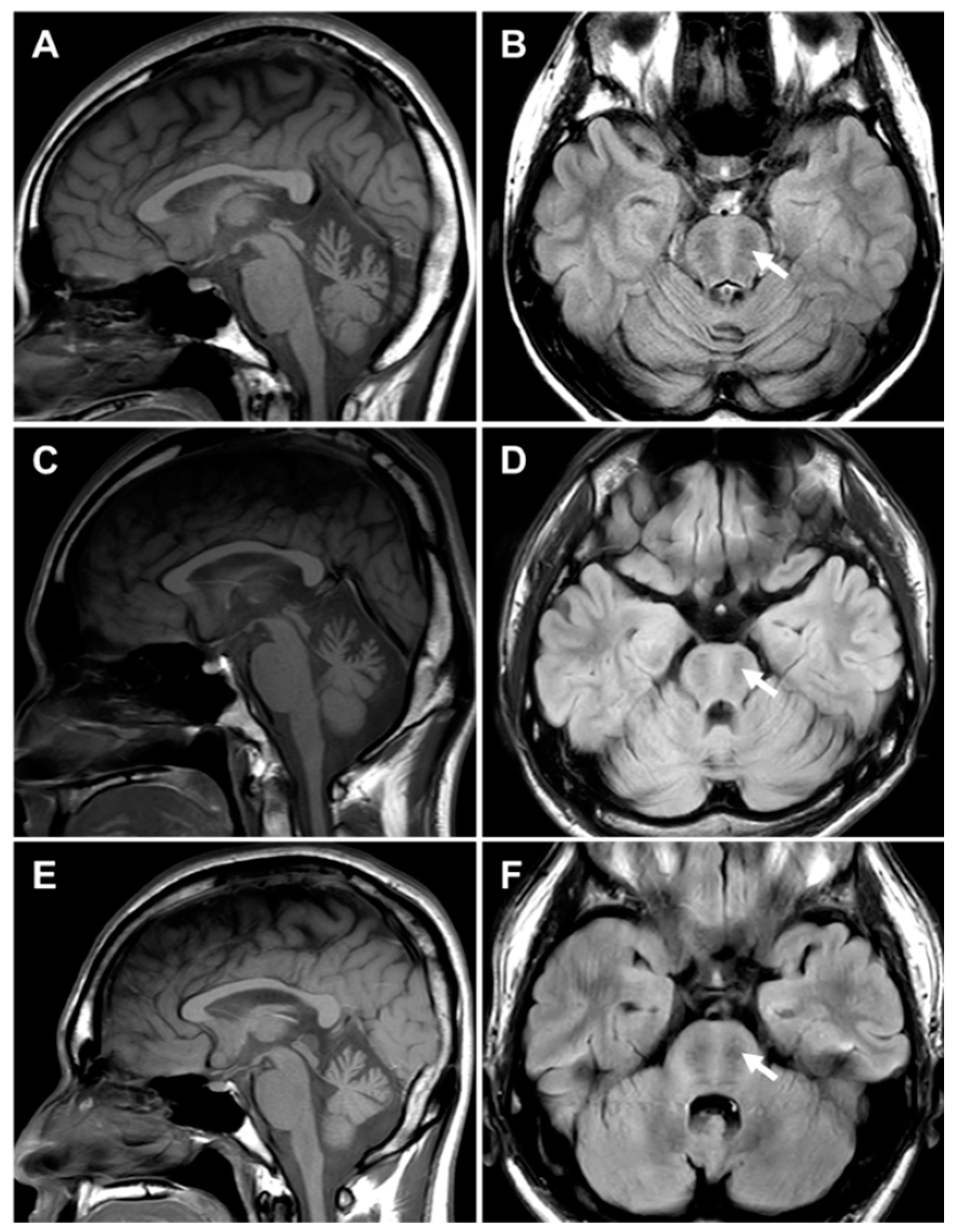
2.5. MRI Features of the Brain and Lower Extremity
3. Discussion
4. Materials and Methods
4.1. Patients
4.2. Molecular Genetic Analysis
4.3. Conservation, Conformational Change, and In Silico Prediction of Mutant Proteins
4.4. Clinical Assessment
4.5. Electrophysiological Examination
4.6. Brain and Lower Extremity MRI
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| ACMG/AMP | American College of Medical Genetics/Genomics/ Association for Molecular Pathology |
| ARSACS | Autosomal recessive spastic ataxia of Charlevoix-Saguenay disease |
| BAEP | Brainstem auditory evoked potential |
| CMAP | Compound muscle action potential |
| CMT | Charcot-Marie-Tooth disease |
| CMTNSv2 | CMT neuropathy score version 2 |
| DTR | Deep tendon reflex |
| FDS | Functional disability scale |
| gnomAD | Genome Aggregation Database |
| GERP | Genomic evolutionary rate profiling score |
| HL | Hearing loss |
| HMSN | Hereditary motor and sensory polyneuropathy |
| IGSR | International Genome Sample Resource |
| KRGDB | Korean Reference Genome Database |
| LP | Likely pathogenic |
| MNCV | Motor nerve conduction velocity |
| MRC | Medical Research Council scale |
| MRI | Magnetic resonance imaging |
| P | Pathogenic |
| PDB | Protein Data Bank |
| SNAP | Sensory nerve action potential |
| SNCV | Sensory nerve conduction velocity |
| WES | Whole exome sequencing |
References
- Pipis, M.; Rossor, A.M.; Laura, M.; Reilly, M.M. Next-generation sequencing in Charcot-Marie-Tooth disease: Opportunities and challenges. Nat. Rev. Neurol. 2019, 15, 644–656. [Google Scholar] [CrossRef] [PubMed]
- Pareyson, D.; Scaioli, V.; Laura, M. Clinical and electrophysiological aspects of Charcot-Marie-Tooth disease. Neuromol. Med. 2006, 8, 3–22. [Google Scholar] [CrossRef] [PubMed]
- Engert, J.C.; Bérubé, P.; Mercier, J.; Doré, C.; Lepage, P.; Ge, B.; Bouchard, J.P.; Mathieu, J.; Melançon, S.B.; Schalling, M.; et al. ARSACS, a spastic ataxia common in northeastern Québec, is caused by mutations in a new gene encoding an 11.5-kb ORF. Nat. Genet. 2000, 24, 120–125. [Google Scholar] [CrossRef] [PubMed]
- El Euch-Fayache, G.; Lalani, I.; Amouri, R.; Turki, I.; Ouahchi, K.; Hung, W.Y.; Belal, S.; Siddique, T.; Hentati, F. Phenotypic features and genetic findings in SACSin-related autosomal recessive ataxia in Tunisia. Arch. Neurol. 2003, 60, 982–988. [Google Scholar] [CrossRef] [PubMed]
- Souza, P.V.S.; Bortholin, T.; Naylor, F.G.M.; Pinto, W.B.V.R.; Oliveira, A.S.B. Early-onset axonal Charcot-Marie-Tooth disease due to SACS mutation. Neuromuscul. Disord. 2018, 28, 169–172. [Google Scholar] [CrossRef] [PubMed]
- Zaman, Q.; Khan, M.A.; Sahar, K.; Rehman, G.; Khan, H.; Rehman, M.; Najumuddin; Ahmad, I.; Tariq, M.; Muthaffar, O.Y.; et al. Novel variants in MPV17, PRX, GJB1, and SACS cause Charcot-Marie-Tooth and spastic ataxia of Charlevoix-Saguenay type diseases. Genes 2023, 14, 328. [Google Scholar] [CrossRef] [PubMed]
- Feely, S.M.; Laura, M.; Siskind, C.E.; Sottile, S.; Davis, M.; Gibbons, V.S.; Reilly, M.M.; Shy, M.E. MFN2 mutations cause severe phenotypes in most patients with CMT2A. Neurology 2011, 76, 1690–1696. [Google Scholar] [CrossRef] [PubMed]
- Kumar, K.R.; Blair, N.F.; Sue, C.M. An update on the hereditary spastic paraplegias: New genes and new disease models. Mov. Disord. Clin. Pract. 2015, 2, 213–223. [Google Scholar] [CrossRef] [PubMed]
- Toft, A.; Birk, S.; Ballegaard, M.; Duno, M.; Hjermind, L.E.; Nielsen, J.E.; Svenstrup, K. Peripheral neuropathy in hereditary spastic paraplegia caused by REEP1 variants. J. Neurol. 2019, 266, 735–744. [Google Scholar] [CrossRef]
- Bouchard, J.P.; Barbeau, A.; Bouchard, R.; Bouchard, R.W. Autosomal recessive spastic ataxia of Charlevoix-Saguenay. Can. J. Neurol. Sci. 1978, 5, 61–69. [Google Scholar] [CrossRef]
- Engert, J.C.; Dore, C.; Mercier, J.; Ge, B.; Betard, C.; Rioux, J.D.; Owen, C.; Berube, P.; Devon, K.; Birren, B.; et al. Autosomal recessive spastic ataxia of Charlevoix-Saguenay (ARSACS): High-resolution physical and transcript map of the candidate region in chromosome region 13q11. Genomics 1999, 62, 156–164. [Google Scholar] [CrossRef] [PubMed]
- Aly, K.A.; Moutaoufik, M.T.; Zilocchi, M.; Phanse, S.; Babu, M. Insights into SACS pathological attributes in autosomal recessive spastic ataxia of Charlevoix-Saguenay (ARSACS). Curr. Opin. Chem. Biol. 2022, 71, 102211. [Google Scholar] [CrossRef] [PubMed]
- Bagaria, J.; Bagyinszky, E.; An, S.S.A. Genetics of autosomal recessive spastic ataxia of Charlevoix-Saguenay (ARSACS) and role of SACSin in neurodegeneration. Int. J. Mol. Sci. 2022, 23, 552. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Li, X.B.; Zi, X.H.; Shen, L.; Hu, Z.M.; Huang, S.X.; Yu, D.L.; Li, H.B.; Xia, K.; Tang, B.S.; et al. A novel hemizygous SACS mutation identified by whole exome sequencing and SNP array analysis in a Chinese ARSACS patient. J. Neurol. Sci. 2016, 362, 111–114. [Google Scholar] [CrossRef]
- Parfitt, D.A.; Michael, G.J.; Vermeulen, E.G.; Prodromou, N.V.; Webb, T.R.; Gallo, J.M.; Cheetham, M.E.; Nicoll, W.S.; Blatch, G.L.; Chapple, J.P. The ataxia protein SACSin is a functional co-chaperone that protects against polyglutamine-expanded ataxin-1. Hum. Mol. Genet. 2009, 18, 1556–1565. [Google Scholar] [CrossRef]
- Lariviere, R.; Gaudet, R.; Gentil, B.J.; Girard, M.; Conte, T.C.; Minotti, S.; Leclerc-Desaulniers, K.; Gehring, K.; McKinney, R.A.; Shoubridge, E.A.; et al. SACS knockout mice present pathophysiological defects underlying autosomal recessive spastic ataxia of Charlevoix-Saguenay. Hum. Mol. Genet. 2015, 24, 727–739. [Google Scholar] [CrossRef]
- Murtinheira, F.; Migueis, M.; Letra-Vilela, R.; Diallo, M.; Quezada, A.; Valente, C.A.; Oliva, A.; Rodriguez, C.; Martin, V.; Herrera, F. SACSin deletion induces aggregation of glial intermediate filaments. Cells 2022, 11, 299. [Google Scholar] [CrossRef]
- Girard, M.; Lariviere, R.; Parfitt, D.A.; Deane, E.C.; Gaudet, R.; Nossova, N.; Blondeau, F.; Prenosil, G.; Vermeulen, E.G.; Duchen, M.R.; et al. Mitochondrial dysfunction and Purkinje cell loss in autosomal recessive spastic ataxia of Charlevoix-Saguenay (ARSACS). Proc. Natl. Acad. Sci. USA 2012, 109, 1661–1666. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Gehring, K. Structural studies of parkin and SACSin: Mitochondrial dynamics in neurodegenerative diseases. Mov. Disord. 2015, 30, 1610–1619. [Google Scholar] [CrossRef]
- Baets, J.; Deconinck, T.; Smets, K.; Goossens, D.; Van den Bergh, P.; Dahan, K.; Schmedding, E.; Santens, P.; Rasic, V.M.; Van Damme, P.; et al. Mutations in SACS cause atypical and late-onset forms of ARSACS. Neurology 2010, 75, 1181–1188. [Google Scholar] [CrossRef]
- Synofzik, M.; Soehn, A.S.; Gburek-Augustat, J.; Schicks, J.; Karle, K.N.; Schule, R.; Haack, T.B.; Schoning, M.; Biskup, S.; Rudnik-Schoneborn, S.; et al. Autosomal recessive spastic ataxia of Charlevoix Saguenay (ARSACS): Expanding the genetic, clinical and imaging spectrum. Orphanet J. Rare Dis. 2013, 8, 41. [Google Scholar] [CrossRef] [PubMed]
- Miyatake, S.; Miyake, N.; Doi, H.; Saitsu, H.; Ogata, K.; Kawai, M.; Matsumoto, N. A novel SACS mutation in an atypical case with autosomal recessive spastic ataxia of Charlevoix-Saguenay (ARSACS). Intern. Med. 2012, 51, 2221–2226. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, F.; Doi, H.; Kunii, M. Autosomal recessive spinocerebellar ataxias in Japan. Rinsho Shinkeigaku 2016, 56, 395–399. [Google Scholar] [CrossRef] [PubMed]
- Aida, I.; Ozawa, T.; Fujinaka, H.; Goto, K.; Ohta, K.; Nakajima, T. Autosomal recessive spastic ataxia of Charlevoix-Saguenay without spasticity. Intern. Med. 2021, 60, 3963–3967. [Google Scholar] [CrossRef] [PubMed]
- Zeng, H.; Tang, J.G.; Yang, Y.F.; Tan, Z.P.; Tan, J.Q. A novel homozygous SACS mutation identified by whole-exome sequencing in a consanguineous family with autosomal recessive spastic ataxia of Charlevoix-Saguenay. Cytogenet. Genome Res. 2017, 152, 16–21. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Lu, X.; Jin, Y.; Li, D.; Ye, X.; Tao, C.; Zhou, M.; Jiang, H.; Yu, H. A novel SACS variant identified in a Chinese patient: Case report and review of the literature. Front. Neurol. 2022, 13, 845318. [Google Scholar] [CrossRef] [PubMed]
- Kwon, K.Y.; Huh, K.; Eun, B.L.; Yoo, H.W.; Kamsteeg, E.J.; Scheffer, H.; Koh, S.B. A probable Korean case of autosomal recessive spastic ataxia of Charlevoix-Saguenay. Can. J. Neurol. Sci. 2015, 42, 271–273. [Google Scholar] [CrossRef] [PubMed]
- Cho, H.; Lyoo, C.H.; Park, S.E.; Seo, Y.; Han, S.H.; Han, J. Optical coherence tomography findings facilitate the diagnosis of autosomal recessive spastic ataxia of Charlevoix-Saguenay. Korean J. Ophthalmol. 2021, 35, 330–331. [Google Scholar] [CrossRef] [PubMed]
- Bong, J.B.; Kim, S.W.; Lee, S.-T.; Choi, J.R.; Shin, H.Y. Autosomal recessive spastic ataxia of Charlevoix-Saguenay. J. Korean Neurol. Assoc. 2019, 37, 69–72. [Google Scholar] [CrossRef]
- Hammer, M.B.; Eleuch-Fayache, G.; Gibbs, J.R.; Arepalli, S.K.; Chong, S.B.; Sassi, C.; Bouhlal, Y.; Hentati, F.; Amouri, R.; Singleton, A.B. Exome sequencing: An efficient diagnostic tool for complex neurodegenerative disorders. Eur. J. Neurol. 2013, 20, 486–492. [Google Scholar] [CrossRef]
- Shakya, S.; Kumari, R.; Suroliya, V.; Tyagi, N.; Joshi, A.; Garg, A.; Singh, I.; Kalikavil Puthanveedu, D.; Cherian, A.; Mukerji, M.; et al. Whole exome and targeted gene sequencing to detect pathogenic recessive variants in early onset cerebellar ataxia. Clin. Genet. 2019, 96, 566–574. [Google Scholar] [CrossRef] [PubMed]
- Panwala, T.F.; Garcia-Santibanez, R.; Vizcarra, J.A.; Garcia, A.G.; Verma, S. Childhood-onset hereditary spastic paraplegia (HSP): A case series and review of literature. Pediatr. Neurol. 2022, 130, 7–13. [Google Scholar] [CrossRef] [PubMed]
- Karakaya, M.; Storbeck, M.; Strathmann, E.A.; Delle Vedove, A.; Holker, I.; Altmueller, J.; Naghiyeva, L.; Schmitz-Steinkruger, L.; Vezyroglou, K.; Motameny, S.; et al. Targeted sequencing with expanded gene profile enables high diagnostic yield in non-5q-spinal muscular atrophies. Hum. Mutat. 2018, 39, 1284–1298. [Google Scholar] [CrossRef] [PubMed]
- Goutallier, D.; Postel, J.M.; Bernageau, J.; Lavau, L.; Voisin, M.C. Fatty muscle degeneration in cuff ruptures: Pre- and postoperative evaluation by CT scan. Clin. Orthop. Relat. Res. 1994, 304, 78–83. [Google Scholar] [CrossRef]
- Kim, H.S.; Kim, H.J.; Nam, S.H.; Kim, S.B.; Choi, Y.J.; Lee, K.S.; Chung, K.W.; Yoon, Y.C.; Choi, B.O. Clinical and neuroimaging features in Charcot-Marie-Tooth patients with GDAP1 mutations. J. Clin. Neurol. 2021, 17, 52–62. [Google Scholar] [CrossRef] [PubMed]
- Kwon, H.M.; Kim, H.S.; Kim, S.B.; Park, J.H.; Nam, D.E.; Lee, A.J.; Nam, S.H.; Hwang, S.; Chung, K.W.; Choi, B.O. Clinical and neuroimaging features in Charcot-Marie-Tooth patients with GNB4 mutations. Life 2021, 11, 494. [Google Scholar] [CrossRef] [PubMed]
- Naz, S.; Imtiaz, A.; Mujtaba, G.; Maqsood, A.; Bashir, R.; Bukhari, I.; Khan, M.R.; Ramzan, M.; Fatima, A.; Rehman, A.U.; et al. Genetic causes of moderate to severe hearing loss point to modifiers. Clin. Genet. 2017, 91, 589–598. [Google Scholar] [CrossRef] [PubMed]
- Kanwal, S.; Choi, Y.J.; Lim, S.O.; Choi, H.J.; Park, J.H.; Nuzhat, R.; Khan, A.; Perveen, S.; Choi, B.O.; Chung, K.W. Novel homozygous mutations in Pakistani families with Charcot-Marie-Tooth disease. BMC Med. Genom. 2021, 14, 174. [Google Scholar] [CrossRef] [PubMed]
- Choi, H.J.; Kanwal, S.; Hameed, R.; Tamanna, N.; Perveen, S.; Mahreen, H.; Son, W.; Lee, K.S.; Chung, K.W. Biallelic mutations in Pakistani families with autosomal recessive prelingual nonsyndromic hearing loss. Genes Genom. 2023, 45, 145–156. [Google Scholar] [CrossRef]
- Lee, J.; Jung, S.C.; Hong, Y.B.; Yoo, J.H.; Koo, H.; Lee, J.H.; Hong, H.D.; Kim, S.B.; Chung, K.W.; Choi, B.O. Recessive optic atrophy, sensorimotor neuropathy and cataract associated with novel compound heterozygous mutations in OPA1. Mol. Med. Rep. 2016, 14, 33–40. [Google Scholar] [CrossRef]
- Lee, A.J.; Nam, S.H.; Park, J.M.; Kanwal, S.; Choi, Y.J.; Lee, H.J.; Lee, K.S.; Lee, J.E.; Park, J.S.; Choi, B.O.; et al. Compound heterozygous mutations of SH3TC2 in Charcot-Marie-Tooth disease type 4C patients. J. Hum. Genet. 2019, 64, 961–965. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.S.; Lee, H.J.; Park, J.M.; Hong, Y.B.; Park, K.D.; Yoo, J.H.; Koo, H.; Jung, S.C.; Park, H.S.; Lee, J.H.; et al. Proximal dominant hereditary motor and sensory neuropathy with proximal dominance association with mutation in the TRK-fused gene. JAMA Neurol. 2013, 70, 607–615. [Google Scholar] [CrossRef] [PubMed]
- Paternostro-Sluga, T.; Grim-Stieger, M.; Posch, M.; Schuhfried, O.; Vacariu, G.; Mittermaier, C.; Bittner, C.; Fialka-Moser, V. Reliability and validity of the Medical Research Council (MRC) scale and a modified scale for testing muscle strength in patients with radial palsy. J. Rehabil. Med. 2008, 40, 665–671. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Family ID | Mutations | Clinical Phenotype | ACMG/AMP | |
|---|---|---|---|---|
| Nucleotide 1 | Amino Acid 1 | |||
| FC591 | [c.1966_1967insT] + [c.4138C>G] | [p.S656Ffs*1] + [p.P1380A] | CMT, cerebellar ataxia, spasticity, and HL | P p |
| FC937 | [c.2439_2440delAT] + [c.10897T>G] | [p.V815Gfs*2] + [p.F3633V] | CMT, cerebellar ataxia, and spasticity, HL | P LP |
| FC1157 | [c.2903_2906delACAG] + [c.13217delC] | [p.D968Vfs*13] + [p.T4406Rfs*45] | CMT, cerebellar ataxia, spasticity, and HL | P P |
| FC1176 | [c.1596T>A] + [c.3159_3160delCT] | [p.Y532X] + [p.F1054X] | CMT, cerebellar ataxia, spasticity, and HL | P P |
| Variants 1 | dbSNP Acc. No. | Mutant Allele Frequencies | GERP | In Silico Analyses 2 | References | |||||
|---|---|---|---|---|---|---|---|---|---|---|
| IGSR | gnomAD | KRGDB | MutT | REVEL | PP2 | MU | ||||
| p.Y532X | rs2137720760 | - | - | - | −2.77 | 200/0 * | - | - | - | |
| p.S656Ffs*1 | - | - | - | - | 5.14 | 200/0 * | - | - | - | |
| p.V815Gfs*2 | rs775059063 | - | 0.0000142 | - | −1.5 | 200/0 * | - | - | - | [30,31,32] |
| p.D968Vfs*13 | rs1259615333 | - | 0.0000099 | - | 3.71 | 199/1 * | - | - | - | [33] |
| p.F1054X | rs2137637877 | - | - | - | 5.09 | 200/0 * | - | - | - | |
| p.P1380A | - | - | - | - | 6.06 | 66/34 * | 0.637 * | 0.965 * | −0.757 * | |
| p.F3633V | rs1382541188 | - | 0.000013 | - | 6.04 | 72/28 * | 0.626 * | 0.999 * | −0.292 * | |
| p.T4406Rfs*45 | - | - | - | - | 5.85 | 198/2 * | - | - | - | |
| Mutation | Protein Stability Prediction Tools 1 | ||
|---|---|---|---|
| PremPS | MAESTROweb | DynaMut2 | |
| p.F3633V | 0.68 * | 0.283 * | −0.73 * |
| p.P1380A | 0.91 * | 0.121 * | −1.10 * |
| Family ID | FC591 | FC937 | FC1157 | FC1176 | ||
|---|---|---|---|---|---|---|
| Patients | III-1 | III-2 | II-1 | II-2 | II-4 | II-1 |
| Mutations | p.S656Ffs*1 + p.P1380A | p.V815Gfs*2 + p.F3633V | p.D968Vfs*13 + p.T4406Rfs*45 | p.Y532X + p.F1054X | ||
| Sex | Male | Male | Female | Female | Man | Man |
| Examined age (years) | 35 | 33 | 27 | 26 | 25 | 22 |
| Onset age (years) | 4 | 5 | 15 | 17 | 15 | 3 |
| Muscle weakness | ||||||
| Upper limb (MRC) | ||||||
| Proximal (Right/Left) 1 | 4+/4+ | 4+/4+ | 4/4 | 4+/4+ | 4+/4+ | 4/4 |
| Distal (Right/Left) 2 | 4+/4+ | 4+/4+ | 4/4 | 4+/4+ | 4+/4+ | 4/4 |
| Lower limb (MRC) | ||||||
| Proximal (Right/Left) 3 | 4/4 | 4+/4+ | 4-/4- | 4+/4+ | 4/4 | 4/4 |
| Distal (Right/Left) 4 | 4/4 | 4+/4+ | 4-/4- | 4+/4+ | 4/4 | 4/4 |
| Muscle atrophy | Mild | Minimal | Moderate | Minimal | Mild | Mild |
| Sensory disturbance | Yes | Yes | Yes | Yes | Yes | Yes |
| DTR 5 | ||||||
| Biceps jerk reflex | ++ | ++ | +++ | +++ | +++ | +++ |
| Knee jerk reflex | +++ | +++ | +++ | +++ | +++ | +++ |
| Disability score | ||||||
| FDS | 2 | 2 | 4 | 2 | 4 | 4 |
| CMTNSv2 | 13 | 11 | 24 | 17 | 22 | 23 |
| Pyramidal sign | Yes | Yes | Yes | Yes | Yes | Yes |
| Spasticity | Yes | Yes | Yes | Yes | Yes | Yes |
| Cerebellar ataxia | Yes | Yes | Yes | Yes | Yes | Yes |
| Dysarthria | No | No | Yes | Yes | No | Yes |
| Nystagmus | Yes | Yes | Yes | Yes | Yes | Yes |
| Foot deformity | Yes | Yes | Yes | Yes | Yes | Yes |
| Intellectual disability | No | No | No | No | No | No |
| Hearing loss | Yes | Yes | Yes | Yes | Yes | Yes |
| Electrophysiology | ||||||
| Nerve conduction | HMSN | HMSN | HMSN | HMSN | HMSN | HMSN |
| BAEP | NA | NA | Abnormal | Abnormal | NA | Abnormal |
| Brain MRI | NA | NA | NA | Cerebellar atrophy | Cerebellar atrophy | Cerebellar atrophy |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pi, B.K.; Chung, Y.H.; Kim, H.S.; Nam, S.H.; Lee, A.J.; Nam, D.E.; Park, H.J.; Kim, S.B.; Chung, K.W.; Choi, B.-O. Compound Heterozygous Mutations of SACS in a Korean Cohort Study of Charcot-Marie-Tooth Disease Concurrent Cerebellar Ataxia and Spasticity. Int. J. Mol. Sci. 2024, 25, 6378. https://doi.org/10.3390/ijms25126378
Pi BK, Chung YH, Kim HS, Nam SH, Lee AJ, Nam DE, Park HJ, Kim SB, Chung KW, Choi B-O. Compound Heterozygous Mutations of SACS in a Korean Cohort Study of Charcot-Marie-Tooth Disease Concurrent Cerebellar Ataxia and Spasticity. International Journal of Molecular Sciences. 2024; 25(12):6378. https://doi.org/10.3390/ijms25126378
Chicago/Turabian StylePi, Byung Kwon, Yeon Hak Chung, Hyun Su Kim, Soo Hyun Nam, Ah Jin Lee, Da Eun Nam, Hyung Jun Park, Sang Beom Kim, Ki Wha Chung, and Byung-Ok Choi. 2024. "Compound Heterozygous Mutations of SACS in a Korean Cohort Study of Charcot-Marie-Tooth Disease Concurrent Cerebellar Ataxia and Spasticity" International Journal of Molecular Sciences 25, no. 12: 6378. https://doi.org/10.3390/ijms25126378
APA StylePi, B. K., Chung, Y. H., Kim, H. S., Nam, S. H., Lee, A. J., Nam, D. E., Park, H. J., Kim, S. B., Chung, K. W., & Choi, B.-O. (2024). Compound Heterozygous Mutations of SACS in a Korean Cohort Study of Charcot-Marie-Tooth Disease Concurrent Cerebellar Ataxia and Spasticity. International Journal of Molecular Sciences, 25(12), 6378. https://doi.org/10.3390/ijms25126378